

Abstract
Loss of function of the FMR1 gene leads to fragile X syndrome (FXS), the most common form of intellectual disability. The loss of FMR1 function is usually caused by epigenetic silencing of the FMR1 promoter leading to expansion and subsequent methylation of a CGG repeat in the 5′ untranslated region. Very few coding sequence variations have been experimentally characterized and shown to be causal to the disease. Here, we describe a novel FMR1 mutation and reveal an unexpected nuclear export function for the C-terminus of FMRP. We screened a cohort of patients with typical FXS symptoms who tested negative for CGG repeat expansion in the FMR1 locus. In one patient, we identified a guanine insertion in FMR1 exon 15. This mutation alters the open reading frame creating a short novel C-terminal sequence, followed by a stop codon. We find that this novel peptide encodes a functional nuclear localization signal (NLS) targeting the patient FMRP to the nucleolus in human cells. We also reveal an evolutionarily conserved nuclear export function associated with the endogenous C-terminus of FMRP. In vivo analyses in Drosophila demonstrate that a patient-mimetic mutation alters the localization and function of Dfmrp in neurons, leading to neomorphic neuronal phenotypes.
Keywords: axon guidance, Drosophila, fragile X syndrome, nuclear export, nucleolus
Introduction
Fragile X syndrome (FXS [MIM 300624]) is a highly prevalent, inherited disorder in humans causing intellectual disability accompanied by a spectrum of behavioral and physical abnormalities (Penagarikano et al, 2007). FXS patients typically show developmental delay and display an IQ below 70 and may suffer from significant decline in short-term memory, executive function, visuo-spatial abilities, and linguistic processing (Crowe & Hay, 1990; Belser & Sudhalter, 2001; Fisch et al, 2002; Cornish et al, 2004; Loesch et al, 2004). In most cases, the cognitive defects are accompanied by autistic behavioral phenotypes, including hyper-reactivity, social anxiety, aggression, stereotypic movements, mood disturbance, and attention deficiency (Lachiewicz & Dawson, 1994; Lachiewicz et al, 1994). Sleep disorders and vulnerability to epileptic seizures have also been reported in association with FXS (Berry-Kravis, 2002; Kronk et al, 2010). In the diagnosis of FXS, clinicians routinely screen for large expansions of the polymorphic CGG repeat elements in the FMR1 locus (Xq27.3 [MIM 309550]). In FXS, the naturally occurring CGG repeats are expanded to numbers above 200, which usually leads to hypermethylation of the repeat itself and the upstream FMR1 promoter (Fu et al, 1991; Oberle et al, 1991; Pieretti et al, 1991; Verkerk et al, 1991). As a consequence, the FMR1 is transcriptionally silenced, and no protein product (FMRP) is formed.
FMRP is an RNA-binding protein that regulates many aspects of RNA biology, including RNA transport, stability and, most importantly, mRNA translation (Bagni & Greenough, 2005; Bassell & Warren, 2008). FMRP is ubiquitously expressed and is particularly abundant in the brain, ovaries and testes (Devys et al, 1993; Bakker et al, 2000), and a large number of potential FMRP mRNA and noncoding RNA targets exist (Fernandez et al, 2013). Collective evidence from mouse and fruit fly models indicates that local translational dysregulation in the absence of FMRP can impair early neuronal development, circuit formation, neurotransmission, and synaptic plasticity (Bassell & Warren, 2008; Zhang et al, 2001; Reeve et al, 2005).
Expansion of promoter-proximal CGG repeats and the consequent epigenetic suppression of FMR1 expression is the leading genetic mechanism underlying FMRP deficiency in FXS patients. Deletions within or across the FMR1 locus can also lead to the loss of FMRP, as reported in several fragile X case studies in the literature (Gedeon et al, 1992; Wohrle et al, 1992; Tarleton et al, 1993; Lugenbeel et al, 1995; Coffee et al, 2008). Although these two genetic mechanisms account for the majority of FXS patients, they do not usually yield insight into the functional properties of the different FMRP domains in a clinically relevant context. Pathogenic FMR1 sequence variants that affect FMRP expression, localization, and function may also result in FXS. However, only a few potentially pathogenic point mutations in FMR1 have so far been described (De Boulle et al, 1993; Collins et al, 2010a,b; Gronskov et al, 2011), and there is a need for detailed genetic and functional analyses to fully characterize these intragenic mutations. One notable exception that has yielded important information on the RNA-binding activity of FMRP is the I304N point mutation, identified in the FMR1 coding sequence of a FXS patient with severe symptoms (De Boulle et al, 1993). This particular FMR1 allele has been investigated extensively in numerous studies, and the mutation was found to profoundly alter many aspects of FMRP function via its effect on one of the RNA-binding domains called the K homology (KH) domain (Siomi et al, 1994; Feng et al, 1997; Tamanini et al, 1999; Laggerbauer et al, 2001; Schrier et al, 2004).
Despite its seemingly low occurrence in the FXS patient population (Collins et al, 2010b), searching and screening for potentially pathogenic FMR1 sequence variants is essential. A segment of the patient population could otherwise remain undiagnosed and therefore may not benefit from future therapies. Moreover, elucidating the functional consequences of these mutations will provide the opportunity to study FMRP domains by identifying critical residues and characterizing the function of different domains in a clinically relevant context, as demonstrated by the I304N point mutation. The fact that genetic screens in the D. melanogaster dfmr1 identified loss-of-function mutations in conserved residues (Reeve et al, 2008) lends credence to this notion.
Here, we report a novel FMR1 frameshift mutation found in a patient with FXS symptoms. This mutation alters the open reading frame, creating a short novel amino acid sequence in the C-terminus followed by a premature stop codon. Functional characterization of this patient FMR1 allele reveals that the mutation targets the protein to the nucleolus in cultured human and mouse cells. Furthermore, we observe the nuclear retention of the patient FMR1 protein only when the C-terminus is truncated, hinting at the presence of a novel nuclear export function in the C-terminus of FMRP. A genetically versatile model for FMR1 loss of function has been established in the fruit fly Drosophila melanogaster. The fly has a single homologue of FMR1, and its loss of function causes many phenotypes similar to those associated with FXS patients (Zhang et al, 2001; Morales et al, 2002; Pan et al, 2004; Reeve et al, 2005; Bassell & Warren, 2008; McBride et al, 2012). Using this model for in vivo analyses, we find that the NLS signal identified in the patient FMRP can target fly dfmr1 protein to the nucleus in fly neurons, also in a C-terminus dependent fashion. This suggests that the nuclear export function of the FMRP C-terminus is evolutionarily conserved. Interestingly, the change in Dfmrp localization alters its function in neurons and leads to neomorphic phenotypes in vivo. Taken together, our results provide evidence for changes in FMRP regulation and function brought on by this novel fragile X patient mutation.
Results
Novel frameshift mutation identified in FXS patient
We selected 16 individuals from our in-house cohort of male patients with intellectual disability, for sequencing analysis of the FMR1. The selection was based on the presence of typical neurodevelopmental features of fragile X syndrome including moderate intellectual disability (IQ < 60), autistic and/or stereotypic behavior, and impaired social interaction, associated with at least one of the following physical features: elongated face, macroorchidism, and/or large ears. The typical CGG repeat expansion in the FMR1 locus was absent in all 16 males.
Among screened individuals, we found seven with previously unreported variations in the FMR1 (Supplementary Fig S1A). In 5 of these individuals, single base polymorphisms were identified in intronic regions. In the other 2, single mutations were present in coding exons. In one of these individuals, we recovered a silent base substitution in exon 15. In the other, we identified a guanine insertion in exon 15 [1457insG] (Supplementary Fig S1A and B). This G-insertion mutation alters the open reading frame to one that is not used by any of the alternative FMR1 isoforms (Ensembl Genome Browser), and is predicted to create a novel peptide sequence followed by a premature stop codon, which results in the truncation of the C-terminus of FMRP (Fig1A). This modification also leads to the disruption of the RGG box, one of the three RNA-binding domains characterized in FMRP (Darnell et al, 2001; Ramos et al, 2003; Bagni & Greenough, 2005; Blackwell et al, 2010).
Figure 1. Novel G-insertion mutation identified in male with FXS symptoms.
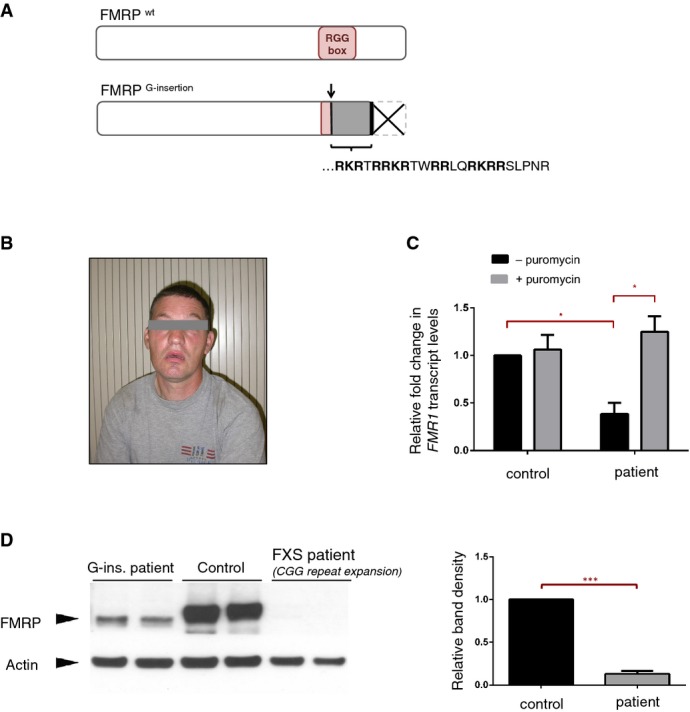
- Predicted effects of the G-insertion mutation on FMRP are schematized. The insertion site corresponds to the beginning of the RNA-binding RGG Box, where it alters the open reading frame and thus disrupts the domain entirely. The frameshift creates a novel peptide sequence [RKRTRRKRTWRRLQRKRRSLPNR] followed by a premature stop codon, causing the truncation of the FMRP C-terminus.
- Photograph of male patient with FMR1G-ins. allele. Typical physical and behavioral features of FXS were noted in the patient, who shows moderate to severe intellectual disability.
- FMR1 mRNA levels were analyzed via RT–qPCR in EBV-transformed lymphocyte cells derived from the patient's blood samples. Patient cells showed a ˜60% decrease in FMR1 mRNA compared to control cells (*P = 0.010, 0.383 ± 0.110 SD, n = 3). Treatment with translational blocker puromycin restored FMR1 mRNA levels in patient cells (*P = 0.048, 0.383 ± 0.110 SD versus 1.25 ± 0.166 SD, n = 3), suggesting that the reduction of FMR1 mRNA in these cells is primarily due to nonsense-mediated decay. RT-qPCR reactions were run in triplicate in three independent experiments. Fold changes in FMR1 expression, normalized to HRPT expression, were calculated using the ΔΔCT method, and analyzed statistically with a two-tailed t-test (GraphPad). Error bars represent mean values with SD.
- Western blot analysis shows a significant decrease (> 90%) in FMRP protein levels in patient-derived cells (***P = 0.008, 0.13 ± 0.031 SD, n = 2), and the patient FMRP migrates slightly lower than the wild-type protein. Band intensities for the different cell lines were quantified, normalized for actin and analyzed statistically with a twot-tailed t-test (GraphPad). Error bars represent mean values with SD.
The FMR1 allele with the G-insertion mutation was identified in a male patient with moderate to severe intellectual disability, first seen at the age of 36 years in a residential care setting (Fig1B). He was born to healthy unrelated parents—now deceased—and has one healthy brother. He went to a specialized school for disabled children, and IQ measured at adult age was 41. Clinical examination at age 36 showed a man with normal build and growth parameters (50th percentile). He made almost no eye contact and had macroorchidism with bilateral testicular volume of 30 ml. His behavior was rather calm, yet periodic aggressive outbursts were noted. Overall, he presented with a typical fragile X clinical phenotype, although the CGG repeat number was reported normal (41 repeats) in multiple testing rounds.
FMR1 mRNA and protein levels are decreased in patient-derived cells
To determine whether FMR1 mRNA and protein levels were affected by the G-insertion mutation, we established an immortalized EBV lymphocyte line from a blood sample of the patient and measured FMR1 mRNA and protein levels in these cells. We found both to be significantly decreased (by ∼60 and > 90%, respectively) compared to the levels in control cells (Fig1C). We determined that the reduction of the FMR1 mRNA in patient cells is primarily due to nonsense-mediated decay (NMD)—a translation-coupled endogenous mechanism to degrade faulty mRNAs with premature stop codons—since FMR1 mRNA levels in the patient cells were restored to control levels upon treatment with the translational inhibitor puromycin (Fig1C). Western blot analysis indicated that FMRP protein levels in patient-derived cells were significantly decreased compared to control cells from a healthy individual with no CGG repeat expansion in the FMR1 locus (Fig1D).
Patient FMRP shows altered cellular localization
To functionally characterize the patient FMRP protein, we began by expressing this novel allele in human cells. We transfected HEK293 cells with constructs expressing either GFP-tagged wild-type FMRP (GFP-FMR1WT) or GFP-tagged patient FMRP (GFP-FMR1G-ins) under the control of a β-actin promoter. While wild-type FMRP was localized predominantly in the cytoplasm as expected, the patient FMRP showed a surprising localization pattern that was not observed with the wild-type protein (Fig2A). Specifically, the patient FMRP forms bright nuclear inclusions with 100% penetrance. Given their size and appearance, we speculated that these inclusions might correspond to a nucleolar aggregation of FMRP. To test this idea, HEK293 cells expressing either GFP-FMR1WT or GFP-FMR1G-ins were stained for the detection of a nucleolar specific protein, nucleophosmin (NPM1). We found that the patient FMRP, unlike its wild-type counterpart, colocalizes with nucleophosmin, confirming the novel nucleolar localization of the patient FMRP. Finally, expression in SH-Sy5y human neuroblastoma cells (Fig2B) as well as primary cultures of FMR1-KO mouse neurons resulted in a similar nuclear and nucleolar localization pattern for the patient FMRP, albeit at lower levels (Fig2C).
Figure 2. Patient FMRP aggregates in the nucleolus.
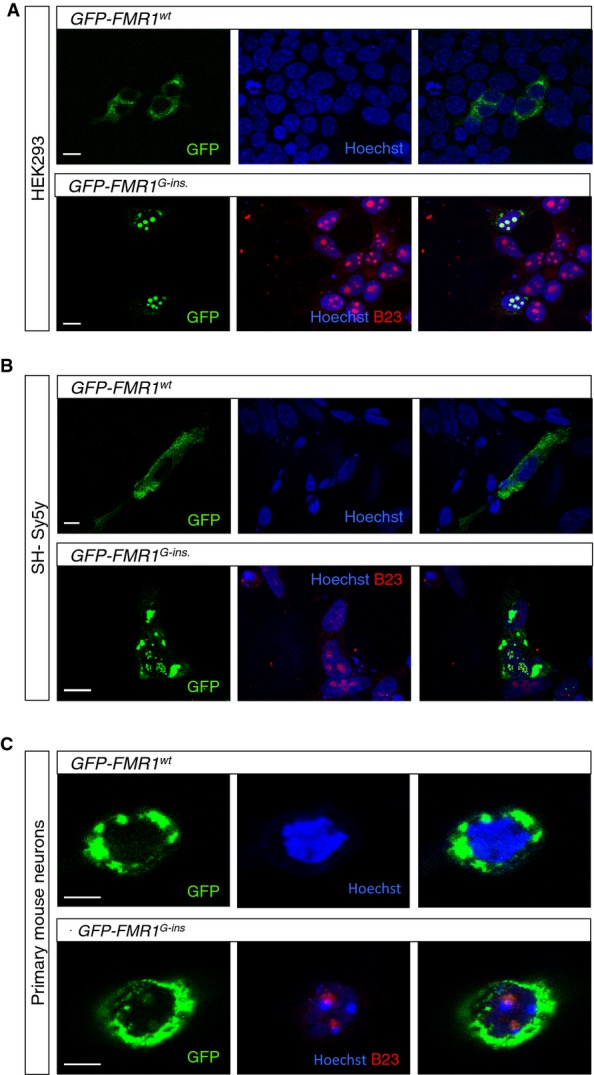
- A–C HEK293 (A) and SH-Sy5y (B) cells, as well as primary neurons from FMR1 KO mice (C), were transfected with vectors encoding the GFP-tagged wild-type FMRP (GFP-FMR1WT) or the GFP-tagged patient FMRP (GFP-FMR1G-insertion) under the control of a β-actin promoter. Wild-type FMRP localizes predominantly in the cytoplasm, whereas the patient FMRP forms nuclear and nucleolar aggregates that colocalize with nucleophosmin. Scale bars in (A, B) represent 10 μm. Scale bar in (C) represents 5 μm.
The frameshifted C-terminus contains a nuclear localization signal
We sought to explore the peptide changes in the patient FMRP that could target the protein to the nucleus. The G-insertion mutation and the accompanying frameshift lead to two significant modifications of the FMR1 protein (Fig1A): (i) the truncation of the C-terminus and (ii) the presence of a novel, 22 amino acid peptide sequence—both of which may contribute to the altered localization pattern. We therefore dissected and characterized the contribution of each of the two components separately. We found that the C-terminus truncation (GFP-FMR1ΔCt) alone did not result in nuclear retention of FMRP (Fig3A), suggesting that the novel amino acid sequence in the patient FMRP directs the change in localization observed. Interestingly, this novel peptide sequence is predicted to be a bipartite nuclear localization signal (NLS) when analyzed with the ScanProsite tool from Expasy (de Castro et al, 2006). This motif scanner identifies the NLS based on the adjacent stretches of Arg and Lys (Fig1A). When we mutated adjacent Arg and Lys residues of the putative NLS to Ala (GFP-FMR1G-ins [NLS mutated]), the nuclear localization of the protein was no longer visible (Fig3B). These results indicate that the novel peptide sequence in the patient FMRP is a functional nuclear localization signal. This was further confirmed by showing that the putative NLS could also target the cytoplasmic protein profilin to the nucleus (Supplementary Fig S2A). Importantly, the change in subcellular localization of the patient protein compared to the various controls does not correlate with levels of protein expression (Supplementary Fig S2B). Specifically, the patient mutation exhibits lower expression levels than the wild-type protein and the C-terminal truncation alone, but comparable levels to the controls where the NLS is mutated or the C-terminus is restored. Together, these data suggest that the nucleolar localization is due to the mutation and not to an increase in expression level.
Figure 3. Patient FMRP contains a novel nuclear localization signal in the C-terminus.
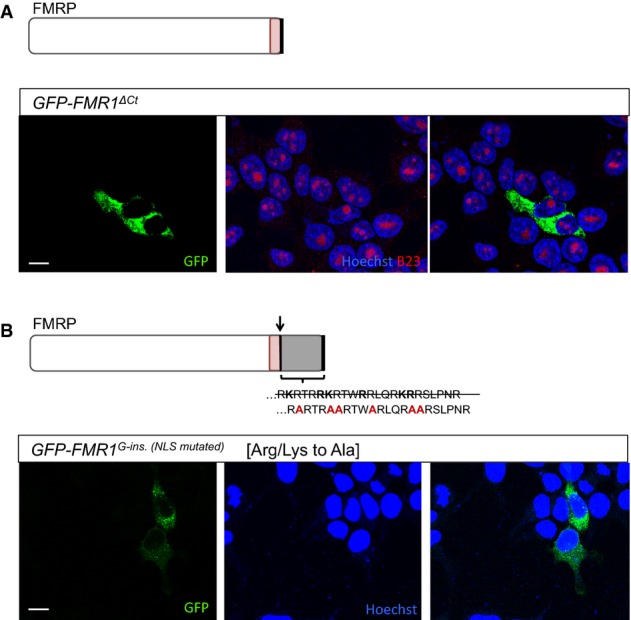
- Transfected HEK293 cells expressing a GFP-tagged FMRP in which the C-terminus is truncated following the G-ins. mutation site (GFP-FMR1ΔCt). GFP-FMR1ΔCt protein is exclusively cytoplasmic, suggesting that the truncation of the C-terminus alone is not sufficient to explain the localization change observed for the patient FMRP.
- Transfected HEK293 cells expressing a modified version of GFP-tagged patient FMRP (GFP-FMR1G-ins. [NLSmutated]). The novel amino acid sequence in the C-terminus of patient FMRP [RKRTRRKRTWRRLQRKRRSLPNR] contains stretches of arginines and lysines predicted to function as a nuclear localization signal (based on Expasy protein motif scanner, ScanProsite). Mutating adjacent R and K residues into alanines [RARTRAARTWARLQRAARSLPNR] abolishes the nucleolar localization of the patient FMRP, strongly suggesting that the novel amino acid sequence present in the patient FMRP C-terminus is a functional nuclear localization signal (NLS).
An intact C-terminus exports patient FMRP out of the nucleus
Our results demonstrate that the frameshift caused by the G-insertion mutation creates a novel C-terminal peptide that targets the patient FMRP to the nucleus. However, it is not clear whether the truncation of the C-terminus contributes to the nuclear retention of the patient FMRP. To test this, we created an allele where the patient NLS motif is fused to the C-terminus of the full-length, wild-type FMR1 protein (GFP-FMR1wt+NLS). Surprisingly, we did not observe any nuclear FMRP in cells expressing this specific FMR1 allele (Fig4A). One possible explanation for this observation is that an intact C-terminus drives the efficient export of the patient FMRP from the nucleus. To test this, we studied the expression of GFP-FMR1wt+NLS in HEK293 cells treated with leptomycin B (LMB), a potent and specific inhibitor of nuclear export (Wolff et al, 1997). It has been shown that full-length wild-type FMRP does not show nuclear retention upon LMB treatment (Dury et al, 2013). In contrast, we find that nucleolar inclusions of the GFP-FMR1wt+NLS protein became visible upon treatment with LMB (Fig4A), indicating that the presence of an intact C-terminus enables the nuclear export of FMRP bearing the patient NLS motif.
Figure 4. An intact C-terminus facilitates nuclear export of patient FMRP.
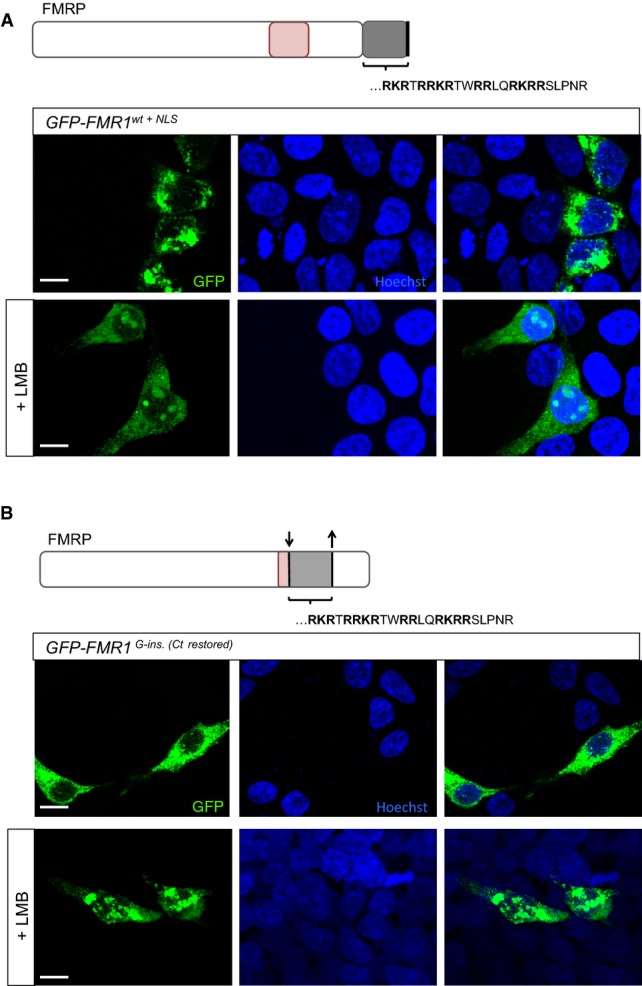
- Transfected HEK293 cells expressing GFP-tagged wild-type FMRP fused to the patient NLS motif (GFP-FMR1wt+NLS). Unlike the patient FMRP, GFP-FMR1wt+NLS protein is predominantly cytoplasmic and does not aggregate in the nucleus. However, treatment of HEK293 cells expressing GFP-FMR1wt+NLS with leptomycin B—an inhibitor of nuclear export—resulted in the appearance of nucleolar inclusions. This suggests that the presence of a full-length C-terminus facilitates the nuclear export of FMRP in this context.
- Transfected HEK293 cells expressing a modified version of GFP-tagged patient FMRP, in which the truncation of the C-terminus is reverted by restoring the open reading frame (GFP-FMR1G-ins.[Ct restored]). The patient protein is not detected in the nucleus when the C-terminus is intact; however, nucleolar retention of the protein is observed upon leptomycin B treatment of the transfected cells. In line with results from (A), this suggests that an intact C-terminus enables nuclear export of the FMR1 protein.
We also created another relevant variant of the patient FMR1 allele, where the frameshift caused by the G-insertion was restored—via the deletion of a single base pair—immediately before the premature stop codon (GFP-FMR1G-ins[Ct revert]). At the protein level, the RGG box is almost entirely disrupted and replaced by the NLS sequence as in the case of patient FMRP, though the C-terminus truncation is avoided in this case. In line with our previous observations, we did not detect FMRP in the nucleus of cells expressing GFP-FMR1G-ins[Ct revert] (Fig4B). However, treatment of these cells with LMB revealed nucleolar inclusions of FMRP (Fig4B), once again demonstrating that the C-terminus truncation is critical for the nuclear retention of patient FMRP.
Taken together, our cell culture experiments demonstrate that the patient's G-insertion mutation leads to profound changes in the FMR1 protein product. The mutation leads to changes in subcellular localization mediated by the novel sequence and the truncation of the C-terminus. The data also suggest a potential nuclear export mechanism associated with the C-terminus of the FMRP protein in this context.
Patient FMRP sequence changes cause novel neuronal phenotypes in vivo
FMRP is a member of protein family known as the FXR protein family (Siomi et al, 1995; Zhang et al, 1995; Kirkpatrick et al, 2001). Although highly similar in their N-terminus, the C-terminus of the FXR protein family (Supplementary Fig S3) is not conserved at the sequence level (Kirkpatrick et al, 2001). We therefore wondered whether the effects of the mutation on the human protein are conserved in other FMRP homologs, or whether they are human specific. Therefore, we exploited the Drosophila melanogaster model, given its genetic tractability and success as a tool to study neurodevelopmental processes and related disorders (Okray & Hassan, 2013). The fruit fly has a single FMR1 homolog—dfmr1—that equally resembles human FMR1 and FXR genes at the amino acid level (Wan et al, 2000; Morales et al, 2002). Various morphological, molecular, and behavioral phenotypes relevant to dfmr1 protein function have been described in Drosophila (Zhang et al, 2001; Morales et al, 2002; Pan et al, 2004; Reeve et al, 2005; Bassell & Warren, 2008; McBride et al, 2012). We selected a well-characterized neuronal population termed the Lateral Neurons ventral (LNv)—the fly circadian pacemaker neurons—whose connectivity phenotype is strongly affected by Dfmrp activity (Reeve et al, 2005). Specifically, the overexpression of dfmr1 in a wild-type background causes a consistent phenotype where the terminal axonal branches of sLNv neurons collapse (Reeve et al, 2005, 2008) (Fig5A).
Figure 5. sLNV neurons are sensitive to changes in Dfmrp function.

- The morphology of small lateral ventral neurons (sLNvs) is sensitive to dfmr1 activity. Overexpression of wild-type dfmr1 in the sLNv neurons results in a consistent “axonal collapse” phenotype, where the branching of axonal termini of sLNvs is reduced. Scale bars represent 50 and 10 μm (magnified images).
- Transgenic fly lines were created bearing UAS-dfmr1 variants, in order to assay in vivo functional changes associated with the effects of the patient mutation: the truncation of the C-terminus (dfmr1ΔCt), presence of the patient NLS sequence (dfmr1wt+NLS), or both (dfmr1ΔCt+NLS).
We took advantage of this robust assay and used the UAS-Gal4 binary expression system (Brand & Perrimon, 1993) to overexpress various dfmr1 alleles and examined changes in the dfmr1 gain-of-function phenotype in the LNv neurons. We created transgenic fly lines with different UAS-dfmr1 variants that dissect the different effects of the G-insertion mutation on the human FMRP protein (Fig5B): wild-type dfmr1 (dfmr1wt); C-terminus truncated dfmr1 (dfmr1ΔCt); dfmr1 with the patient NLS only (dfmr1wt+NLS); dfmr1 with both the patient NLS; and a truncated C-terminus mimicking the patient mutation (dfmr1ΔCt+NLS). These variants were all inserted into the same genomic locus to minimize position effects on levels of transcription.
Overexpression of wild-type dfmr1wt, dfmr1ΔCt, and dfmr1wt+NLS leads to collapse of axonal branches in LNv neurons (Fig6A and B). These findings suggest that there is no substantial loss in Dfmrp function associated with the truncation of the C-terminus alone, or with the presence of the NLS motif alone, despite significant differences in protein levels (Supplementary Fig S4). On the other hand, overexpression of dfmr1ΔCt+NLS—where both modifications are present simultaneously—fails to induce the collapse of LNv axonal branches (Fig6A and B). Instead, we detected novel axonal misguidance phenotypes with the overexpression of this allele, including aberrant bifurcations of axonal bundle termini, “tangles” of axons failing to extend medially, and misguided projections of single axons that appear to form loops (Fig6C). Importantly, the de novo axonal phenotypes appear despite the fact that DfmrpΔCt+NLS is expressed at lower levels than the wild-type or DfmrpΔCt controls, and at comparable levels to the Dfmrpwt+NLS control (Supplementary Fig S4), consistent with the data for the human counterparts (Supplementary Fig S2B). Together, these data suggest that the defects caused by the DfmrpΔCt+NLS are an intrinsic property of the compound mutations, rather than expression levels of the mutant protein.
Figure 6. Compound effects of the patient mutation confer a neomorphic function for Dfmrp in sLNV neurons.

- Overexpression of wild-type dfmr1 (UAS-dfmr1wt) using a LNv neuron-specific driver line (Pdf-Gal4) causes reduction of branching area—“axonal collapse”—in the sLNv axonal termini. dfmr1ΔCt and dfmr1wt+NLS overexpression phenocopies overexpression of dfmr1wt, suggesting that the truncation of the C-terminus or the presence of the patient NLS alone does not impair or alter the functional efficacy of dfmr1 in this context. However, overexpression of dfmr1ΔCt+NLS fails to cause collapse of sLNv termini, indicating a significant loss or change of Dfmrp protein function. Axonal morphology is visualized by expressing CD8-GFP under the control of the same neuronal driver. Scale bar represents 10 μm.
- Quantification of axonal termini branching in LNv neurons overexpressing various dfmr1 alleles. Overexpression of dfmr1wt, dfmr1ΔCt, or dfmr1wt+NLS significantly reduced axonal branching area [****P < 0.0001; control 101.3 ± 29.76 SD, n = 30; UAS-dfmrwt 45.41 ± 21.69 SD, n = 29; UAS-dfmrΔCt 52.31 ± 14.67 SD, n = 32; UAS-dfmrwt+NLS 72.45 ± 21.83 SD, n = 29; UAS-dfmrΔCt+NLS 117.8 ± 36.96 SD, n = 28]. dfmr1ΔCt+NLS overexpression does not result in a significant change of axonal branching area compared to the background control [Pdf-Gal4, UAS-GFP] (P = 0.672). For quantification purposes, each branching area (n) was manually outlined, starting from the first point of defasculation of the axonal bundle. These measurements were normalized for variability in brain size across samples using LNv commissure length measurements for each brain. Two-tailed t-tests using Welch's correction were then used to compare controls with mutant phenotypes (GraphPad). Error bars represent mean values with SD. Dots visible for box plots of UAS-dfmrwt and UAS-dfmrΔCt represent samples that outlier the whiskers.
- Overexpression of dfmr1ΔCt+NLS results in aberrant axonal termini morphology. Axonal guidance defects were frequently observed (arrowheads) and scored manually for bifurcations of the axon bundle (9/48), tangling of axons in the termini (13/48), and apparent looping of axons (7/48).
Finally, we investigated whether the neomorphic axonal phenotypes associated with dfmr1ΔCt+NLS correlate with a change in subcellular localization of the transgenic protein. We found that the Dfmrp which shows predominantly cytoplasmic localization in fly neurons (Wan et al, 2000), is targeted to and retained in the nucleus only when both the NLS motif is present and the C-terminus is truncated (dfmr1ΔCt+NLS) (Fig7A). Neither the truncation of the C-terminus (dfmr1ΔCt) nor the presence of the NLS sequence alone (dfmr1wt+NLS) is sufficient to retain Dfmrp in the nucleus (Fig7B–D). Altogether, these data suggest that the gain of function observed for the patient-like form of Dfmrp is linked to the nuclear retention of the protein. Consistent with this idea, further increasing expression of Dfmrp using two copies of UAS-dfmr1 does not cause nuclear localization nor axonal looping or bifurcation (Supplementary Fig S5).
Figure 7. Subcellular localization of the Dfmrp variants in sLNv neurons.
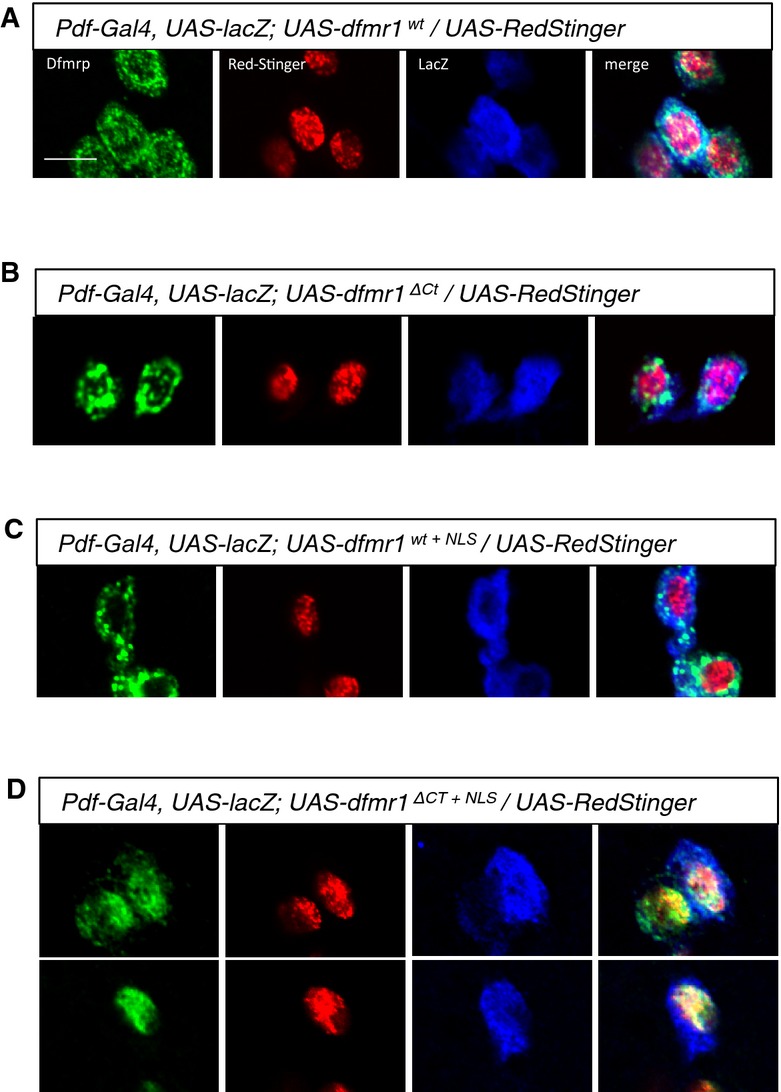
- A–D Subcellular localization of transgenically expressed dfmr1wt, dfmr1ΔCt, dfmr1wt+NLS, and dfmr1ΔCt+NLS proteins in LNv neurons. DfmrpΔCt+NLS shows nuclear localization (A), colocalizing with the genetically encoded nuclear marker RedStinger, while Dfmrpwt, DfmrpΔCt, and Dfmrpwt+NLS are predominantly cytoplasmic (B–D). These results suggest that an intact C-terminus can mediate the nuclear export of the Dfmrp protein. Scale bar represents 5 μm.
Discussion
Here, we have identified and characterized the effects of a novel, intragenic FMR1 frameshift mutation discovered in a patient with typical FXS symptoms. To our knowledge, the G-insertion mutation reported here is the first naturally occurring, clinically relevant mutation to significantly alter the localization of FMRP.
The frameshift leads to profound changes in the peptide sequence: A premature stop codon results in the truncation of the C-terminus, abolishing the RNA-binding RGG Box, and creates a novel amino acid sequence encoding a nuclear localization signal (NLS). This NLS sequence can target the FMRP protein to the nucleolus. Interestingly, restoring the C-terminus enables efficient nuclear export of the protein in this context. Finally, using a Drosophila model, we show that the presence of the NLS sequence together with the truncation of the C-terminus alters FMRP function in neurons in vivo.
Our findings strongly support the notion that genetic mechanisms other than CGG repeat expansions and deletions in the FMR1 locus can underlie fragile X syndrome. Although it is likely that the patient's symptoms arise from the decrease in FMRP levels, it cannot be ruled out that impaired and aberrant functions associated with the remaining mutant protein also contribute. It is worth noting that males with reduced FMRP levels yet normal range IQs have been described, indicating that reduction—as opposed to total absence—of FMRP is not necessarily always causal to FXS (Hagerman et al, 1994).
The striking change in localization observed for the patient FMRP appears to confer a change in function for the protein. The nucleolar aggregation and retention of the mutant protein could lead to an exaggeration of a previously proposed (Willemsen et al, 1996) and recently confirmed (Taha et al, 2014) molecular function for FMRP: Specific endogenous isoforms have been detected in trace amounts in the nucleolus, where they biochemically interact with nucleolin, a multi-functional nucleolar protein required for rRNA transcription and several steps of ribosome biogenesis. Despite its subtlety, it is conceivable that localization of endogenous FMRP to the nucleolus has significant functional consequences. For example, it is proposed that the combined action of FMRP and nucleolin in this context can potentially impact ribosomal biology (Kim et al, 2009; Taha et al, 2014). The patient FMR1 allele identified in this study may provide a unique opportunity to gain insight into this unexplored nucleolar function of FMRP. It is worth noting that in a sense the patient mutation mimics a reduction of the diversity of FMRP isoforms to a single, predominantly nucleolar form. Future experiments at endogenous expression levels in vivo will help ascertain whether these interpretations are true.
Interestingly, the mammalian paralogs of FMR1—the FXR1 [MIM 600819] and FXR2 [MIM 605339] protein products—have been shown to shuttle between the cytoplasm and nucleolus (Tamanini et al, 1999, 2000). Moreover, cells appear to regulate the nucleolar shuttling of the FXR1 protein by generating multiple isoforms with different C-termini, where some isoforms of FXR1 generate a C-terminal nucleolar localization signal, as a result of a frameshift induced by alternative splicing of the 3′ end of FXR1 mRNA (Tamanini et al, 2000). The fact that the C-terminus is highly variable across FMR/FXR proteins might suggest that changes in the C-terminus underlie the functional diversification of the protein family.
Our findings also highlight the possibility that the FMRP C-terminus regulates nuclear/nucleolar shuttling of the endogenous protein. Nuclear shuttling of FMRP has been widely studied, although the exact mechanisms and protein motifs involved still remain somewhat unclear (Kim et al, 2009). Despite the fact that the C-terminus is highly divergent across FMR1 homologs (Wan et al, 2000; Kirkpatrick et al, 2001), our data suggest that the nuclear export function mediated by this domain is evolutionarily conserved.
At any rate, the unique effects of the G-insertion mutation on the FMR1 protein suggest that future molecular and functional analyses of the patient allele identified herein can yield crucial insight into FMRP function in a clinically relevant context.
Materials and Methods
Ethical considerations and patient consents
The clinical screening protocol was approved by the appropriate Institutional Review Board of the University Hospitals of Leuven (Belgium), which operates in agreement with the principles in the WMA Declaration of Helsinki. Informed consent was obtained from the parents/guardians of the affected patients, and permission to publish photos of the patient was granted. Mouse housing conditions and experiments were approved by the Dutch Ethical Committee (DEC) under Erasmus MC Permit Number EMC 140-09-06.
Data deposition
The patient FMR1 variant has been submitted to the Leiden Open Variation Database (LOVD), with the accession ID #00025860.
CGG repeat analysis and FMR1 sequencing
Genomic DNA from patients was isolated from peripheral blood according to standard procedures and stored at 4°C. Molecular FMR1 CGG repeat expansion analyses were performed with an in-house PCR method and with Southern blot, using probe StB12.3 after a combined EcoRI and EagI digestion, as described in Rousseau et al (1994). FMR1 locus was amplified from genomic DNA for Sanger sequencing, performed by VIB Genetic Service Facility (University of Antwerp, Belgium).
Culture and treatment of EBV-transformed lymphoblastoid cell lines
EBV-transformed lymphoblastoid cell lines were generated from patient peripheral blood using standard protocols. Cells were propagated as a suspension culture in DMEM/F12 medium containing 10% fetal bovine serum. For experiments involving puromycin, cells were treated with 200 μg/ml puromycin (Sigma-Aldrich) overnight (15 h). Cells were washed twice with PBS before RNA extraction.
RNA extraction and RT–qPCR
Total RNA was extracted using TRIzol reagent (Life Technologies), following the manufacturer's protocol. The RNA extract was cleaned up using RNeasy kit (Qiagen). One μg of total RNA was used for cDNA synthesis (Quantitect Reverse Transcription kit, Qiagen), providing template for the qPCR. qPCR mixes were prepared using LightCycler® 480 SYBR Green I Master kit (Roche Life Science), following the manufacturer's instructions. The qPCR was carried out using the Roche LightCycler® 480 Real-Time PCR System, and software recommended standard 3-step cycles were used (95°C, 10 s; 60°C, 10 s, and 72°C, 10 s) for 40 cycles. Reactions were run in triplicate in three independent experiments, where each experiment was analyzed independently, with control (−puromycin) FMR1 levels were set to 1. The mean of expression values of the housekeeping gene HPRT was used as an internal control to normalize for loading variability. Fold changes in FMR1 expression were calculated using the ΔΔCT method and analyzed statistically with a two-tailed t-test (GraphPad). Error bars represent mean values with SD.
Western blotting
Protein from total cell lysates was resolved in NuPAGE 4–12% Bis–Tris polyacrylamide gels (Life technologies) under denaturing conditions and transferred to nitrocellulose membranes (Whatman, GE Healthcare Life Sciences). The blots were probed using anti-FMRP antibody (1C3 MAB2160 Millipore), diluted 1:500 in 3% bovine serum albumin, anti-GFP 1:750 (AB3080; Millipore), and anti-actin (JLA20 Hybridoma bank or MAB1501 Chemicon) diluted 1:500 or 1:10,000, respectively, in 5% milk. Anti-Dfmrp (5D7) antibody was specifically generated by EMBL Monoclonal Antibodies Core Facility and used in 1:500 dilution in 5% milk. ECL IgG horseradish peroxidase-linked antibodies (Amersham, GE Healthcare Life Sciences) or Li-Cor IRDye antibodies were used as secondary antibodies. Bands were visualized using the ECL Western Blotting Detection System (GE Healthcare Life Sciences) or the Odyssey system (Li-Cor). Band intensities for the different cell lines were quantified using ImageJ software, normalized for actin and analyzed statistically with a two-tailed t-test (GraphPad). Error bars represent mean values with SD.
Cloning
We acquired a plasmid in which the GFP-FMR1wt construct was cloned in a standard mammalian expression vector under a beta-actin promoter (Levenga et al, 2009). We used this plasmid as template for generating all FMR1 variant alleles. We modified the FMR1wt insert using site-directed mutagenesis (QuickChange II Site-directed Mutagenesis Kit, Stratagene) or (overlap extension) PCR (Phusion polymerase, NEB) with classical restriction enzyme cloning.
The fly overexpression constructs were created using the pUAST-attp-vector backbone. The dfmr1 variants were cloned from template wt dfmr1 cDNA. We modified the wild-type dfmr1 insert by using (overlap extension) PCR (Phusion polymerase, NEB) with classical restriction enzyme cloning.
Cell culture and transfection experiments
Human embryonic kidney (HEK) 293T cells and human neuroblastoma SH-SY5Y cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Lonza, Verviers, Belgium) supplemented with 10% fetal bovine serum, 1% penicillin, and 1% streptomycin at 37°C in a 5% CO2 humidified incubator. The cell lines used were standard and tested regularly for mycoplasma contamination and never found positive. Primary hippocampal neurons of Fmr1 KO(2) mice were prepared and cultured as described in de Vrij et al (2008). For each experiment, one pregnant Fmr1 KO(2) female mouse in the C57Bl/6 background was sacrificed, when embryos were at day E17. Neurons were then pooled from all embryos per litter, with average litter size being 7. The mice were housed at the Erasmus MC animal facility (Rotterdam, the Netherlands), under standard housing and husbandry conditions, approved by the local animal welfare committee.
Cultured cells were transfected with the appropriate expression constructs using polyethylenimine (PEI) (Polysciences Inc., Warrington, PA, USA) for 293T cells and Lipofectamine 2000/Plus Reagent (Invitrogen) for SH-SY5Y cells and 21-day-old primary mouse neurons according to standard manufacturers protocols. One day after transfection, cells were fixed with 4% formaldehyde, washed in PBS, and then washed in a Hoechst solution (0.67 mg/ml) (Invitrogen) before mounting in Mowiol mounting solution (Mowiol 4–88) after a final PBS wash step. For visualization of nucleoli, cells were incubated overnight with primary nucleophosmin (NPM1) antibody (Santa Cruz SC-6013) 1:100 in staining buffer containing 0.05 M Tris, 0.9% NaCl, 0.25% gelatin, and 0.5% Triton X-100, pH 7.4 at 4°C, followed by standard incubation with secondary anti-goat Cy3 1:200 (Jackson Immunoresearch). Then, the cells were fixed, stained with Hoechst and mounted in Mowiol. Imaging was done using a Leica SP5 confocal microscope and LAS AF lite software (Leica Microsystems). For the leptomycin B (LMB) experiments, 293T cells were seeded in 12-well plates and transfected with the appropriate constructs using PEI as well. Two days after transfection, 50 ng/ml LMB (Sigma) was added to the cells for four hours. For cell culture experiments, wild-type and mutant FMRP constructs were transfected in parallel, in duplicate, in at least three independent experiments. Cells were transfected in random order. During imaging, the experimenter was blinded to the transfection conditions.
Fly stocks and husbandry
Flies were raised at 25°C, in standard rearing conditions. Following fly strains were used for experiments:
yw; Pdf-Gal4, UAS-CD8-GFP; UAS-CD8-GFP (Ayaz et al, 2008)
w;; UAS-dfmr1wt/TM6C
w;; UAS-dfmr1ΔCt/TM6C
w;; UAS-dfmr1wt+NLS/TM6C
w;; UAS-dfmr1ΔCt+NLS/TM6C
Canton S 10 (w)
w;Pdf-Gal4 UAS-lacZ/Cyo;
UAS-RedStinger (Bloomington #8545)
ElavC155-Gal4 (Bloomington #458).
The UAS-dfmr1 strains were created using PhiC31 mediated transgenesis in the VK33 docking site (3L, 65B2) (Venken et al, 2006). Injection of the embryos was done in-house.
Immunochemistry on fly tissue
Brains were dissected during morning hours (with genotypes in random order) from 0- to 7-day-old adult flies and stained with primary antibodies using the standard protocol described in Soldano et al (2013). Primary antibodies anti-GFP (A-11222, Life Technologies, dil. 1:500), anti-βGal (A-11132, Life Technologies, dil. 1:1,000), anti-Dfmrp (20E4, specifically generated for our lab by EMBL-MACF Hybridoma, dil. 1:50) and Alexa Fluor® secondary antibodies (Life Technologies) were used. Images of the stained fly brains were acquired using confocal microscopy, Nikon AIR confocal unit mounted on a TI2000 inverted microscope (Nikon Corporations).
Quantification of axonal branching of LNv neurons
Maximum projections of the confocal stacks were analyzed using ImageJ software. During analysis, the experimenter was blinded to the genotypic conditions. Brains with gross mechanical damage from dissections/staining procedure were excluded from the analysis. A minimum sample size of 20 was analyzed for each genotype, deemed sufficient to detect changes in Dfmrp activity based on previous studies (Reeve et al, 2005, 2008). The branching areas of small LNv axonal termini were calculated based on parameters defined by Reeve et al (2005). The branching area was manually outlined, starting from the first point of defasciculation of the axonal bundle. Branching area measurements were normalized for variability in brain size across samples by dividing branching area values by total LNv commissure length for each brain. The Shapiro test for normality indicated that all were normally distributed (P > 0.05) except for the UAS-dFMR1wt sample, where the data, apart from one outlier, were also normally distributed. ANOVA indicated that genotype was a significant variable explaining variation in normalized axon branching values (F = 42.32, P < 0.0001). Two-tailed t-tests using Welch's correction were then used to compare controls with mutant phenotypes. Similar results were obtained when a nonparametric Wilcoxon test was used (not shown). Normalized branching area values for each genotype were then analyzed based on a two-tailed t-test with Welch's correction, using GraphPad Prism software. Error bars represent mean values with SD.
The paper explained
Problem
Fragile X syndrome (FXS) is the most common genetic cause of intellectual disability and includes features such as autistic-like behaviors, and distinctive craniofacial phenotypes. FXS is almost always caused by epigenetic silencing of the FMR1 gene. While the FMRP protein is heavily studied, there is only a single confirmed disease-causing point mutation reported thus far, making the link between functional analysis of the FMRP domains and the disease difficult to analyze.
Results
We sequenced patients with FXS symptoms but no epigenetic silencing of FMR1. We found a point mutation in one patient, which causes a frameshift in the FMRP sequence and a subsequent deletion of the C-terminal domain. This mutation changed the localization of FMRP from cytoplasmic to nuclear. Further analyses in human cells, and using Drosophila as an in vivo model, demonstrated a novel and evolutionarily conserved role for FMRP C-terminus in nuclear export. This change in localization caused the gain of a novel function in neurons in vivo.
Impact
Our study suggests that several FXS patients may remain undiagnosed because clinics only screen for the epigenetic silencing of FMR1 when FXS is suspected. Sequencing the coding region would be important to determine whether a patient has FXS, and may therefore benefit from future treatments. Furthermore, we identify the C-terminal domain of the FMRP as a nuclear export domain, perhaps explaining why naturally occurring nuclear isoforms of FMRP are generated by alternative splicing in the C-terminus.
Acknowledgments
We thank Dr. Aaron New, Dr. Alessia Soldano, Simon Weinberger, other members of the Hassan lab and Canmert Koral for helpful discussions and comments on the manuscript. We would like to thank M. Baghdadi and Layka Abbasi for technical assistance. This work was supported by VIB (to BAH), the Belgian Science Policy Interuniversity Attraction Pole (BELSPO IUAP) networks (P7/20-WiBrain to BAH and P7/43-BeMGI to HVE, KD, and GF), Fonds Wetenschappelijke Onderzoeks (FWO) Grants G.0543.08, G.0680.10, G.0681.10, G.0682.10, and G.0503.12 (to BAH), by grants from the Geconcerteerde Onderzoeks Acties (GOA) of the University of Leuven (GOA/12/015 to HVE, KD, and GF), by the Netherlands Organization for Health Research and Development (RW; ZonMw; 912-07-022), and FRAXA Research Organization (to RW and FMSdV). HVE and KD are clinical investigators of the FWO. The Nikon AIR confocal used in the study was acquired through the Hercules Type 1 AKUL.09.037 grant.
Author contributions
ZO, CEFdE, AC, JY, JV, GF, and FMSdV performed the experiments. HVE and KD provided clinical data and EBV cell lines. GF, FMSdV, RW, and BAH supervised the work. ZO and BAH wrote the manuscript with comments and edits from CEFdE, HVE, GF, FMSdV, and RW.
Conflict of interest
The authors declare that they have no conflict of interest.
For more information
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Ensembl Genome Browser, http://www.ensembl.org
ScanProsite Tool provided by Expasy, http://prosite.expasy.org/scanprosite/
Leiden Open Variation Database (LOVD), http://www.lovd.nl/3.0/home
Supporting Information
Supplementary Figures S1–S5
Review Process File
References
- Ayaz D, Leyssen M, Koch M, Yan J, Srahna M, Sheeba V, Fogle KJ, Holmes TC, Hassan BA. Axonal injury and regeneration in the adult brain of Drosophila. J Neurosci. 2008;28:6010–6021. doi: 10.1523/JNEUROSCI.0101-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Bakker CE, de Diego Otero Y, Bontekoe C, Raghoe P, Luteijn T, Hoogeveen AT, Oostra BA, Willemsen R. Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp Cell Res. 2000;258:162–170. doi: 10.1006/excr.2000.4932. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belser RC, Sudhalter V. Conversational characteristics of children with fragile X syndrome: repetitive speech. Am J Ment Retard. 2001;106:28–38. doi: 10.1352/0895-8017(2001)106<0028:CCOCWF>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44:724–728. doi: 10.1017/s0012162201002833. [DOI] [PubMed] [Google Scholar]
- Blackwell E, Zhang X, Ceman S. Arginines of the RGG box regulate FMRP association with polyribosomes and mRNA. Hum Mol Genet. 2010;19:1314–1323. doi: 10.1093/hmg/ddq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee B, Ikeda M, Budimirovic DB, Hjelm LN, Kaufmann WE, Warren ST. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: a case report and review of the literature. Am J Med Genet A. 2008;146A:1358–1367. doi: 10.1002/ajmg.a.32261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME, Warren ST. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A. 2010a;152A:2512–2520. doi: 10.1002/ajmg.a.33626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SC, Coffee B, Benke PJ, Berry-Kravis E, Gilbert F, Oostra B, Halley D, Zwick ME, Cutler DJ, Warren ST. Array-based FMR1 sequencing and deletion analysis in patients with a fragile X syndrome-like phenotype. PLoS ONE. 2010b;5:e9476. doi: 10.1371/journal.pone.0009476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish K, Sudhalter V, Turk J. Attention and language in fragile X. Ment Retard Dev Disabil Res Rev. 2004;10:11–16. doi: 10.1002/mrdd.20003. [DOI] [PubMed] [Google Scholar]
- Crowe SF, Hay DA. Neuropsychological dimensions of the fragile X syndrome: support for a non-dominant hemisphere dysfunction hypothesis. Neuropsychologia. 1990;28:9–16. doi: 10.1016/0028-3932(90)90082-y. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B, Van den Bos F, de Graaff E, Oostra BA, Willems PJ. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet. 1993;3:31–35. doi: 10.1038/ng0193-31. [DOI] [PubMed] [Google Scholar]
- Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4:335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- Dury AY, El Fatimy R, Tremblay S, Rose TM, Cote J, De Koninck P, Khandjian EW. Nuclear fragile X mental retardation protein is localized to cajal bodies. PLoS Genet. 2013;9:e1003890. doi: 10.1371/journal.pgen.1003890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1:109–118. doi: 10.1016/s1097-2765(00)80012-x. [DOI] [PubMed] [Google Scholar]
- Fernandez E, Rajan N, Bagni C. The FMRP regulon: from targets to disease convergence. Front Neurosci. 2013;7:191. doi: 10.3389/fnins.2013.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch GS, Simensen RJ, Schroer RJ. Longitudinal changes in cognitive and adaptive behavior scores in children and adolescents with the fragile X mutation or autism. J Autism Dev Disord. 2002;32:107–114. doi: 10.1023/a:1014888505185. [DOI] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Jr, Warren ST, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- Gedeon AK, Baker E, Robinson H, Partington MW, Gross B, Manca A, Korn B, Poustka A, Yu S, Sutherland GR, et al. Fragile X syndrome without CCG amplification has an FMR1 deletion. Nat Genet. 1992;1:341–344. doi: 10.1038/ng0892-341. [DOI] [PubMed] [Google Scholar]
- Gronskov K, Brondum-Nielsen K, Dedic A, Hjalgrim H. A nonsense mutation in FMR1 causing fragile X syndrome. Eur J Hum Genet. 2011;19:489–491. doi: 10.1038/ejhg.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Hull CE, Safanda JF, Carpenter I, Staley LW, O'Connor RA, Seydel C, Mazzocco MM, Snow K, Thibodeau SN, et al. High functioning fragile X males: demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am J Med Genet. 1994;51:298–308. doi: 10.1002/ajmg.1320510404. [DOI] [PubMed] [Google Scholar]
- Kim M, Bellini M, Ceman S. Fragile X mental retardation protein FMRP binds mRNAs in the nucleus. Mol Cell Biol. 2009;29:214–228. doi: 10.1128/MCB.01377-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick LL, McIlwain KA, Nelson DL. Comparative genomic sequence analysis of the FXR gene family: FMR1, FXR1, and FXR2. Genomics. 2001;78:169–177. doi: 10.1006/geno.2001.6667. [DOI] [PubMed] [Google Scholar]
- Kronk R, Bishop EE, Raspa M, Bickel JO, Mandel DA, Bailey DB., Jr Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep. 2010;33:679–687. doi: 10.1093/sleep/33.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachiewicz AM, Dawson DV. Behavior problems of young girls with fragile X syndrome: factor scores on the Conners' Parent's questionnaire. Am J Med Genet. 1994;51:364–369. doi: 10.1002/ajmg.1320510413. [DOI] [PubMed] [Google Scholar]
- Lachiewicz AM, Spiridigliozzi GA, Gullion CM, Ransford SN, Rao K. Aberrant behaviors of young boys with fragile X syndrome. Am J Ment Retard. 1994;98:567–579. [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Levenga J, Buijsen RA, Rife M, Moine H, Nelson DL, Oostra BA, Willemsen R, de Vrij FM. Ultrastructural analysis of the functional domains in FMRP using primary hippocampal mouse neurons. Neurobiol Dis. 2009;35:241–250. doi: 10.1016/j.nbd.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch DZ, Huggins RM, Hagerman RJ. Phenotypic variation and FMRP levels in fragile X. Ment Retard Dev Disabil Res Rev. 2004;10:31–41. doi: 10.1002/mrdd.20006. [DOI] [PubMed] [Google Scholar]
- Lugenbeel KA, Peier AM, Carson NL, Chudley AE, Nelson DL. Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat Genet. 1995;10:483–485. doi: 10.1038/ng0895-483. [DOI] [PubMed] [Google Scholar]
- McBride SM, Bell AJ, Jongens TA. Behavior in a Drosophila model of fragile X. Results Probl Cell Differ. 2012;54:83–117. doi: 10.1007/978-3-642-21649-7_6. [DOI] [PubMed] [Google Scholar]
- Morales J, Hiesinger PR, Schroeder AJ, Kume K, Verstreken P, Jackson FR, Nelson DL, Hassan BA. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron. 2002;34:961–972. doi: 10.1016/s0896-6273(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- Okray Z, Hassan BA. Genetic approaches in Drosophila for the study neurodevelopmental disorders. Neuropharmacology. 2013;68:150–156. doi: 10.1016/j.neuropharm.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Pan L, Zhang YQ, Woodruff E, Broadie K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14:1863–1870. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- Penagarikano O, Mulle JG, Warren ST. The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet. 2007;8:109–129. doi: 10.1146/annurev.genom.8.080706.092249. [DOI] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- Ramos A, Hollingworth D, Pastore A. G-quartet-dependent recognition between the FMRP RGG box and RNA. RNA. 2003;9:1198–1207. doi: 10.1261/rna.5960503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SP, Bassetto L, Genova GK, Kleyner Y, Leyssen M, Jackson FR, Hassan BA. The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr Biol. 2005;15:1156–1163. doi: 10.1016/j.cub.2005.05.050. [DOI] [PubMed] [Google Scholar]
- Reeve SP, Lin X, Sahin BH, Jiang F, Yao A, Liu Z, Zhi H, Broadie K, Li W, Giangrande A, et al. Mutational analysis establishes a critical role for the N terminus of fragile X mental retardation protein FMRP. J Neurosci. 2008;28:3221–3226. doi: 10.1523/JNEUROSCI.5528-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Heitz D, Tarleton J, MacPherson J, Malmgren H, Dahl N, Barnicoat A, Mathew C, Mornet E, Tejada I, et al. A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: the first 2,253 cases. Am J Hum Genet. 1994;55:225–237. [PMC free article] [PubMed] [Google Scholar]
- Schrier M, Severijnen LA, Reis S, Rife M, van't Padje S, van Cappellen G, Oostra BA, Willemsen R. Transport kinetics of FMRP containing the I304N mutation of severe fragile X syndrome in neurites of living rat PC12 cells. Exp Neurol. 2004;189:343–353. doi: 10.1016/j.expneurol.2004.05.039. [DOI] [PubMed] [Google Scholar]
- Siomi H, Choi M, Siomi MC, Nussbaum RL, Dreyfuss G. Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell. 1994;77:33–39. doi: 10.1016/0092-8674(94)90232-1. [DOI] [PubMed] [Google Scholar]
- Siomi MC, Siomi H, Sauer WH, Srinivasan S, Nussbaum RL, Dreyfuss G. FXR1, an autosomal homolog of the fragile X mental retardation gene. EMBO J. 1995;14:2401–2408. doi: 10.1002/j.1460-2075.1995.tb07237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldano A, Okray Z, Janovska P, Tmejova K, Reynaud E, Claeys A, Yan J, Atak ZK, De Strooper B, Dura JM, et al. The Drosophila homologue of the amyloid precursor protein is a conserved modulator of Wnt PCP signaling. PLoS Biol. 2013;11:e1001562. doi: 10.1371/journal.pbio.1001562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha MS, Nouri K, Milroy LG, Moll JM, Herrmann C, Brunsveld L, Piekorz RP, Ahmadian MR. Subcellular fractionation and localization studies reveal a direct interaction of the fragile X mental retardation protein (FMRP) with nucleolin. PLoS ONE. 2014;9:e91465. doi: 10.1371/journal.pone.0091465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamanini F, Bontekoe C, Bakker CE, van Unen L, Anar B, Willemsen R, Yoshida M, Galjaard H, Oostra BA, Hoogeveen AT. Different targets for the fragile X-related proteins revealed by their distinct nuclear localizations. Hum Mol Genet. 1999;8:863–869. doi: 10.1093/hmg/8.5.863. [DOI] [PubMed] [Google Scholar]
- Tamanini F, Kirkpatrick LL, Schonkeren J, van Unen L, Bontekoe C, Bakker C, Nelson DL, Galjaard H, Oostra BA, Hoogeveen AT. The fragile X-related proteins FXR1P and FXR2P contain a functional nucleolar-targeting signal equivalent to the HIV-1 regulatory proteins. Hum Mol Genet. 2000;9:1487–1493. doi: 10.1093/hmg/9.10.1487. [DOI] [PubMed] [Google Scholar]
- Tarleton J, Richie R, Schwartz C, Rao K, Aylsworth AS, Lachiewicz A. An extensive de novo deletion removing FMR1 in a patient with mental retardation and the fragile X syndrome phenotype. Hum Mol Genet. 1993;2:1973–1974. doi: 10.1093/hmg/2.11.1973. [DOI] [PubMed] [Google Scholar]
- Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Dockendorff TC, Jongens TA, Dreyfuss G. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol Cell Biol. 2000;20:8536–8547. doi: 10.1128/mcb.20.22.8536-8547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R, Bontekoe C, Tamanini F, Galjaard H, Hoogeveen A, Oostra B. Association of FMRP with ribosomal precursor particles in the nucleolus. Biochem Biophys Res Commun. 1996;225:27–33. doi: 10.1006/bbrc.1996.1126. [DOI] [PubMed] [Google Scholar]
- Wohrle D, Kotzot D, Hirst MC, Manca A, Korn B, Schmidt A, Barbi G, Rott HD, Poustka A, Davies KE, et al. A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am J Hum Genet. 1992;51:299–306. [PMC free article] [PubMed] [Google Scholar]
- Wolff B, Sanglier JJ, Wang Y. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem Biol. 1997;4:139–147. doi: 10.1016/s1074-5521(97)90257-x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, O'Connor JP, Siomi MC, Srinivasan S, Dutra A, Nussbaum RL, Dreyfuss G. The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J. 1995;14:5358–5366. doi: 10.1002/j.1460-2075.1995.tb00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1–S5
Review Process File