

Abstract
Accumulation of misfolded proteins in proteinaceous inclusions is a prominent pathological feature common to many age-related neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease, Huntington's disease, and amyotrophic lateral sclerosis. In cultured cells, when the production of misfolded proteins exceeds the capacity of the chaperone refolding system and the ubiquitin-proteasome degradation pathway, misfolded proteins are actively transported to a cytoplasmic juxtanuclear structure called an aggresome. Aggresome formation is recognized as a cytoprotective response serving to sequester potentially toxic misfolded proteins and facilitate their clearance by autophagy. Recent evidence indicates that aggresome formation is mediated by dynein/dynactin-mediated microtubule-based transport of misfolded proteins to the centrosome and involves several regulators, including histone deacetylase 6, E3 ubiquitin-protein ligase parkin, deubiquitinating enzyme ataxin-3, and ubiquilin-1. Characterization of the molecular mechanisms underlying aggresome formation and its regulation has begun to provide promising therapeutic targets that may be relevant to neurodegenerative diseases. In this review, we provide an overview of the molecular machinery controlling aggresome formation and discuss potential useful compounds and intervention strategies for preventing or reducing the cytotoxicity of misfolded and aggregated proteins.
Keywords: Neurodegenerative diseases, aggresome, HDAC6, parkin, ataxin-3, ubiquilin-1, inclusion body, protein misfolding
Introduction
Pathological inclusions containing misfolded proteins are a frequent feature of age-related neurodegenerative diseases, including Parkinson's disease (PD), Alzheimer's disease (AD), Huntington's disease (HD), spinocerebellar ataxias (SCA), amyotrophic lateral sclerosis (ALS), and many others [1]. The pathogenesis of these diseases involves the misfolding of disease-specific proteins, and these disorders are sometimes referred to as “conformational diseases” [2]. Genetic mutations or environmental insults can induce many different proteins to misfold and aggregate, suggesting that a common pathological mechanism may link these clinically distinct neurodegenerative diseases [3-6]. While it is clear that the presence of misfolded proteins is integrally linked with the pathology of these diseases, the molecular mechanisms underlying the cellular management of misfolded proteins are not fully understood.
Accumulating evidence indicates that one way cells handle excess misfolded proteins, which could result from UPS impairment or increased oxidative stress, is to collect and compartmentalize misfolded proteins in specialized inclusions called aggresomes [7-9]. Aggresomes are thought to be cytoprotective because they sequester toxic, aggregated proteins and may facilitate their elimination by autophagy [10-14]. Although aggresomes are a cell culture phenomenon and do not necessarily represent inclusion bodies found in neurodegenerative diseases, recent studies of aggresome formation have yielded important insights into the molecular mechanisms by which cells manage misfolded protein stress [8,15]. In this review we discuss our current understanding of the molecular machinery involved in aggresome formation and the potential targeting of this pathway to generate mechanism-based therapies for the treatment of neurodegenerative disease.
Aggresome Formation is a Protective Cellular Response Against Misfolded Protein Stress
Within the cell, protein folding occurs both co-translationally as the nascent polypeptide exits the large ribosomal subunit and posttranslationally after trafficking and import into specific subcellular compartments, such as the mitochondria [16,17]. Based in large part on the primary amino acid sequence, protein folding is driven by the formation of hydrophilic interactions, collapse of hydrophobic regions, and burial of electrostatic interactions, seeking the lowest free energy state, which usually corresponds to the native conformation of the protein [16,18]. There is a constant competition between multiple potential folding pathways, some terminating in kinetically trapped and incorrectly folded proteins, referred to as ‘dead end’ conformations. Improper protein folding can occur as the result of incomplete protein synthesis, missense mutations, high levels of protein expression, postsynthetic damage, or a shortage of necessary co-factors or components of multimeric complexes [19]. Protein misfolding is not a rare event and it is estimated that approximately 30% of newly synthesized proteins are misfolded [20]. Protein misfolding not only can result in a functionally inactive protein, but can also lead to protein aggregation, which refers to the abnormal association of proteins or protein fragments and is usually defined biochemically by detergent insolubility [1,15]. Protein aggregates visible by light microscopy are called inclusion bodies [1,15]. Recent reports indicate that misfolded and aggregated proteins are able to disrupt cellular function through a variety of mechanisms, including pore formation, inhibition of proteasomal degradation, and sequestration of critical cellular factors [1,15]. Together these findings emphasize the importance of highly vigilant protein quality control systems to prevent the cytotoxic accumulation of misfolded proteins.
Protein quality control is particularly important to neuronal homeostasis and normal function because neurons are post-mitotic and unable to dilute cytotoxic misfolded proteins through cell division [1]. As outlined in Fig. (1), the molecular chaperone and the ubiquitin-proteasome systems (UPS) compose the initial cellular defense against misfolded protein accumulation (Fig. (1), steps 2 and 3) [21-23]. However, when these systems fail or are overwhelmed, misfolded proteins have the potential to form cytotoxic oligomers, which are small aggregates composed of approximately 3-50 monomers 3-50 monomers (Fig. (1), step 4) [15]. The aggresome-autophagy pathway represents a third cellular defense system, in which misfolded and aggregated proteins are recognized and coupled to the retrograde microtubule motor dynein for transport to a perinuclear aggresome (Fig. (1), steps 5 and 6) [8,9]. The aggresome acts to sequester cytotoxic proteins and also to facilitate their clearance by autophagy (Fig. (1), step 7) [8,9].
Fig. 1. Aggresome formation is a cellular defense against the accumulation of aggregated proteins.
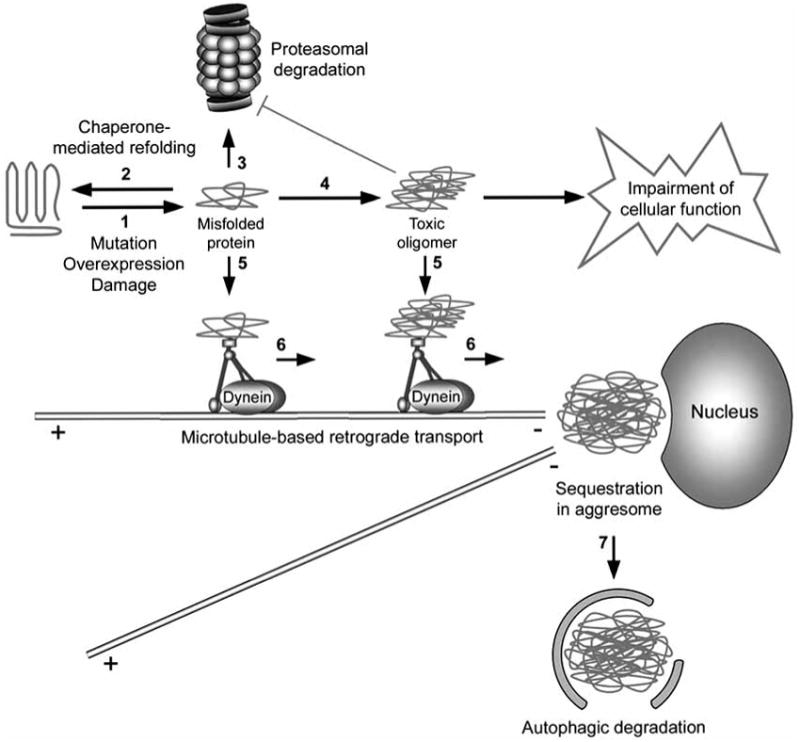
Genetic mutation, increased protein levels, and oxidative stress can induce protein misfolding (step 1). Once formed, misfolded proteins may be refolded/stabilized by chaperones (step 2) or degraded by the 26S proteasome (step 3). However, when the chaperone and proteasome systems are damaged or overwhelmed, misfolded proteins have the potential to aggregate (step 4) and impair cellular function, such as the inhibition of the proteasomal function. The cell recognizes misfolded and aggregated proteins and transports these proteins to aggresomes in a process mediated by the dynein motor complex (steps 5 and 6). Aggresomes not only sequester potentially harmful aggregated proteins, but also concentrate aggregated proteins for more efficient autophagic degradation (step 7).
The Chaperone System
Molecular chaperones are a highly conserved class of proteins that assist in protein folding (Fig. (1), step 2). This class includes the ATP-dependent HSP70 proteins, HSP90 proteins, and cylindrical chaperonin complexes, and also the ATP-independent small chaperone proteins [24]. Several of the chaperones are referred to as heat shock proteins (HSPs) and their expression is induced by temperature increases and other types of stress as a way to increase the cellular capacity for the handling of misfolded proteins [21]. Chaperones function by transiently binding exposed hydrophobic regions and unstructured backbone regions, which are normally buried within the properly folded protein and are a feature of nonnative conformations [17,24]. Chaperones increase the efficiency of protein folding by stabilizing particular folding intermediates and preventing non-specific protein interactions, protein misfolding, and protein aggregation [16]. In some cases, chaperones promote the solubilization and refolding of damaged or aggregated proteins produced during times of cell stress [16,25]. The importance of the chaperone system to neuronal survival is highlighted by recent genetic evidence showing that neurodegenerative disease can be caused by mutations in chaperones, such as HSP22 [26], HSP27 [27], and HSP60 [28].
The Ubiquitin-Proteasome System
The ubiquitin-proteasome system (UPS) is a major intracellular proteolytic pathway for eliminating misfolded proteins (Fig. (1), step 3). In this system, substrates are first tagged by covalent linkage to multiple molecules of ubiquitin, a 76-amino-acid polypeptide. The ubiquitinated substrate proteins are subsequently recognized and degraded by the 26S proteasome (Fig. (2)). Conjugation of ubiquitin to a substrate is a multi-step process that requires sequential action of three enzymes. First, ubiquitin is activated by the ubiquitin-activating enzyme (E1) at the expense of ATP. The activated ubiquitin is then transferred to an ubiquitin-conjugating enzyme (E2). The ubiquitin-protein ligase (E3) binds the substrate and catalyzes the transfer of the activated ubiquitin from the E2 to the substrate via formation of a covalent isopeptide linkage between the ubiquitin carboxyl-terminal glycine residue and a lysine residue within the substrate protein [29]. It is estimated that the mammalian genome contains two E1 ubiquitin-activating enzyme, several E2 ubiquitin-conjugating enzymes, and hundreds of E3 ubiquitin-conjugating enzymes [29-31]. E3 enzymes play a particularly important role in conferring specificity to the ubiquitination reaction by selectively recognizing distinctive sets of substrates and facilitating the final transfer of the ubiquitin molecule to the substrate [29]. Additional ubiquitin molecules can be covalently attached to the preceding ubiquitin molecule to form a polyubiquitin chain, which targets the substrate for degradation by the 26S proteasome [32]. The 26S proteasome is composed of a barrel-shaped 20S catalytic core, capped on either end by a 19S regulatory complex [19,33]. The 19S complex recognizes polyubiquitinated substrates and assists in unfolding and translocation of the substrate into the proteolytic chamber of the 20S core for degradation into small peptides. The polyubiquitin chain is removed from the substrate prior to entering the proteolytic core, and is recycled to free ubiquitin by the action of a deubiquitinating enzyme [19,33]. A direct link between impaired UPS and neurodegeneration is provided by recent identification of mutations in the E3 enzymes parkin [34] and SIMPLE [35] and deubiubiquitinating enzymes UCH-L1 [36] and ataxin-3 [37] as the cause for familial forms of neurodegenerative diseases.
Fig. 2. The ubiquitin-proteasome system.
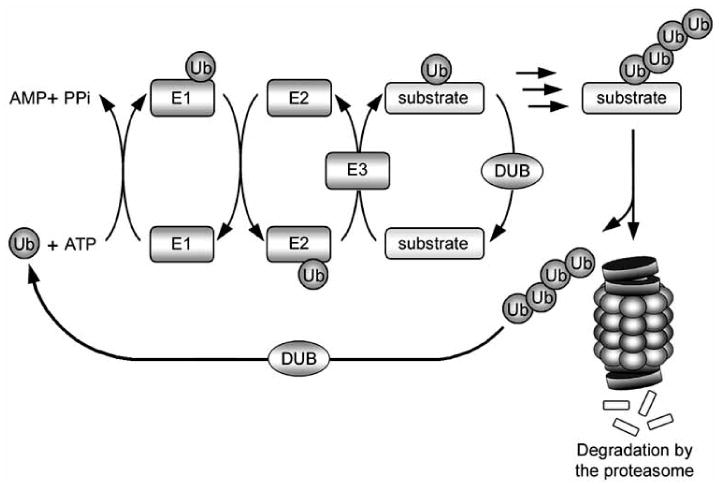
Ubiquitin is covalently attached to a substrate protein through a series of sequential reactions involving three enzymes: an E1 ubiquitin-activating enzyme, which forms a thiol-ester linkage with ubiquitin; an E2 ubiquitin-conjugating enzyme, which transiently carries ubiquitin via a thiol-ester linkage; and finally an E3 ubiquitin-protein ligase, which facilitates the transfer of ubiquitin from the E2 enzyme to the substrate. Successive reactions result in the attachment of a polyubiquitin chain, which targets the substrate for degradation by the 26S proteasome. The polyubiquitin chain is removed from the substrate and recycled by deubiquitinating enzymes (DUBs).
The Aggresome-Autophagy Pathway
The aggresome-autophagy pathway sequesters misfolded proteins and facilitates their clearance when the chaperone and ubiquitin proteasome systems are overwhelmed. The formation of the aggresome is a multi-step process (Fig. (1), steps 5 and 6), involving recognition of misfolded and aggregated proteins, coupling to the dynein motor complex, and retrograde transport along microtubules to the centrosome [8,9]. Aggresome formation is invariably accompanied by a distinctive collapse of the intermediate filament cytoskeleton into a cage-like structure that encircles the aggresome [7,12,38-45]. Intermediate filaments types are cell type specific. In cultured non-neuronal cells, aggresomes are encapsulated by vimentin intermediate filaments [7,40,41,46], whereas in neurons, aggresomes are encapsulated by neurofilaments [11]. The purpose of the cage-like intermediate filament structure is unclear, although it may promote the stability of the aggresome or aid in the prevention of non-specific interactions [47]. In addition to misfolded and aggregated proteins, molecular chaperones, UPS components, ubiquitinated proteins, autophagic machinery, and centrosomal markers also localize to aggresomes [7,12,38-45]. Furthermore, autophagosomes and lysosomes have been found to accumulate around the periphery of the aggresome, consistent with a role for autophagy in the clearance of aggresomes [10,11]. Ultrastructural analyses indicate that aggresomes consist of electron dense particles that surround or are in close proximity to the centrioles and that the intermediate filaments are rearranged into parallel bundles around the aggresome [7,10,13,42]. Recent reports indicate that some pathological inclusion bodies, particularly Lewy bodies, display similarities to aggresomes in morphology and protein composition [48,49]. However, it remains to be determined whether Lewy bodies or any other pathological inclusions are bona fide in vivo correlates of aggresomes.
Aggresome formation may protect the cell by sequestering toxic protein species. During the process of protein aggregation several distinct intermediates can be formed, and accumulating evidence suggests that these intermediates may be the primary cytotoxic species (Fig. (1), step 4), while the mature forms found in aggresomes are inert [3,50-53]. The smaller intermediate forms display a higher amount of exposed surface area compared to larger aggregates, increasing the potential for aberrant interactions with cellular membranes, proteins, or other macromolecules [2]. Thus the transport of these intermediates to the aggresome reduces the exposed surface area and also removes them from sites of action, such as nerve terminals [8,54]. In support of a protective role for aggresome formation, recent studies have shown that, blocking the formation of aggresomes by inhibiting microtubule polymerization [11] or impairing dynein motor function [11] leads to decreased viability in cells expressing disease proteins. Conversely, promoting the formation of α-synuclein aggresomes, by expression of synphilin-1 [43,55] or incubation with small molecules [56], is protective. Moreover, by using a sophisticated automated microscopy system that allows tracking of single cells and their intracellular proteins, Arrasate et al. found that inclusion formation reduced the amount of mutant huntingtin protein present in other areas of the cell and was associated with increased cell survival [57,58].
Aggresome formation also promotes the degradation and clearance of aggregated proteins (Fig. (1), step 7). The colocalization of proteasomes with aggresomes at the centrosome [39,59] has led to the hypothesis that aggresomes might be centers for proteasomal degradation [60]. However, aggregated proteins are poor substrates for proteasomal degradation [61,62] and can actually impair UPS function [63,64], possibly by binding and blocking the axial pore or by prolonged occupation of the 20S proteasome compartment [62,65,66]. Although aggregated proteins are poorly degraded by the UPS, cultured cells are able to clear aggresomes if the production of misfolded proteins is blocked. Accumulating evidence suggests that aggresomes are substrates for autophagy [10-14]. Autophagy is a degradation pathway that mediates bulk clearance of cytosolic proteins and organelles by the lysosome in a highly regulated process involving the coordinated actions of a large number of autophagy-related (Atg) genes [67,68]. In response to particular stimuli, such as proteasomal dysfunction [13], an isolation membrane forms and expands to sequester portions of cytoplasm into double membrane structures called autophagosomes [67,68]. The autophagosomes eventually fuse with lysosomes and their contents are degraded by lysosomal hydrolases [67,68]. One hypothesis is that aggresomes may concentrate aggregated proteins for more efficient autophagic degradation [13,69,70]. In contrast to proteasomal degradation, which requires that proteins are first unfolded for entry into the 20S core particle, autophagy is able to degraded completely folded proteins and aggregated proteins [67,68]. In support of a role for autophagy in the clearance of aggresomes and potentially pathological inclusion bodies, autophagic machinery has been found to localize to aggresomes [12,13,71], inclusions formed in mouse models of polyglutamine disease [12], and Lewy bodies in PD [12]. Moreover, recent studies show that aggresome clearance can be facilitated by the induction of autophagy and blocked by the inhibition of autophagy [10,14,71,72]. Thus the formation of the aggresome does not appear to be an endpoint, but instead is an intermediate in a pathway destined for autophagic degradation [10,14,71-75]. There is also evidence that aggresomes may have an additional function in the induction of autophagy by sequestering mammalian target of rapamycin (mTOR), a phosphatidylinositol kinase-related kinase that acts as a key inhibitor of autophagy [72]. Thus, it appears that aggresomes play multiple roles that are important in the cellular defense against misfolded protein stress.
The Molecular Machinery of Aggresome Formation
Although our understanding of the molecular mechanisms underlying aggresome formation remains limited, recent studies have implicated several proteins in the recognition and transport of misfolded proteins to the aggresome, including the dynein motor complex, HDAC6, parkin, ataxin-3, and ubiquilin-1 (Table 1).
Table 1. Proteins Implicated in Aggresome Formation.
| Protein | Function | Wild-type protein localized to inclusion bodies | Mutations associated with disease | Ref. |
|---|---|---|---|---|
| Histone deacetylase 6 | Deacetylase, adaptor protein | Lewy bodies | Unknown | [46] |
| Parkin | E3 ubiquitin-protein ligase | Lewy bodies | Parkinson's disease | [34, 38] |
| Ataxin-3 | Deubiquitinating enzyme | SCA type-1 and 2 DRPLA intranuclear inclusions | SCA type-3 | [37] |
| Dynein motor complex | Retrograde microtubule motor | Unknown | Motor neuron degeneration | [87, 88] |
| Ubiquilin-1 | Protein turnover, intracellular trafficking | Lewy bodies and neurofibrillary tangles | Alzheimer's disease (potential risk factor) | [147, 154] |
SCA, spinocerebellar ataxia; DRPLA, dentatorubral-pallidoluysian atrophy; ALS, amyotrophic lateral sclerosis.
The Dynein Motor Complex
The cytoplasmic dynein motor complex is responsible for the retrograde transport of misfolded and aggregated proteins to the aggresome [7,41,76]. Dynein is a large protein complex that drives retrograde transport along microtubules [77,78]. The core of dynein is formed by two heavy chains, which each contain a protruding microtubule binding site, a large motor domain with 6 AAA motifs that act as the site of ATP-dependent force generation, and an N-terminal stalk that homo-dimerizes to produce a two-headed molecule [77,78]. In addition, associated with the heavy chains is a diverse array of light, light intermediate, and intermediate chains involved in cargo binding [77,78]. Dynactin is an accessory or activating complex that is also made up of several distinct protein subunits [77,78]. p150GLUED is a particularly important subunit of dynactin that binds directly to the intermediate chain of dynein and also contains a microtubule-binding site, providing an additional microtubule contact for the motor complex [79,80]. During retrograde transport, the dynein motor generates the force necessary for movement through the hydrolysis of ATP, while dynactin increases processivity and binds cargo [77,78].
Initial studies found that aggresomes form at the centrosome and that an intact microtubule cytoskeleton is necessary for the aggresome formation, suggesting the involvement of dynein-mediated retrograde transport [7]. Using time-lapse fluorescence microscopy, Garcia-Mata et al. found that small protein aggregates form in the periphery of the cell and are transported to the centrosome at rates comparable to those measured for dynein-mediated transport of membrane-bound organelles [41]. Furthermore, overexpression of the dynamitin (p50) subunit of the dynactin complex, which causes the dissociation of the dynactin complex and inhibits dynein-mediated transport [81,82], disrupts aggresome formation and results in the accumulation of peripherally distributed small protein aggregates [41,76]. Interestingly, impairment of dynein function not only leads to decreased aggresome formation, but also to an increase in the levels of aggregated proteins [83,84], which is in agreement with the role for aggresomes in autophagic degradation [10,11]. In addition, in support of a key role for the dynein motor complex in neurons, it has been found that disruption of dynein function by mutations in the dynein heavy chain [85] or by overexpression of the dynactin subunit dynamitin [86] are sufficient to cause progressive motor neurodegeneration in mice. Furthermore, a G59S mutation in the p150GLUED subunit of dynactin has been associated with distal spinal bulbar and muscular atrophy in humans [87,88]. The G59S mutation disrupts folding of the cytoskeleton-associated protein, glycine-rich (CAP-Gly) domain, reducing microtubule binding and EB1 binding and also causing aggregation of p150GLUED [87,89]. In addition, heterozygous mutations in the gene encoding p150GLUED have been found in patients with sporadic and familial ALS [90] and also in a family presenting with both ALS and frontotemporal dementia (FTD) [91], although it remains to be determined whether these mutations represent the primary causative factor or allelic variants. Despite ample evidence linking dynein to aggresome formation, the molecular factors regulating dynein function and mediating the specific coupling to misfolded cargo remain poorly understood and are important areas for investigation.
Histone Deacetylase 6 (HDAC6)
HDAC6 is a key protein involved in aggresome formation that may act as an adaptor protein linking polyubiquitinated proteins to the dynein motor complex for transport [46,92,93]. The histone deacetylase (HDAC) family consists of eighteen different proteins, most of which function in the removal of acetyl groups from acetylated lysine residues of histones and are involved in gene regulation. HDAC6 is a 1215 amino-acid, class IIb deacetylase that is unique among HDACs in that it localizes to the cytoplasm, mediates the deacetylation of non-histone proteins, including α-tubulin [94], HSP90 [95], and cortactin [96], and has been implicated in the regulation of microtubule dynamics [97], microtubule-based transport [46,98], and processing of misfolded proteins [46]. HDAC6 is comprised of two independently functional deacetylase domains [99,100], a dynein motor binding domain [46], and a polyubiquitin-binding motif referred to as a bound to ubiquitin zinc finger (BUZ domain) [101,102].
Converging lines of evidence have implicated HDAC6 as a key regulator of aggresome formation [46,103]. HDAC6 localizes to aggresomes formed in cell culture [13,40,46] and Lewy bodies in PD [46]. One mechanism by which HDAC6 modulates aggresome formation is by linking polyubiquitinated proteins, including the misfolded and aggregation-prone ΔF508 mutant cystic fibrosis transmembrane conducting regulator (CFTR), to the dynein motor complex by simultaneously binding polyubiquitinated proteins through the bound to BUZ domain and dynein through the dynein motor binding domain [46]. Consistent with a fundamental role in aggresome formation, siRNA-mediated depletion of HDAC6 profoundly attenuated the formation of aggresomes induced by either expression of ΔF508 CFTR [46] or by proteasomal impairment [46,104]. Interestingly, this phenotype was rescued by overexpression of the full length HDAC6 protein, but not truncated versions lacking either the BUZ domain or catalytic domains, indicating that the deacetylase activity of HDAC6 may also be important in the formation of aggresomes [46,92].
It is currently unclear precisely how HDAC6 deacetylase activity relates to aggresome formation. HDAC6 may regulate aggresome formation via the deacetylation of one of its identified substrates (α-tubulin, Hsp90, cortactin) or of an as yet unidentified substrate. Given the role of dynein-mediated transport in aggresome formation, the recent finding that inhibition of HDAC6 results in high levels of α-tubulin acetylation at lysine 40 and a consequent increase in motor protein binding and microtubule-dependent transport is particularly noteworthy [98,105]. However, an increase in microtubule-dependent transport would be expected to facilitate dynein-mediated aggresome formation, and studies have shown that inhibition or deletion of HDAC6 blocks aggresome formation [46,103]. Dompierre et al. suggest that the mechanism of HDAC6-mediated regulation of microtubule-dependent transport is distinct from its role in aggresome formation [98], and further studies will be necessary to determine if the acetylation state of tubulin is involved in aggresome formation. HDAC6 also deacetylates the chaperone Hsp90, and deletion of HDAC6 results in hyperacetylation of Hsp90, disruption of the interaction between Hsp90 and its cochaperone p23, and inactivation of Hsp90 chaperone activity [95,106,107]. Through its client proteins Hsp90 is involved in a wide variety of cellular process, including cell signaling and gene expression [108]. However, whether these signaling pathways or other Hsp90 client proteins influence aggresome formation remains to be determined.
There is also evidence suggesting that HDAC6 could modulate aggresome formation through its regulation of ubiquitin-dependent protein degradation. HDAC6 has been found to bind ubiquitin with a calculated equilibrium constant (KD) of 60 nM [101,102,109], which is much higher than the reported affinity of other ubiquitin binding proteins (KD between 2 μM and 500 μM [110]). This high affinity binding promotes polyubiquitin chain stability, inhibiting the proteasomal degradation of ubiquitinated substrates by preventing their recognition and facilitating their accumulation into aggresomes [102]. Furthermore, HDAC6 interacts with two proteins involved in handling ubiquitinated proteins, phospholipase A2 inactiving protein (PLAP) and valosin containing protein (VCP), the mammalian homologues of yeast ubiquitin fusion degradation protein 3 (UFD3) and cdc48 [102,109]. VCP, an AAA-ATPase chaperone that plays a vital role in ubiquitin-dependent endoplasmic reticulum-associated degradation (ERAD), enables dissociation of the HDAC6-polyubiquitin complex and promotes proteasomal degradation [102]. The interaction between HDAC6 and VCP appears to be a critical decision point in which polyubiquitinated proteins are either targeted for proteasomal degradation by VCP or for sequestration into the aggresome by HDAC6 [102].
The importance of HDAC6 in neurodegeneration and as a potential therapeutic target was highlighted in a recent study in which Pandey et al. showed that expression of HDAC6 in Drosophila protects against neurodegeneration associated with UPS dysfunction or expression of a spinobulbar muscular atrophy (SBMA)-associated mutant androgen receptor via an autophagy-dependent mechanism [111]. This study also found that a catalytically dead mutant of HDAC6 was no longer able to suppress the degenerative phenotype, providing further support for the importance of HDAC6 deacetylase activity in facilitating autophagic degradation of misfolded proteins [111]. Although the precise mechanism underlying HDAC6-mediated protection remains to be determined, the current findings are consistent with a critical role for HDAC6 regulating aggresome formation and autophagy.
Parkin
The E3 ligase parkin has recently been implicated in aggresome formation. Parkin is a 465 amino-acid RING-type E3 ligase that contains an amino-terminal ubiquitin like (Ubl) domain and two really interesting new gene (RING) finger domains [112,113]. Loss of function mutations in parkin are the most common cause of autosomal-recessive, juvenile onset PD [34,114,115]. Parkin has recently been found to mediate multiple forms of ubiquitination, including monoubiquitination [116-118] and K48- and K63-linked polyubiquitination [93,119-121]. Wild-type parkin localizes to Lewy bodies in sporadic PD patients [122-124] and to aggresomes formed in cultured cells induced by proteasomal impairment [44,45,125,126]. However, parkin-associated PD is devoid of Lewy bodies [122], raising the possibility that parkin-mediated ubiquitination may be directly involved in the formation of Lewy bodies and that the inability to form these protective inclusion bodies may underlie the rapid disease onset and progression observed in patients with mutations in parkin [113,127].
Parkin has been found to polyubiquitinate synphilin-1 [120,128], a protein known to form aggresomes when co-expressed with its binding partner α-synuclein [43,55]. Parkin polyubiquitination of synphilin-1 is primarily K63-linked, which promotes the redistribution of synphilin-1 into a detergent insoluble pool [121] and into ubiquitin-positive aggresomes [121,128]. However, the folding state of synphilin-1 and other parkin substrates is unknown. We have previously shown that the PD-linked L166P mutation in the protein DJ-1 disrupts its intrinsic folding, yielding a misfolded protein that is prone to aggregation [129]. In a recent study, we employed L166P mutant DJ-1 as a model misfolded protein to investigate the role of parkin in the cellular management of misfolded proteins [93]. We found that parkin selectively binds and mediates K63-linked polyubiquitination of misfolded mutant DJ-1 but not correctly folded wild-type DJ-1. K63-linked polyubiquitination of misfolded DJ-1 promotes the binding of misfolded DJ-1 to the dynein linker protein HDAC6, resulting in increased transport of misfolded DJ-1 to aggresomes and its consequent accumulation in a detergent insoluble pool [93]. Conversely, the expression of ubiquitin mutants unable to form K63-linked polyubiquitin chains resulted in decreased recruitment of the misfolded DJ-1 to aggresomes [93]. These findings link parkin to aggresome formation and further suggest that K63-linked polyubiquitination may act as an important signal in the regulation of dynein-mediated retrograde transport. Thus by mediating a form of ubiquitination that is not associated with proteasomal degradation, parkin may act to channel misfolded proteins away from impaired proteasomes and facilitate their transport to the aggresome for eventual autophagic degradation.
Ataxin-3
Ataxin-3 has been implicated in aggresome formation [130], although its precise role remains unclear. Alternative splicing yields two major ataxin-3 isoforms (360 or 373 amino-acids), each containing a Josephin domain, a coiled-coil domain, multiple ubiquitin interacting motifs (UIMs), and a stretch of polyglutamines that is expanded in spinocerebellar ataxia (SCA) type 3 (Machado-Joseph disease) [37]. Although there is little sequence similarity between the Josephin domain and the catalytic domain of known deubiquitinating enzymes, recent structural analyses indicate that the Josephin domain exhibits structural similarity to papain-like cysteine proteases and the arrangement of the Cys-His-Asn catalytic triad is conserved [131-133]. Ataxin-3 displays Cys14-dependent deubiquitinating activity in vitro and may act as a polyubiquitin-editing enzyme [130-132,134,135]. The ataxin-3 UIMs have been found to specifically bind polyubiquitin chains, but not mono- or diubiquitin, and may recruit polyubiquitinated proteins and facilitate their presentation to the Josephin domain [132,134,136,137].
Accumulating evidence suggests that ataxin-3 plays several roles in protein quality control, including regulation of endoplasmic reticulum-associated degradation [135,138-140] and proteasomal degradation [138,141]. Wild-type ataxin-3 protein is found in aggresomes formed by expression of CFTRΔ508 [130] and to inclusion bodies in dentatorubral-pallidoluysian atrophy (DRPLA) and SCA types 1 and 2 [142]. Burnett et al. reported that knockdown of ataxin-3 reduces CFTRΔ508 aggresome formation [130]. The ability of ataxin-3 to promote aggresome formation requires a functional deubiquitinating domain and the presence of its UIMs [130], suggesting that ataxin-3 may contribute to aggresome formation via multiple mechanisms. Since deubiquitinating enzymes can inhibit the proteasomal degradation of substrate proteins by removal of the ubiquitin tag [143,144], one possibility is that under certain conditions ataxin-3 reduces the polyubiquitination of misfolded proteins, channeling them away from an impaired proteasome. In support of this hypothesis, ataxin-3 is able to trim K48-linked polyubiquitin chains [130,134] and to sequester polyubiquitinated proteins from proteasomal degradation via its UIMs [130]. Burnett et al. also found that ataxin-3 interacts with HDAC6 and dynein motor [130], raising the possibility that ataxin-3 could serve as an adaptor linking polyubiquitinated proteins to the dynein motor for retrograde transport to the aggresomes. Further studies will be important to further our understanding of the mechanisms by which ataxin-3 regulates aggresome formation.
Ubiquilin-1/PLIC-1
Ubiquilin-1 (also known as PLIC-1) is another protein involved in aggresome formation, although its mechanism of action remains unknown. Ubiquilin-1 is a 595 amino-acid protein that contains an amino-terminal ubiquitin-like (Ubl) domain and a carboxyl-terminal ubiquitin-associated (UBA) domain. Ubiquilin-1 has also been implicated in the regulation of protein turnover [145-147], intracellular trafficking [148-150], and receptor surface expression [151-153]. Two recent studies have found that polymorphisms in ubiquilin-1 are associated with the development of AD [154,155], although this finding has been controversial [156-159] and requires further investigation. Functional studies have also provided support for a role for ubiquilin-1 in AD, and demonstrate that ubiquilin-1 modulates the turnover of presenilin-1 and 2 [146,147,160] and regulates the trafficking of amyloid-β precursor protein [148]. Furthermore, ubiquilin-1 immunoreactivity has been observed in neurofibrillary tangles in AD [147], Lewy bodies in PD [147], and aggresomes formed in cell culture [149,150] (Table 1).
Although the precise mechanisms are unknown, recent studies found that Ubiquilin-1 mediates the recruitment of epidermal growth factor receptor pathway substrate 15 (Eps15) to aggresomes [149,150]. Transport of Eps15 was dependent upon the interaction between the Ubl domain of ubiquilin-1 and the UIM domain of Eps15, and deletion of either domain abolished Eps15 transport to aggresomes [149,150]. Interestingly, ubiquilin-1 also interacts with many other UIM domain-containing proteins via its Ubl domain, including hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) [149], Hrs binding protein (Hbp) [149], ataxin-3 [150], and human neuron-specific DnaJ-like protein 1a (HSJ1a) [150], and several UIM domain-containing proteins have been observed in aggresomes and inclusion bodies in disease [137,149,150]. It will be important to determine whether ubiquilin-1 also mediates the recruitment of these proteins to the aggresome. One possibility is that ubiquilin-1 is involved in the transport of UIM-containing proteins or polyubiquitinated proteins via interactions with its Ubl and UIM domains, respectively. It has also been found that small interfering RNA-mediated depletion of ubiquilin-1 reduces the formation of aggresomes by an AU1-tagged polyglutamine fragment [150]. This protein fragment does not contain a UIM domain or any lysine residues for ubiquitination, suggesting that in addition to its recruitment of polyubiquitinated or UIM-containing proteins, ubiquilin-1 may play additional roles in the formation of the aggresome. In order to understand the role of ubiquilin-1 in aggresome formation, it will be important identify ubiquilin-1 binding partners and to determine the molecular mechanism coupling ubiquilin-1 to retrograde transport.
Aggresome Formation as a Potential Thereapeutic Target for Neurodegenerative Diseases
Recently, investigators have invested considerable resources to develop automated high-throughput screening platforms for the identification of compounds that inhibit aggresome formation and also compounds that promote aggresome formation with the hopes that these compounds may be used to better understand the mechanisms of inclusion formation and could potentially be used to develop therapeutics for disease [56,161-163]. These high throughput screens have mostly employed large universal screening libraries, which are useful when performing a screen in which the target is unknown or no structural information is available [56,161-163]. These screens play an essential role in generating new leads in drug discovery and chemical biology [161]. However, because protein aggregation and aggresome formation are complex multi-step processes [1,15], care must be taken in the interpretation of the results, especially when using the presence of aggresomes as a reporter. For example, as depicted in Fig. (3), inhibiting early steps in the aggresome-autophagy pathway would lead to reduced aggresome formation and lower levels of toxic protein species. In contrast, inhibiting later steps in this pathway would also lead to reduced aggresome formation, but would increase the accumulation of soluble-toxic protein species. This complication underscores the importance of target validation and characterization of the site(s) and mechanism(s) of action of the identified compounds.
Fig. 3. Potential steps in the aggresome-autophagy pathway for therapeutic intervention.
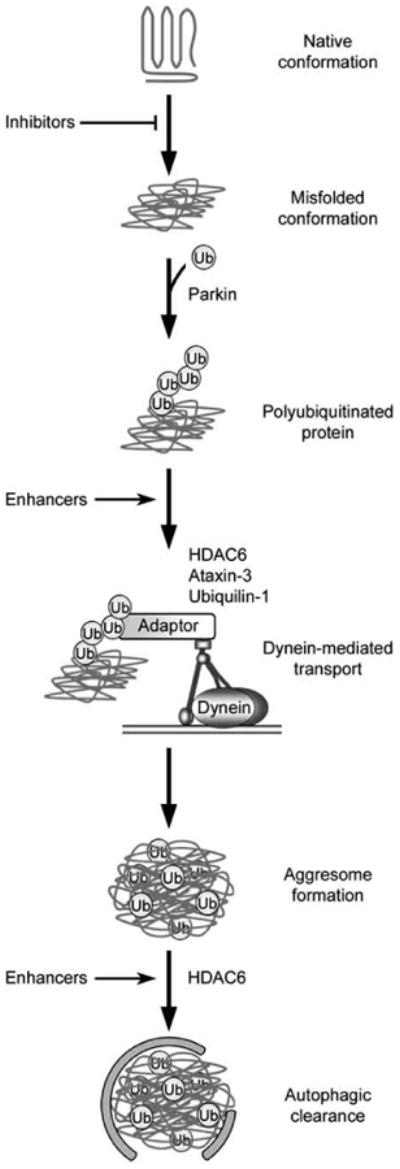
In this hypothetical model, an initiating event results in the generation of misfolded proteins, which are recognized and polyubiquitinated by an E3 ubiquitin-protein ligase such as parkin. Adaptor proteins, which may include HDAC6, ataxin-3, and ubiquilin-1, link the polyubiquitinated proteins to the dynein motor complex for retrograde transport to the aggresome. Autophagic machinery is recruited to the aggresome in a process involving HDAC6, and aggresomes are degraded. Multiple steps of this pathway could be targeted for treatment of neurodegenerative disease, including the use of small molecules to inhibit protein misfolding, enhance the coupling of misfolded proteins to dynein for retrograde transport, or enhance autophagic clearance of aggresomes. Furthermore, additional intervention strategies could potentially target any step along this pathway.
Small Molecule Inhibitors of Aggresome Formation
In the hopes of identifying small molecules that would facilitate the study of the role of inclusion bodies in disease, Corcoran et al. recently performed a screen of 20,000 compounds and identified several small molecules that impair aggresome formation (Fig. (4)) [162,163]. In this study three libraries of compounds were screened, including the ChemBridge Diverset E, the National Cancer Institute Structural Diversity Set, Version 1, and the National Institute of Neurological Disorders and Strokes Custom collection [162]. Corcoran et al. employed a high throughput screening protocol in which COS1 cells expressing a green fluorescent protein (GFP)-tagged ALS-linked G85R mutant superoxide dismutase 1 (SOD1) were incubated for 4 hours with compounds from the libraries, treated for 16 hours with the proteasomal inhibitor lactacystin to induce aggresome formation, and fluorescence microscopy images automatically captured [162]. Aggresome formation was manually scored from these images [162]. While this is a robust screening platform for the identification of compounds that affect aggresome formation, manually scoring the effect of 20,000 small molecules is a daunting task and is prone to human error. Combining this screening platform with automated image analysis software would provide more consistent data, such as the number of cells containing aggresomes and the size of the aggresome. The primary drawback in this approach is that the target of the small molecule eventually must be identified, and this can be challenging in some cases [161].
Fig. 4.

Chemical structure of aggresome inhibitors and their analogues.
The screen has led to the identification of several molecules that inhibit aggresome formation by the GFP-tagged G85R mutant SOD1 (Fig. (4)) [162], including the cardiac glycoside 54K09 (1) and a flavin-like compound 5-(3-Dimethyl amino-propylamino)-3,10-dimethyl-10H-pyrimidol[4, 5-b]quinoline-2,4-dione (DPD) (2). The mechanism by which these compounds inhibit aggresome formation is unknown. However, it is interesting to note that cardiac glycosides were previously shown to reduce polyglutamine-dependent activation of caspase-3, in manner that may be independent of their Na+K+-ATPase inhibitor activity [164]. Further study will be necessary to determine the inhibitory role of 54K09 and DPD in aggresome formation. Another potent inhibitor of aggresome formation identified from the screen is Scriptaid (3), a broad spectrum-HDAC inhibitor that displays significant structural similarity to previously described hydroxamic-containing HDAC inhibitors [165,166]. It is likely that Scriptaid binds to HDACs in a manner similar to the hydroxamic-containing HDAC inhibitor trichostatin A (TSA). Crystal structure analysis of TSA bound to a bacterial HDAC1 homologue revealed that the 5 carbon aliphatic chain of TSA inserts into a long tube-like groove on the surface of the HDAC, with the hydroxamic acid group coordinating a zinc atom within a pocket at the end of the grove and the bulky aromatic end group making contacts at the entrance to the groove [167]. The length of the aliphatic chain is important for spanning the length of the groove and enables contacts at the entrance to the grove and within the pocket [167]. Consistent with this mechanism of action, a chemical analogue of Scriptaid, termed Nullscript (4), which contains a shorter 3 carbon aliphatic chain, lacks HDAC inhibitory activity [166]. In addition, Scriptaid reduced aggresome formation induced by expression of G85R mutant SOD1 in concert with proteasomal impairment or by expression of a misfolded thiopurine S-methyltransferase (TPMT) polymorphic variant [40,162], suggesting that deacetylation is a common and important event underlying aggresome formation. Although Scriptaid blocked the formation of mutant SOD1 and TPMT-containing aggresomes, it did not affect their aggregation, indicating that it specifically disrupts transport of aggregated proteins. The known involvement of HDAC6 in the transport of aggregated proteins to the aggresome suggests that Scriptaid could be acting by inhibiting HDAC6 activity. However, Scriptaid is a broad spectrum-HDAC inhibitor and it is difficult to determine if its actions are due to inhibition of HDAC6 or due to inhibition of other histone deacetylases.
In a recent screen of a 7,392 deacetylase-biased 1,3-dioxane library, Haggarty et al. identified a selective small-molecule inhibitor of HDAC6, called tubacin (5) (Fig. (4)) [168], and it has been found that tubacin impairs aggresome formation and recruitment of autophagic machinery to the aggresome [13,103]. Similar to Scriptaid, tubacin also contains a hydroxamic acid head group that is expected to coordinate the HDAC zinc ion. A chemical analogue of tubacin, termed niltubacin (6), which lacks the hydroxamic acid head group, no longer displays deacetylase inhibitory activity [168]. However, the precise mechanism underlying tubacin HDAC6 selectivity remains unclear. Importantly, tubacin does not affect histone acetylation, gene expression, or cell-cycle progression [168]. Furthermore, it was also found that by preventing the sequestration of misfolded and aggregated proteins into the aggresome, tubacin increased the cytotoxic effects of bortezomib [103], a proteasome inhibitor used to treat multiple myeloma [103,104,169,170]. Interestingly, in addition to inhibiting the deacetylase activity of HDAC6, both Scriptaid and tubacin also disrupt the association between aggregated proteins and the dynein motor complex, possibly by interfering with the interaction between HDAC6 and dynein [103,162]. Therefore these studies are unable to distinguish between importance of HDAC6 as an adaptor and HDAC6 deacetylase activity, and further investigation will be necessary to resolve precisely how HDAC6-mediated deacetylation is involved in regulating aggresome formation. Although the identification of selective HDAC6 inhibitors are important research tools and may potentially be useful in supplementing cancer treatments [103,104,169,170], the identification of molecules that promote HDAC6 activity and the sequestration of misfolded proteins into aggresomes may provide important treatments for neurodegenerative disease.
Small Molecule Enhancers of Aggresome Formation
Accumulating evidence indicates that aggresome formation is a protective cellular response [11,43,46,103] and therefore Bodner et al. screened 37,000 compounds for small molecules that enhance the formation of aggresomes by a mutant huntingtin fragment [56,171]. The libraries of compounds used in the screen include ChemBridge Diverse and CNS sets, an unspecified Maybridge library, and a TimTec Natural products library [171]. Bodner et al. employed the 14A2.6 cell line, which expresses a GFP-tagged huntingtin fragment with 97 glutamine residues under the control of an ecdysone inducible promoter [171]. Cells were incubated for 72 hours with compounds from the libraries, lysed, and GFP fluorescence levels measured using a plate reader [171]. It should be noted that this study uses a huntingtin fragment, which although it is well characterized and provides a useful model system for studying aggresome formation, may not be representative of the aggregation properties of full-length huntingtin protein [171]. In addition, the authors used increases in total fluorescence levels as the reporter, a phenotypic output that is not specific to aggresome (could be caused by alterations in expression or degradation), and only the positive hits were analyzed by microscopy [171].
This screen has identified several molecules that increased the size and number of aggresomes, of which the compounds B2 (7) and B5 (8) were the most effective (Fig. (5)). Structure-activity relationship analyses of B2 found that the related compound B21 (9), which lacks the chloride at position 4 on the benzene ring and contains an additional fluorine group at position 2 on the benzene ring, displayed greatly diminished ability to promote aggresome formation [171]. Moreover, compound B22 (10), which switches the position of the nitrogen in the nitroquinoline group, completely abolished this activity [171]. B2 (7) was also found to promote the formation of α-synuclein containing aggresomes [171], suggesting that B2 (7) may act on a common cellular target involved in aggresome formation. Furthermore, enhancing aggresome formation by incubation with B2 or B5 increased cell survival and reduced proteasomal impairment as measured by the accumulation of the fluorescent degron GFPu [171]. The mechanism by which B2 and B5 promotes aggresome formation is unknown, although it does not appear to be due to global changes in transcription or alterations in the levels or activity of chaperones [171]. It will be important for future studies to identify the cellular target(s) of B2 and B5. This study provides further support for a protective role for aggresomes and suggests that small molecules that promote the sequestration of misfolded proteins into aggresomes may be beneficial in the treatment of neurodegenerative disease.
Fig. 5.

Chemical structure of aggresome enhancers and their analogues.
Mechanism-Based Therapeutic Strategies
The recent advances in our understanding of the molecular mechanisms and cellular machinery involved in aggresome formation have implicated the dynein motor, HDAC6, ataxin-3, parkin, and ubiquilin-1 as key proteins in the aggresome-autophagy pathway and have identified several stages in this pathway that could be targeted for the development of mechanism-based therapeutics (Fig. (3)). The enzymatic activities of dynein, HDAC6, ataxin-3, and parkin play important roles in aggresome formation, and the discovery of small molecules that enhance their activity would be expected to promote aggresome formation, but could represent a considerable challenge. Still, multiple steps in this pathway show potential for the treatment of neurodegenerative disease.
Several of the proteins implicated in aggresome formation are involved in coupling misfolded aggregated proteins to dynein for transport to the aggresome. This transport step is clearly a significant initial step that could potentially be targeted with small molecules. Recent advances have demonstrated the feasibility of modulating protein-protein interactions with small molecules, and one possibility would be the synthesis of small molecule adaptors that directly link aggregated proteins to dynein. Bifunctional small molecules (Fig. (6)) such as rapamycin (11) [172], cyclosporin (12) [173,174], and FK506 (13) [173,174] have been shown to function by promote the formation of protein complexes. Furthermore, using a similar strategy, Gestwicki et al. recently generated the bifunctional small molecule SLF-CR (14) (synthetic ligand for FK506-binding protein – Congo Red), which inhibits amyloid-β aggregation by increasing the steric bulk of Congo Red via simultaneous recruitment of a prevalent chaperone protein [175] (Fig. (6)). An analogous approach could be envisioned to enhance the transport of aggregated proteins, and would require a bifunctional small molecule containing a dynein or dynactin binding element, a linker element, and a recruiting element. In principle, this recruiting element could target a common structure presented by misfolded proteins, or like the known adaptor proteins, target polyubiquitin chains. Ubistatin A (15) and ubistatin B (16) are symmetrical linear molecules that selectively bind the ubiquitin-ubiquitin interface of K48-linked polyubiquitin chains [176] (Fig. (7)), and similar small molecules that bind K48-linked or K63-linked polyubiquitin chains could potentially serve as useful recruiting elements. An alternative possibility would be the use of small molecules that stabilize known adaptor proteins, such as HDAC6. One approach successfully used to stabilize the protein p53 for cancer treatments has been the generation of small molecules that disrupt the interaction of p53 with its cognate E3 enzyme human double minute-2 (HDM2), thereby impairing its proteasomal degradation [177-179]. However, in this case, the availability of high resolution structural information and a detailed understanding of the mechanism of p53 degradation greatly facilitated the structure-based design and identification of inhibitory compounds [180,181]. Structural analyses and characterization of the mechanisms underlying the turnover of proteins involved in aggresome formation will be important to the identification of compounds that selectively regulate their stability.
Fig. 6.

Chemical structure of bifunctional molecules that promote protein complex formation.
Fig. 7.

Chemical structure of ubistatins, a class of small molecules that selectively bind K48-linked polyubiquitin chains.
Targeting the clearance of aggresomes using small molecules that promote autophagy is also an important step that displays potential for therapeutic intervention (Reviewed in [68]). Rapamycin, a compound that induces autophagy by inhibiting mTOR, reduces the levels of aggregated proteins and is protective in both cell and animal models of neurodegenerative disease [72]. However, mTOR is involved in multiple cellular processes, and in addition to inducing autophagy, inhibition of mTOR also results in immunosuppression and cell-cycle inhibition [68]. The generation of more specific inducers of autophagy could be more clinically viable. To this end, a recent study identified several compounds that induce autophagy in a mTOR-independent manner, and enhance the autophagic clearance of aggregated proteins in cell-based models of HD and PD [182] and a Drosophila model of HD, suggesting therapeutic potential [182]. It will be important to characterize the mechanism of action for these compounds and to determine if these compounds are able to facilitate the clearance of already formed inclusion bodies. Another potential means for inducing autophagy was identified in a recent genetic screen, which found that activation of insulin receptor substrate-2 (IRS2) increases the autophagic clearance of mutant huntingtin aggresomes, independent of mTOR activation [71]. Insulin receptor activation also activates a neuroprotective signaling cascade mediated by phosphoinositide 3-kinase (PI3K) and Akt [183,184], suggesting that small molecules that modulate insulin receptor signaling pathways could have pleiotropic effects beneficial in the treatment of neurodegenerative disease.
Conclusions and Perspectives
Approaches that promote chaperone mediated refolding or proteasomal degradation of misfolded proteins remain valid strategies for the treatment of neurodegenerative disease [21-23]. However, because soluble oligomers or aggregates may be resistant to refolding or proteasomal degradation, combinatorial therapeutic strategies designed to also promote sequestration and degradation of soluble oligomers or aggregates via the aggresome-autophagy pathway may be beneficial [68]. The aggresome-autophagy pathway has emerged as an important cellular defense system against the accumulation of misfolded proteins [8,9,68], and recently the proteins dynein, HDAC6, ataxin-3, parkin, and ubiquilin-1 have been implicated in aggresome formation. The identification of these proteins is a significant advance, but their role in aggresome formation remains unclear and is an important area of investigation. Moreover, our understanding of the precise molecular mechanisms involved in aggresome formation remains limited, and further studies are essential to elucidate the underlying cellular machinery and means of regulation. We suggest that the aggresome-autophagy pathway is a viable target for the treatment of neurodegenerative disease, and could potentially increase the sequestration of toxic protein species, autophagic clearance of degradation-resistant proteins, and overall cellular viability.
Acknowledgments
This work was supported by National Institutes of Health grants NS054597 (JAO), NS050650 (LSC), AG021489 (LL), and NS047199 (LL).
Abbreviations
- AD
Alzheimer's disease
- ALS
Amyotrophic lateral sclerosis
- Atg
Autophagy-related
- BUZ
Bound to ubiquitin zinc finger
- CAP-Gly
Cytoskeleton-associated protein, glycine-rich
- CFTR
Cystic fibrosis transmembrane conducting regulator
- DLB
Dementia with Lewy bodies
- DRPLA
Dentatorubral-pallidoluysian atrophy
- DUB
Deubiquitinating enzymes
- Eps15
Epidermal growth factor receptor pathway substrate 15
- ERAD
Endoplasmic reticulum-associated degradation
- FTD
Frontotemporal dementia
- GFP
Green fluorescent protein
- Hbp
Hrs binding protein
- HD
Huntington's disease
- HDAC6
Histone deacetylase 6
- HDM2
Human double minute-2
- Hrs
Hepatocyte growth factor-regulated tyrosine kinase substrate
- HSJ1a
Human neuron-specific DnaJ-like protein 1a
- HSP
Heat shock protein
- IRS2
Insulin receptor substrate-2
- mTOR
Mammalian target of rapamycin
- PD
Parkinson's disease
- PI3K
Phosphoinositide 3-kinase
- PLAP
Phospholipase A2 inactiving protein
- PLIC-1
Protein linking IAP to the cytoskeleton
- RING
Really interesting new gene
- SCA
Spinocerebellar ataxias
- SIMPLE
Small integral membrane protein of the lysosome/late endosome
- SLF-CR
Synthetic ligand for FK506-binding protein – Congo Red
- SOD1
Superoxide dismutase 1
- TPMT
Thiopurine S-methyltransferase
- TSA
Trichostatin A
- UBA
Ubiquitin-associated
- Ubl
Ubiquitin-like
- UCH-L1
Ubiquitin carboxyl-terminal hydrolase L1
- UIM
Ubiquitin interacting motif
- UPS
Ubiquitin-proteasome system
- VCP
Valosin containing protein
References
- 1.Ross CA, Poirier MA. Nat Med. 2004;10:S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 4.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 5.Fandrich M, Fletcher MA, Dobson CM. Nature. 2001;410:165–166. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- 6.Glabe CG. Trends Biochem Sci. 2004;29:542–547. doi: 10.1016/j.tibs.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 7.Johnston JA, Ward CL, Kopito RR. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kopito RR. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Mata R, Gao YS, Sztul E. Traffic. 2002;3:388–396. doi: 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- 10.Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. J Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Hum Mol Genet. 2003;12:749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- 12.Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR. Proc Natl Acad Sci USA. 2005;102:13135–13140. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwata A, Riley BE, Johnston JA, Kopito RR. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 14.Ravikumar B, Duden R, Rubinsztein DC. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 15.Ross CA, Poirier MA. Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 16.Dobson CM. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 17.Hartl FU, Hayer-Hartl M. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 18.Schroder M, Kaufman RJ. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 19.Goldberg AL. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 20.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 21.Muchowski PJ, Wacker JL. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 22.Ciechanover A. Biochem Soc Trans. 2003;31:474–481. doi: 10.1042/bst0310474. [DOI] [PubMed] [Google Scholar]
- 23.Nalepa G, Rolfe M, Harper JW. Nat Rev Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 24.Frydman J. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 25.Sherman MY, Goldberg AL. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 26.Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, Michalik A, De Vriendt E, Jacobs A, Van Gerwen V, Vennekens K, Mazanec R, Tournev I, Hilton-Jones D, Talbot K, Kremensky I, Van Den Bosch L, Robberecht W, Van Vandekerckhove J, Van Broeckhoven C, Gettemans J, De Jonghe P, Timmerman V. Nat Genet. 2004;36:597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- 27.Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, Schagina O, Verpoorten N, Van Impe K, Fedotov V, Dadali E, Auer-Grumbach M, Windpassinger C, Wagner K, Mitrovic Z, Hilton-Jones D, Talbot K, Martin JJ, Vasserman N, Tverskaya S, Polyakov A, Liem RK, Gettemans J, Robberecht W, De Jonghe P, Timmerman V. Nat Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- 28.Hansen JJ, Durr A, Cournu-Rebeix I, Georgopoulos C, Ang D, Nielsen MN, Davoine CS, Brice A, Fontaine B, Gregersen N, Bross P. Am J Hum Genet. 2002;70:1328–1332. doi: 10.1086/339935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pickart CM. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 30.Jin J, Li X, Gygi SP, Harper JW. Nature. 2007;447:1135–1138. doi: 10.1038/nature05902. [DOI] [PubMed] [Google Scholar]
- 31.Pelzer C, Kassner I, Matentzoglu K, Singh RK, Wollscheid HP, Scheffner M, Schmidtke G, Groettrup M. J Biol Chem. 2007;282:23010–23014. doi: 10.1074/jbc.C700111200. [DOI] [PubMed] [Google Scholar]
- 32.Pickart CM, Fushman D. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 33.Wolf DH, Hilt W. Biochim Biophys Acta. 2004;1695:19–31. doi: 10.1016/j.bbamcr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 35.Street VA, Bennett CL, Goldy JD, Shirk AJ, Kleopa KA, Tempel BL, Lipe HP, Scherer SS, Bird TD, Chance PF. Neurology. 2003;60:22–26. doi: 10.1212/wnl.60.1.22. [DOI] [PubMed] [Google Scholar]
- 36.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 37.Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, Kimura J, Narumiya S, Kakizuka A. Nat Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 38.Marx FP, Soehn AS, Berg D, Melle C, Schiesling C, Lang M, Kautzmann S, Strauss KM, Franck T, Engelender S, Pahnke J, Dawson S, von Eggeling F, Schulz JB, Riess O, Kruger R. FASEB J. 2007;8:1750–1767. doi: 10.1096/fj.06-6734com. [DOI] [PubMed] [Google Scholar]
- 39.Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ. J Cell Biol. 1999;145:481–490. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Nguyen TV, McLaughlin RW, Sikkink LA, Ramirez-Alvarado M, Weinshilboum RM. Proc Natl Acad Sci USA. 2005;102:9394–9399. doi: 10.1073/pnas.0502352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. J Cell Biol. 1999;146:1239–1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM. J Biol Chem. 2004;279:4625–4631. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- 44.Muqit MM, Davidson SM, Payne Smith MD, MacCormac LP, Kahns S, Jensen PH, Wood NW, Latchman DS. Hum Mol Genet. 2004;13:117–135. doi: 10.1093/hmg/ddh012. [DOI] [PubMed] [Google Scholar]
- 45.Ardley HC, Scott GB, Rose SA, Tan NG, Markham AF, Robinson PA. Mol Biol Cell. 2003;14:4541–4556. doi: 10.1091/mbc.E03-02-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 47.Mayer RJ, Lowe J, Lennox G, Landon M, MacLennan K, Doherty FJ. Biochem Soc Symp. 1989;55:193–201. [PubMed] [Google Scholar]
- 48.McNaught KS, Shashidharan P, Perl DP, Jenner P, Olanow CW. Eur J Neurosci. 2002;16:2136–2148. doi: 10.1046/j.1460-9568.2002.02301.x. [DOI] [PubMed] [Google Scholar]
- 49.Olanow CW, Perl DP, DeMartino GN, McNaught KS. Lancet Neurol. 2004;3:496–503. doi: 10.1016/S1474-4422(04)00827-0. [DOI] [PubMed] [Google Scholar]
- 50.Conway KA, Lee SJ, Rochet JC, Ding TT, Harper JD, Williamson RE, Lansbury PT., Jr Ann N Y Acad Sci. 2000;920:42–45. doi: 10.1111/j.1749-6632.2000.tb06903.x. [DOI] [PubMed] [Google Scholar]
- 51.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe D. J Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 52.Goldberg MS, Lansbury PT., Jr Nat Cell Biol. 2000;2:E115–119. doi: 10.1038/35017124. [DOI] [PubMed] [Google Scholar]
- 53.Caughey B, Lansbury PT. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 54.Kramer ML, Schulz-Schaeffer WJ. J Neurosci. 2007;27:1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, Dawson VL, Dawson TM, Ross CA. Nat Genet. 1999;22:110–114. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- 56.Bodner RA, Housman DE, Kazantsev AG. Cell Cycle. 2006;5:1477–1480. doi: 10.4161/cc.5.14.2929. [DOI] [PubMed] [Google Scholar]
- 57.Arrasate M, Finkbeiner S. Proc Natl Acad Sci USA. 2005;102:3840–3845. doi: 10.1073/pnas.0409777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 59.Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. J Biol Chem. 2000;275:409–413. doi: 10.1074/jbc.275.1.409. [DOI] [PubMed] [Google Scholar]
- 60.Corboy MJ, Thomas PJ, Wigley WC. Methods Mol Biol. 2005;301:305–327. doi: 10.1385/1-59259-895-1:305. [DOI] [PubMed] [Google Scholar]
- 61.Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Mol Cell. 2004;14:95–104. doi: 10.1016/s1097-2765(04)00151-0. [DOI] [PubMed] [Google Scholar]
- 62.Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI. EMBO J. 2004;23:4307–4318. doi: 10.1038/sj.emboj.7600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bence NF, Sampat RM, Kopito RR. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 64.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Mol Cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 65.Verhoef LG, Lindsten K, Masucci MG, Dantuma NP. Hum Mol Genet. 2002;11:2689–2700. doi: 10.1093/hmg/11.22.2689. [DOI] [PubMed] [Google Scholar]
- 66.Diaz-Hernandez M, Valera AG, Moran MA, Gomez-Ramos P, Alvarez-Castelao B, Castano JG, Hernandez F, Lucas JJ. J Neurochem. 2006;98:1585–1596. doi: 10.1111/j.1471-4159.2006.03968.x. [DOI] [PubMed] [Google Scholar]
- 67.Reggiori F, Klionsky DJ. Eukaryot Cell. 2002;1:11–21. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 69.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bjorkoy G, Lamark T, Johansen T. Autophagy. 2006;2:138–139. doi: 10.4161/auto.2.2.2405. [DOI] [PubMed] [Google Scholar]
- 71.Yamamoto A, Cremona ML, Rothman JE. J Cell Biol. 2006;172:719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 73.Klionsky DJ. Nature. 2006;441:819–820. doi: 10.1038/441819a. [DOI] [PubMed] [Google Scholar]
- 74.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 75.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 76.Johnston JA, Illing ME, Kopito RR. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- 77.Vale RD. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 78.Vallee RB, Hook P. J Struct Biol. 2006;156:175–181. doi: 10.1016/j.jsb.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 79.Vaughan KT, Vallee RB. J Cell Biol. 1995;131:1507–1516. doi: 10.1083/jcb.131.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Waterman-Storer CM, Karki S, Holzbaur EL. Proc Natl Acad Sci USA. 1995;92:1634–1638. doi: 10.1073/pnas.92.5.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. J Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 83.Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, Brown SD, Rubinsztein DC. Nat Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 84.Rubinsztein DC, Ravikumar B, Acevedo-Arozena A, Imarisio S, O'Kane CJ, Brown SD. Autophagy. 2005;1:177–178. doi: 10.4161/auto.1.3.2050. [DOI] [PubMed] [Google Scholar]
- 85.Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y, Augustin M, Korthaus D, Wattler S, Wabnitz P, Dickneite C, Lampel S, Boehme F, Peraus G, Popp A, Rudelius M, Schlegel J, Fuchs H, Hrabe de Angelis M, Schiavo G, Shima DT, Russ AP, Stumm G, Martin JE, Fisher EM. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- 86.LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 87.Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 88.Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, Finnegan K, Holzbaur EL, Fischbeck KH, Ludlow CL. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, Wallace KE, LaMonte BH, Harmison GG, Puls I, Fischbeck KH, Holzbaur EL. J Cell Biol. 2006;172:733–745. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- 91.Munch C, Rosenbohm A, Sperfeld AD, Uttner I, Reske S, Krause BJ, Sedlmeier R, Meyer T, Hanemann CO, Stumm G, Ludolph AC. Ann Neurol. 2005;58:777–780. doi: 10.1002/ana.20631. [DOI] [PubMed] [Google Scholar]
- 92.Kopito RR. Mol Cell. 2003;12:1349–1351. doi: 10.1016/s1097-2765(03)00498-2. [DOI] [PubMed] [Google Scholar]
- 93.Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, Chin LS. J Cell Biol. 2007;178:1025–1038. doi: 10.1083/jcb.200611128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 95.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 96.Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS, Seto E. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, Horinouchi S, Yoshida M. EMBO J. 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grozinger CM, Hassig CA, Schreiber SL. Proc Natl Acad Sci USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang Y, Gilquin B, Khochbin S, Matthias P. J Biol Chem. 2006;281:2401–2404. doi: 10.1074/jbc.C500241200. [DOI] [PubMed] [Google Scholar]
- 101.Hook SS, Orian A, Cowley SM, Eisenman RN. Proc Natl Acad Sci USA. 2002;99:13425–13430. doi: 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, Matthias P, Muller CW, Khochbin S. EMBO J. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC. Proc Natl Acad Sci USA. 2005;102:8567–8572. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K, Jr, Pal A, Bornmann WG, Chiao PJ, Huang P, Xiong H, Abbruzzese JL, McConkey DJ. Cancer Res. 2006;66:3773–3781. doi: 10.1158/0008-5472.CAN-05-2961. [DOI] [PubMed] [Google Scholar]
- 105.Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Curr Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 106.Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. Mol Cell. 2007;25:151–159. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 108.Aoyagi S, Archer TK. Trends Cell Biol. 2005;15:565–567. doi: 10.1016/j.tcb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 109.Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S, Khochbin S. Mol Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hicke L, Schubert HL, Hill CP. Nat Rev Mol Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 111.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. Nature. 2007;447:860–864. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 112.Cookson MR. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 113.von Coelln R, Dawson VL, Dawson TM. Cell Tissue Res. 2004;318:175–184. doi: 10.1007/s00441-004-0924-4. [DOI] [PubMed] [Google Scholar]
- 114.Hattori N, Mizuno Y. Lancet. 2004;364:722–724. doi: 10.1016/S0140-6736(04)16901-8. [DOI] [PubMed] [Google Scholar]
- 115.Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 116.Hampe C, Ardila-Osorio H, Fournier M, Brice A, Corti O. Hum Mol Genet. 2006;15:2059–2075. doi: 10.1093/hmg/ddl131. [DOI] [PubMed] [Google Scholar]
- 117.Joch M, Ase AR, Chen CX, Macdonald PA, Kontogiannea M, Corera AT, Brice A, Seguela P, Fon EA. Mol Biol Cell. 2007;18:3105–3118. doi: 10.1091/mbc.E05-11-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Matsuda N, Kitami T, Suzuki T, Mizuno Y, Hattori N, Tanaka K. J Biol Chem. 2006;281:3204–3209. doi: 10.1074/jbc.M510393200. [DOI] [PubMed] [Google Scholar]
- 119.Doss-Pepe EW, Chen L, Madura K. J Biol Chem. 2005;280:16619–16624. doi: 10.1074/jbc.M413591200. [DOI] [PubMed] [Google Scholar]
- 120.Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. J Neurosci. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lim KL, Dawson VL, Dawson TM. Neurobiol Aging. 2006;27:524–529. doi: 10.1016/j.neurobiolaging.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 122.Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, Langston JW, Mizuno Y, Hyman BT, Selkoe DJ, Kosik KS. Am J Pathol. 2002;160:1655–1667. doi: 10.1016/S0002-9440(10)61113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Murakami T, Shoji M, Imai Y, Inoue H, Kawarabayashi T, Matsubara E, Harigaya Y, Sasaki A, Takahashi R, Abe K. Ann Neurol. 2004;55:439–442. doi: 10.1002/ana.20064. [DOI] [PubMed] [Google Scholar]
- 124.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- 125.Junn E, Lee SS, Suhr UT, Mouradian MM. J Biol Chem. 2002;277:47870–47877. doi: 10.1074/jbc.M203159200. [DOI] [PubMed] [Google Scholar]
- 126.Zhao J, Ren Y, Jiang Q, Feng J. J Cell Sci. 2003;116:4011–4019. doi: 10.1242/jcs.00700. [DOI] [PubMed] [Google Scholar]
- 127.Sulzer D. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 128.Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- 129.Olzmann JA, Brown K, Wilkinson KD, Rees HD, Huai Q, Ke H, Levey AI, Li L, Chin LS. J Biol Chem. 2004;279:8506–8515. doi: 10.1074/jbc.M311017200. [DOI] [PubMed] [Google Scholar]
- 130.Burnett BG, Pittman RN. Proc Natl Acad Sci USA. 2005;102:4330–4335. doi: 10.1073/pnas.0407252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 132.Mao Y, Senic-Matuglia F, Di Fiore PP, Polo S, Hodsdon ME, De Camilli P. Proc Natl Acad Sci USA. 2005;102:12700–12705. doi: 10.1073/pnas.0506344102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nicastro G, Menon RP, Masino L, Knowles PP, McDonald NQ, Pastore A. Proc Natl Acad Sci USA. 2005;102:10493–10498. doi: 10.1073/pnas.0501732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Burnett B, Li F, Pittman RN. Hum Mol Genet. 2003;12:3195–3205. doi: 10.1093/hmg/ddg344. [DOI] [PubMed] [Google Scholar]
- 135.Zhong X, Pittman RN. Hum Mol Genet. 2006;15:2409–2420. doi: 10.1093/hmg/ddl164. [DOI] [PubMed] [Google Scholar]
- 136.Chai Y, Berke SS, Cohen RE, Paulson HL. J Biol Chem. 2004;279:3605–3611. doi: 10.1074/jbc.M310939200. [DOI] [PubMed] [Google Scholar]
- 137.Donaldson KM, Li W, Ching KA, Batalov S, Tsai CC, Joazeiro CA. Proc Natl Acad Sci USA. 2003;100:8892–8897. doi: 10.1073/pnas.1530212100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Doss-Pepe EW, Stenroos ES, Johnson WG, Madura K. Mol Cell Biol. 2003;23:6469–6483. doi: 10.1128/MCB.23.18.6469-6483.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stirling CJ, Lord JM. Curr Biol. 2006;16:R1035–1037. doi: 10.1016/j.cub.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 140.Wang Q, Li L, Ye Y. J Cell Biol. 2006;174:963–971. doi: 10.1083/jcb.200605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang G, Sawai N, Kotliarova S, Kanazawa I, Nukina N. Hum Mol Genet. 2000;9:1795–1803. doi: 10.1093/hmg/9.12.1795. [DOI] [PubMed] [Google Scholar]
- 142.Uchihara T, Fujigasaki H, Koyano S, Nakamura A, Yagishita S, Iwabuchi K. Acta Neuropathol (Berl) 2001;102:149–152. doi: 10.1007/s004010100364. [DOI] [PubMed] [Google Scholar]
- 143.Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Nature. 2002;416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- 144.Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Dijke PT, Chantry A. Oncogene. 2005;24:8080–8084. doi: 10.1038/sj.onc.1208944. [DOI] [PubMed] [Google Scholar]
- 145.Ko HS, Uehara T, Tsuruma K, Nomura Y. FEBS Lett. 2004;566:110–114. doi: 10.1016/j.febslet.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 146.Massey LK, Mah AL, Monteiro MJ. Biochem J. 2005;391:513–525. doi: 10.1042/BJ20050491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mah AL, Perry G, Smith MA, Monteiro MJ. J Cell Biol. 2000;151:847–862. doi: 10.1083/jcb.151.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hiltunen M, Lu A, Thomas AV, Romano DM, Kim M, Jones PB, Xie Z, Kounnas MZ, Wagner SL, Berezovska O, Hyman BT, Tesco G, Bertram L, Tanzi RE. J Biol Chem. 2006;281:32240–32253. doi: 10.1074/jbc.M603106200. [DOI] [PubMed] [Google Scholar]
- 149.Regan-Klapisz E, Sorokina I, Voortman J, de Keizer P, Roovers RC, Verheesen P, Urbe S, Fallon L, Fon EA, Verkleij A, Benmerah A, van Bergen en Henegouwen PM. J Cell Sci. 2005;118:4437–4450. doi: 10.1242/jcs.02571. [DOI] [PubMed] [Google Scholar]
- 150.Heir R, Ablasou C, Dumontier E, Elliott M, Fagotto-Kaufmann C, Bedford FK. EMBO Rep. 2006;7:1252–1258. doi: 10.1038/sj.embor.7400823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bedford FK, Kittler JT, Muller E, Thomas P, Uren JM, Merlo D, Wisden W, Triller A, Smart TG, Moss SJ. Nat Neurosci. 2001;4:908–916. doi: 10.1038/nn0901-908. [DOI] [PubMed] [Google Scholar]
- 152.Chen ZW, Olsen RW. J Neurochem. 2007;100:279–294. doi: 10.1111/j.1471-4159.2006.04206.x. [DOI] [PubMed] [Google Scholar]
- 153.N'Diaye EN, Brown EJ. J Cell Biol. 2003;163:1157–1165. doi: 10.1083/jcb.200307155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, Mullin K, Menon R, Sampson AJ, Hsiao MY, Elliott KJ, Velicelebi G, Moscarillo T, Hyman BT, Wagner SL, Becker KD, Blacker D, Tanzi RE. N Engl J Med. 2005;352:884–894. doi: 10.1056/NEJMoa042765. [DOI] [PubMed] [Google Scholar]
- 155.Kamboh MI, Minster RL, Feingold E, DeKosky ST. Mol Psychiatry. 2006;11:273–279. doi: 10.1038/sj.mp.4001775. [DOI] [PubMed] [Google Scholar]
- 156.Bensemain F, Chapuis J, Tian J, Shi J, Thaker U, Lendon C, Iwatsubo T, Amouyel P, Mann D, Lambert JC. Neurobiol Dis. 2006;22:691–693. doi: 10.1016/j.nbd.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 157.Brouwers N, Sleegers K, Engelborghs S, Bogaerts V, van Duijn CM, De Deyn PP, Van Broeckhoven C, Dermaut B. Neurosci Lett. 2006;392:72–74. doi: 10.1016/j.neulet.2005.08.064. [DOI] [PubMed] [Google Scholar]
- 158.Slifer MA, Martin ER, Bronson PG, Browning-Large C, Doraiswamy PM, Welsh-Bohmer KA, Gilbert JR, Haines JL, Pericak-Vance MA. Am J Med Genet B Neuropsychiatr Genet. 2006;141:208–213. doi: 10.1002/ajmg.b.30298. [DOI] [PubMed] [Google Scholar]
- 159.Smemo S, Nowotny P, Hinrichs AL, Kauwe JS, Cherny S, Erickson K, Myers AJ, Kaleem M, Marlowe L, Gibson AM, Hollingworth P, O'Donovan MC, Morris CM, Holmans P, Lovestone S, Morris JC, Thal L, Li Y, Grupe A, Hardy J, Owen MJ, Williams J, Goate A. Ann Neurol. 2006;59:21–26. doi: 10.1002/ana.20673. [DOI] [PubMed] [Google Scholar]
- 160.Massey LK, Mah AL, Ford DL, Miller J, Liang J, Doong H, Monteiro MJ. J Alzheimers Dis. 2004;6:79–92. doi: 10.3233/jad-2004-6109. [DOI] [PubMed] [Google Scholar]
- 161.Eggert US, Mitchison TJ. Curr Opin Chem Biol. 2006;10:232–237. doi: 10.1016/j.cbpa.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 162.Corcoran LJ, Mitchison TJ, Liu Q. Curr Biol. 2004;14:488–492. doi: 10.1016/j.cub.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 163.Zhang X, Smith DL, Meriin AB, Engemann S, Russel DE, Roark M, Washington SL, Maxwell MM, Marsh JL, Thompson LM, Wanker EE, Young AB, Housman DE, Bates GP, Sherman MY, Kazantsev AG. Proc Natl Acad Sci USA. 2005;102:892–897. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Piccioni F, Roman BR, Fischbeck KH, Taylor JP. Hum Mol Genet. 2004;13:437–446. doi: 10.1093/hmg/ddh045. [DOI] [PubMed] [Google Scholar]
- 165.Monneret C. Eur J Med Chem. 2005;40:1–13. doi: 10.1016/j.ejmech.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 166.Su GH, Sohn TA, Ryu B, Kern SE. Cancer Res. 2000;60:3137–3142. [PubMed] [Google Scholar]
- 167.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 168.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Catley L, Weisberg E, Kiziltepe T, Tai YT, Hideshima T, Neri P, Tassone P, Atadja P, Chauhan D, Munshi NC, Anderson KC. Blood. 2006;108:3441–3449. doi: 10.1182/blood-2006-04-016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Suzuki T, Kouketsu A, Itoh Y, Hisakawa S, Maeda S, Yoshida M, Nakagawa H, Miyata N. J Med Chem. 2006;49:4809–4812. doi: 10.1021/jm060554y. [DOI] [PubMed] [Google Scholar]
- 171.Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, McLean PJ, Young AB, Housman DE, Kazantsev AG. Proc Natl Acad Sci USA. 2006;103:4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Choi J, Chen J, Schreiber SL, Clardy J. Science. 1996;273:239–242. doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- 173.Ke H, Huai Q. Biochem Biophys Res Commun. 2003;311:1095–1102. doi: 10.1016/s0006-291x(03)01537-7. [DOI] [PubMed] [Google Scholar]
- 174.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 175.Gestwicki JE, Crabtree GR, Graef IA. Science. 2004;306:865–869. doi: 10.1126/science.1101262. [DOI] [PubMed] [Google Scholar]
- 176.Verma R, Peters NR, D'Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW. Science. 2004;306:117–120. doi: 10.1126/science.1100946. [DOI] [PubMed] [Google Scholar]
- 177.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Nat Med. 2004;10:1321–1328. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 178.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 179.Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, Safiran YJ, Oberoi P, Kenten JH, Phillips AC, Weissman AM, Vousden KH. Cancer Cell. 2005;7:547–559. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 180.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 181.Vassilev LT. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 182.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O'Kane CJ, Schreiber SL, Rubinsztein DC. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Vincent AM, Feldman EL. Growth Horm I G F Res. 2002;12:193–197. doi: 10.1016/s1096-6374(02)00017-5. [DOI] [PubMed] [Google Scholar]
- 184.Russo VC, Gluckman PD, Feldman EL, Werther GA. Endocr Rev. 2005;26:916–943. doi: 10.1210/er.2004-0024. [DOI] [PubMed] [Google Scholar]