

Abstract
HIV-1Tat (trans-acting activator of transcription) plays essential roles in the replication through viral mRNA and genome transcription from the HIV-1 LTR promoter. However, Tat undergoes continuous amino acid substitutions. As a consequence, the virus escapes from host immunity indicating that genetic diversity of Tat protein in major HIV-1 subtypes is required to be continuously monitored. We analyzed available full-length HIV-1 sequences of subtypes B (n=493) and C (n=280) strains circulating worldwide. We observed 81% and 84% nucleotide sequence identities of HIV-1 Tat for subtypes B and C, respectively. Based on phylogenetic and mutation analyses, global diversity of subtype B was apparently higher compared to that of subtype C. Positively selected sites, such as positions Ser68 and Ser70 in both subtypes, were located in the Tat-transactivation responsive RNA (TAR) interaction domain. We also found positively selected sites in exon 2, such as positions Ser75, Pro77, Asp80, Pro81 and Ser87 for both subtypes. Our study provides useful information on the full-length HIV-1 Tat sequences in globally circulating strains.
Keywords: full-length HIV-1 Tat, Tat, molecular evolution, Tat genetic diversity, Tat genetics
Background
The human immune deficiency virus type 1(HIV-1) Tat (trans acting activator of transcription), is one of the essential proteins, which directly enhances HIV-1 replication through interaction with HIV-1 long terminal repeat (LTR) promoter [1]. Tat is therefore a promising target for developing HIV-1 vaccines and anti-HIV-1 drugs [2– 3]. Unlike viral essential enzymes, such as protease and reverse transcriptase, Tat undergoes continuous substitutions due to host selection pressure [4], leading to viral escape from Tat-specific CD8- positive T-lymphocyte responses [5– 6]. The sequence variation of target epitopes in Tat reduces antibody recognition and neutralization [1, 7]. Molecular characterization of full-length Tat in globally circulating strains is therefore imperative.
is a 101-amino acid protein encoded by two exons (exon 1: 1 to 72 residues, and the exon 2: 73 to 101 residues) in most of the clinical isolates [8]. As shown in Figure 1a, Tat has been categorized into six different functional domains [1, 8]. The first domain (residues 1 to 21) is the N-terminal acidic domain consisting of a Pro-rich tract and a conserved Trp residue at the position 20 (Trp20) [1]. The second domain (residues 22 to 37) contains a highly conserved seven Cys tract at positions 22, 25, 27, 30, 31, 34, and 37 [1, 8]. The third domain (residues 38 to 48) contains a hydrophobic core sequence: 43LeuGlyIleSerTry Gly48 [4]. The fourth domain (residues 49 to 57) is a positively charged region composed of a well-conserved arginine-rich motif, 49-ArgLysLysArgArgGlnArgArgArg-57, and acts as a transactivation response element (TAR) binding domain [9]. This domain has an extra ordinary property for nuclear localization [10, 11] and protein transduction, thus it has also been used to deliver various molecules inside the cells in vitro [12–15]. The fifth domain (residues 58 to 72) is a Gln rich region [4, 8], and the fourth and fifth domains (residues 49 to 72) are known as basic domains for transactivation [1, 4, 8]. The sixth domain (residues 73 to 101) encoded by the exon 2 is known as RDG domain; this domain contains the highly conserved Glu Ser Lys Lys Lys Val Glu motif, which is related to optimal HIV-1 replication in vivo [16], thereby, the region may contribute to viral infectivity and binding to cell-surface integrins [17, 18]. It is therefore worthwhile to investigate the genetic diversity of HIV-1 full-length Tat in commonly circulating subtypes, which has an impact on clinical outcome of HIV disease as well as success of Tat-based vaccination and Tat-targeted antagonists. However, updated information regarding the evolution of HIV-1 focusing Tat protein at the global level is still unavailable. We therefore performed phylogenetic, selection pressure, and mutation analyses to understand diversity of Tat and its phylogenetic relationships between the subtypes using global data sets of sequences in subtypes B and C, the two major subtypes of HIV-1 circulating worldwide [19].
Figure 1.
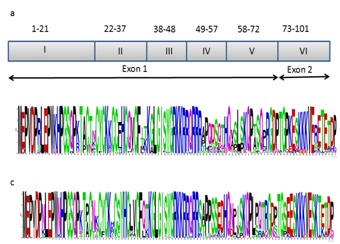
Functional domains of Tat and its genetic diversity. Schematic presentation of the domains of tat exon 1 and 2 were highlighted (1a): domain I (residues 1 to 21), an acidic/Pro-rich region; domain II (residues 22 to 37), a Cys-rich/Zn2 Finger domain; domain III (residues 38 to 48), containing conserved Phe (F); domain IV (residues 49–57, the basic domain); domain V (residues 58–72, a Glu rich domain); and domain VI (residues 3–101, encoded by the second exon). Sequence logo showing the Tat amino acid diversity observed at positions 1 to 100 in both subtype B (1b) and subtype C (1c)
Methodology
Sequence data:
Full-length HIV-1 Tat sequences were collected from the ‘Web alignment’ in the Los Alamos National Laboratory (LANL) HIV sequence database [20]. The sequences were downloaded on February 2, 2014. Notably, the sequences ‘up to 2012’ were available on the accessed date. A total of 2156 sequences were initially downloaded. Sequence data of the other subtypes than subtype B or C, and of circulating recombinant forms (CRFs) were then excluded. Consequently, totals of 713 and 353 sequences for subtype B and subtype C, respectively, were obtained. The sequence data for full-length coding regions were only used after eliminating the problematic sequences.
The reference strains for subtype B and C sequences were also obtained from the Los Alamos HIV-1 sequence data base. The reference sequences were also downloaded on February 2, 2014. As the reference sequences, accession numbers AY423387 (Europe), AY173951 (Asia), and AY331295 (North America) for subtype B and AF067155 (India), U52953 (South America), and AY772699 (Africa) for subtype C were used for alignment. Finally, we prepared a data set of totals of 493 and 280 sequences for subtype B and subtype C, respectively, which contained 100 amino acids encoded by 300 nucleotides (nt) from the positions 5831 to 6045 nt in exon 1 and 8379 to 8463 nt in exon 2, respectively of HXB2 genome (GenBank accession No. K03455), the details information of the sequences (the isolation year, isolated country etc), were mentioned in Table 1 (Available with authors). A multiple-sequence alignment of the nucleotide sequences (without any gap) was made using the ClustalW [21]. The divergence of sequences was schematically visualized using Weblogo [22].
Estimating Phylogenetic tree:
Maximum likelihood method was employed to built the phylogenetic trees. Notably, the method was selected as the best model by model test performed in MEGA 6 [23]. A discrete gamma distribution was used to measure the evolutionary rate differences among sites (5 categories) and the analyses were done using 1,000 bootstrap replicates. The tree was rooted by using following reference strain: simian immune deficiency virus (SIV) sequence, CPZ.US.85.US_ Marilyn.AF103.
Selection pressure analysis:
Global (ω) value of relative rates of non-synonymous (dN) and synonymous (dS) substitutions were calculated to measure the positive selection strength [24]. All analyses were carried out using the online Datamonkey facility [24– 26] after identifying the best fit model from every possible time-reversible model. Positive selection pressure analysis was performed at whole gene and site-by-site codon level using three likelihood methods: single-likelihood ancestor counting (SLAC), fixed effects likelihood (FEL), and interior branches Fixed Likelihood (iFEL). Briefly, in the SLAC method, the mean ratio of nonsynonymous changes per non-synonymous site (dN) and the synonymous changes per synonymous site (dS) were measured using SLAC which considered inferred ancestral sequences for each internal node in a phylogeny using a codon model and then, calculated the synonymous and nonsynonymous mutations by comparing each codon to its immediate ancestor. The FEL method is based on maximumlikelihood estimates. This method estimates the ratio of nonsynonymous to synonymous substitutions on a site-by-site basis for the entire tree. iFEL is principally the same as FEL, except that selection is only tested along the internal branches of the phylogeny. To detect co-evolving sites from multiple alignments of amino acid sequence data and to identify significant associations among sites, we applied the Bayesian graphical models (BGM) method implemented in Spidermonkey through the Datamonkey web-based interface [27].
Results
Sequence homology and Mutation analysis:
Nucleotide sequence identities of HIV-1 Tat subtypes B and C were 81% and 84%, respectively. In comparison to subtype C, amino acid substitutions were more frequently observed in subtype B, as shown in Table 2 (see supplementary material). Amino acid sequences diversity in both subtypes B and C were illustrated by sequence logo (Figure 1b & c). The detailed substitution positions in each domain were described below following HXB2 numbering in both subtypes (the position of amino acid, aa, which had major changes).
In domain I, we found that positions 1, 11, 14, 15, and 16were completely conserved in both subtypes; in addition, Asp5 in subtype C was also completely conserved. Notably, Lys12Asn (90%) and Ala21Pro (53%) were frequently observed only in subtype C.
In domain II, among the conserved Cys positions, Cys27 in subtype B and Cys22 and Cys34 in subtype C were completely conserved.Lys28 in subtype B and His33 in subtype C were also found as completely conserved in our analysis. Cys31Ser was observed in subtype C (83%), and all sequences of subtype C in our data sets were substituted as Phe32Tyr.
In domain III, we found mutations in all positions in this domain except Lys41 in subtype C and Gly48 in subtype B. In subtype C, 80% sequences were substituted as Ile39Gln. In both subtypes, most of the sequences contained Ala42Gly (99%).
In domain IV, the consensus Arg-rich motif provided two of the key functions of Tat, nuclear localization and membrane transduction [10– 15], a total of 6 Arg at the positions 49, 52, 53, 55, 56, and 57. We did not find any conserved Arg position in subtype B. Only positions 49 and 55 were found conserved in subtype C accompanied with Arg57Ser substitution predominantly (88%). In domain V, only Gln66 was conserved in both subtypes; besides, Gln72 was almost completely conserved in subtype Band subtype C. For other sites, His59Pro substitution was apparent in both subtypes (81% and 95% in subtype B and C, respectively). In addition, Asn61Asp (64%) and Asn67Val (44%) in subtype B, and also Gln60Pro (89%), Asn61Ser (92%), Gln63Glu (75%), Thr64Asp (87%), Ala67Asn (73%), and Lys69Ile (61%) substitutions were frequently found in subtype C.
The domain VI also known as the RGD (Arg-Gly-Asp) domain, and RGD is a ligand for several integrins, which play important roles in HIV replication and cell surface binding [1, 28, 29]. Even though representative conserved domain in Tat was well maintained in both subtypes, we observed genetic variation in the sixth domain of our data set. In subtype B, 87Ser (77%) was most frequently found with many different substitutions in small percentages atother positions of exon 2. In subtype C, high frequencies of substitutions were observed, includingThr74Leu (92%), Pro77Thr (79%), Pro84Ser (80%), Lys85Glu (89%) and 87Ser (95%) indicating that this domain is also variable. In subtype C Tat exon 2, we mostly observed substitutions, 93Ser (98%) and 94Lys (97%).Overall, we found genetic variation in domains IV, V, and VI, which may have an impact on the functional properties of Tat, such as Tat-TAR interactions, protein transductions as well as cell surface binding and replication. The summary of substitutions was shown in Table 2 (see supplementary material).
Phylogenetic inference:
Analysis of the phylogenetic relationship was performed using the maximum likelihood (ML) tree based on the nucleotide sequences of subtypes B and C (Figure 2a & b). The phylogeny of Tat in subtype B featured with well intermixing of sequences among the different continents. The wide genetic diversity and several poorly defined clusters were observed in the sequences particularly in the strains from USA, South America and Europe. However, Asian strains, especially Thai and Korean strains clustered together distinctly, may result from different single introduction. In subtype C, it was hard to define clear clustering (Figure 2b).In the tree, the majority of subtype C strains were found in Africa and those African stains did not show any monophyletic distribution, and there was a interspersing of Asian, South American and European strains with African stains. Overall, we observed a slow and continuous introduction of new HIV-1 strains in different parts of the world with repeated cross-border transmission as reflected by diffuse distributions and intermixing of the different HIV-1 variants in both subtypes B and C.
Figure 2.
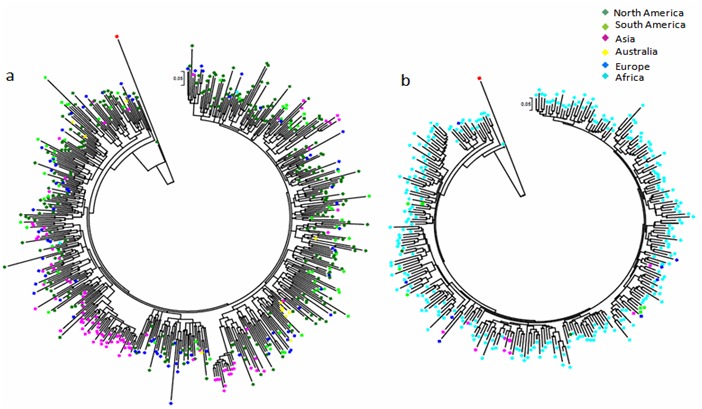
Phylogenetic trees of HIV-1 subtypes B and C. Maximum likelihood (ML) phylogenetic tree of HIV-1 subtypes B (a) and C (b) sequences based on 300 nucleotide sites of Tat gene sequence generated through the Los Alamos database. GTR+I+γ5nucleotide substitution model was employed with 1000 bootstrapped data by MEGA 6. The reference Tat sequences were downloaded from the Los Alamos database showed in round bullet. SIV sequence (Ref.CPZ.US.85.US_Marilyn.AF103) was used to root the tree showed in red square bullet
Analysis for selection pressure:
Global ω values for the nucleotide sequences of Tat in subtypes B and C were 0.883 and 0.760, respectively (less than 1), indicating that there is no detectable positive selection on the entire Tat genes. We found that a total of 23 and 18 amino acid positions in subtypes B and C, respectively, were under significant positive selection based on the FEL, iFEL and SLAC methods. The detailed positively selected codons with statistical significance were calculated with SLAC, FEL, and iFEL methods as shown in Table 3, 4, 5, & 6 (see supplementary material). Position Thr40 in the third domain was positively selected in both subtypes by all three methods. We found several positively selected sites in the basic region of Tat, such as Ser68 and Ser70 in both subtypes; His59, Asn61, Ser62, and Thr64, His65 in subtype B; and Ala58, Ala67, and Leu69 in subtype C. Remarkably, in both subtypes of exon 2, Ser75, Pro77, Asp80, Pro81, and Ser87 were positively selected by all 3 methods.
Discussion
We observed high genetic diversity in HIV-1 full length Tat in both subtypes B and C irrespective of their region of origin as revealed by the phylogenetic tree and mutation analysis. Genetic diversity reflected by relative branch lengths in the phylogenetic tree, particularly in subtype B suggests that the clustering occurs due to the transmission network at individual or at local level. Previous study also showed high genetic divergence of HIV-1 Tat exon-1 in both subtypes [30]. Furthermore, we found several positively selected sites located in the sixth domain of Tat encoded in exon 2. We found substitutions even in highly conserved Cys-rich and49Arg Lys Lys Arg Arg Gln Arg Arg Arg57 domains. In addition, we found a Ser31Cys substitution (both belong to nucleophilic amino acid group) in HIV-1 subtype C as described previously [31]. Absence of a critical Cys31 in the Cys-rich domain has been reported only in subtype C [31]; this position may play a role in evolution of subtype C. In fact, we observed substitutions such asArg57Gly or Thr in subtype B, Arg57Ser in subtype C, and Gln63Asn in both subtypes B and C, which are within and close to the basic domains, respectively. Interestingly, we found position 63 was under significant diversifying selection for subtype B. However, position 57 was not positively selected in either subtype. Rather, it showed purifying selection in subtype B (data not shown). Previous study showed that mutated Tat in HIV-1 subtype C in those sites exhibited greater transcriptional activity in Jurk at cells compared with subtypes B and E, without LTR sequence dependency [32, 33]. Overall, the amino acid diversity that we found in well conserved positions is likely to have an impact on Tat mediated viral transcription as described previously [31, 33, 34].
We have found that Ser68 and Ser70 positions in the basic domains were positively selected in both subtypes which were previously reported as genetically variable region [4], and we also observed similar results. This result implies that changes in amino acids in basic region may have a functional impact, and those changes may fix in the virus population. However, further in vitro experiment is needed to validate this hypothesis. We found that positions encoded in exon 2 such as Ser75, Pro77, Asp80, Pro81, and Ser87 were positively selected. As reported previously, exon 2 plays a role in the kappa-lightchain enhancer of activated B cell-(NF-κB) dependent control of HIV-1 transcription in T cells [8, 35]. It has been previously reported that unlike laboratory-passaged strains, such as HIV- 1HXB2 with premature stop codon at position 87, majority of HIV-1 strains encode 101 amino acids without any truncation beyond the position 86 [1]. We found Ser87 in subtypes B and C which was positively selected. As previously noted, the existence of two exons is essential to maintain stability of Tat in vivo [36]; therefore, this position may be crucial to maintain the functional stability of Tat. Again, mutations of the exon 2 were found particularly at intimately networked coevolving sites with exon1 in the fourth, fifth, and sixth domains (data not shown). This may also have some impact on HIV mRNA transcription through Tat-TAR interaction and initiation of reverse transcription, which were previously reported as influenced by genetic variation of Tat [8, 37].
Developing antivirals targeting the interaction site between HIV-1 Tat and TAR has been under process [38]. In addition, Tat based vaccine development is also underway [39]. Examining the molecular diversity of full-length Tat gene in globally circulating strains is therefore imperative. Thus, our study findings accomplish to understand the genetic diversity of full-length Tat in common HIV-1 subtypes like B and C.
Supplementary material
Acknowledgments
We would like to thank Ms. Karen Lewis for her careful reading the manuscript. We also thank Drs. Yasuhiro Suzuki Junji Imamura and Eiichi N. Kodama for their guidance, inspiration and critical comments regarding writing the manuscript.
Footnotes
Citation:Roy et al, Bioinformation 11(3): 151-160 (2015)
References
- 1.Jeang KT, et al. J Biol Chem. 1999;274:28837. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- 2.Ensoli B, et al. AIDS. 2006;20:2245. doi: 10.1097/QAD.0b013e3280112cd1. [DOI] [PubMed] [Google Scholar]
- 3.Hamy F, et al. Chem Biol. 2000;7:669. doi: 10.1016/s1074-5521(00)00012-0. [DOI] [PubMed] [Google Scholar]
- 4.Allen TM, et al. Nature. 2000;407:386. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- 5.Mason RD, et al. Virology. 2009;388:315. doi: 10.1016/j.virol.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein G, et al. Vaccine. 2001;19:1738. doi: 10.1016/s0264-410x(00)00393-5. [DOI] [PubMed] [Google Scholar]
- 7.Ruckwardt TJ, et al. J Virol. 2004;78:13190. doi: 10.1128/JVI.78.23.13190-13196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, et al. Adv Virol. 2012;2012:123605. doi: 10.1155/2012/123605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rana TM, Jeang KT. Arch Biochem Biophys. 1999;365:175. doi: 10.1006/abbi.1999.1206. [DOI] [PubMed] [Google Scholar]
- 10.Truant R, Cullen BR. Mol Cell Biol. 1999;19:1210. doi: 10.1128/mcb.19.2.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Fuente JM, Berry CC. Bioconjug Chem. 2005;16:1176. doi: 10.1021/bc050033+. [DOI] [PubMed] [Google Scholar]
- 12.Ziegler A, Seelig J. Biophys J. 2004;86:254. doi: 10.1016/S0006-3495(04)74101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy S, et al. Genes Dev. 1990;4:1365. doi: 10.1101/gad.4.8.1365. [DOI] [PubMed] [Google Scholar]
- 14.Fawell S, et al. Proc Natl Acad Sci U S A. 1994;91:664. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hidema S, et al. J Biosci Bioeng. 2012;113:5. doi: 10.1016/j.jbiosc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 16.Smith SM, et al. J Biol Chem. 2003;278:44816. doi: 10.1074/jbc.M307546200. [DOI] [PubMed] [Google Scholar]
- 17.Brake DA, et al. J Cell Biol. 1990;111:1275. doi: 10.1083/jcb.111.3.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orsini MJ, et al. J Neurosci. 1996;16:2546. doi: 10.1523/JNEUROSCI.16-08-02546.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buonaguro L, et al. J Virol. 2007;81:10209. doi: 10.1128/JVI.00872-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Los alamos National Laboratory (LANL) HIV sequence database. p. 2014. http://www.Hiv.Lanl.Gov/content/sequence /newalign /align.Html.
- 21.p. 2014. http://www.genome.jp/tools/clustalw/
- 22.p. 2014. http://weblogo.berkeley.edu/logo.cgi;2008.
- 23.Tamura K, et al. Mol Biol Evol. 2013;30:2725. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pond SL, Frost SD. Bioinformatics. 2005;21:2531. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- 25.Kosakovsky Pond SL, Frost SD. Mol Biol Evol. 2005;22:1208. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 26.Delport W, et al. Bioinformatics. 2010;26:2455. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poon AF, et al. Bioinformatics. 2008;24:1949. doi: 10.1093/bioinformatics/btn313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Sayed A, Futaki S. AAPS J. 2009;11:13. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sood V, Ranjan R. AIDS. 2008;22:1683. doi: 10.1097/QAD.0b013e3282f56114. [DOI] [PubMed] [Google Scholar]
- 30.Kandathil AJ, et al. Bioinformation. 2009;4:237. doi: 10.6026/97320630004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurosu T, et al. Microbiol Immunol. 2002;46:787. doi: 10.1111/j.1348-0421.2002.tb02766.x. [DOI] [PubMed] [Google Scholar]
- 32.Rossenkhan R, et al. J Virol. 2013;87:5732. doi: 10.1128/JVI.03297-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desfosses Y, et al. J Virol. 2005;79:9180. doi: 10.1128/JVI.79.14.9180-9191.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Opi S, et al. J Biol Chem. 2002;277:35915. doi: 10.1074/jbc.M204393200. [DOI] [PubMed] [Google Scholar]
- 35.Mahlknecht U, et al. J Leukoc Biol. 2008;83:718. doi: 10.1189/jlb.0607405. [DOI] [PubMed] [Google Scholar]
- 36.Campbell GR, Loret EP. Retrovirology. 2009;6:50. doi: 10.1186/1742-4690-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrich D, et al. EMBO J. 1997;16:1224. doi: 10.1093/emboj/16.6.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamasaki T, et al. Antimicrob Agents Chemother. 2013;57:1323. doi: 10.1128/AAC.01711-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein G, Chicca JJ. Hum Vaccin Immunother. 2012;8:479. doi: 10.4161/hv.19184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.