

Abstract
Genotoxic drugs constitute a major treatment modality for human cancers; however, cancer cells' intrinsic DNA repair capability often increases the threshold of lethality and renders these drugs ineffective. The emerging roles of HDACs in DNA repair provide new opportunities for improving traditional genotoxic drugs. Here, we report the development and characterization of CY190602, a novel bendamustine-derived drug with significantly enhanced anticancer potency. We show that CY190602's enhanced potency can be attributed to its newly gained ability to inhibit HDACs. Using this novel DNA/HDAC dual-targeting drug as a tool, we further explored HDAC's role in DNA repair. We found that HDAC activities are essential for the expression of several genes involved in DNA synthesis and repair, including TYMS, Tip60, CBP, EP300, and MSL1. Importantly, CY190602, the first-in-class example of such DNA/HDAC dual-targeting drugs, exhibited significantly enhanced anticancer activity in vitro and in vivo. These findings provide rationales for incorporating HDAC inhibitory moieties into genotoxic drugs, so as to overcome the repair capacity of cancer cells. Systematic development of similar DNA/HDAC dual-targeting drugs may represent a novel opportunity for improving cancer therapy.
Keywords: DNA repair, dual-targeting anticancer drug, HDAC, nitrogen mustard
Introduction
A significant challenge for treating cancer is to enhance the efficacy of existing drugs. Genotoxic drugs represent an important branch of chemotherapy, which kill cancer cells by attacking cellular DNA. Double-strand breaks (DSBs) resulting from such attacks are extremely toxic, and one irreparable DSB is sufficient to induce cell death (Jackson & Bartek, 2009). Despite such potent lethality, DNA damage caused by anticancer drugs can be mitigated by cellular DNA repair machinery, thus enabling some cancer cells to survive and ultimately cause treatment failure. Drugs that inhibit DNA repair, such as ATM and DNAPK inhibitors, have been intensely investigated as potential means to improve chemotherapy (Hickson et al, 2004; Helleday et al, 2008; Willmore et al, 2008; Jiang et al, 2009).
Nitrogen mustards are a major type of genotoxic anticancer drugs. They work by attacking the N7 position of guanines on opposing DNA strands, thereby causing DNA interstrand cross-linkings (ICLs), which impede DNA replication forks and ultimately cause DSBs. Several pathways exist in mammalian cells to repair such damage, including the Fanconi anemia (FA) pathway, translesional synthesis (TLS), and homologous recombination (HR) (Knipscheer et al, 2009; Moldovan & D'Andrea, 2009). As a result, nitrogen mustards are generally well tolerated by cancer cells. Consequently, these drugs exhibit poor potency and limited treatment success. An emerging approach is to suppress DNA repair in order to enhance the efficacy of nitrogen mustards. Several lines of evidence support the potential benefits of this approach. For example, human patients with inherited deficiencies in FA pathway are extremely sensitive to ICLs (Taniguchi & D'Andrea, 2006), and tumors with mutations in the FA and HR pathways are hypersensitive to ICL agents (Van der Heijden et al, 2005; Kennedy & D'Andrea, 2006; Byrski et al, 2010). Efforts to screen for inhibitors of these repair pathways have yielded interesting results that may benefit future anticancer therapy (Chirnomas et al, 2006).
Bendamustine, a nitrogen mustard originally synthesized in the 1960s, recently regained great clinical interest due to its beneficial outcome in treating cancers (Keating et al, 2008; Cheson & Rummel, 2009; Knauf, 2009; Knauf et al, 2009; Garnock-Jones, 2010; Flinn et al, 2014). The recently approved clinical indications for bendamustine include chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), follicular lymphoma (FL), and mantle-cell lymphoma (MCL). However, the efficacy of bendamustine is limited by its poor drug potency. Therefore, bendamustine represents an interesting candidate for drug optimization. Here, we report our effort in developing and characterizing a more potent bendamustine derivative and argue for a knowledge-based redesign of existing genotoxic drugs.
Results
To improve bendamustine's anticancer activity, a series of chemical derivatives were synthesized. Among these, CY190602 (Fig1A, and referred to as CY thereafter) displayed 50- to 100-fold enhanced anticancer toxicity. Treatment of the NCI60 cell lines (http://dtp.nci.nih.gov/branches/btb/ivclsp.html) indicated a median growth inhibitor concentration (GI50) of 2.2 μM for CY, in contrast to the median GI50 of 77 μM for bendamustine (Fig1B). Of note, the GI50 for CY in MEFs was 57 μM, about 20-fold higher than its average GI50 (3.2 μM) in NCI60 cell lines, suggesting that it may preferentially kill cancer cells. Comparing the GI50 data of CY and bendamustine, such a significant increase in drug potency is rather surprising, given that CY differs from bendamustine only on its side chain, furthest away from the purported nitrogen mustard functional group. To understand CY's mechanism of action, we utilized a functional genetic approach for drug characterization (Jiang et al, 2011). Using this approach, we previously reported that despite the dramatic increase in potency, CY's primary mode of cell death induction is still nitrogen mustard-mediated DNA damage (Jiang et al, 2011).
Figure 1. The side chain structure of CY enhances nitrogen mustard activity in cancer cells.
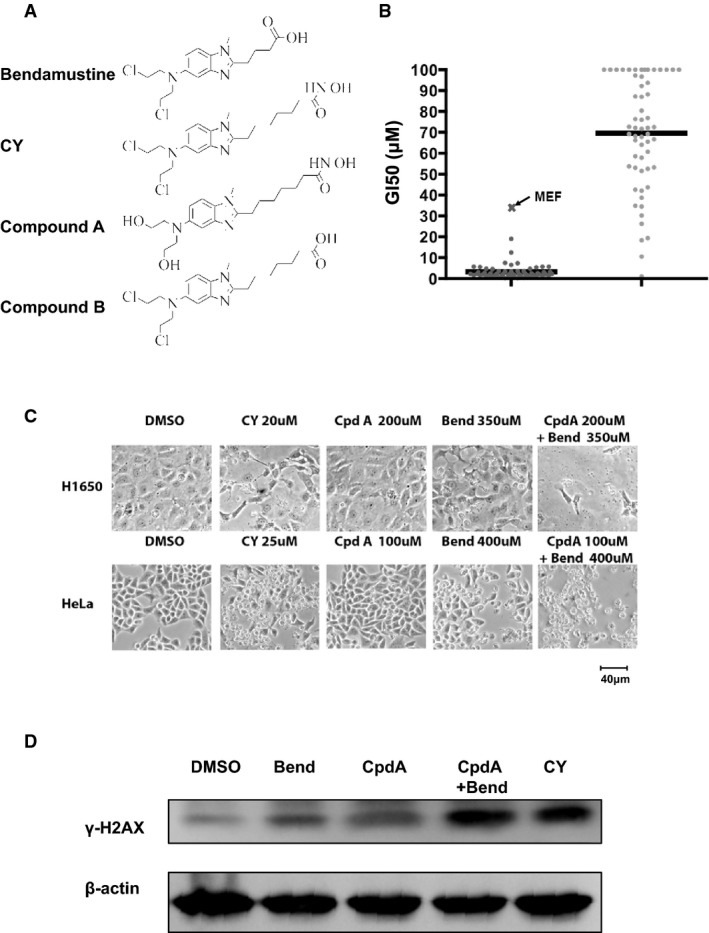
- Chemical structures of bendamustine, CY, compound A, and compound B (referred to as Cpd A and Cpd B hereafter).
- Growth inhibitory concentration (GI50) of CY and bendamustine in the NCI60 cell line panel.
- Nontoxic dose of Cpd A enhances bendamustine cytotoxicity in cancer cells. Images were taken at 48 h posttreatment.
- Cpd A augments DNA damage caused by bendamustine. H1650 cells were treated with 100 μM Bend, 50 μM Cpd A, 100 μM Bend plus 50 μM Cpd A, or 10 μM CY for 12 h, and cell lysates were blotted for γ-H2AX. Actin was used as a loading control.
Source data are available online for this figure.
To further understand how CY's chemical modifications might have resulted in such an increased potency, we synthesized compound A (Fig1A), in which the chloride atoms of CY's nitrogen mustard group were substituted with hydroxyl groups. As a result, this compound retains the side chain modification of CY but lacks ability to attack DNA. Study of this compound would therefore enable us to focus on the side chain of CY. Consistent with the notion that CY primarily kills cancer cells through its nitrogen mustard group, compound A is very ineffective in killing cancer cells, even at 200 μM concentration (Fig1C). Interestingly, despite its inability to kill cancer cells, compound A significantly synergized with bendamustine to kill cancer cells (Fig1C). This suggests that compound A, and therefore the side chain of CY, is capable of enhancing the activity of bendamustine. Moreover, although compound A alone did not cause DNA damage, it significantly increased the level of γ-H2AX in bendamustine-treated cells (Fig1D). Taken together, these results suggest that although the side chain group of CY is ineffective in killing cancer cells by itself, it is capable of enhancing the action of CY's nitrogen mustard group, which may explain CY's significantly increased anticancer potency.
Reexamination of CY's side chain suggested that it conforms to several rules of hydroxamic acid-based HDAC inhibitors (Suzuki et al, 2005). First, aromatic rings of CY facilitate its interaction with the surface of HDAC's enzyme pocket. Second, CY's 7-carbon side chain mimics the length of a lysine residue, which is optimal for inserting into HDAC's enzyme pocket. Third, CY's terminal hydroxamic acid group serves to chelate the zinc atom inside HDAC's enzyme pocket, thereby inhibiting HDAC's enzymatic activity. Indeed, when tested in cell-free HDAC activity assays, CY exhibited an IC50 of 10–100 nM against HDAC1-6 in vitro (Fig2A). In intact cells, CY and Cpd A also induced significant accumulation of acetylated histone H3 (Fig2B and Supplementary Fig S1). In contrast, bendamustine lacks both the proper side chain length and the zinc-chelating hydroxamic acid group, which explains its inability to inhibit HDAC (Fig2A and 2B, Supplementary Fig S1). Lastly, molecular docking of CY with the active sites of HDAC1 and HDAC2's X-ray structure suggested that CY is indeed capable of fitting into HDAC active sites (Fig2C).
Figure 2. CY harbors HDAC inhibitory activity that enhances nitrogen mustard's anticancer efficacy.
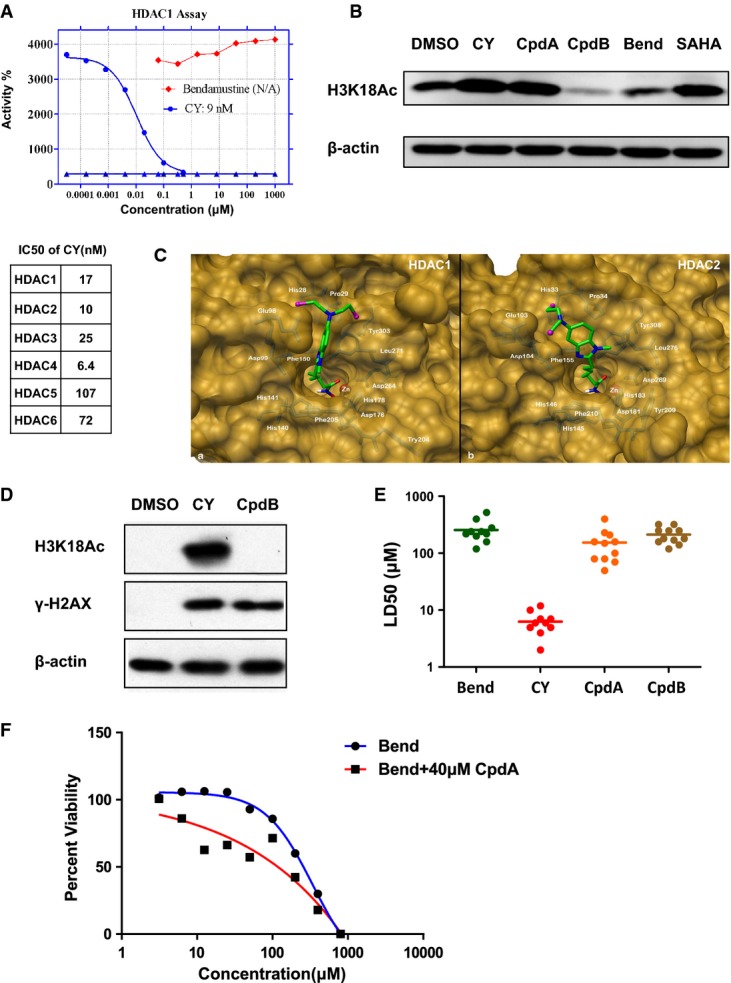
- Upper panel, in vitro HDAC1 inhibition assay using bendamustine and CY. Lower panel, summary of CY IC50 against other HDACs.
- CY and CpdA, but not bendamustine or CpdB, inhibited HDAC activities in cancer cells. H1650 cells were treated with 20 μM CY, 20 μM CpdA, 200 μM CpdB, 200 μM Bend, or 10 μM SAHA for 12 h, and cell lysates were blotted for histone acetylation markers. Actin was used as a loading control.
- Molecular docking of CY with the structure of HDAC1/2 enzyme pocket.
- Alteration of CY side chain (Cpd B) caused loss of HDAC inhibitory activity and resulted in reduced ability to induce DNA damage. Cells were treated for 12 h. Cells treated with 200 μM Cpd B and 20 μM CY exhibited similar levels of γ-H2AX. Actin was used as a loading control.
- Summary of lethal dose 50 (LD50) of bendamustine, CY, Cpd A (HDAC inhibition only), and Cpd B (nitrogen mustard only) against 11 human cancer cell lines.
- Cpd A enhances the anticancer activity of bendamustine. H1650 cells were treated with various doses of bendamustine with or without 40 μM Cpd A (nontoxic dose, see Fig1C and Supplementary Fig S2) for 48 h and subjected to MTT viability assays.
Source data are available online for this figure.
Taken together, these results suggest that CY is a dual-targeting drug, with both the DNA-damaging nitrogen mustard group and an HDAC inhibitory moiety. To address whether CY's HDAC inhibitory activity is essential for its anticancer activity, we synthesized compound B (Fig1A), in which CY's hydroxamic acid group was substituted with a carboxylic acid group. Since carboxylic acid is far less effective in chelating zinc, this compound harbors little HDAC inhibitory activity (Fig2D). We found that this compound is 10- to 20-fold less potent than CY (Fig2E). This suggests that HDAC inhibitory group is essential for CY's enhanced anticancer activity. Given that both compound A (HDAC inhibitor only) and compound B (nitrogen mustard only) are far inferior in their ability to kill cancer cells, these results also suggested that these two functional groups synergize with each other to confer significantly enhanced anticancer potency. Such a synergy between nitrogen mustard and HDAC inhibitors was confirmed by treating cells with bendamustine and nontoxic dose of HDAC inhibitor Cpd A (Fig2F and Supplementary Fig S2). To our knowledge, CY represents the first-in-class example of such DNA/HDAC dual-targeting drug that utilizes such intra-molecular synergy.
Given that nontoxic doses of the HDAC inhibitor compound A were capable of enhancing DNA damage caused by bendamustine (Fig2E), we hypothesized that the function of CY's nitrogen mustard group may be greatly potentiated by its HDAC inhibitory group. Next, we performed several experiments to address the mechanism of such potentiation.
Several recent publications argue for HDAC's crucial involvement in DNA repair (Miller et al, 2010; Robert et al, 2011). Although several mechanisms have been proposed, the full range of HDAC's function in DNA repair remains undefined. Given HDAC's important roles in regulating gene expression, we asked whether expressions of certain DNA repair genes are deregulated in CY-treated cells. According to current model, there are several core groups of ICL repair genes, including those participating in the ATR-Chk1, FA, TLS, and HR pathways (Knipscheer et al, 2009). In addition, since TLS and HR require active DNA synthesis, genes involved in dNTP production may also significantly impact ICL repair. To address how these groups of genes might be affected by HDAC inhibition, we treated cells with bendamustine, CY or HDAC inhibitor SAHA, and compared the expression level of more than 50 genes belonging to these aforementioned gene groups (Supplementary Tables S1 and S2) as well as many other genes that have been reported to regulate DNA repair. Among these genes, we found that the expression levels of three histone acetyltransferases Tip60, CBP, and MORF, and MSL1, a gene associated with the histone acetyltransferases MOF (Smith et al, 2005; Huang et al, 2012), were all greatly suppressed in CY-treated cells as early as 6 h (Fig3A), and such downregulation was not caused by drug-induced cell death (Fig3B). Tip60 has been reported to regulate ATM-mediated DNA repair (Sun et al, 2005; Kaidi & Jackson, 2013), whereas CBP have been shown to regulate ATR-Chk1 pathway and chromatin remodeling at DNA lesions (Hasan et al, 2001; Stauffer et al, 2007). MSL1 has also been shown to affect DNA repair (Gironella et al, 2009; Aguado-Llera et al, 2013). Importantly, shRNA suppression of these four genes (Fig3C, Supplementary Fig S3) each sensitized cells to bendamustine (Fig3D, Supplementary Tables S3 and S4). Of note, suppression of these genes alone did not affect cellular viability without drug treatment (Supplementary Fig S4). Taken together, our data demonstrated that in addition to other reported mechanisms (Miller et al, 2010; Robert et al, 2011), HDAC inhibition could increase the potency of nitrogen mustards through downregulation of genes that regulate DNA repair, including Tip60, CBP, MORF, and MSL1. Our study of CY as a prototype of DNA/HDAC dual-targeting drug demonstrates that by incorporating HDAC inhibitory moiety into traditional DNA-damaging drugs, it is indeed possible to achieve much higher toxicity against cancer cells.
Figure 3. HDAC inhibition by CY leads to downregulation of several histone acetyltransferases involved in DNA repair.
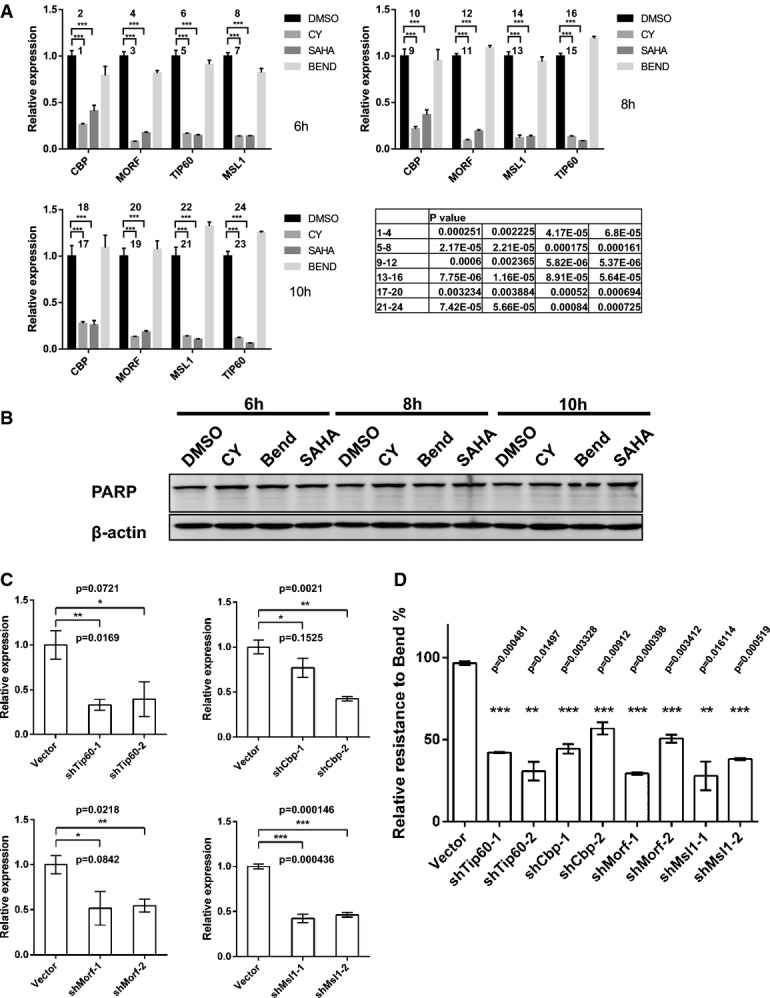
- CY, but not bendamustine treatment, led to suppression of CBP, TIP60, MORF, and MSL1 in H1650 cells. Cells were treated with DMSO, bendamustine (350 μM), CY (15 μM), or SAHA (10 μM) for 6 h, and mRNA was extracted for qPCR analysis. Data represent mean ± SEM from three independent experiments, and statistical significance was determined by unpaired two-tailed t-test. ***P < 0.01.
- Drug treatment up to 10 h at the concentrations indicated in (A) did not cause apoptosis as judged by PARP cleavage. Actin was used as a loading control.
- ShRNA-mediated suppression of CBP, TIP60, MORF, and MSL1 at mRNA level. Data represent mean ± SEM from three independent experiments, and statistical significance was determined by unpaired two-tailed t-test. *P < 0.1, **P < 0.05, ***P < 0.01.
- Suppression of CBP, TIP60, MORF, and MSL1 sensitized cells to bendamustine. Y-axis represents relative resistance calculated from results of GFP competition assays, and statistical significance was determined by unpaired two-tailed t-test. Data represent mean ± SEM from two independent experiments. **P < 0.05, ***P < 0.01.
Source data are available online for this figure.
Lastly, we studied the antitumor activity of this dual-targeting drug in vivo. We first determined that the MTD (maximally tolerated doses) of CY is 60 mg/kg in mice (Fig4A). Next, we used a transplantable BCR-ABL-driven acute lymphoblastic leukemia (ALL) mouse model to assess CY's in vivo activity. BCR-ABL-positive ALL accounts for about 1/3 of adult human ALL cases and is traditionally treated with many types of chemotherapeutics. Despite the use of heavy chemotherapy regimen, patients with this disease have a very poor survival rate (Stock, 2010). Treatment with targeted therapeutics that inhibit BCR-ABL, such as dasatinib, is an emerging therapy approach for this disease (Yanada et al, 2009). We chose this mouse model therefore in order to compare CY with a wide range of traditional chemotherapeutics as well as targeted therapeutics in an in vivo setting.
Figure 4. CY exhibited enhanced anticancer activity in vivo.

- A Determination of maximally tolerated dose (MTD) of CY at 60 mg/kg. Other drugs were used at MTD according to the literature. For each drug, n = 5 and all mice survived treatment. Data represent mean ± SEM.
- B CY exhibited enhanced activity against BCR-ABL Arf−/− murine ALL cells in vitro.
- C, D CY showed superior antitumor activity compared with bendamustine, SAHA, and other chemotherapeutic drugs in mice transplanted with BCR-ABL ALL cells. P-values of CY versus other drugs: NT (P = 5.02E-06) (C), SAHA (P = 4.45E-06), Bend (P = 5.18E-06), NT (P = 1.20E-05) (D), VCR (P = 1.13E-05), Dox (P = 8.08E-06), AraC (P = 7.62E-06), and CTX (P = 1.13E-05). Survival statistical analysis was done with the Mantel–Cox (log-rank) test of GraphPad Prism.
- E Therapeutic effects of CY (weekly dose) and targeted drugs PP242 (daily dose) and dasatinib (daily dose) in mice transplanted with BCL-ABL ALL cells. P-values of CY versus other drugs: NT (P = 3.94E-05), PP242 (P = 1.20E-05), and dasatinib (P = 0.99). Survival statistical analysis was done with the Mantel–Cox (log-rank) test of GraphPad Prism.
- F, G Antitumor activity of CY and bendamustine in nude mice models. Mice were inoculated with human NSCLC cell line H460. When tumor volume reached 200 mm3, mice were treated with single dose of bendamustine (40 mg/kg, MTD) or CY (20, 40, 60 mg/kg). In (G), tumors were dissected out after 15 days posttreatment.
In cultured BCR-ABL ALL cells, CY is more active than bendamustine (Fig4B). Mice transplanted with BCR-ABL cells died around 12 days after the injection of 40,000 leukemia cells without treatment (Fig4C). Treatment with SAHA or bendamustine at MTD extended survival by approximately 2 and 5 days, respectively. In contrast, mice treated with CY showed average survival extension of 14 days (Fig4C). Moreover, when compared with other chemotherapeutic agents commonly used in BCR/ABL ALL human patients, including cyclophosphamide, doxorubicin, cytarabine, and vincristine, CY's survival extension was also superior to these drugs (Fig4D). This indicated that the DNA/HDAC dual-targeting approach confers better therapeutic outcome and may have possible clinical advantages.
In both mouse models and human patients, BCR-ABL ALL can be effectively managed by continuous administration of the BCR-ABL/Src inhibitor dasatinib (Gruber et al, 2009; Boulos et al, 2011). In addition, a recent report indicated potential efficacy of mTOR inhibitor PP242 in BCR-ABL ALL mouse model (Janes et al, 2010). Next, we compared CY's efficacy with targeted therapeutics dasatinib and PP242 in this ALL model. To access the long-term clinical benefit of CY treatment, we developed a weekly CY treatment schedule that was well tolerated. Consistent with existing report (Janes et al, 2010), daily treatment of the mTOR inhibitor PP242 extended the mice survival by an average of 8 days (Fig4E). In contrast, daily doses of dasatinib extended the survival up to 2 months. Importantly, weekly doses of CY prolonged the survival to a similar extent as dasatinib (Fig4E), further demonstrating CY's potent anticancer effects in vivo. Lastly, in xenograft models using the human lung cancer cell line H460, CY also showed potent anticancer activity that was superior to bendamustine (Fig4F and G). Taken together, these data showed that the DNA/HDAC dual-targeting drug CY has potent anticancer activity in vivo.
Given that bendamustine has shown significant clinical efficacies in treating chronic lymphocytic leukemia (CLL), we further tested CY's efficacy in freshly isolated human CLL cells. CY killed nearly all CLL cells at 5 μM. In contrast, bendamustine, and fludarabine, another CLL frontline drug, at 100 μM could only kill about 80% CLL cells (Fig5A). Moreover, despite CY's high efficacy in killing CLL cells at 5 μM, healthy primary B cells still remain about 40% viable after treatment with 20 μM CY (Fig5B). These data show that CY potently kills CLL cells, and there is a preferential killing of transformed cells over healthy, non-transformed B cells.
Figure 5. CY exhibited activity with CLL samples.

- Dose curve of CY, fludarabine, and bendamustine using fresh CLL samples. Data represent mean ± SD.
- CY preferentially killed CLL cells over healthy B cells. Data represent mean ± SD.
- Toxicity of CY190602 in patients harboring deletion of chromosome 17p (including p53 locus) compared to patients without del17p. Viability of samples was normalized to background apoptosis for individual patient samples. For all experiments, cell viability was determined after 72 h of drug treatment. Data represent mean ± SD.
In clinics, bendamustine is ineffective in treating CLL cells with 17p deletion. CLL cells harboring this deletion lose p53 and become refractory to bendamustine treatment (Zaja et al, 2013). Interestingly, CY kills 17p-deleted and 17p-retaining CLL cells with similar efficacy (Fig5C), suggesting that CY may represent a potential new choice for exploring treatment strategies for 17p-deleted CLLs.
Discussion
In this report, we described the development and characterization of CY, a bendamustine-derived, DNA/HDAC dual-targeting anticancer drug. In our previous report (Jiang et al, 2011), we tested CY in several cell lines and found it to exhibit significantly enhanced anticancer activity in vitro; however, the source of such enhanced activity remained undefined. One important question is whether there are generally applicable rules that we can derive from CY, so that we can apply such rules to improve other existing anticancer drugs. In this report, we dissected this phenomenon and provided mechanistic explanation. Biochemically, CY is capable of both inducing DNA damage and inhibiting HDACs (Figs1D and 2B). In a panel of 60 commonly used cancer cell lines, CY exhibited 50- to 100-fold increased activity compared to the parental compound bendamustine. In vivo, CY also exhibited far-superior anticancer efficacy compared to bendamustine. Given that bendamustine has recently been shown to exert strong clinical activity in several types of human malignancies, it should be interesting to test CY's efficacy in these disease settings.
One interesting question remains whether CY would be superior to the combination treatment of bendamustine and HDAC inhibitors. Due to both activities being tethered in the same molecule of CY, it is possible that by delivering both DNA damaging and HDAC inhibitory ability at the same time to tumor cells in vivo, CY may work better than the combinatory treatment of bendamustine and HDAC inhibitor, whose pharmacodynamics and pharmacokinetics differ in vivo. Unfortunately due to the difference in treatment schedule (bendamustine, single dose; SAHA, daily dose) and toxicity issues, we could not establish an effective treatment schedule that involves both bendamustine and SAHA. Considering that there is currently no protocol using both bendamustine and SAHA in clinics and that bendamustine is effective in several cancer types as a single drug, we therefore focused on comparing the efficacy between CY and bendamustine in vivo. Our results suggested that CY had significantly enhanced in vivo anticancer activity compared to bendamustine in several transplanted and xenograft cancer models. Moreover, the BCR-ABL mouse ALL model enabled us to compare the efficacy of CY with several other commonly used anticancer drugs. Our results showed that CY has superior anticancer activity over several first-line chemotherapeutic drugs, and in this model CY's efficacy is even comparable to the targeted drug, BCR-ABL inhibitor dasatinib. Taken together, the data suggested that this novel DNA/HDAC dual-targeting drug CY has significant anticancer efficacy in vivo.
Although HDAC inhibitors have been investigated as radio-sensitizing agents (Ree et al, 2010), the complete picture of HDAC in DNA repair remained elusive. Several recent publications shed light on this topic and suggested that HDAC may function directly at sites of DNA damage by altering local histone code (Miller et al, 2010; Robert et al, 2011). In addition, a recent proteomic study found that many DNA repair proteins including MDC1, BLM, and Rad50 are modified by acetylation after HDAC inhibition (Choudhary et al, 2009). It is possible that HDAC inhibition may negatively impact the stability and/or activity of these repair proteins. For example, it was shown that upon HDAC inhibition, the HR nuclease Sae2/CtIP is acetylated and degraded (Robert et al, 2011). In addition, HDAC inhibition can suppress the ATM signaling pathway, thereby sensitizing cancer cells to DNA damage (Thurn et al, 2013).
In this report, we focused on HDAC's role in DNA repair by demonstrating its rapid transcriptional control of other groups of important DNA repair genes. We examined gene expression level after 6-h treatment with CY or SAHA in multiple types of cancer cell lines. Interestingly, among genes whose expression is significantly suppressed upon HDAC inhibition, Tip60, CBP, MORF, and MSL1 are all histone acetyltransferases or histone acetyltransferase-associated protein, and shRNA suppression of these genes sensitized cells to DNA damage. Moreover, we also found that upon HDAC inhibition, another histone acetyltransferase EP300, and TYMS, a gene involved in nucleotide synthesis, are both transcriptionally downregulated upon HDAC inhibition (Supplementary Fig S5), and shRNA suppression of either gene by itself is lethal to cancer cells even without DNA damage (Supplementary Fig S6). Because cells with TYMS or EP300 shRNAs were rapidly eliminated in cell culture, we could not analyze whether loss of these two genes further sensitized cells to DNA damage. However, given their important roles in nucleotide pool maintenance and DNA repair (Hasan et al, 2001), it is rather possible that downregulation of these two genes upon HDAC inhibition could cause further impairment in DNA repair.
Of note, previous reports have suggested that BRCA1 and the NHEJ repair genes Ku70, Ku80, and DNAPKcs are suppressed by HDAC inhibitors (Zhang et al, 2007, 2009). In our hand, unlike the case for Tip60, CBP, MORF, MSL1, and EP300, suppression of BRCA1, Ku70, Ku80, and DNAPKcs did not occur at 6 h upon HDAC inhibition, suggesting that it may not be an early response upon HDAC inhibitor treatment. To further examine this discrepancy, we searched the connectivity map (Lamb et al, 2006), a consortium of microarray data, and identified 12 microarray datasets from cells treated by HDAC inhibitors. Importantly, none of the previously reported genes (BRCA1, Ku70, Ku80, and DNAPKcs) were among the top 200 downregulated genes in any of these HDAC inhibitor-treated cells. In contrast, Tip60, CBP, MORF, MSL1, and EP300 were among the top 200 downregulated genes in 5, 6, 11, 9, and 5 of the 12 HDAC inhibitor-treated cells, suggesting that this is a highly potent and consistent effect of HDAC inhibitors. Importantly, given our finding that HDAC inhibition caused rapid and significant downregulation of these histone acetyltransferases or histone acetyltransferase-associated protein, it is possible that upon HDAC inhibition, a transcriptional feedback mechanism is activated to downregulate cellular acetyltransferase activity, which subsequently caused impairment of cellular DNA repair capacity. This may constitute a major transcription-related mechanism that contributes to HDAC inhibitor-mediated repression of DNA repair.
Given the potent lethality of irreparable DNA strand breaks, approaches to suppressing DNA repair have been intensively investigated, as they may bring significant benefits to cancer therapy. The recent success of PARP inhibitors in treating BRCA-deficient tumors showcases the therapeutic potential of such approach. Inhibitors of kinases with long-established roles in DNA repair, such as ATM, ATR, and DNAPKcs, are in various stages of preclinical and clinical investigations for their potential benefits in improving traditional chemotherapy. Moreover, with the recent advances in our understanding of DNA repair, several additional groups of enzymes have been recognized for their involvement of DNA repair, including HDACs (Miller et al, 2010; Robert et al, 2011), histone acetyltransferases (Sun et al, 2009; Niida et al, 2010), ubiquitin ligases (Kolas et al, 2007; Stewart et al, 2009), deubiquitinases (Nakada et al, 2010), SUMO ligases (Galanty et al, 2009; Morris et al, 2009), and histone methyltransferases (Liu et al, 2010). Pharmacological targeting of these enzymes may also enhance the efficacy of traditional genotoxic anticancer drugs. Our study of CY as a prototype of DNA/HDAC dual-targeting drug demonstrates that by incorporating small enzyme inhibitory moiety into traditional DNA-damaging drugs, we can achieve higher toxicity against cancer cells. Importantly, our result showed that it is structurally compatible to incorporate small enzyme inhibitory chemical moieties into DNA damage drugs, and such modifications can significantly enhance nitrogen mustard's anticancer activity. On the basis of this rationale, we have developed a novel nitrogen mustard derivative that targets both DNA and CDK, and it also showed about 100-fold increases in anticancer efficacy. This suggests that it may be generally applicable to incorporate various enzyme inhibitory moieties into traditional genotoxic drug to achieve better efficacy. Taken together, it is interesting to develop other types of HDAC/DNA dual-targeting drugs, as well as other types of drugs that target both DNA and some of the enzymes involved in DNA repair. Such drugs may by itself improve cancer treatment, and their much-improved anticancer efficacy also offers new possibilities for antibody-coupled, tumor-targeted drug delivery research. This may provide several novel categories of anticancer drugs for clinical investigation.
Materials and Methods
Cell culture and reagents
Cell lines were cultured using standard protocols provided by ATCC. Myc Arf−/− cells were cultured as described (Jiang et al, 2009). BCR-ABL mouse ALL cells were a kind gift from Dr. Richard Williams. CY190602 and its derivatives compound A and B were synthesized by Dr. Wang's Laboratory (U. New Mexico). Other chemicals were purchased from Selleck, EMD, or VWR. Antibodies against γ-H2AX (Millipore), actin (Sigma), H3K18Ac, H3K9Ac, and H3K56Ac (Cell Signaling) were used for Western blotting.
shRNA construct cloning, RNA preparation and qPCR
Retroviral pMSCV-IRES-GFP vector and the cloning procedures have been previously described (Jiang et al, 2009, 2011). Target-gene knockdown efficiency was analyzed by qPCR. Total mRNA of cells was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instruction. Total mRNA was transcribed to cDNA with the SuperScriptIII First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA).
Cell viability assays and determination of relative drug resistance
Cells were seeded in 96-well plates, treated with different concentrations of drugs for 48 h, and cell viability was analyzed by MTT assays according to manufacturer's protocols. Experiments to determine GI50 with the NCI60 panel cell lines (http://dtp.nci.nih.gov/branches/btb/ivclsp.html) were performed at NCI (Alley et al, 1988; Shoemaker, 2006).
To test how shRNA suppression of certain genes affects cellular sensitivity to drugs, we used a GFP-based competition experimental system previously described by Jiang et al (Jiang et al, 2011). Briefly, shRNA and GFP were stably transduced into cells via retroviral vectors; therefore, cells in which targeted genes were knocked down also express GFP. A mixture of knockdown cells (GFP positive) and control cells (no viral infection, GFP negative) was treated with drugs. If knockdown of target gene sensitizes cells to drug, then in the survival cell population, the percentage of GFP-positive (gene knockdown) cells will decrease. By comparing GFP percentages with and without drug treatment, we can calculate relative resistance or sensitivity caused by target-gene knockdown, using a method previously described.
Mouse experiments
Experimental procedures were approved by the Animal Care and Use Committee of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. In all, 200 wild-type C57BL/6 mice (6 weeks old, female) were used for determining maximally tolerated dose of CY, and testing CY and other drugs' efficacy in the BCR-ABL ALL model. BCR-ABL cells have been previously described (Williams et al, 2006). For in vivo experiments, 1 million cells were injected into mice via tail vein (Williams et al, 2006). At 7 days postinjection, mice were treated with indicated drugs at their MTDs. Mice were monitored daily for survival after drug treatment. Survival curve and statistical analysis were done using the Prism software. For xenograft H460 model, in all, nude mice (8 weeks old, female) were used to test CY and bendamustine's in vivo efficacy. The numbers of mice used in each experimental group are labeled on Fig4.
CLL patients and cells
This study was approved by the ethics committee of the University of Cologne (approval 01-163). Blood samples were obtained from patients fulfilling diagnostic criteria for CLL with informed consent according to the Helsinki protocol. Only patients without prior therapy or at least 12 months without prior chemotherapy were included in this study. Fresh blood samples were enriched by applying B-RosetteSep (StemCell Technologies, Vancouver, Canada) to aggregate unwanted cells with erythrocytes and Ficoll-Hypaque (Seromed, Berlin, Germany) density gradient purification resulting in purity > 98% of CD19+/CD5+ CLL cells. CLL cells were characterized for CD19, CD5, CD23, FMC7, CD38, ZAP-70, slgM, slgG, CD79a, and CD79b expression on a FACSCanto flow cytometer (BD PharMingen, Heidelberg, Germany). Controls were isolated from healthy blood donors using anti-CD19 MACS beads (Miltenyi, Bergisch Gladbach, Germany) resulting in at least 95% CD19+ B cells.
Apoptosis and cell viability assays
Apoptosis was determined by flow cytometry using Annexin V-FITC/7AAD staining (BD PharMingen) after 72 h. The cellular potency of compounds as defined by half-maximal induction of apoptosis in primary CLL cells was determined using concentrations up to 100 μM. The fraction of viable cells was determined by counting annexin V/7-AAD double-negative cells for each individual dosage. Median values were subsequently applied for regression analysis and calculation of the half-maximal dosage effect (IC50). Curve fitting was performed using SigmaPlot (SPSS, Chicago). IC50 values were determined by fitting data to the Hill equation y = y0 + (axb)/(cb+xb).
The paper explained
Problem
Most existing anticancer drugs attack single targets in cancer cells, which, however, activate various strategies to repair the damage induced by the drugs or to counter their efficacy. This significantly limits the efficacy of existing drugs and can cause treatment failure and illustrates the need for developing novel, more potent anticancer drugs.
Results
We developed and characterized a novel dual-targeting drug based on side chain modification of the nitrogen mustard drug bendamustine, with high potency against cancer. This drug not only attacks DNA, but also inhibits HDACs, a group of enzymes important for DNA repair, so that cancer cells cannot readily repair the damage it causes. We also show that this dual-targeting drug has significantly improved efficacy over drugs that have single targets in many cancer cell lines and various cancer mouse models.
Impact
Our results indicate that by incorporating two cooperating anticancer chemical groups into the same compound, dual-targeting drugs with higher efficacy can be generated. This approach can therefore be used to systematically increase the potency of traditional DNA-damaging drugs. Such dual-targeting drugs may provide new categories of anticancer drugs for cancer treatment.
Acknowledgments
We thank the Developmental Therapeutics Department of the NCI/NIH for testing CY190602 (NSC#751447) in their NCI60 anticancer screening program and for supplying bendamustine HCl salt. This work was supported by the major scientific research project (2013CB910404), the National Natural Science Foundation of China (31371418; 81372854; 81102010; 81202096), and Changhai Hospital 1255 discipline construction projects (No. CH125530400). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. M.T.H. is the Eisen and Chang Career Development Associate Professor of Biology and is supported by NIH RO1 CA128803 and the Ludwig Center for Molecular Oncology at MIT.
Author contributions
HJ, MTH and YC were involved in study conception and design. HJ and MTH contributed to the development of methodology. CL, HD, XL, LH, CPP, DG, YC, DW and WW were involved in acquisition of data such as provision of animals and facilities, and acquisition and management of patients. HJ, CL, HD and CPP were involved in analysis and interpretation of data, such as statistical analysis, biostatistics, and computational analysis. HJ, MTH and YW participated in the writing, review, and/or revision of the manuscript. HJ, MTH and YW participated in the study supervision.
Conflict of interest
Yi Chen is an employee of Crystal Biopharmaceutical LLC, a cancer drug discovery and development company located in Pleasanton, CA, USA. Dianwu Guo is the CSO of Hangzhou Minsheng Pharma Research Institute Ltd, who is developing CY190602 in China market. Liya Hong currently works in Zhejiang Institute for Food and Drug Control.
Supporting Information
Supplementary Figures and Tables
Source Data for Supplementary Figure S3
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
References
- Aguado-Llera D, Hamidi T, Doménech R, Pantoja-Uceda D, Gironella M, Santoro J, Velázquez-Campoy A, Neira JL, Iovanna JL. Deciphering the binding between Nupr1 and MSL1 and their DNA-repairing activity. PLoS ONE. 2013;8:e78101. doi: 10.1371/journal.pone.0078101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- Boulos N, Mulder HL, Calabrese CR, Morrison JB, Rehg JE, Relling MV, Sherr CJ, Williams RT. Chemotherapeutic agents circumvent emergence of dasatinib-resistant BCR-ABL kinase mutations in a precise mouse model of Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;117:3585–3595. doi: 10.1182/blood-2010-08-301267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, Mierzwa T, Szwiec M, Wisniowski R, Siolek M, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28:375–379. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- Cheson BD, Rummel MJ. Bendamustine: rebirth of an old drug. J Clin Oncol. 2009;27:1492–1501. doi: 10.1200/JCO.2008.18.7252. [DOI] [PubMed] [Google Scholar]
- Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M, Hartman A-R, Kennedy R, Foster R, Mahoney J, Seiden MV, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006;5:952–961. doi: 10.1158/1535-7163.MCT-05-0493. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Flinn IW, van der Jagt R, Kahl BS, Wood P, Hawkins TE, Macdonald D, Hertzberg M, Kwan Y-L, Simpson D, Craig M, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123:2944–2952. doi: 10.1182/blood-2013-11-531327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935–939. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnock-Jones KP. Bendamustine: a review of its use in the management of indolent non-Hodgkin's lymphoma and mantle cell lymphoma. Drugs. 2010;70:1703–1718. doi: 10.2165/11205860-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Gironella M, Malicet C, Cano C, Sandi MJ, Hamidi T, Tauil RMN, Baston M, Valaco P, Moreno S, Lopez F, et al. p8/nupr1 regulates DNA-repair activity after double-strand gamma irradiation-induced DNA damage. J Cell Physiol. 2009;221:594–602. doi: 10.1002/jcp.21889. [DOI] [PubMed] [Google Scholar]
- Gruber F, Mustjoki S, Porkka K. Impact of tyrosine kinase inhibitors on patient outcomes in Philadelphia chromosome-positive acute lymphoblastic leukaemia. Br J Haematol. 2009;145:581–597. doi: 10.1111/j.1365-2141.2009.07666.x. [DOI] [PubMed] [Google Scholar]
- Hasan S, Hassa PO, Imhof R, Hottiger MO. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature. 2001;410:387–391. doi: 10.1038/35066610. [DOI] [PubMed] [Google Scholar]
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NMB, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GCM. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- Huang J, Wan B, Wu L, Yang Y, Dou Y, Lei M. Structural insight into the regulation of MOF in the male-specific lethal complex and the non-specific lethal complex. Cell Res. 2012;22:1078–1081. doi: 10.1038/cr.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–1909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Pritchard JR, Williams RT, Lauffenburger DA, Hemann MT. A mammalian functional-genetic approach to characterizing cancer therapeutics. Nat Chem Biol. 2011;7:92–100. doi: 10.1038/nchembio.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidi A, Jackson SP. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. 2013;498:70–74. doi: 10.1038/nature12201. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Keating MJ, Bach C, Yasothan U, Kirkpatrick P. Bendamustine. Nat Rev Drug Discov. 2008;7:473–474. doi: 10.1038/nrd2596. [DOI] [PubMed] [Google Scholar]
- Kennedy RD, D'Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24:3799–3808. doi: 10.1200/JCO.2005.05.4171. [DOI] [PubMed] [Google Scholar]
- Knauf W. Bendamustine in the treatment of chronic lymphocytic leukemia. Expert Rev Anticancer Ther. 2009;9:165–174. doi: 10.1586/14737140.9.2.165. [DOI] [PubMed] [Google Scholar]
- Knauf WU, Lissichkov T, Aldaoud A, Liberati A, Loscertales J, Herbrecht R, Juliusson G, Postner G, Gercheva L, Goranov S, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol. 2009;27:4378–4384. doi: 10.1200/JCO.2008.20.8389. [DOI] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Liu H, Takeda S, Kumar R, Westergard TD, Brown EJ, Pandita TK, Cheng EH-Y, Hsieh JJ-D. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature. 2010;467:343–346. doi: 10.1038/nature09350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan G-L, D'Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature. 2009;462:886–890. doi: 10.1038/nature08593. [DOI] [PubMed] [Google Scholar]
- Nakada S, Tai I, Panier S, Al-Hakim A, Iemura S-I, Juang Y-C, O'Donnell L, Kumakubo A, Munro M, Sicheri F, et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature. 2010;466:941–946. doi: 10.1038/nature09297. [DOI] [PubMed] [Google Scholar]
- Niida H, Katsuno Y, Sengoku M, Shimada M, Yukawa M, Ikura M, Ikura T, Kohno K, Shima H, Suzuki H, et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev. 2010;24:333–338. doi: 10.1101/gad.1863810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ree AH, Dueland S, Folkvord S, Hole KH, Seierstad T, Johansen M, Abrahamsen TW, Flatmark K. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010;11:459–464. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Smith ER, Cayrou C, Huang R, Lane WS, Côté J, Lucchesi JC. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol. 2005;25:9175–9188. doi: 10.1128/MCB.25.21.9175-9188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer D, Chang B, Huang J, Dunn A, Thayer M. p300/CREB-binding protein interacts with ATR and is required for the DNA replication checkpoint. J Biol Chem. 2007;282:9678–9687. doi: 10.1074/jbc.M609261200. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- Stock W. Current treatment options for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Leuk Lymphoma. 2010;51:188–198. doi: 10.3109/10428190903452834. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11:1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Nagano Y, Kouketsu A, Matsuura A, Maruyama S, Kurotaki M, Nakagawa H, Miyata N. Novel inhibitors of human histone deacetylases: design, synthesis, enzyme inhibition, and cancer cell growth inhibition of SAHA-based non-hydroxamates. J Med Chem. 2005;48:1019–1032. doi: 10.1021/jm049207j. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, D'Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107:4223–4233. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- Thurn KT, Thomas S, Raha P, Qureshi I, Munster PN. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther. 2013;12:2078–2087. doi: 10.1158/1535-7163.MCT-12-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Heijden MS, Brody JR, Dezentje DA, Gallmeier E, Cunningham SC, Swartz MJ, DeMarzo AM, Offerhaus GJA, Isacoff WH, Hruban RH, et al. In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin Cancer Res. 2005;11:7508–7515. doi: 10.1158/1078-0432.CCR-05-1048. [DOI] [PubMed] [Google Scholar]
- Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2006;103:6688–6693. doi: 10.1073/pnas.0602030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmore E, Elliott SL, Mainou-Fowler T, Summerfield GP, Jackson GH, O'Neill F, Lowe C, Carter A, Harris R, Pettitt AR, et al. DNA-dependent protein kinase is a therapeutic target and an indicator of poor prognosis in B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2008;14:3984–3992. doi: 10.1158/1078-0432.CCR-07-5158. [DOI] [PubMed] [Google Scholar]
- Yanada M, Ohno R, Naoe T. Recent advances in the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia. Int J Hematol. 2009;89:3–13. doi: 10.1007/s12185-008-0223-z. [DOI] [PubMed] [Google Scholar]
- Zaja F, Mian M, Volpetti S, Visco C, Sissa C, Nichele I, Castelli M, Ambrosetti A, Puglisi S, Fanin R, et al. Bendamustine in chronic lymphocytic leukemia: outcome according to different clinical and biological prognostic factors in the everyday clinical practice. Am J Hematol. 2013;88:955–960. doi: 10.1002/ajh.23546. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Carr T, Dimtchev A, Zaer N, Dritschilo A, Jung M. Attenuated DNA damage repair by trichostatin A through BRCA1 suppression. Radiat Res. 2007;168:115–124. doi: 10.1667/RR0811.1. [DOI] [PubMed] [Google Scholar]
- Zhang F, Zhang T, Teng Z-H, Zhang R, Wang J-B, Mei Q-B. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol Ther. 2009;8:823–831. doi: 10.4161/cbt.8.9.8143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Tables
Source Data for Supplementary Figure S3
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3