

SUMMARY
A prion is an infectious protein horizontally transmitting a disease or trait without a required nucleic acid. Yeast and fungal prions are nonchromosomal genes composed of protein, generally an altered form of a protein that catalyzes the same alteration of the protein. Yeast prions are thus transmitted both vertically (as genes composed of protein) and horizontally (as infectious proteins, or prions). Formation of amyloids (linear ordered β-sheet-rich protein aggregates with β-strands perpendicular to the long axis of the filament) underlies most yeast and fungal prions, and a single prion protein can have any of several distinct self-propagating amyloid forms with different biological properties (prion variants). Here we review the mechanism of faithful templating of protein conformation, the biological roles of these prions, and their interactions with cellular chaperones, the Btn2 and Cur1 aggregate-handling systems, and other cellular factors governing prion generation and propagation. Human amyloidoses include the PrP-based prion conditions and many other, more common amyloid-based diseases, several of which show prion-like features. Yeast prions increasingly are serving as models for the understanding and treatment of many mammalian amyloidoses. Patients with different clinical pictures of the same amyloidosis may be the equivalent of yeasts with different prion variants.
INTRODUCTION
The ability of DNA or RNA to template its own sequence by complementary base pairing to make an identical copy enables the inheritance of traits by organisms or viruses. The notion that proteins can do something similar first arose from findings of the extreme UV resistance of the scrapie agent (1), the cause of a fatal infectious neurodegenerative disease of sheep, transmissible spongiform encephalopathy (TSE). Griffith suggested that a protein multimer in an altered conformation might incorporate a monomer of the normal form and induce the normal form to change into the same altered conformation (2). How this—the equivalent of the base-pairing scheme for DNA or RNA—could occur was not evident. The discovery of yeast prions (3) and the elucidation of the architecture of the amyloids that underlie them (4–6) have suggested such a mechanism (7), as discussed below. Yeast and fungal prions are units of inheritance (i.e., genes) transmitting traits or diseases, much as DNA genes can determine phenotypes or inherited disorders. As genes, yeast prions rather stably propagate but can change (mutate), presumably by a failure of accurate structural templating, much as DNA replication can produce mutations by occasional inaccurate nucleotide templating.
The various mammalian TSEs, including human Creutzfeldt-Jakob disease, chronic wasting disease of deer and elk, bovine spongiform encephalopathy (mad cow disease), and scrapie of sheep, are uniformly fatal, and all involve altered forms of the host PrP protein (reviewed in references 8 and 9). Although PrP is essential for infectivity (10) and the specificity of TSE transmission clearly resides in the PrP sequence (e.g., see reference 11), attempts to show that amyloids of recombinant PrP are infectious led to evidence that one or more other components are involved (e.g., see reference 12). The lethal and near-lethal forms of yeast prions were only recently detected (13). The original studies (14, 15) detecting what proved later to be yeast prions (3) could not have detected lethal variants. The mild effects of some yeast prion variants and the existence of the clearly functional [Het-s] prion of Podospora anserina (16, 17; reviewed in reference 18) have led to suggestions that yeast prions may actually benefit their hosts (19, 20). We discuss the evidence for and against this notion.
Cells have developed an array of components and organelles that deal with denatured and aggregated proteins, and prions have provided both a key tool for detecting and studying these components and an important target of these systems. Chaperones, cochaperones, ubiquitin-proteasomes, vacuoles/lysosomes, autophagy, aggresomes, the Btn2p and Cur1p systems, and various sites of aggregate accumulation may have roles in prion propagation and defense against prions, many of which have already been demonstrated.
The classical mammalian prion diseases are the TSEs based on the PrP protein (9, 21), but recently evidence has emerged of infectious (prion or prion-like) aspects of many amyloid diseases, including Alzheimer's disease, Parkinson's disease, and serum amyloidosis A (22; reviewed in references 23 and 8). The expanding horizon of prion diseases increases the importance of the study of yeast prions. Some of the many means of interfering with yeast prion propagation may find analogy or homology in human systems and may lead to treatments of the many amyloid-based human diseases.
HISTORY OF YEAST PRIONS
In 1965, Brian Cox discovered a nonchromosomal gene, which he named [PSI], that elevated the efficiency of readthrough of translation termination codons (14). Soon thereafter, Francois Lacroute found another nonchromosomal genetic element, dubbed [URE3], that allowed cells to take up ureidosuccinate to allow growth of a ura2 mutant that was blocked in the production of this intermediate (15). Careful studies of these systems by Cox, Lacroute, Michel Aigle, Michael Ter-Avanesyan, Mick Tuite, Fred Sherman, Sue Liebman, and Yury Chernoff, along with our own experiments, led us to propose that [URE3] and [PSI+] are prions of the Ure2 and Sup35 proteins, respectively (3) (Fig. 1).
FIG 1.

Prions [URE3], [PSI+], and [PIN+] of S. cerevisiae and [Het-s] of Podospora anserina. These prions are based on self-propagating amyloids of Ure2p, Sup35p, Rnq1p, and HET-s, respectively. The prion domains of Ure2p and Sup35p have nonprion functions, explaining their retention in evolution despite detrimental prion formation.
Genetic Criteria for a Yeast Prion
Although mammalian spongiform encephalopathies are uniformly fatal, it was clear that if yeast prions were always as lethal, they would not have been detected, and thus that lethality could no more be an essential part of being a prion than it is of being a virus. (As we discuss below, yeast prions are often lethal, but some variants only slightly impair growth.) We inferred three properties that would be expected for a nonchromosomal genetic element that was a yeast prion but not for a nucleic acid replicon (3).
Reversible curability.
Curing of a nucleic acid replicon, such as the mitochondrial genome cured by ethidium bromide, is an irreversible event. Short of geologic time, the mitochondrial genome will not spontaneously reappear in a strain from which it was cured. However, a prion can arise de novo at some low frequency, because the protein from which it arises is still being produced in the cell. Note that it is the reversibility of the curing, not the curing itself, that suggests a prion.
Prion appearance is induced by overproduction of the prion protein.
Once the change from the normal form to the prion form has occurred in a cell, this self-catalyzing alteration should propagate through most of the molecules in the cell. Thus, the more there is of the protein that is capable of this change, the more frequently it will occur in a given cell. Overproduction of prion protein should increase the frequency of the prion change. This is particularly striking in cases (such as [URE3] and [PSI+]) where the phenotype is produced by deficiency of the normal form. Prion formation may be the only way to explain obtaining a stable phenotype of deficiency of protein A as a result of transient overproduction of the same protein A.
Prion phenotype mimics prion protein gene mutation.
Before one knows one has a prion, one may have a nonchromosomal gene (such as [URE3]) and a chromosomal gene on which the nonchromosomal gene depends for propagation (URE2). The phenotype of cells carrying [URE3] is essentially the same as that of cells with a recessive ure2 mutation. This is in marked contrast to nucleic acid replicon-based nonchromosomal genes. For example, the mitochondrial genome makes cells respiratory sufficient, but pet mutants that lose the mitochondrial genome are respiration insufficient.
[URE3] and [PSI+] Have Genetic Properties of Prions of Ure2p and Sup35p
[URE3] can be cured by low concentrations of guanidine but can arise again at a low frequency from the cured clones (3). Overproduction of Ure2p increases the frequency with which [URE3] arises de novo by over 100-fold (3). Propagation of [URE3] depends on an intact URE2 gene, and the [URE3] phenotype is similar to that of ure2 mutants (15, 24).
[PSI+] can be cured by high-osmolarity conditions (25), but the cured strains can again acquire [PSI+] (26). Overproduction of Sup35p results in increased generation of [PSI+] clones (27), and the [PSI+] phenotype resembles that of sup35 mutants (14). Thus, it was concluded that [URE3] is a prion of Ure2p and that [PSI+] is a prion of Sup35p (3) (Fig. 1).
PRION DOMAINS
The part of Ure2p whose overproduction induced the formation of [URE3] was found to be the N-terminal 65 residues (28), and this region proved to be sufficient to propagate [URE3] in the absence of the remainder of the molecule (29). Ure2p is a negative regulator of the enzymes and transporters necessary for the utilization of poor nitrogen sources, acting by binding to the positive transcription factor Gln3p and keeping it in the cytoplasm (30, 31). The C-terminal part of Ure2p is sufficient to carry out the nitrogen regulation function of Ure2p if overexpressed, while the N-terminal prion domain normally functions to stabilize Ure2p against degradation (32).
Sup35p is a subunit of the translation termination factor (33, 34), and residues 254 to 685 (Sup35C) are sufficient to carry out the essential translation termination function (35). Residues 1 to 253 (Sup35NM) regulate general mRNA turnover through interactions with the poly(A) binding protein and the poly(A)-degrading enzyme (36–40), and they direct protein synthesis to the tubulin cytoskeleton (41). Sup35p residues 1 to 114 (Sup35N) are sufficient to propagate the original [PSI+] (35), but residues 1 to 61 are sufficient to propagate several variants of this prion (42, 43).
Parts of the Sup35M domain (residues 115 to 253), up to residue 137, are needed for propagation of some strong and weak [PSI+] variants (44), and deletions and substitutions within the M domain alter the character of [PSI+] in profound ways (45). Differences in the Sup35p M domain among wild Saccharomyces cerevisiae isolates are also partially responsible for an intraspecies [PSI+] transmission barrier between such strains (see below) (46). The importance of Sup35M in faithful propagation of many [PSI+] variants correlates with the existence of some structure within this region. Solid-state nuclear magnetic resonance (ssNMR) experiments with Sup35NM filaments showed that Tyr residues, all of which are within N, are in an in-register parallel β-sheet structure (5). There are eight Leu residues, i.e., residues 110, 126, 144, 146, 154, 212, 218, and 238. The ssNMR data suggest that four of these have an in-register parallel structure (47). Solution NMR experiments with Sup35NM filaments showed that four Leu residues are mobile, consistent with the other four being in a β-sheet structure (48). Taken together, the biological and structural data imply some structure within the M domain, but also considerable unstructured regions. Since four of the Leu residues are within or near the Sup35N domain, it is likely that most of M is unstructured, or at least not in a parallel in-register form as implied by its highly charged nature.
THE MENAGERIE OF YEAST PRIONS
[URE3] and [PSI+] Are Amyloids of Ure2p and Sup35p
Sup35p is aggregated specifically in [PSI+] strains (49, 50), and a self-propagating aggregation of Sup35p, primed by extracts of prion-carrying cells, can be demonstrated (51). Either the prion domain (52) or the full-length protein (53) can form amyloids in vitro, and such amyloids infect cells, transmitting the [PSI+] prion (43, 54).
Ure2p is protease resistant (28) and aggregated (55) specifically in [URE3] cells, and the prion domain peptide or the full-length protein forms amyloids in vitro (56). Such amyloids are infectious for yeast, transmitting [URE3] to the cells (57). The prion domains of Ure2p and Sup35p are both rich in Q and N residues and poor in charged residues, properties that proved true of most, but not all, yeast prions.
Extending the Yeast Prion World
A list of yeast and fungal prions is given in Table 1. Although overproduction of Sup35p increased the de novo generation of [PSI+] in Chernoff et al.'s early study (27), Derkatch et al. found that this effect largely depended on what strain was being used. A nonchromosomal genetic element, named [PIN+], for [PSI+] inducibility, was found to be the basis for this difference (58). Soon thereafter, a protein rich in N and Q residues (Rnq1p) was found to be aggregated in some strains, and this aggregation behaved like a prion (59). Neither the [PIN+] prion state nor deletion of RNQ1 showed a noticeable phenotype (59). Derkatch et al. then found that overproduction of any of an array of Q/N-rich proteins or the [URE3] prion could produce the Pin+ phenotype. Examination of Rnq1p showed that it was the protein forming the original [PIN+] prion (60).
TABLE 1.
Yeast and fungal prionsa
| Prion | Prion protein | Protein normal function | Reference |
|---|---|---|---|
| [URE3] | Ure2 | Nitrogen catabolism regulation | 3 |
| [PSI+] | Sup35 | Translation termination | 3 |
| [PIN+] | Rnq1 | None known | 60, 58, 59 |
| [Het-s] | HET-s | Heterokaryon incompatibility in Podospora anserina for the prion form; the nonprion form has no known function | 16 |
| [BETA] | Prb1 | Active vacuolar protease B is the prion form and is needed for sporulation and survival in stationary phase | 68 |
| [SWI+] | Swi1 | Chromatin remodeling component | 61 |
| [OCT+] | Cyc8 | Transcription repressor subunit | 62 |
| [MOT+] | Mot3 | Transcription regulator | 64 |
| [ISP+] | Sfp1 | Transcription factor | 65 |
| [MOD+] | Mod5 | tRNA isopentenyltransferase | 63 |
Except for [Het-s], which was found in Podospora anserina, the prions listed were found in Saccharomyces cerevisiae. Some prions have also been found in some other species. All but [BETA] involve amyloids. Only [Het-s] and [BETA] have normal functions as prions.
Several of the proteins whose overproduction produced the Pin+ phenotype were found to form prions themselves, including Swi1p, forming the [SWI+] prion (61), and Cyc8p, forming the [OCT+] prion (62). A similar approach was used by Tanaka's group to detect a prion of Mod5p, called [MOD+] (63). Proteins whose overexpression allowed the inducibility of the appearance of [PSI+] by overexpression of a modified Sup35p protein were candidates to be prions. Mod5p is a tRNA isopentenyltransferase, and the reported phenotype of the prion was that of a deficiency of Mod5p, including resistance to fluconazole. Interestingly, the prion domain of Mod5p is not rich in N or Q residues (63). An extensive survey of proteins with Q/N-rich domains uncovered a prion of Mot3p, called [MOT+] (64).
[ISP+], so named because its phenotype is the opposite of that of [PSI+], is a largely intranuclear amyloid of Sfp1p (65). The antisuppression produced by [ISP+] results from increased expression of Sup35p (66).
Nonamyloid Prions
The definition of a prion is simply an “infectious protein,” and it need not involve amyloids. The vacuolar protease B of S. cerevisiae (Prb1p) can activate its own inactive precursor protein by specific cleavages (67) (Fig. 2). Normally this is done by protease A, but in mutants lacking this enzyme, the active mature protease B acts as a prion, called [BETA] (68). Cells lacking the active enzyme largely remain so, but about 1 in 105 cells spontaneously acquires activity. Once active, the protease B continues to activate its precursor, and the progeny of this cell continue to maintain the activity. Protease B, and thus the [BETA] prion, is important for survival in stationary phase and for the ability to undergo meiosis and spore formation.
FIG 2.

[BETA] prion based on autocatalysis of Prb1p activation. Prb1p (vacuolar protease B) is made as an inactive precursor which can be activated by the active form of the same protein (67). Thus, a cell starting with no active enzyme remains so, while a cell with active enzyme continues to activate the precursor as it is synthesized. Transmission of active Prb1p to a cell lacking the active form “infects” it with the [BETA] prion (68). The active form of protease B (red) can cleave inactive (blue) precursor molecules (at sites labeled “autocleavage”) to activate them.
[GAR] is a nonchromosomal genetic element determining resistance to glucosamine (69, 70). While overproduction of Std1p dramatically increases the appearance of [GAR+], deletion of the gene does not result in loss of [GAR+] (71). However, [GAR+] is lost by combined deletion of STD1 and the N terminus of Pma1p, encoded by PMA1, which is an essential gene. Std1p is involved in glucose-regulated gene expression, while Pma1p is the major plasma membrane H+-ATPase. Std1p and Pma1p are found in a complex whose amount is larger in [GAR+] cells, and it was suggested that [GAR+] is a prion and involves this complex in some way, but apparently not as an amyloid (71).
A Podospora anserina Prion Controls Heterokaryon Incompatibility
Like most other filamentous fungi, Podospora anserina can fuse cellular processes (hyphae) in a nonmeiotic process called heterokaryon formation (72). This process is controlled by the het genes, with identical alleles of each such gene between the two fusing colonies required for the process to produce viable heterokaryons. Nonidentity results in death of the trial heterokaryon cells and formation of a barrier to further fusions (heterokaryon incompatibility). Heterokaryon incompatibilty protects cells from the spread of fungal viruses and mitochondrial senility factors. The het-s gene, with het-s and het-S alleles, produces the proper death of heterokaryons produced between het-s and het-S strains only if the HET-s protein (product of the het-s allele) is in an amyloid form (Fig. 1). This amyloid is infectious and is called the [Het-s] prion, while its absence is indicated by [Het-s*] (16, 73). [Het-s] is remarkable in that it is clearly a functional prion (17), and it provides a model for what properties should be expected for such a prion both in biology and in structure.
PRION GENETICS
Interspecies and Intraspecies Transmission Barriers
Sheep scrapie can be transmitted to goats, but only after a long incubation period (74). Because subsequent passages from goat to goat show a shorter incubation period, the infection appears to be slowed by the transition from one species to another. This phenomenon, seen in TSE transmission between any pair of species, is called the “species barrier,” and in some cases is apparently absolute. The basis of the species barrier is sequence differences between the PrPs of the two species (11). Transmission barriers can, in some cases, result from even a single amino acid difference between the PrPs of the donor and recipient (75).
The yeast prions [PSI+] and [URE3] also show interspecies transmission barriers (76–81). Experiments have generally been carried out with S. cerevisiae and the SUP35 or URE2 gene from the species to be tested. For example, if one strain with the Saccharomyces paradoxus URE2 gene and carrying the [URE3] prion donates cytoplasm to another S. cerevisiae strain with the URE2 gene of S. cerevisiae by cytoplasmic mixing (cytoduction [82]) and then the cytoductants are scored for the [URE3] prion, only ∼25% of cytoductants are found to carry [URE3] (83). If both the donor and recipient have the URE2 gene of the same species, transmission is nearly 100%. Species barriers need not be symmetrical. For example, in the reverse of the above-described experiment, [URE3] of S. cerevisiae Ure2p is transmitted to Ure2p of S. paradoxus with 100% efficiency (83).
Sup35p proteins of wild strains of S. cerevisiae have an array of sequences (polymorphs) that fall roughly into three groups (46, 84). Sup35ps of each of the three groups can form [PSI+], but transmission of [PSI+] from one group to another is partially blocked (46). Rnq1p also has an array of sequences in wild strains, with some showing a partial block of transmission of [PIN+] from the reference sequence (85). Among these wild RNQ1 sequences are five with premature termination codons at different places in the gene, all of which prevent propagation of [PIN+] (85). The rapid variation seen in the prion domains of Ure2p, Sup35p, and Rnq1p may be selected to produce these barriers to prion transmission (46, 83, 85), much as polymorphism at residue 129 of PrP is proposed to be selected to produce resistance to Creutzfeldt-Jakob disease (86).
Prion Variants
Originally defined by the incubation period of mouse scrapie, prion variants (prion “strains” in mammalian systems) are prion isolates with different properties despite being based on a prion protein with the same sequence. Yeast prion variants were first noted as strong and weak isolates of [PSI+] isolated from the same yeast strain (87), and prion variants of [URE3] (57, 88) and [PIN+] (89) have now been described. They may also differ in the stability of propagation and interactions with other prions (89, 90), in sensitivity to overproduction or deficiency of chaperones (91–93), and in sensitivity to interspecies barriers (83, 94) or intraspecies barriers (46, 95) to transmission. Lethal and near-lethal variants of [PSI+] and [URE3] (13) and variants of [URE3] that are hypersensitive to the Btn2/Cur1 antiprion system(s) (96) are eliminated as soon as they arise in a wild-type strain. A strong [PSI+] variant may be any of four different intraspecies transmission types, as may a weak [PSI+] variant (see below) (46). Thus, strong [PSI+] is not a single variant. It is possible that prion variants may be similarly divided further based on responses to chaperones or other properties, implying a very large number of possible variants. Different prion variants are due to different amyloid structures (e.g., see references 97, 42, and 48), but the detailed structure of a prion variant has not yet been obtained.
Prion Clouds
Under selective conditions, prions can change their properties in a heritable way. For example, infection of hamsters with certain mouse prions results in selection of a prion variant that, when transferred back to mice, shows an altered incubation time (98). It has been suggested that the mouse prion extract is actually a mixture of variants, or a “prion cloud,” from which one is selected that can propagate with hamster PrP (99). However, it is difficult to distinguish this selection model from a change in amyloid form produced by the presence of hamster PrP, with its different sequence.
The variants affecting transmission of [PSI+] across intraspecies barriers mentioned above provided an opportunity to test this notion in a manner that involved no selection. Extensive propagation of [PSI+] in a strain with Sup35p sequence A resulted in segregation of cells with each of four different transmission phenotypes: (i) high transmission to sequence B and to sequence C, (ii) high transmission to B but not to C, (iii) high transmission to C but not to B, and (iv) low transmission to both B and C. Further extensive propagation of any one of these purified variants resulted in the same four variants again segregating out. The data showed that segregation of variants was occurring as well as mutation, both under nonselective conditions (95). Although the strain had been propagated extensively before the experiments began, [PSI+] was present as a mixture of variants in a single cell, which could be separated under nonselective conditions, indicating the existence of a prion cloud.
The prion cloud phenomenon implies that there will be difficulties in treating prion diseases, as resistant variants are potentially present before the treatment even begins (100). It also implies difficulties in studies of prion amyloid structures, since one inevitably is dealing with a mixture of structures.
PRION AMYLOID STRUCTURES
Shuffleable Prion Domains Suggest an In-Register Parallel Architecture
Despite the presence of oligopeptide repeats in the Sup35p prion domain, reminiscent of octapeptide repeats in PrP, it was found that shuffling the prion domain sequences of Sup35p or Ure2p did not impair the ability of either to form prions (101–103). This made it clear that the composition of the prion domain, not the sequence, was the critical factor in determining prion-forming ability. The compositional requirements for prion formation favor hydrophobic residues, while charged residues or prolines inhibit prion formation (104). However, as mentioned above, transmission barriers result from sequence differences between donor and receptor, and even a single amino acid difference can produce a barrier (46, 105, 106). The sequence identity requirement for propagation suggests a positive interaction between amino acid side chains which is not affected by the shuffling, consistent with an in-register parallel amyloid but not with an antiparallel or β-helix structure (107) (Fig. 3).
FIG 3.
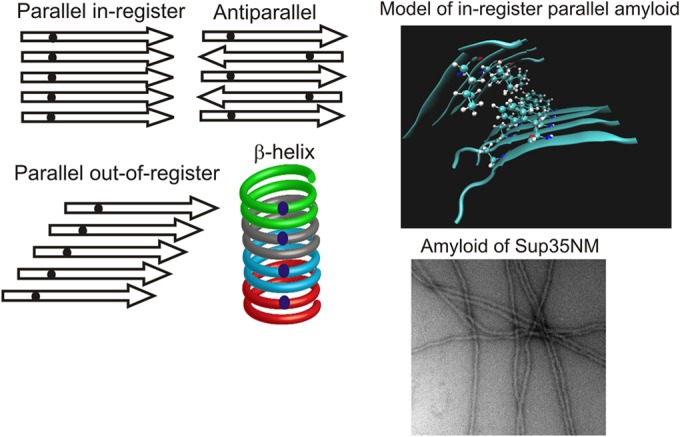
β-Helix versus in-register parallel β-sheet for amyloid structure. (Left) Four types of β-sheet. Small dark dots represent a single 13C-labeled atom in each protein molecule. Only for the in-register parallel architecture will the labeled atoms have an ∼0.5-nm spacing, while the spacing (measured by solid-state NMR) will be much greater for the other structures. (Top right) Model of an in-register parallel structure based on data from reference 215. The only side chains shown are those of a single residue in the top sheet and a single residue in the bottom sheet. (Bottom right) Electron microscopic image of amyloid formed from recombinant Sup35NM. Magnification, ×56,000.
HET-s Infectious Amyloid Is a β-Helix
An amyloid cannot form crystals and is not soluble, so it cannot be studied by X-ray crystallography or solution NMR. However, useful information can be obtained by X-ray fiber diffraction, electron microscopy, and electron spin resonance, and detailed structures may be obtained by solid-state NMR in favorable cases (for reviews, see references 108 and 109). Homogeneity of amyloid structure is the key to obtaining high-resolution solid-state NMR data, as with any method of structure determination. Amyloids of recombinant HET-s protein are so far unique in producing a single conformation, as judged by sharp (∼0.25 to 1 ppm) lines in two-dimensional solid-state NMR experiments (110), probably correlated with this protein having only a single prion variant in vivo (18).
Amyloid of the prion domain of HET-s has a β-helix structure, with each monomer contributing two turns to the β-helix (110–112). The sequence has partial repeats which comprise the β-strands.
Infectious Prion Domains of Sup35p, Ure2p, and Rnq1p Are Folded, In-Register Parallel β-Sheets
X-ray fiber diffraction, circular dichroism, solid-state NMR, and Fourier transform infrared spectroscopy (FTIR) studies have all shown that amyloids of the prion domains of Sup35p, Ure2p, and Rnq1p are all in a β-sheet conformation (4–6, 52, 53, 56, 113–115).
In an in-register parallel β-sheet, a single labeled atom in each molecule should be ∼0.5 nm from the same atom in another molecule, because this is the distance between peptide chains in a β-sheet (Fig. 3). This distance should be about twice as far for an antiparallel β-sheet or a β-helix. This distance, measured rather accurately using a dipolar recoupling solid-state NMR experiment, is consistently about 0.5 nm for labeled Sup35p, Ure2p, and Rnq1p, and dilution with unlabeled molecules shows that the nearest neighbor is on another molecule (4–6, 47, 116, 117). Confirmation of this result for Ure2p comes from analogous experiments using electron spin resonance instead of NMR (118). Such experiments also suggest the locations of some of the folds (119).
Recently, we confirmed the in-register parallel architecture of the Sup35 prion domain (Sup35NM) and found evidence for the locations of some of the folds in the sheet for the amyloid variant(s) formed under the conditions used (120). We labeled 16 single residues in Sup35NM and found that 10 of them showed the 0.5-nm spacing expected for an in-register parallel β-sheet architecture. We suggest that those residues showing wider spacing are in turns/loops in the molecule, the locations of folds in the sheets (120).
An alternative view for Sup35NM, based on chemical modification with a large fluorescent probe, is that Sup35NM forms a β-helix with N-terminal-to-N-terminal and C-terminal-to-C-terminal intermolecular hydrogen bonding to join monomers (121). The N-ethylmaleimide pyrene probes used in these experiments were larger than the fundamental structure being probed, suggesting that the probe may alter the structure and bias the results. Moreover, deletion of the C-terminal region, roughly residues 90 to 110, would be predicted to prevent formation of filaments in vitro or the [PSI+] prion in vivo. In fact, Sup35 residues 1 to 60 are sufficient for filament formation in vitro, with propagation of [PSI+] infectivity (43). It is proposed that residues 30 to 90 comprise the helical part (121), but we find that in our infectious Sup35NM amyloid, 6 of 10 residues in that domain have the ∼0.5-nm spacing diagnostic of an in-register parallel structure, ruling out the β-helix model (120).
Mass-per-length measurements of infectious amyloid filaments of these proteins consistently show a single protein molecule per ∼0.5 nm (122–124). This is also the expected result for an in-register parallel β-sheet, as each molecule comprises a single layer along the long axis of the filament, and the distance between such layers is just the distance between β-strands (Fig. 3). This result also argues against a β-helix because, in that structure, each molecule constitutes two or more layers, so the mass per length would be one-half or less of the observed value. Only if there were a compensating requirement for several β-helices per filament could the observed results be obtained. The β-sheets of which these amyloids are composed must be folded along the long axis of the filament, because the filament diameter in all cases is severalfold smaller than it would be for a simple flat, unfolded β-sheet (6, 52, 56) (Fig. 3 and 4).
FIG 4.

Proposed mechanism of conformational templating by prion protein amyloids. Energetically favorable interactions between identical side chains enforce the in-register architecture of these amyloids. H-bonds between the side chains of identical Gln residues, for example, can form only if the residues are aligned in register. Interactions between charged side chains would be unfavorable, and charged side chains are rare in yeast prion domains. Similarly, in order to form these favorable interactions, a new molecule being added to the end of the filament must assume the same conformation as that of molecules previously added to the filament. Thus, the protein can template its own conformation, just as a DNA or RNA can template its own sequence (7, 216).
Solid-state NMR studies of amyloid filaments formed from full-length Ure2p showed that the C-terminal domain is largely immobile (125) and confirmed the lack of change of conformation of this part of the molecule previously shown by maintenance of the glutathione peroxidase activity on amyloid formation (126).
In-Register Parallel Folded Architecture Explains Conformational Templating
If the difference between prion variants is the conformation of the protein in the amyloid fiber, then the fiber must act as a template, directing the monomer joining the fiber end to adopt the same conformation as the other molecules in that fiber. Such a mechanism is necessary to explain the rather stable inheritance of prion variant characteristics. The architecture of the yeast prion amyloid naturally suggests a mechanism that can explain this templating (7, 127) (Fig. 4). We proposed that the locations of the folds in the sheet are at least one characteristic that distinguishes one variant from another; the extent of the β-sheet may be a further difference between variants. The positive interactions between identical side chains, such as hydrogen bonds along a row of glutamine, asparagine, serine, or threonine side chains or hydrophobic interactions along a row of hydrophobic amino acid side chains, keep the structure in register. Monomers (at least in the case of Sup35 [128]) are added to the end of the filament, and in order to form these favorable interactions, each monomer newly joining the end of the filament must have its turns (the folds in the sheet) at the same places as in the molecules already in the filament. In this way, the molecules in the filament transmit/template their conformation to the monomer newly joining the end (7, 127). This allows a protein to be a gene and to have many different “alleles,” or self-propagating conformations (Fig. 4).
BIOLOGY OF YEAST AND FUNGAL PRIONS
[Het-s] Is a Known Beneficial Prion
[Het-s], the Podospora anserina prion controlling heterokaryon incompatibility (16), was the first prion proposed to be carrying out a normal function and not a disease (17). However, [Het-s] also produces a meiotic drive phenomenon in crosses of het-s ([Het-s]) with het-S strains that results in lethality of meiotic spores with the het-S allele and preferential survival of the het-s prion-forming allele (129, 130). Thus, the [Het-s] prion has both a normal function for the cells and a pathological role. It is difficult to decide which is primary and which a side effect, if there is such a distinction to be made, but by any interpretation, the het-s allele is certainly evolved/selected to be a prion, and that prion has two specific roles to play. There is only one prion variant known for [Het-s] (18), consistent with the very uniform structure of HET-s filaments formed in vitro (110).
Reports of Evidence for Benefits of Yeast Prions
The [PSI+] prion was proposed to make cells more resistant to high temperatures or high ethanol concentrations (19), but this was not reproduced in another study (20). The latter study reported that when there was a growth difference under one of the many stress conditions tested, [psi−] strains were more fit than [PSI+] strains in 75% of cases (20). Nonetheless, it was proposed that [PSI+] may help yeasts to evolve by helping cells to resist stress (20). However, in direct tests, there was no consistent effect of [PSI+] on experimental adaptation to stress (131). Moreover, these occasional favorable effects of [PSI+] were not reproduced in another lab using the same strains (132). In another report, certain stress conditions somewhat increased the frequency of [PSI+] appearance in a strain with an altered prion domain (133), but two other groups were unable to reproduce these results (85, 134). Moreover, [PSI+] was detrimental to cell survival under most of the stress conditions reported to induce [PSI+] (133), indicating that [PSI+] induction was not an adaptive response. We have been unable to reproduce the recent report of growth advantages of the prion in some of the rare wild [PSI+] strains (135), although we did confirm a slight [PSI+]-dependent fluconazole resistance of one strain (see Fig. S1 to S9 in the supplemental material). In contrast to this prion-dependent slight stress resistance, the beauty of the yeast “stress response” is that it responds to stress, occurring specifically when the stress occurs and helping the cell to survive the stress. Prions arise in a stochastic manner, and [PSI+] and [URE3] are often lethal or severely toxic (see below) (13). The notion that yeast is “hedging its bets” by becoming [PSI+] under stress (136) is untenable unless some [PSI+] variant can be shown to reproducibly relieve some stress. Moreover, the quality of the bet hedge is adversely affected by the fact that many prions are toxic or lethal.
[MOD+] is a prion of Mod5p (63), a tRNA isopentenyltransferase, whose inactivation by mutation results in partial fluconazole resistance because dimethylallyl pyrophosphate is diverted from tRNA modification to sterol biosynthesis (137). The presence of the prion has the same effect (63). In the presence of fluconazole, [MOD+] cells are rapidly enriched in a mixed culture, but in the absence of the drug, [mod−] cells are about as rapidly enriched (63). The prion is quite stable over this time frame, and the loss of [MOD+] cells without the drug is a result of their lower growth rate. S. cerevisiae is only rarely a human pathogen and is not a plant pathogen, but azoles with an action similar to that of fluconazole are used as antifungals in agriculture, so S. cerevisiae could conceivably be exposed to the synthetic azole drugs. Because [MOD+] substantially slows growth in the absence of the drug, acquisition of [MOD+] will generally be detrimental to the cells. Nonetheless, these results are important as the first mechanistically established advantage for a yeast prion.
Like swi1 mutants, [SWI+] prion-carrying cells grow poorly on nonfermentable carbon sources, including raffinose, galactose, and glycerol (61). Such defects are probably a disadvantage in the wild, and indeed, a survey of 70 wild strains showed that none carried this prion (46). The [OCT+] prion inactivates Cyc8p (62) and thus, like cyc8 mutants, presumably slows growth and impairs mating and sporulation due to inappropriate derepression of an array of genes. These are not likely to be advantageous.
Prions Rarely Found in Wild Strains Have a Net Detrimental Effect
Detrimental human pathogens are certainly easily found in the population: most humans carry the tuberculosis bacillus in dormant form and get several viral infections each year. The uniformly lethal chronic wasting disease prion is found in 10 to 50% of wild deer and elk in parts of Wyoming and Colorado. Infectious agents spread in spite of debilitating the host because they are not restricted by the rules of meiosis. A beneficial infectious agent would certainly spread rapidly through the susceptible population, because infection and advantage to the host would be working together instead of in opposition. The mitochondrial genome began as a bacterial invader and is now nearly universal in eukaryotes because it is beneficial and non-Mendelian (infectious in the same way that yeast prions are infectious). Likewise, the [Het-s] prion is found in >95% of wild het-s strains of Podospora anserina (138).
Surveys by four groups have consistently found yeast prions to be rare in wild populations. Two small early surveys found no wild [PSI+] strains, but two wild [PIN+] strains were observed (78, 84). A larger survey of 70 wild strains showed that none had [PSI+] or [URE3], but 11 (∼16%) had [PIN+] (139). A fourth, even larger survey, consistent with the earlier work, found that 9 of 700 strains had [PSI+] and ∼6% had [PIN+] (135). In contrast, the mildly detrimental 2μm DNA plasmid was found in 38 of the 70 strains surveyed by Nakayashiki et al. (139). Because 2μm DNA is known to slow the growth of yeast by ∼1 to 3% (85, 140–142), one can conclude that the mildest variants of [URE3], [PSI+], and [PIN+] each impart a >1% growth/survival detriment on their host, because they are less frequently found in wild strains and their mode of spread is the same as that of 2μm DNA (85).
Although rare, the [PIN+] prion is found in the wild at a higher frequency than its rate of generation. This spreading of [PIN+] may be a consequence of a benefit to the cells resulting from carrying [PIN+] or a result of spread of [PIN+] by outcross mating (mating with unrelated cells) in spite of the prion being mildly detrimental. Detailed studies of the wild strains carrying [PIN+] showed that they were enriched for strains with evidence of outcross mating, suggesting the latter scenario (143).
Fully one-third of wild strains have a guanidine-curable trait according to one report (135), which is interpreted as evidence for prions being common in the wild. Kryndushkin et al. developed a biochemical method to detect amyloids based on their SDS insolubility and mass spectrometry (144). This method detects amyloids of Ure2p, Sup35p, and Rnq1p but did not find any amyloids in 5 strains reported to have guanidine-curable traits (144). Because guanidine induces mutations in mitochondrial DNA, we excluded guanidine-induced rho− clones (defective in the mitochondrial genome) from the analysis and were then unable to reproduce the reported guanidine-curable traits in any of the strains tested (see Fig. S10 in the supplemental material).
[PSI+] and [URE3] Are Often Lethal or Severely Toxic
The commonly studied variants of yeast prions are of course those that are mildest. Sup35p is an essential protein, and a prion variant that converted essentially all of the soluble Sup35p to the amyloid form would be lethal. To detect lethal variants of [PSI+], Sup35C, the essential domain lacking the prion domain, was expressed from a URA3 plasmid at low levels, i.e., high enough to allow cell growth but low enough that substantial readthrough of termination codons allowed cells to be Ade+ (using the ade1-14 nonsense [UGA] allele) if the chromosomally encoded full-length Sup35 protein was all present as an amyloid (13). [PSI+] isolates were tested for the ability to lose the plasmid, and many were found to be unable to do so unless they were first cured of the prion (13). Over half of all isolates were either dead or severely growth impaired without the Sup35C plasmid.
Ure2p is not essential, and in some strains a ure2Δ mutation does not even slow growth. Using such a strain, it was found that many [URE3] isolates have severely slow growth, implying a toxic effect of the prion that is not attributable to deficiency of Ure2p (13). These lethal and near-lethal effects are part of the burden of prion formation in these cases. The risk of development of such a lethal prion must be weighed against any possible benefit of the prion. These toxic prions may serve as a model for pathological mammalian amyloidoses.
[URE3] and [PSI+] Prion-Forming Ability Is Not Conserved
S. cerevisiae has been used widely as a test bed for prion-forming ability by proteins from other species. Notable successes include the ability of HET-s from Podospora anserina to form the [Het-s] prion in S. cerevisiae (145). Schizosaccharomyces pombe Hsp104 can replace that of S. cerevisiae in propagating [PSI+], as can S. pombe Hsp70 (146). Even Escherichia coli Hsp104 (ClpB) can serve this function if E. coli Hsp70 (DnaK) and the nucleotide exchange factor (GrpE) are provided (147). Nonetheless, it is possible that differences between species in some of the many prion-handling factors may restrict propagation of some prion variants or allow propagation in a foreign host of some prions that could not propagate in the native environment.
The presence of [PSI+]-forming ability by the N-terminal domains of Sup35 proteins from Pichia methanolica, Kluyveromyces lactis, Candida albicans, and several different Saccharomyces species fused to Sup35MC of S. cerevisiae, and tested in S. cerevisiae (77, 78, 80, 81, 148, 149), has been interpreted to mean that prion formation is conserved, i.e., selected to be present during evolution (150). However, a wider survey of species by similar methods showed that many Sup35 proteins are unable to form [PSI+] in S. cerevisiae (151). Specifically, no evidence of [PSI+] formation could be found for the NM domains of Ashbya gossypii, Schizosaccharomyces pombe, Aspergillus nidulans, Aspergillus fumigatus, Magnaporthe grisea, Ustilago maydis, and Cryptococcus neoformans (151).
[URE3] can be formed by Ure2p from most Saccharomyces species (76, 79, 83) but not by that of Saccharomyces castellii (83). Kluyveromyces lactis Ure2p cannot form [URE3] in either S. cerevisiae or K. lactis itself (152), although Ure2p of K. lactis is very closely related to that of S. cerevisiae. Ure2p of Candida albicans can form [URE3] as tested in either S. cerevisiae or Candida glabrata, but that of C. glabrata, which is much more closely related to S. cerevisiae Ure2p, cannot form [URE3] in either S. cerevisiae or its own species (153–155). Thus, the ability to form the [URE3] and [PSI+] prions appears to be distributed sporadically rather than conserved. Even conservation of occasional broken limbs (or neurodegenerative diseases) among vertebrates does not suggest that this ability is advantageous.
The cells themselves consider infection with [URE3] or [PSI+] to be a stressful event, as shown by their induction of heat shock proteins (156). The prion domains of Ure2p and Sup35p have much more variation than the C-terminal domains, resulting in barriers to transmission: interspecies barriers in both cases and intraspecies barriers in the case of [PSI+] (see above). These barriers may have been selected as a defense against prion infection. The prion domain of Ure2p is important for stabilizing the full-length protein against degradation (32), while the prion domain of Sup35p is involved in regulation of mRNA turnover (38). The presence of these domains does not imply that they are preserved for forming prions but for their normal, nonprion functions.
CELLULAR PRION-HANDLING SYSTEMS
Cellular systems deal with aggregates by resolubilization, degradation, selective segregation, and sequestration. These systems include (separately and in combinations) chaperones, the ubiquitin-proteasome system, autophagy, aggresomes, vacuoles (the yeast lysosome), the Btn2-Cur1 system(s), asymmetric segregation of damaged proteins, the GET pathway (157), and an array of variously named sites. It is evident from many studies that aggregates of different proteins are handled differently (e.g., see reference 158).
Chaperones and Prions
Seed production by the Hsp104-Hsp70-Hsp40 system.
Hsp104 is a disaggregase, working with Hsp70s, Hsp40s, and nucleotide exchange factors to extract monomers from aggregates and give them an opportunity to refold (159, 160). Hsp70s direct Hsp104 to suitable substrates and assist in their disaggregation and refolding (161). When this mechanism works on an amyloid filament, a single molecule drawn from an in-register parallel β-sheet filament interrupts the continuity of the fiber and produces two filaments (Fig. 5). Thus, a series of studies have shown that Hsp104 is essential for propagation of yeast amyloid-based prions (e.g., see reference 162), and its inhibition by guanidine (163–165) results in a loss of yeast prions by failure of new seed formation (166–168).
FIG 5.

Mechanism of seed generation by Hsp104-Hsp70-Hsp40. Hsp104, working with Hsp40 (mainly Sis1p) and Hsp70 (Ssa proteins), extracts a monomer from an amyloid filament, resulting in the formation of two filaments, with two more growing ends. (Adapted from reference 217.)
Hsp104, Hsp70, and a nucleotide exchange factor (Fes1p) from Schizosaccharomyces pombe can each substitute for their S. cerevisiae homologs in their prion propagation functions (146). Even ClpB, the E. coli homolog of Hsp104, can do so if its cognate Hsp70 (DnaK) and nucleotide exchange factor (GrpE) are supplied (147). These findings suggest that prions utilize common chaperone functions provided in a wide range of organisms for other purposes and that the chaperones are not specifically adapted to propagate prions.
Cytoplasmic Hsp70s, the Ssa proteins, are critical for propagation of [PSI+] and [URE3], and strikingly, Ssa1 and Ssa2, which are 98% identical, nonetheless differentially affect these two prions. [PSI+] requires Ssa1, while [URE3] requires Ssa2 for stable propagation (169–171). Remarkably, a single methyl group, at Ala83 for Ssa1 and Gly83 for Ssa2, determines this difference (172). The balance between the ATP and ADP forms of the Ssa proteins also critically affects prion propagation, with increased ATP-bound Ssa1p stabilizing [PSI+] and the increased ADP-bound form destabilizing the same prion (173). Optimal levels of Sse1p are necessary for [URE3] propagation, probably through its nucleotide exchange activity on Hsp70s (174). An sse1Δ strain also cannot propagate some [PSI+] variants (174, 175).
Of the many Hsp40s in yeast, only Sis1p and Ydj1 have been shown to be critical for prion propagation (147, 176). Although Sis1p is essential, it may be depleted, resulting in a rapid loss of [URE3] and [PIN+] and a slower loss of [PSI+] (176). Deletion of all but the J and GF domains of Sis1 leaves cells able to grow if they are [psi−], but they are killed by the presence of the [PSI+] prion (177). Ydj1 is essential for the [SWI+] prion (178).
Hsp104 overproduction curing of [PSI+].
Several lines of evidence indicate that the mechanism of curing of [PSI+] by overproduction of Hsp104 is quite distinct from the fiber breakage reaction that generates new seeds (reviewed in reference 179). Hsp90 and its cofactor Sti1 are necessary for Hsp104 overproduction curing of [PSI+] but not for its propagation (180, 181). Deletion of the N-terminal domain of Hsp104 abolishes overproduction curing of [PSI+] but not the ability to support [PSI+] propagation (182). Hsp104 overproduction only cures [PSI+], perhaps because Hsp104 specifically recognizes a site in Sup35M (183).
Ssb1/2 antiprion activity.
While the Ssa cytoplasmic Hsp70s are necessary for prion propagation (see above), overproduction of the ribosome-associated Hsp70 Ssb1 or Ssb2 helps overproduced Hsp104 to cure [PSI+], and ssb1Δ ssb2Δ strains show an increased frequency of spontaneous or induced [PSI+] prion formation (184). Replacing the SSB1 gene in [PSI+] strains generated in an ssb1Δ ssb2Δ background does not cure the prion, indicating that the Ssb chaperones act to prevent prion formation during the generation phase, presumably by ensuring proper folding of Sup35p during synthesis (184, 185).
Sgt2p, the GET Pathway, and [PSI+]
Kiktev et al. found that get2 mutation or deletion impairs curing of [PSI+] by overexpression of Hsp104 (186). The GET pathway (guided entry of tail-anchored proteins) prevents aggregation of proteins with hydrophobic tails destined for membrane sites and includes the Get1 and Get2 membrane components, cytoplasmic Get3, Get4, and Get5, and the cochaperone Sgt2p (157). Deletion of any of the five get genes impairs Hsp104 overproduction curing of [PSI+] without affecting Hsp104 levels or its other actions (186). An sgt2Δ mutation interferes with the effects of get mutations or overproduction of Ssa1p in preventing [PSI+] curing by elevated Hsp104 (186). Sgt2p directly associates with Sup35p and Rnq1p in vivo (186) and with Get proteins and chaperones (157), suggesting that Sgt2p has a role in bringing these components together. Neither sgt2Δ mutation nor overproduction of Sgt2p appears to affect [PSI+] propagation in an otherwise wild-type cell (186). However, infection by [PSI+] and/or [PIN+] results in up to a 4-fold increase in Sgt2p level, suggesting that Sgt2p serves as a signal to the cell of the presence of prions (186).
Btn2 and Cur1 Are Components of Antiprion Systems
Btn2 and Cur1 are homologous proteins identified in a screen for proteins whose overproduction cures [URE3] (187). Remarkably, in cells being cured of [URE3] by overproduced Btn2, the Ure2p aggregates were collected in a single site coincident with Btn2 (187, 188). These observations and a requirement for cell growth for curing suggested that Btn2 cures by collecting prion aggregates, preventing their distribution to progeny cells. The fact that Btn2 and Cur1 work at normal levels to lower the prion seed number was indicated at first by the larger seed number of [URE3] measured in btn2Δ cur1Δ cells than in isogenic wild-type strains (187). Furthermore, btn2Δ cur1Δ strains show a 5-fold higher rate of spontaneous [URE3] emergence, and most of the [URE3] prions arising are cured by returning to normal levels of Btn2 and Cur1 (96). These Btn2-Cur1-hypersensitive [URE3-bcs] isolates all have smaller seed numbers than those of several [URE3] isolates that are cured only by overproduction of Btn2 or Cur1 (96). This correlation of seed number and level of Btn2/Cur1 needed for curing supports the prion seed sequestration model (Fig. 6).
FIG 6.
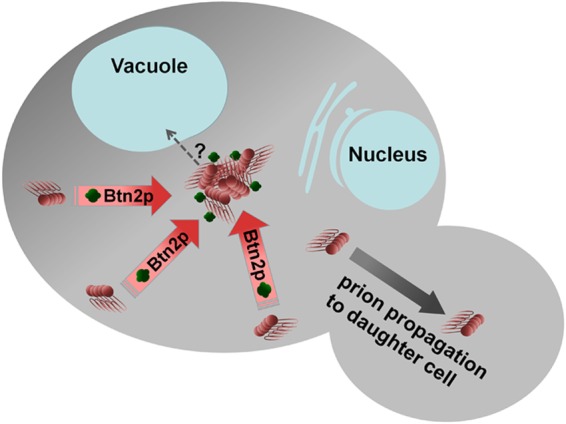
Model of Btn2 curing of the [URE3] prion. Btn2p sequesters amyloid filaments at a single site, increasing the probability of prion loss on cell division (96, 187).
Other studies have examined sites for accumulation of nonspecific protein aggregates, but prion aggregates were not tested. While overproduced Ure2p, Sup35p, and Rnq1p form aggregates in cells without prions, and this overproduction increases prion formation by hundreds of times, only a tiny minority of cells actually develop a prion (e.g., see references 27, 28, 60, and 3). Kaganovich et al. showed that some overproduced aggregated proteins tended to be localized at one of two sites: a perinuclear site and a peripheral site (189). Overproduced Ure2p and Rnq1p aggregated and went to the peripheral site, but this was likely not an amyloid/prion form of either protein. Specht et al. showed that the small heat shock protein Hsp42 is involved in partitioning of nonamyloid aggregates to a peripheral site but not a perinuclear site (190). Aggregates of overproduced Rnq1p localized to a peripheral site in spite of an hsp42Δ mutation, leading these authors to suggest that amyloids were handled differently from other aggregates (190), but there was no indication that the strain carried the [PIN+] prion. Malinovska et al. confirmed that overproduction of Btn2p or Cur1p could cure a prion, in this case an artificial prion comprised of the Sup35 C-terminal translation termination domain fused to a domain of Nrp1 that can act as a prion domain (191). This group showed that localization of Btn2p to a peripheral compartment required Hsp42 and that Hsp42 and Btn2p coimmunoprecipitated and showed colocalization by microscopy (191). However, Cur1p did not colocalize or coimmunoprecipitate with Hsp42 (191). Malinovska et al. saw no colocalization of Ure2p, Rnq1p, or Sup35p with Btn2p and inferred that the prion curing by Btn2p must be indirect (191), but again, there was no indication that any of these strains carried the corresponding prion, so this inference is not justified. Using cells which carried the respective prion, Kryndushkin et al. did see colocalization of prion amyloids of Ure2p and Sup35p with Btn2p, although not with Cur1p (187), and Btn2p colocalization with nonprion aggregates has also been found (144).
Prompted by these studies showing a role of Hsp42 in handling of nonamyloid aggregates and its association with Btn2 (190, 191), we found that overproduction of Hsp42 also cures [URE3] (96). Hsp42 overproduction curing requires Cur1p but not Btn2p, and Btn2 overproduction curing requires Hsp42 (but not another small heat shock protein, Hsp26), but Cur1 overproduction curing of [URE3] does not require Hsp42 (or Hsp26) (96). Curing by Btn2 or Cur1 overproduction is not dependent on the other (96). Thus, Btn2, Cur1, and Hsp42 appear to be interacting/overlapping factors, but each has distinct features.
Malinovska et al. made the case that Btn2p and Cur1p cure prions indirectly by sequestering Sis1p (191), an Hsp40 family chaperone needed by several prions for propagation (176). Engineered sequestration of Sis1p in the nucleus can cure the Nrp1 hybrid prion (see above), and overexpression of Sis1p can stabilize it (191). Sis1p overproduction also prevents curing of [URE3] by Btn2, Cur1, or Hsp42 (96). However, the Sis1p depletion model does not explain the curing of most [URE3] isolates by normal levels of Btn2 and Cur1, which are 20- to 400-fold lower than the Sis1p level (192). Nor does that model explain the colocalization of Btn2 and Ure2 aggregates when Btn2 overproduction is curing [URE3] (187). It is possible that Sis1p binds to the amyloid and that Btn2p does not directly bind to the amyloid but does bind to the Sis1p bound to the amyloid and then is moved to the sequestration site. This model (amyloid-Sis1p-Btn2p-transporter), first suggested by Kryndushkin et al. (193), can explain the results without invoking Sis1p depletion. Excess Sis1p may saturate the Btn2 binding sites, competing with amyloid-bound Sis1p for transport to the sequestration site.
In a study examining the cell's handling of nonamyloid aggregates of optineurin, a protein associated with amyotrophic lateral sclerosis, Kryndushkin et al. showed perfect colocalization of these aggregates, as well as those of PrP and a fragment of huntingtin, with Btn2p (193). Excess Btn2p decreased the number of aggregates, and deficiency of Btn2p had the opposite effect (193). These observations parallel those on the handling of [URE3] aggregates and support the sequestration model for nonamyloid aggregates as well.
It is possible that curing by Btn2 or Cur1 involves one of the sites or systems that deal with apparently nonprion aggregates. Btn2 and Cur1 overproduction curing of [URE3] occurs even in autophagy-deficient atg1Δ cells, and inducing autophagy does not cure [URE3] (187). Wang et al. showed that huntingtin aggregates are collected near the yeast centrosome (spindle pole body) in a process dependent on microtubule function (194), a site that they argue is equivalent to the mammalian aggresome (195). They found that the 14-3-3 protein Bmh1p was associated with huntingtin in these aggregates and that bmh1Δ cells failed to form the aggresome (194). However, bmh1Δ cells are not resistant to curing of [URE3] by overproduced Btn2, Cur1, or Hsp42, separating the aggresome from the action of these curing agents (96). The findings of Specht et al. (190) and Malinovska et al. (191) indicate that Btn2 brings nonamyloid aggregates to a peripheral site, and that may prove to be the destination in its handling of amyloid aggregates as well.
The Btn2p gene was originally identified as a gene upregulated in mutants of the Batten's disease gene homolog, BTN1 (196), and it is known to be involved in late endosome-Golgi protein sorting (197), but how this activity relates to its effects on protein aggregates is not yet clear. Btn3p is an inhibitor of Btn2 activities, both its protein trafficking role and its prion curing function (188). Overproduced Btn3p binds to Btn2 and changes its localization (188).
Btn2 and Cur1 have substantial homology to each other, but there are several differences in their properties, so whether they act through the same system in curing is not yet clear. Btn2 has modest but significant homology to human Hook1, a member of the Hook family of proteins involved in microtubule-dependent transport. Remarkably, Hook2 is involved in aggresome formation (198), but there is no significant homology of Btn2 with Hook2.
YEAST PRIONS AS MODELS FOR HUMAN AMYLOIDOSES
Many human amyloidoses have prion-like aspects, including Alzheimer's disease, Parkinson's disease, serum protein A amyloidosis, Huntington's disease, and the nonamyloid aggregate accumulation disease amyotrophic lateral sclerosis (22; reviewed in references 199, 23, and 8). While as yet there is no epidemiological evidence for spread of these diseases among humans, in some cases spread of amyloids among tissue culture cells or infection of humanized mice with patient brain material has been shown. Studies of an epidemic of amyloidosis A among cheetahs in captivity revealed amyloids of serum amyloid protein A in animals' feces which could induce the homologous disease in mice, suggesting an infectious component of the epidemic (200). Mouse senile amyloidosis is due to spontaneous deposition of apolipoprotein A-II, but this amyloid is found in feces and milk and can transmit the disease to young mice ingesting it (201).
The parallel between the yeast and mammalian prion systems was recently highlighted by the important finding that Sup35NM amyloid can infect tissue culture lines or primary neuron isolates expressing soluble Sup35NM and induce a self-propagating aggregated state in these cells (202). Notably, this aggregated state of Sup35NM can be transmitted not only vertically to offspring but also horizontally to neighboring cells by cell-to-cell contact, showing that aggregated Sup35NM has prion properties in mammalian cells that are similar to its behavior in yeast (203).
A common argument for the hypothesis that oligomers are the toxic species in amyloid diseases, such as Alzheimer's disease, is that many die without detectable mental problems but with extensive Aβ amyloid plaques; it is thus concluded that amyloids are not the toxic species. The problem with this argument is that there was no damage done by these persons' oligomers either! Recent structural data suggest that different Alzheimer's patients have distinct amyloid structural variants (204). In analogy with the lethal and nearly harmless yeast prion variants (13), it is likely that there are some Aβ amyloid “variants” that are not toxic to neurons, while other variant amyloids are neurotoxic and produce clinical disease.
There are many ways to cure yeast prions, and it is conceivable that some of these may be applicable to humans, because the amyloid-handling systems are conserved in some cases. Yeast prions may be cured by under- or overproduction of the Hsp104 or Sse1 chaperone, by deficiency of Ssa1 or -2 (Hsp70s), by overproduction of Ydj1 or deficiency of Sis1 (Hsp40s), and by overproduction of the small Hsps (Hsp26 and Hsp42). Overproduction of Btn2 or Cur1 cures [URE3], apparently by sequestering the prion fibers at one site in the cell, and may be an analog of the mammalian aggresome. Expressing fragments of a prion protein cures the prion in some cases, as does shutting off synthesis of the prion protein. In yeast, a prion that does not segregate to both daughter cells or whose filaments are not split to form new seeds will be cured as the cells divide. Thus, curing a yeast prion may be easier than curing a mammalian prion, but the fundamentals learned about how yeast cells handle amyloid prions are already aiding in the understanding of amyloidoses in general. Screens for compounds effective at curing yeast prions have revealed some that appear to also cure mammalian prions in tissue culture cells (205).
Yeast has been used as a testing ground for the effects of human amyloids and other pathogenic misfolded proteins (193, 206–209). Such studies can apply the full range of yeast genetic methods to discover interactions with cellular components that would be difficult to identify in mammalian systems.
PERSPECTIVE
The discovery of yeast prions in 1994 made plausible the “protein-only” model of the infectious agent responsible for the mammalian spongiform encephalopathies then under intense debate (reviewed in references 210 and 211). More recently, description of the folded, in-register parallel β-sheet architecture of the amyloid that underlies the yeast prions provided an explanation for how proteins can template their own conformation and thus act as genes. Amyloids of PrP have a similar architecture (212–214), although the preparations studied thus far have limited infectivity. The detailed knowledge of the mechanisms of interactions of yeast prions with chaperones and with the aggregate-handling Btn2 and Cur1 systems will doubtless find applications in human prion or amyloid diseases, as there are clear human homologs of these yeast components. There are numerous known methods of curing yeast prions, and antiprion drug screening methods using yeast prions have produced some promising results.
Supplementary Material
ACKNOWLEDGMENTS
We thank Riley Sennett for help in performing growth assays. We thank Michael Reidy and Dan Masison (both of NIH) for thoughtful comments on the manuscript.
This work was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MMBR.00041-14.
REFERENCES
- 1.Alper T, Haig DA, Clarke MC. 1966. The exceptionally small size of the scrapie agent. Biochem Biophys Res Commun 22:278–284. doi: 10.1016/0006-291X(66)90478-5. [DOI] [PubMed] [Google Scholar]
- 2.Griffith JS. 1967. Self-replication and scrapie. Nature 215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 3.Wickner RB. 1994. [URE3] as an altered URE2 protein: evidence for a prion analog in S. cerevisiae. Science 264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 4.Baxa U, Wickner RB, Steven AC, Anderson D, Marekov L, Yau W-M, Tycko R. 2007. Characterization of β-sheet structure in Ure2p1-89 yeast prion fibrils by solid state nuclear magnetic resonance. Biochemistry 46:13149–13162. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- 5.Shewmaker F, Wickner RB, Tycko R. 2006. Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc Natl Acad Sci U S A 103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wickner RB, Dyda F, Tycko R. 2008. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register β-sheet structure. Proc Natl Acad Sci U S A 105:2403–2408. doi: 10.1073/pnas.0712032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wickner RB, Edskes HK, Bateman DA, Kelly AC, Gorkovskiy A, Dayani Y, Zhou A. 2013. Amyloids and yeast prion biology. Biochemistry 52:1514–1527. doi: 10.1021/bi301686a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kraus A, Groveman BR, Caughey B. 2013. Prions and the potential transmissibility of protein misfolding diseases. Annu Rev Microbiol 67:543–564. doi: 10.1146/annurev-micro-092412-155735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lloyd S, Mead S, Collinge J. 2013. Genetics of prion diseases. Curr Opin Genet Dev 23:345–351. doi: 10.1016/j.gde.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bueler H, Aguzzi A, Sailer A, Greiner R-A, Autenried P, Aguet M, Weissmann C. 1993. Mice devoid of PrP are resistant to scrapie. Cell 73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 11.Prusiner SB, Scott M, Foster D, Pan K-M, Groth D, Mirenda C, Torchia M, Yang S-L, Serban D, Carlson GA, Hoppe PC, Westaway D, DeArmond SJ. 1990. Transgenic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63:673–686. doi: 10.1016/0092-8674(90)90134-Z. [DOI] [PubMed] [Google Scholar]
- 12.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. 2012. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A 109:8546–8551. doi: 10.1073/pnas.1204498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGlinchey R, Kryndushkin D, Wickner RB. 2011. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A 108:5337–5341. doi: 10.1073/pnas.1102762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox BS. 1965. PSI, a cytoplasmic suppressor of super-suppressor in yeast. Heredity 20:505–521. doi: 10.1038/hdy.1965.65. [DOI] [Google Scholar]
- 15.Lacroute F. 1971. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J Bacteriol 106:519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coustou V, Deleu C, Saupe S, Begueret J. 1997. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci U S A 94:9773–9778. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wickner RB. 1997. A new prion controls fungal cell fusion incompatibility. Proc Natl Acad Sci U S A 94:10012–10014. doi: 10.1073/pnas.94.19.10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saupe SJ. 2011. The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Sem Cell Dev Biol 22:460–468. doi: 10.1016/j.semcdb.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 19.Eaglestone SS, Cox BS, Tuite MF. 1999. Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism. EMBO J 18:1974–1981. doi: 10.1093/emboj/18.7.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.True HL, Lindquist SL. 2000. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- 21.Aguzzi A, Falsig J. 2012. Prion propagation, toxicity and degradation. Nat Neurosci 15:936–939. doi: 10.1038/nn.3120. [DOI] [PubMed] [Google Scholar]
- 22.Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM-Y. 2012. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jucker M, Walker LC. 2011. Pathogenic protein seeding in Alzheimer's disease and other neurodegenerative disorders. Ann Neurol 70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aigle M, Lacroute F. 1975. Genetical aspects of [URE3], a non-Mendelian, cytoplasmically inherited mutation in yeast. Mol Gen Genet 136:327–335. doi: 10.1007/BF00341717. [DOI] [PubMed] [Google Scholar]
- 25.Singh AC, Helms C, Sherman F. 1979. Mutation of the non-Mendelian suppressor y+ in yeast by hypertonic media. Proc Natl Acad Sci U S A 76:1952–1956. doi: 10.1073/pnas.76.4.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lund PM, Cox BS. 1981. Reversion analysis of [psi−] mutations in Saccharomyces cerevisiae. Genet Res 37:173–182. doi: 10.1017/S0016672300020140. [DOI] [PubMed] [Google Scholar]
- 27.Chernoff YO, Derkach IL, Inge-Vechtomov SG. 1993. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr Genet 24:268–270. doi: 10.1007/BF00351802. [DOI] [PubMed] [Google Scholar]
- 28.Masison DC, Wickner RB. 1995. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 29.Masison DC, Maddelein M-L, Wickner RB. 1997. The prion model for [URE3] of yeast: spontaneous generation and requirements for propagation. Proc Natl Acad Sci U S A 94:12503–12508. doi: 10.1073/pnas.94.23.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper TG. 2002. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol Rev 26:223–238. doi: 10.1111/j.1574-6976.2002.tb00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magasanik B, Kaiser CA. 2002. Nitrogen regulation in Saccharomyces cerevisiae. Gene 290:1–18. doi: 10.1016/S0378-1119(02)00558-9. [DOI] [PubMed] [Google Scholar]
- 32.Shewmaker F, Mull L, Nakayashiki T, Masison DC, Wickner RB. 2007. Ure2p function is enhanced by its prion domain in Saccharomyces cerevisiae. Genetics 176:1557–1565. doi: 10.1534/genetics.107.074153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frolova L, Le Goff X, Zhouravleva G, Davydova E, Philippe M, Kisselev L. 1996. Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA 2:334–341. [PMC free article] [PubMed] [Google Scholar]
- 34.Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, Nierras CR, Cox BS, Ter-Avanesyan MD, Tuite MF. 1995. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J 14:4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.TerAvanesyan A, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. 1994. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 137:671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cosson B, Couturier A, Chabelskaya S, Kiktev D, Inge-Vechtomov S, Philippe M, Zhouravleva G. 2002. Poly(A)-binding protein acts in translation termination via eukaryotic release factor 3 interaction and does not influence [PSI+] propagation. Mol Cell Biol 22:3301–3315. doi: 10.1128/MCB.22.10.3301-3315.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Funakoshi Y, Doi Y, Hosoda N, Uchida N, Osawa M, Shimada I, Tsujimoto M, Suzuki T, Katada T, Hoshino S. 2007. Mechanism of mRNA deadenylation: evidence for a molecular interplay between translation termination factor eRF3 and mRNA deadenylases. Genes Dev 21:3135–3148. doi: 10.1101/gad.1597707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoshino S, Imai M, Kobayashi T, Uchida N, Katada T. 1999. The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3′-poly(A) tail of mRNA. J Biol Chem 274:16677–16680. doi: 10.1074/jbc.274.24.16677. [DOI] [PubMed] [Google Scholar]
- 39.Hosoda N, Kobayashii T, Uchida N, Funakoshi Y, Kikuchi Y, Hoshino S, Katada T. 2003. Translation termination factor eRF3 mediates mRNA decay through the regulation of deadenylation. J Biol Chem 278:38287–38291. doi: 10.1074/jbc.C300300200. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi T, Funakoshi Y, Hoshino S, Katada T. 2004. The GTP-binding release factor eRF3 as a key mediator coupling translation termination to mRNA decay. J Biol Chem 279:45693–45700. doi: 10.1074/jbc.M405163200. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Rayman JB, Kandel ER, Derkatch IL. 2014. Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol Cell 55:305–318. doi: 10.1016/j.molcel.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang H-Y, Lin J-Y, Lee H-C, Wang H-L, King C-Y. 2008. Strain-specific sequences required for yeast prion [PSI+] propagation. Proc Natl Acad Sci U S A 105:13345–13350. doi: 10.1073/pnas.0802215105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King CY, Diaz-Avalos R. 2004. Protein-only transmission of three yeast prion strains. Nature 428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 44.Bradley ME, Liebman SW. 2004. The Sup35 domains required for maintenance of weak, strong or undifferentiated yeast [PSI+] prions. Mol Microbiol 51:1649–1659. doi: 10.1111/j.1365-2958.2003.03955.x. [DOI] [PubMed] [Google Scholar]
- 45.Liu J-J, Sondheimer N, Lindquist S. 2002. Changes in the middle region of Sup35p profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+]. Proc Natl Acad Sci U S A 99:16446–16453. doi: 10.1073/pnas.252652099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bateman DA, Wickner RB. 2012. [PSI+] prion transmission barriers protect Saccharomyces cerevisiae from infection: intraspecies ‘species barriers.’ Genetics 190:569–579. doi: 10.1534/genetics.111.136655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shewmaker F, Kryndushkin D, Chen B, Tycko R, Wickner RB. 2009. Two prion variants of Sup35p have in-register β-sheet structures, independent of hydration. Biochemistry 48:5074–5082. doi: 10.1021/bi900345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toyama BH, Kelly MJ, Gross JD, Weissman JS. 2007. The structural basis of yeast prion strain variants. Nature 449:233–237. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 49.Patino MM, Liu J-J, Glover JR, Lindquist S. 1996. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 50.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. 1996. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 51.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. 1997. In vitro propagation of the prion-like state of yeast Sup35 protein. Science 277:381–383. doi: 10.1126/science.277.5324.381. [DOI] [PubMed] [Google Scholar]
- 52.King C-Y, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. 1997. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci U S A 94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glover JR, Kowal AS, Shirmer EC, Patino MM, Liu J-J, Lindquist S. 1997. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89:811–819. doi: 10.1016/S0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature 428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 55.Edskes HK, Gray VT, Wickner RB. 1999. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci U S A 96:1498–1503. doi: 10.1073/pnas.96.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. 1999. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 283:1339–1343. doi: 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- 57.Brachmann A, Baxa U, Wickner RB. 2005. Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J 24:3082–3092. doi: 10.1038/sj.emboj.7600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. 1997. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 147:507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sondheimer N, Lindquist S. 2000. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell 5:163–172. doi: 10.1016/S1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 60.Derkatch IL, Bradley ME, Hong JY, Liebman SW. 2001. Prions affect the appearance of other prions: the story of [PIN]. Cell 106:171–182. doi: 10.1016/S0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 61.Du Z, Park K-W, Yu H, Fan Q, Li L. 2008. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat Genet 40:460–465. doi: 10.1038/ng.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patel BK, Gavin-Smyth J, Liebman SW. 2009. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat Cell Biol 11:344–349. doi: 10.1038/ncb1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suzuki G, Shimazu N, Tanaka M. 2012. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 336:355–359. doi: 10.1126/science.1219491. [DOI] [PubMed] [Google Scholar]
- 64.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. 2009. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogoza T, Goginashvili A, Rodionova S, Ivanov M, Viktorovskaya O, Rubel A, Volkov K, Mironova L. 2010. Non-mendelian determinant [ISP+] in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1. Proc Natl Acad Sci U S A 107:10573–10577. doi: 10.1073/pnas.1005949107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Radchenko E, Rogoza RE, Khokhrina M, Drozdova P, Mironova L. 2011. SUP35 expression is enhanced in yeast containing [ISP+], a prion form of the transcription regulator Sfp1. Prion 5:317–322. doi: 10.4161/pri.18426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jones EW. 1991. Three proteolytic systems in the yeast Saccharomyces cerevisiae. J Biol Chem 266:7963–7966. [PubMed] [Google Scholar]
- 68.Roberts BT, Wickner RB. 2003. A class of prions that propagate via covalent auto-activation. Genes Dev 17:2083–2087. doi: 10.1101/gad.1115803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ball AJ, Wong DK, Elliott JJ. 1976. Glucosamine resistance in yeast. I. A preliminary genetic analysis. Genetics 84:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kunz BA, Ball AJ. 1977. Glucosamine resistance in yeast. II. Cytoplasmic determinants conferring resistance. Mol Gen Genet 153:169–177. [DOI] [PubMed] [Google Scholar]
- 71.Brown JC, Lindquist S. 2009. A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev 23:2320–2332. doi: 10.1101/gad.1839109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Glass NL, Dementhon K. 2006. Non-self recognition and programmed cell death in filamentous fungi. Curr Opin Microbiol 9:553–558. doi: 10.1016/j.mib.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 73.Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. 2002. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci U S A 99:7402–7407. doi: 10.1073/pnas.072199199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cuille J, Chelle PL. 1939. Experimental transmission of trembling to the goat. C R Seances Acad Sci 208:1058–1060. [Google Scholar]
- 75.Priola SA, Caughey B, Race RE, Chesebro B. 1994. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol 68:4873–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baudin-Baillieu A, Fernandez-Bellot E, Reine F, Coissac E, Cullin C. 2003. Conservation of the prion properties of Ure2p through evolution. Mol Biol Cell 14:3449–3458. doi: 10.1091/mbc.E03-01-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen B, Newnam GP, Chernoff YO. 2007. Prion species barrier between the closely related yeast proteins is detected despite coaggregation. Proc Natl Acad Sci U S A 104:2791–2796. doi: 10.1073/pnas.0611158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chernoff YO, Galkin AP, Lewitin E, Chernova TA, Newnam GP, Belenkiy SM. 2000. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol Microbiol 35:865–876. doi: 10.1046/j.1365-2958.2000.01761.x. [DOI] [PubMed] [Google Scholar]
- 79.Edskes HK, Wickner RB. 2002. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full-length protein. Proc Natl Acad Sci U S A 99(Suppl 4):16384–16391. doi: 10.1073/pnas.162349599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kushnirov VV, Kochneva-Pervukhova NV, Cechenova MB, Frolova NS, Ter-Avanesyan MD. 2000. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J 19:324–331. doi: 10.1093/emboj/19.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Santoso A, Chien P, Osherovich LZ, Weissman JS. 2000. Molecular basis of a yeast prion species barrier. Cell 100:277–288. doi: 10.1016/S0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 82.Conde J, Fink GR. 1976. A mutant of Saccharomyces cerevisiae defective for nuclear fusion. Proc Natl Acad Sci U S A 73:3651–3655. doi: 10.1073/pnas.73.10.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edskes HK, McCann LM, Hebert AM, Wickner RB. 2009. Prion variants and species barriers among Saccharomyces Ure2 proteins. Genetics 181:1159–1167. doi: 10.1534/genetics.108.099929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Resende CG, Outeiro TF, Sands L, Lindquist S, Tuite MF. 2003. Prion protein gene polymorphisms in Saccharomyces cerevisiae. Mol Microbiol 49:1005–1017. doi: 10.1046/j.1365-2958.2003.03608.x. [DOI] [PubMed] [Google Scholar]
- 85.Kelly AC, Shewmaker FP, Kryndushkin D, Wickner RB. 2012. Sex, prions and plasmids in yeast. Proc Natl Acad Sci U S A 109:E2683–E2690. doi: 10.1073/pnas.1213449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mead S, Stumpf MP, Whitfield J, Beck JA, Poulter M, Campbell T, Uphill JB, Goldstein D, Alpers M, Fisher EM, Collinge J. 2003. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science 300:640–643. doi: 10.1126/science.1083320. [DOI] [PubMed] [Google Scholar]
- 87.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. 1996. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144:1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schlumpberger M, Prusiner SB, Herskowitz I. 2001. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol 21:7035–7046. doi: 10.1128/MCB.21.20.7035-7046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. 2002. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci U S A 99(Suppl 4):16392–16399. doi: 10.1073/pnas.152330699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bradley ME, Liebman SW. 2003. Destabilizing interactions among [PSI+] and [PIN+] yeast prion variants. Genetics 165:1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Borchsenius AS, Muller S, Newnam GP, Inge-Vechtomov SG, Chernoff YO. 2006. Prion variant maintained only at high levels of the Hsp104 disaggregase. Curr Genet 49:21–29. doi: 10.1007/s00294-005-0035-0. [DOI] [PubMed] [Google Scholar]
- 92.Kushnirov VV, Kryndushkin D, Boguta M, Smirnov VN, Ter-Avanesyan MD. 2000. Chaperones that cure yeast artificial [PSI+] and their prion-specific effects. Curr Biol 10:1443–1446. doi: 10.1016/S0960-9822(00)00802-2. [DOI] [PubMed] [Google Scholar]
- 93.Lancaster DL, Dobson CM, Rachubinski RA. 2013. Chaperone proteins select and maintain [PIN+] prion conformations in Saccharomyces cerevisiae. J Biol Chem 288:1266–1276. doi: 10.1074/jbc.M112.377564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vishveshwara N, Liebman SW. 2009. Heterologous cross-seeding mimics cross-species prion conversion in a yeast model. BMC Biol 7:26. doi: 10.1186/1741-7007-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bateman D, Wickner RB. 2013. The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genet 9:e1003257. doi: 10.1371/journal.pgen.1003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wickner RB, Beszonov E, Bateman DA. 2014. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc Natl Acad Sci U S A 111:E2711–E2720. doi: 10.1073/pnas.1409582111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bessen RA, Marsh RF. 1992. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 66:2096–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kimberlin RH, Cole S, Walker CA. 1987. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J Gen Virol 68:1875–1881. doi: 10.1099/0022-1317-68-7-1875. [DOI] [PubMed] [Google Scholar]
- 99.Collinge J, Clarke AR. 2007. A general model of prion strains and their pathogenicity. Science 318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 100.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. 2010. Darwinian evolution of prions in cell culture. Science 327:869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ross ED, Baxa U, Wickner RB. 2004. Scrambled prion domains form prions and amyloid. Mol Cell Biol 24:7206–7213. doi: 10.1128/MCB.24.16.7206-7213.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ross ED, Edskes HK, Terry MJ, Wickner RB. 2005. Primary sequence independence for prion formation. Proc Natl Acad Sci U S A 102:12825–12830. doi: 10.1073/pnas.0506136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Toombs JA, Liss NM, Cobble KR, Ben-Musa Z, Ross ED. 2011. [PSI] maintenance is dependent on the composition, not the primary sequence, of the oligopeptide repeat domain. PLoS One 6:e21953. doi: 10.1371/journal.pone.0021953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Toombs JA, McCarty BR, Ross ED. 2010. Compositional determinants of prion formation in yeast. Mol Cell Biol 30:319–332. doi: 10.1128/MCB.01140-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.DePace AH, Santoso A, Hillner P, Weissman JS. 1998. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 93:1241–1252. doi: 10.1016/S0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 106.Doel SM, McCready SJ, Nierras CR, Cox BS. 1994. The dominant PNM2− mutation which eliminates the [PSI] factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 137:659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ross ED, Minton AP, Wickner RB. 2005. Prion domains: sequences, structures and interactions. Nat Cell Biol 7:1039–1044. doi: 10.1038/ncb1105-1039. [DOI] [PubMed] [Google Scholar]
- 108.Tycko R. 2011. Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem 62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tycko R, Wickner RB. 2013. Molecular structures of amyloid and prion fibrils: consensus versus controversy. Acc Chem Res 46:1487–1496. doi: 10.1021/ar300282r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, Meier BH, Saupe SJ, Riek R. 2005. Correlation of structural elements and infectivity of the HET-s prion. Nature 435:844–848. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Siemer AB, Ritter C, Steinmetz MO, Ernst M, Riek R, Meier BH. 2006. 13C, 15N resonance assignment of parts of the HET-s prion protein in its amyloid form. J Biomol NMR 34:75–87. doi: 10.1007/s10858-005-5582-7. [DOI] [PubMed] [Google Scholar]
- 112.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. 2008. Amyloid fibrils of the HET-s(218-279) prion form a beta solenoid with a triangular hydrophobic core. Science 319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 113.Baxa U, Cheng N, Winkler DC, Chiu TK, Davies DR, Sharma D, Inouye H, Kirschner DA, Wickner RB, Steven AC. 2005. Filaments of the Ure2p prion protein have a cross-beta core structure. J Struct Biol 150:170–179. doi: 10.1016/j.jsb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 114.Frederick KK, Debelouchina GT, Kayatekin C, Dominy T, Jacvone AC, Griffin RG, Lindquist S. 2014. Distinct prion strains are defined by amyloid core structure and chaperone binding site dynamics. Chem Biol 21:1–11. doi: 10.1016/j.chembiol.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Luckgei N, Schutz AK, Bousset L, Habenstein B, Souigues Y, Gardiennet C, Meier BH, Melki R, Bockmann A. 2013. The conformation of the prion domain of Sup35p in isolation and in the full-length protein. Angew Chem Int Ed Engl 52:12741–12744. doi: 10.1002/anie.201304699. [DOI] [PubMed] [Google Scholar]
- 116.Kryndushkin DS, Wickner RB, Tycko R. 2011. The core of Ure2p prion fibrils is formed by the N-terminal segment in a parallel cross-β structure: evidence from solid-state NMR. J Mol Biol 409:263–277. doi: 10.1016/j.jmb.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shewmaker F, Ross ED, Tycko R, Wickner RB. 2008. Amyloids of shuffled prion domains that form prions have a parallel in-register β-sheet structure. Biochemistry 47:4000–4007. doi: 10.1021/bi7024589. [DOI] [PubMed] [Google Scholar]
- 118.Ngo S, Gu L, Guo Z. 2011. Hierarchical organization in the amyloid core of yeast prion protein Ure2. J Biol Chem 286:29691–29699. doi: 10.1074/jbc.M111.269092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ngo S, Chiang V, Guo Z. 2012. Quantitative analysis of spin exchange interactions to identify β strand and turn regions in Ure2 prion domain fibrils with site-directed spin labeling. J Struct Biol 180:374–381. doi: 10.1016/j.jsb.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 120.Gorkovskiy A, Thurber KR, Tycko R, Wickner RB. 2014. Locating the folds of the in-register parallel β-sheet of the Sup35p prion domain infectious amyloid. Proc Natl Acad Sci U S A 111:E4615–E4622. doi: 10.1073/pnas.1417974111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krishnan R, Lindquist S. 2005. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Baxa U, Taylor KL, Wall JS, Simon MN, Cheng N, Wickner RB, Steven A. 2003. Architecture of Ure2p prion filaments: the N-terminal domain forms a central core fiber. J Biol Chem 278:43717–43727. doi: 10.1074/jbc.M306004200. [DOI] [PubMed] [Google Scholar]
- 123.Chen B, Thurber KR, Shewmaker F, Wickner RB, Tycko R. 2009. Measurement of amyloid fibril mass-per-length by tilted-beam transmission electron microscopy. Proc Natl Acad Sci U S A 106:14339–14344. doi: 10.1073/pnas.0907821106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Diaz-Avalos R, King CY, Wall JS, Simon M, Caspar DLD. 2005. Strain-specific morphologies of yeast prion amyloids. Proc Natl Acad Sci U S A 102:10165–10170. doi: 10.1073/pnas.0504599102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Loquet A, Bousset L, Gardiennet C, Sourigues Y, Wasmer C, Habenstein B, Schutz A, Meier BH, Melki R. 2009. Prion fibrils of Ure2p assembled under physiological conditions contain highly ordered, natively folded molecules. J Mol Biol 394:108–118. doi: 10.1016/j.jmb.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 126.Bai M, Zhou JM, Perrett S. 2004. The yeast prion protein Ure2 shows glutathione peroxidase activity in both native and fibrillar forms. J Biol Chem 279:50025–50030. doi: 10.1074/jbc.M406612200. [DOI] [PubMed] [Google Scholar]
- 127.Wickner RB, Shewmaker F, Kryndushkin D, Edskes HK. 2008. Protein inheritance (prions) based on parallel in-register β-sheet amyloid structures. Bioessays 30:955–964. doi: 10.1002/bies.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Collins SR, Douglass A, Vale RD, Weissman JS. 2004. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol 2:e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bernet J. 1965. Mode d'action des gènes de barrage et relation entre l'incompatibilité cellulaire et l'incompatibilité sexuelle chez le Podospora anserina. Ann Sci Natl Bot 6:611–768. [Google Scholar]
- 130.Dalstra HJ, Swart K, Debets AJ, Saupe SJ, Hoekstra RF. 2003. Sexual transmission of the [Het-s] prion leads to meiotic drive in Podospora anserina. Proc Natl Acad Sci U S A 100:6616–6621. doi: 10.1073/pnas.1030058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Joseph SB, Kirkpatrick M. 2008. Effects of [PSI+] prion on rates of adaptation in yeast. J Evol Biol 21:773–780. doi: 10.1111/j.1420-9101.2008.01515.x. [DOI] [PubMed] [Google Scholar]
- 132.Namy O, Galopier A, Martini C, Matsufuji S, Fabret C, Rousset C. 2008. Epigenetic control of polyamines by the prion [PSI+]. Nat Cell Biol 10:1069–1075. doi: 10.1038/ncb1766. [DOI] [PubMed] [Google Scholar]
- 133.Tyedmers J, Madariaga ML, Lindquist S. 2008. Prion switching in response to environmental stress. PLoS Biol 6:e294. doi: 10.1371/journal.pbio.0060294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Westergard L, True HL. 2014. Extracellular environment modulates the formation and propagation of particular amyloid structures. Mol Microbiol 92:698–715. doi: 10.1111/mmi.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Halfmann R, Jarosz DF, Jones SK, Chang A, Lancster AK, Lindquist S. 2012. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 482:363–368. doi: 10.1038/nature10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Halfmann R, Alberti S, Lindquist S. 2010. Prions, protein homeostasis, and phenotypic diversity. Trends Cell Biol 20:125–133. doi: 10.1016/j.tcb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Benko AL, Vaduva G, Martin NC, Hopper AK. 2000. Competition between a sterol biosynthetic enzyme and tRNA modification in addition to changes in the protein synthesis machinery causes altered nonsense suppression. Proc Natl Acad Sci U S A 97:61–66. doi: 10.1073/pnas.97.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Debets AJ, Dalstra HJ, Slakhorst M, Koopmanschap B, Hoekstra RF, Saupe SJ. 2012. High natural prevalence of a fungal prion. Proc Natl Acad Sci U S A 109:10432–10437. doi: 10.1073/pnas.1205333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. 2005. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci U S A 102:10575–10580. doi: 10.1073/pnas.0504882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Futcher AB, Cox BS. 1983. Maintenance of the 2 microns circle plasmid in populations of Saccharomyces cerevisiae. J Bacteriol 154:612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Futcher B, Reid E, Hickey DA. 1988. Maintenance of the 2 micron circle plasmid of Saccharomyces cerevisiae by sexual transmission: an example of selfish DNA. Genetics 118:411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mead DJ, Gardner DCJ, Oliver SG. 1986. The yeast 2 μ plasmid: strategies for the survival of a selfish DNA. Mol Gen Genet 205:417–421. doi: 10.1007/BF00338076. [DOI] [PubMed] [Google Scholar]
- 143.Kelly AC, Busby B, Wickner RB. 2014. Effect of domestication on the spread of the [PIN+] prion in Saccharomyces cerevisiae. Genetics 197:1007–1024. doi: 10.1534/genetics.114.165670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kryndushkin D, Pripuzova N, Burnett B, Shewmaker F. 2013. Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells. J Biol Chem 288:27100–27111. doi: 10.1074/jbc.M113.485359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Taneja V, Maddelein ML, Talarek N, Saupe SJ, Liebman SW. 2007. A non-Q/N-rich prion domain of a foreign prion, [Het-s], can propagate as a prion in yeast. Mol Cell 27:67–77. doi: 10.1016/j.molcel.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Reidy M, Sharma R, Masison DC. 2013. Schizosaccharomyces pombe disaggregation machinery chaperones support Saccharomyces cerevisiae growth and prion propagation. Eukaryot Cell 12:739–745. doi: 10.1128/EC.00301-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Reidy M, Masison DC. 2012. Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics 192:185–193. doi: 10.1534/genetics.112.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Afanasieva EG, Kushnirov VV, Tuite MF, Ter-Avanesyan MD. 2011. Molecular basis for transmission barrier and interference between closely related prion proteins in yeast. J Biol Chem 286:15773–15780. doi: 10.1074/jbc.M110.183889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nakayashiki T, Ebihara K, Bannai H, Nakamura Y. 2001. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell 7:1121–1130. doi: 10.1016/S1097-2765(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 150.Harrison LB, Yu Z, Stajich J E, Dietrich FS, Harrison PM. 2007. Evolution of budding yeast prion-determinant sequences across diverse fungi. J Mol Biol 368:273–282. doi: 10.1016/j.jmb.2007.01.070. [DOI] [PubMed] [Google Scholar]
- 151.Edskes HE, Khamar HJ, Winchester C-L, Greenler AJ, Zhou A, McGlinchey RP, Gorkovskiy A, Wickner RB. 2014. Sporadic distribution of prion-forming ability of Sup35p from yeasts and fungi. Genetics 198:605–616. doi: 10.1534/genetics.114.166538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Safadi RA, Talarek N, Jacques N, Aigle M. 2011. Yeast prions: could they be exaptations? The URE2/[URE3] system in Kluyveromyces lactis. FEMS Yeast Res 11:151–153. doi: 10.1111/j.1567-1364.2010.00700.x. [DOI] [PubMed] [Google Scholar]
- 153.Edskes HK, Engel A, McCann LM, Brachmann A, Tsai H-F, Wickner RB. 2011. Prion-forming ability of Ure2 of yeasts is not evolutionarily conserved. Genetics 188:81–90. doi: 10.1534/genetics.111.127217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Edskes HK, Wickner RB. 2013. The [URE3] prion in Candida. Eukaryot Cell 12:551–558. doi: 10.1128/EC.00015-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Engel A, Shewmaker F, Edskes HK, Dyda F, Wickner RB. 2011. Amyloid of the Candida albicans Ure2p prion domain is infectious and has a parallel in-register β-sheet structure. Biochemistry 50:5971–5978. doi: 10.1021/bi200142x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schwimmer C, Masison DC. 2002. Antagonistic interactions between yeast [PSI+] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol Cell Biol 22:3590–3598. doi: 10.1128/MCB.22.11.3590-3598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Denic V, Dotsch V, Sinning I. 2013. Endoplasmic reticulum targeting and insertion of tail-anchored membrane proteins by the GET pathway. Cold Spring Harb Perspect Biol 5:a013334. doi: 10.1101/cshperspect.a013334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Escusa-Toret S, Vonk WIM, Frydman J. 2013. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat Cell Biol 15:1231–1243. doi: 10.1038/ncb2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Glover JR, Lindquist S. 1998. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94:73–82. doi: 10.1016/S0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 160.Schlieker C, Tews I, Bukau B, Mogk A. 2004. Solubilization of aggregated proteins by ClpB/DnaK relies on the continuous extraction of unfolded polypeptides. FEBS Lett 578:351–356. doi: 10.1016/j.febslet.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 161.Winkler J, Tyedmers J, Bukau B, Mogk A. 2012. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol 198:387–404. doi: 10.1083/jcb.201201074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chernoff YO, Lindquist SL, Ono B-I, Inge-Vechtomov SG, Liebman SW. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 163.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. 2001. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol 40:1357–1369. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 164.Jung G, Jones G, Masison DC. 2002. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci U S A 99:9936–9941. doi: 10.1073/pnas.152333299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jung G, Masison DC. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol 43:7–10. doi: 10.1007/s002840010251. [DOI] [PubMed] [Google Scholar]
- 166.Cox BS, Ness F, Tuite MF. 2003. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 165:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. 2000. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 97:240–244. doi: 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ness F, Ferreira P, Cox BS, Tuite MF. 2002. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol Cell Biol 22:5593–5605. doi: 10.1128/MCB.22.15.5593-5605.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jung G, Jones G, Wegrzyn RD, Masison DC. 2000. A role for cytosolic Hsp70 in yeast [PSI(+)] prion propagation and [PSI(+)] as a cellular stress. Genetics 156:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Roberts BT, Moriyama H, Wickner RB. 2004. [URE3] prion propagation is abolished by a mutation of the primary cytosolic Hsp70 of budding yeast. Yeast 21:107–117. doi: 10.1002/yea.1062. [DOI] [PubMed] [Google Scholar]
- 171.Sharma D, Masison DC. 2008. Functionally redundant isoforms of a yeast Hsp70 chaperone subfamily have different antiprion effects. Genetics 179:1301–1311. doi: 10.1534/genetics.108.089458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sharma D, Masison DC. 2011. Single methyl group determines prion propagation and protein degradation activities of yeast heat shock protein (Hsp)-70 chaperones Ssa1p and Ssa2p. Proc Natl Acad Sci U S A 108:13665–13670. doi: 10.1073/pnas.1107421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jones G, Song Y, Chung S, Masison DC. 2004. Propagation of yeast [PSI+] prion impaired by factors that regulate Hsp70 substrate binding. Mol Cell Biol 24:3928–3937. doi: 10.1128/MCB.24.9.3928-3937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kryndushkin D, Wickner RB. 2007. Nucleotide exchange factors for Hsp70s are required for [URE3] prion propagation in Saccharomyces cerevisiae. Mol Biol Cell 18:2149–2154. doi: 10.1091/mbc.E07-02-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fan Q, Park K-W, Du Z, Morano KA, Li L. 2007. The role of Sse1 in the de novo formation and variant determination of the [PSI+] prion. Genetics 177:1583–1593. doi: 10.1534/genetics.107.077982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Higurashi T, Hines JK, Sahi C, Aron R, Craig EA. 2008. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc Natl Acad Sci U S A 105:16596–16601. doi: 10.1073/pnas.0808934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kirkland PA, Reidy M, Masison DC. 2011. Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics 188:565–577. doi: 10.1534/genetics.111.129460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hines JK, Li X, Du Z, Higurashi T, Li L, Craig EA. 2011. [SWI], the prion formed by the chromatin remodeling factor Swi1, is highly sensitive to alterations in Hsp70 chaperone system activity. PLoS Genet 7:e1001309. doi: 10.1371/journal.pgen.1001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Reidy M, Masison DC. 2011. Modulation and elimination of yeast prions by protein chaperones and co-chaperones. Prion 5:245–249. doi: 10.4161/pri.17749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Moosavi B, Wongwigkam J, Tuite MF. 2010. Hsp70/Hsp90 co-chaperones are required for efficient Hsp104-mediated elimination of the yeast [PSI+] prion but not for prion propagation. Yeast 27:167–179. doi: 10.1002/yea.1742. [DOI] [PubMed] [Google Scholar]
- 181.Reidy M, Masison DC. 2010. Sti1 regulation of Hsp70 and Hsp90 is critical for curing of Saccharomyces cerevisiae [PSI+] prions by Hsp104. Mol Cell Biol 30:3542–3552. doi: 10.1128/MCB.01292-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hung GC, Masison DC. 2006. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 173:611–620. doi: 10.1534/genetics.106.056820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Helsen CW, Glover JR. 2012. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104). J Biol Chem 287:542–556. doi: 10.1074/jbc.M111.302869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD. 1999. Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI+] prion. Mol Cell Biol 19:8103–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Allen KD, Wegrzyn RD, Chernova TA, Muller S, Newnam GP, Winslett PA, Wittich KB, Wilkinson KD, Chernoff YO. 2005. Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. Genetics 169:1227–1242. doi: 10.1534/genetics.104.037168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kiktev DA, Patterson JC, Muller S, Bariar B, Pan T, Chernoff YO. 2012. Regulation of the chaperone effects on a yeast prion by the cochaperone Sgt2. Mol Cell Biol 32:4960–4970. doi: 10.1128/MCB.00875-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kryndushkin D, Shewmaker F, Wickner RB. 2008. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J 27:2725–2735. doi: 10.1038/emboj.2008.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kanneganti V, Kama R, Gerst JE. 2011. Btn3 is a negative regulator of Btn2-mediated endosomal protein trafficking and prion curing in yeast. Mol Biol Cell 22:1648–1663. doi: 10.1091/mbc.E10-11-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kaganovich D, Kopito R, Frydman J. 2008. Misfolded proteins partition between two distinct quality control compartments. Nature 454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Specht S, Miller SBM, Mogk A, Bukau B. 2011. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol 195:617–629. doi: 10.1083/jcb.201106037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S. 2012. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spaciotemporal distribution of protein aggregates. Mol Biol Cell 23:3041–3056. doi: 10.1091/mbc.E12-03-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. 2003. Global analysis of protein expression in yeast. Nature 425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 193.Kryndushkin D, Ihrke G, Piermartiri TC, Shewmaker F. 2012. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol Microbiol 86:1531–1547. doi: 10.1111/mmi.12075. [DOI] [PubMed] [Google Scholar]
- 194.Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, Sherman MY. 2009. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J 23:451–463. doi: 10.1096/fj.08-117614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kopito R. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10:524–530. doi: 10.1016/S0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 196.Pearce DA, Ferea T, Nosel SA, Das B, Sherman F. 1999. Action of BTN1, the yeast ortholog of the gene mutated in Batten disease. Nat Genet 22:55–58. doi: 10.1038/8861. [DOI] [PubMed] [Google Scholar]
- 197.Kama R, Robinson M, Gerst JE. 2007. Btn2, a Hook1 ortholog and potential Batten disease-related protein, mediates late endosome-Golgi protein sorting in yeast. Mol Cell Biol 27:605–621. doi: 10.1128/MCB.00699-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Szebenyi G, Wigley WC, Hall B, Didier A, Yu M, Thomas P, Kramer H. 2007. Hook2 contributes to aggresome formation. BMC Cell Biol 8:19. doi: 10.1186/1471-2121-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cascarina SM, Ross ED. 2014. Yeast prions and human prion-like proteins: sequence features and prediction methods. Cell Mol Life Sci 71:2047–2063. doi: 10.1007/s00018-013-1543-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhang B, Une Y, Fu X, Yan J, Ge F, Yao J, Sawashita J, Mori M, Tomozawa H, Kametani F, Higuchi K. 2008. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proc Natl Acad Sci U S A 105:7263–7268. doi: 10.1073/pnas.0800367105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Qian J, Yan J, Ge F, Zhang B, Fu X, Tomozawa H, Sawashita J, Mori M, Higuchi K. 2010. Mouse senile amyloid fibrils deposited in skeletal muscle exhibit amyloidosis-enhancing activity. PLoS Pathol 6:e1000914. doi: 10.1371/journal.ppat.1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Krammer C, Kryndushkin D, Suhre MH, Kremmer E, Hofmann A, Pfeifer A, Scheibel T, Wickner RB, Schatzl HM, Vorberg I. 2009. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proc Natl Acad Sci U S A 106:462–467. doi: 10.1073/pnas.0811571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hofmann JP, Denner P, Nussbaum-Krammer C, Kuhn P-H, Suhre MH, Scheibel T, Lichtenthaler SF, Schatzl HM, Bano D, Vorberg IM. 2013. Cell-to-cell propagation of infectious cytosolic protein aggregates. Proc Natl Acad Sci U S A 110:5951–5956. doi: 10.1073/pnas.1217321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. 2013. Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell 154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nguyen P, Oumata M, Soubigou F, Evrard J, Desban N, Lemoine P, Bouaziz S, Blondel M, Voisset C. 2014. Evaluation of the antiprion activity of 6-aminophenanthridines and related heterocycles. Eur J Med Chem 82C:363–371. doi: 10.1016/j.ejmech.2014.05.083. [DOI] [PubMed] [Google Scholar]
- 206.Bharadwaj P, Martins R, Macreadie I. 2010. Yeast as a model for studying Alzheimer's disease. FEMS Yeast 10:961–969. doi: 10.1111/j.1567-1364.2010.00658.x. [DOI] [PubMed] [Google Scholar]
- 207.Braun RJ, Buttner S, Ring J, Kroemer G, Madeo F. 2010. Nervous yeast: modeling neurotoxic cell death. Trends Biochem Sci 35:135–144. doi: 10.1016/j.tibs.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 208.Khurana V, Lindquist S. 2010. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nat Rev Neurosci 10:436–449. doi: 10.1038/nrn2809. [DOI] [PubMed] [Google Scholar]
- 209.Winderickx J, Delay C, De Vox A, Klinger H, Pellens K, Vanhelmont T, van Leuven F, Zabrocki P. 2008. Protein folding diseases and neurodegeneration: lessons learned from yeast. Biochim Biophys Acta 1783:1381–1395. doi: 10.1016/j.bbamcr.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 210.Chesebro B. 1997. Human TSE disease—viral or protein only? Nat Med 3:491–492. doi: 10.1038/nm0597-491. [DOI] [PubMed] [Google Scholar]
- 211.Weissmann C, Bueler H, Sailer A, Fischer M, Aguet M, Aguzzi A. 1993. Role of PrP in prion diseases. Br Med Bull 49:995–1011. [DOI] [PubMed] [Google Scholar]
- 212.Cobb NJ, Sonnichsen FD, Mchaourab H, Surewicz WK. 2007. Molecular architecture of human prion protein amyloid: a parallel, in-register β-structure. Proc Natl Acad Sci U S A 104:18946–18951. doi: 10.1073/pnas.0706522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Groveman BR, Dolan MA, Taubner LM, Kraus A, Wickner RB, Caughey B. 2014. Parallel in-register intermolecular β-sheet architectures for prion-seeded prion protein (PrP) amyloids. J Biol Chem 289:24129–24142. doi: 10.1074/jbc.M114.578344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tycko R, Savtchenko R, Ostapchenko VG, Makarava N, Baskakov IV. 2010. The α-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel β-sheet structure in PrP fibrils: evidence from solid state nuclear magnetic resonance. Biochemistry 49:9488–9497. doi: 10.1021/bi1013134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Petkova AT, Yau WM, Tycko R. 2006. Experimental constraints on quaternary structure in Alzheimer's beta-amyloid fibrils. Biochemistry 45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. 2007. Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol 5:611–618. doi: 10.1038/nrmicro1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Masison DC, Kirkland PA, Sharma D. 2009. Influence of Hsp70s and their regulators on yeast prion propagation. Prion 3:65–73. doi: 10.4161/pri.3.2.9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.