

Abstract
Transdermal patches are now widely used as cosmetic, topical and transdermal delivery systems. These patches represent a key outcome from the growth in skin science, technology and expertise developed through trial and error, clinical observation and evidence-based studies that date back to the first existing human records. This review begins with the earliest topical therapies and traces topical delivery to the present-day transdermal patches, describing along the way the initial trials, devices and drug delivery systems that underpin current transdermal patches and their actives. This is followed by consideration of the evolution in the various patch designs and their limitations as well as requirements for actives to be used for transdermal delivery. The properties of and issues associated with the use of currently marketed products, such as variability, safety and regulatory aspects, are then described. The review concludes by examining future prospects for transdermal patches and drug delivery systems, such as the combination of active delivery systems with patches, minimally invasive microneedle patches and cutaneous solutions, including metered-dose systems.
Tables of Links
| LIGANDS | ||
|---|---|---|
| Clonidine | Methylphenidate | Oxybutynin |
| Dihydroergotamine | Methyl salicylate | Rivastigmine |
| Dimenhydrinate (diphenhydramine) | Naltrexone | Rotigotine |
| Ephedrine | Nicotine | Scopolamine |
| Fentanyl | Nitroglycerin | Selegiline |
| Haloperidol | Sumatriptan | Stilboestrol |
| Lidocaine | Oestradiol | Sufentanil |
| Testosterone |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
The skin is the largest organ in the human body by mass, with an area of between 1.5 and 2.0 m2 in adults. Drugs have been applied to the skin to treat superficial disorders, for the transdermal administration of therapeutics to manage systemic ailments and as cosmetics, dating back to the oldest existing medical records of man. For instance, the use of salves, ointments, potions and even patches, consisting of plant, animal or mineral extracts, was already popular in ancient Egypt and in Babylonian medicine (around 3000 BC) (Magner, 2005; Geller, 2010). However, the routine use of transdermal delivery systems only became a common practice in the latter third of the 20th century when delivery technology was developed to enable precise and reproducible administration through the skin for systemic effects.
The goal of this review is to detail the rich history of topical and transdermal delivery that has evolved over thousands of years, focusing particularly on the evolution and current use of transdermal patches. The potential efficacy and suitability of this technology for systemic therapy is normally determined by drug blood level–time profiles, which can be compared to or predicted from p.o. or parenteral administration. These drug concentrations in the blood are, in turn, defined by the amount of drug released into the body from the delivery system and the application area. Transdermal delivery is also used to produce clinical effects, such as local anaesthesia and anti-inflammatory activity, deep within or beneath the skin. In contrast, topical delivery seeks to treat superficial, although at times very serious, skin problems through a relatively local action.
History
Early use of topical therapy (pre-20th century)
Topical remedies anointed, bandaged, rubbed or applied to the skin (Figure 1A) are likely to have been used since the origin of man, with the practices becoming evident with the appearance of written records, such as on the clay tablets used by the Sumerians (Kramer, 1963). Indeed, it has been suggested that a liquefied ochre-rich mixture, made some 100 000 years ago and found at the Blombos Cave in South Africa, may have been used for decoration and skin protection (Henshilwood et al., 2011). Ancient Egyptians used oil (e.g. castor, olive and sesame), fats (mainly animals), perfumes (e.g. bitter almond, peppermint and rosemary) and other ingredients to make their cosmetic and dermatological products (unguents, creams, pomades, rouges, powders, and eye and nail paints) (Forbes, 1955). The mineral ores of copper (malachite: green) and lead (galena: dark grey) were used to prepare kohl, a paste used to paint the eyes. Red ochre was used as a lip or face paint, and a mixture of powdered lime and oil was used as a cleansing cream (Lucas and Harris, 1962). The ancient lead-based products were applied for both appearance and, based upon religious beliefs, for protection against eye diseases (Tapsoba et al., 2010). However, these effects may have been real as recent studies involving incubation of low lead ion concentrations with skin cells produced NO (Tapsoba et al., 2010), which is known to provide defence against infection (Coleman, 2001). On the negative side, it could be asked if these lead products also caused toxicity, noting that high blood levels of lead have been reported in modern kohl users (Hallmann, 2009).
Figure 1.

Historical development of patches. Early topical products: (A) products from ancient times; (B) Galen's cold cream; (C) mercurial ointment; (D) mustard and belladonna plasters; controlled dosing of topical products. (E) First quantitative systemic delivery (Zondek's system). (F) Individualized delivery system: nitroglycerin ointment. (G) Topical delivery device (Wurster & Kramer's system). Passive non-invasive patches. (H) First patch system – the reservoir – introduced for scopolamine, nitroglycerin, clonidine and oestradiol. (I, J, K) Other types of patches – matrix and drug-in-adhesive (e.g. fentanyl and nicotine patches). Next-generation patches. (L) Cutaneous solutions (e.g. Patchless Patch®, Evamist®). (M) Active patches (e.g. iontophoresis, Zecuity®). (N) Minimally invasive patches (e.g. microneedles, Nanopatch®).
The well-known Papyrus Ebers (1550 BC), describing more than 800 prescriptions and about 700 drugs, appears to be the best pharmaceutical record from ancient times (LaWall, 1927). It contains many recipes for treating skin conditions, including burns, wounds, blisters and exudation. Other remedies are to preserve the hair, to make the hair grow, to improve the skin and to beautify the body. A poultice (with 35 ingredients) is reported for the weakness of the male member. Other remedies are the first transdermal delivery of drugs for systemic effects, such as the topical application of frankincense to expel pain in the head and a product applied to the belly of a woman or a man to expel pains caused by tapeworm (Bryan, 1930; Ebbell, 1937). The emphasis on topical treatments at that time is evident by the portrayal of an ointment workroom in an Egyptian tomb painting from 1400 BC (Kremers, 1976).
A millennium and a half later, Galen (AD 129–199), a Greek physician, introduced the compounding of herbal drugs and other excipients into dosage forms. He is widely considered to be the ‘Father of Pharmacy’ and his practices are known as ‘Galenic pharmacy’. Galen's Cerate (Cérat de Galien), a cold cream (Figure 1B), is certainly his most renowned formula with a composition relatively similar to the one used today (Bender and Thom, 1966). Medicated plasters (emplastra), which were generally applied to the skin for local conditions, can be traced back to Ancient China (around 2000 BC) and are the early predecessors of today's transdermal patches (emplastra transcutanea). These early plasters generally contained multiple ingredients of herbal drugs dispersed into an adhesive natural gum rubber base applied to a backing support made of fabric or paper (Chien, 1987). Nicotine, a new-world transdermal agent, was already being used in a plaster (Emplastrum opodeldoch) during the time of Paracelsus (1493–1541) (Aiache, 1984). Unlike the medicated plasters that originated in China, Western-type medicated plasters were much simpler formulations in that they contained only a single active ingredient. Examples of plasters that were listed in the United States Pharmacopoeia (USP) almost 70 years ago included belladonna (used as a local analgesic), mustard (as an effective local irritant) and salicylic acid (as a keratolytic agent) (Pfister, 1997). The concept that certain drugs cross the skin appears to have been applied by Ibn Sina (AD 980–1037), a Persian physician best known as Avicenna within the Western World. In The Canon of Medicine, he proposed that topical drugs have two spirits or states: soft and hard. He suggested that when topical products are applied to the skin, the soft part penetrates the skin whereas the hard part does not. He further proposed that dermally applied drugs not only have local effects but also affect tissues immediately beneath the skin including joints (regional effects) as well as effects in remote areas (systemic effects). One of his topical formulations acting systematically was for conditions where drugs could not be taken orally. One of Avicenna's regional therapies was the use of a plaster-like formulation in which sulphur was mixed with tar and applied to the skin with a piece of paper applied as backing to keep the formulation in place. This product was used to treat sciatica, that is, pain arising from the compression of the sciatic nerve felt in the back, hip and outer side of the leg (Moghimi et al., 2011). Other forerunners of modern transdermal medications include mercurial ointments (Unguentum Hydrargyri) that were used for the treatment of syphilis in the late 15th century (Figure 1C) (Cole et al., 1930). Unguentum Hydrargyri Fortius L. (stronger mercurial ointment), made of purified mercury, lard and suet (Castle, 1828; Coxe, 1830; Pereira, 1839), is one example of these preparations.
The late 19th century as a phase of ‘non-belief’ in transdermal products
The German Pharmacopoeia 1872, a compilation produced in Latin, listed 28 Emplastra formulae. These included adhesive products (e.g. Emplastrum adhaesivum, which contained oleic acid, lead oxide and colophony, and Emplastrum adhaesivum anglicum, a hydrophilic formula); products meant to produce systemic effects [e.g. Emplastrum aromaticum, which contained peppermint and other aromatic oils targeted for the treatment of the stomach; Emplastrum belladonnae (Figure 1D), from Atropa belladonna leaves, which was meant for the treatment of tuberculosis and tumours; Emplastrum opiatum, which was used to reduce stomach movement and associated pain; Emplastrum conii containing Conium maculatum (poison hemlock, as used by Socrates), which was thought useful for treating tuberculosis and tumours]; and products for topical use (e.g. Emplastrum hydrargyri with pure quicksilver for treating topical swellings and infections, Emplastrum cantharidum ordinarium, a vesicant, Emplastrum picis irritans and Emplastrum fuscum for dealing with topical infections). However, many of these disappeared in later formulations so that the German Pharmacopoeia 2 of 1883 had reduced the number of patch monographs to 11 – Leukoplast® [BSN Medical (formely Beiersdorf) Hamburg, Germany], which is still used was invented in 1882. Nevertheless, in 1877, one review still suggested that intact human skin was totally impermeable to all substances (Fleischer, 1877) – even though several cases of systemic poisoning after external application of belladonna (e.g. plaster, liniment and lotion) were reported in the British Medical Journal in the 1860–1870s (Morgan, 1866; Harrison, 1872).
Development of topical products in the 20th century
In 1904, Schwenkenbecker generalized that the skin was relatively permeable to lipid-soluble substances but not to water and electrolytes (Schwenkenbecker, 1904). Various cases of poisoning, mostly in children, were reported in the early 1900s in France after topical application of nitrobenzene or aniline dyes in dyed clothing or shoes (The Lancet annotations, 1902; White, 1909; Muehlberger, 1925), and further supported the notion of the potential systemic absorption of topical products. The death arising from the systemic absorption of phenol from a large body surface in a young man after the accidental spillage of a bottle of phenol over himself (Johnstone, 1948) emphasized the potential lethal consequences associated with accidental ‘overexposure’ to drugs applied to the skin. However, lethality was promoted by the corrosive nature of phenol at higher concentrations, causing a substantial enhancement of human skin penetration (Roberts et al., 1977) and the saturation of the sulphate and glucuronidation pathways present in the body for its detoxification (Mellick and Roberts, 1999). A more recent series of reports described the potential lethal toxicity arising from exposure to hexachlorophene after topical application to babies (Martin-Bouyer et al., 1982).
In the beginning of the 20th century, various in vivo studies demonstrated systemic absorption after topical application by estimating drug levels in blood, urine and faeces (Malkinson and Rothman, 1963). Initial analytical methods were strictly qualitative and substances were detected in the blood or urine by looking at the change in a measured sample with regard to its colour, acidity or density relative to that of a standard sample (Scheuplein and Blank, 1971). Mercury, one of the first therapeutic compounds to be detected and then quantified in human excreta, was initially detected in urine following inunction treatment of syphilis using amalgamation methods (i.e. Reinsch test) (Wile and Elliott, 1917). Later more accurate analytical methods (e.g. using a calibrated capillary tube) enabled the quantitative determination of 5 mg of mercury in 1 L of solution (Cole et al., 1926). Colorimetric methods were commonly used. The concentration of p-chloro-m-xylenol (a halogenated phenol) in biological materials (i.e. urine, blood and minced tissues) was determined using Millon's reagent (an aqueous solution of mercury and nitric acid). The dirty red compound that was formed was then extracted by ether to give a clear yellow solution suitable for photometric measurements (Zondek et al., 1943). The absorption of methyl salicylate from various vehicles in 10 male subjects was studied via excretion in the urine of its salicylate metabolite using a colorimetric titration with ferric alum (Brown and Scott, 1934). The absorption of free iodine, through unbroken dog skin, was investigated by redox titration of the iodine eliminated in the urine with sodium thiosulphate (Nyiri and Jannitti, 1932). The penetration-promoting effect of a polyethylene glycol ointment was investigated in vivo in humans by determining the excreted concentration of phenolsulfonphthalein that was used as a tracer dye using a photoelectric colorimeter (Nadkarni et al., 1951).
In other early studies, characteristic pharmacological or physiological end points were used as proof of absorption of compounds into the systemic circulation (Gemmell and Morrison, 1957). For instance, sex hormones were widely investigated using experimental animals as subjects. Testosterone or testosterone propionate applied as an ointment to the skin of castrated male guinea pigs was shown to be readily absorbed as the accessory reproductive organs remained functional (Moore et al., 1938). Similarly, the application of oestrogen to the shaven back skin of ovariectomized female mice, using vehicles containing ethanol and/or benzol, led to oestrus (Zondek, 1938). The occurrence of convulsions in mice, rats and guinea pigs was observed following external application of the highly toxic strychnine alkaloids (Macht, 1938). The percutaneous absorption of another alkaloid, eserine, was studied using the amount and colour of secretion of tears in rats in response to ACh potentiated by the topically applied eserine. This method was used as a physiological end point for different ointment bases (Hadgraft and Somers, 1954). One questionable method used to determine the amount of mercury absorbed following application of mercurial ointment made with different bases was based upon the amount of mercurial ointment recovered after scraping a defined skin surface area with a pre-weighed razor blade, that is, the difference in applied and recovered weight represented the amount of ointment absorbed by the skin (Wild, 1911; Wild and Roberts, 1926).
The introduction of radioactive trace substances later offered a new approach for studying the systemic absorption through the skin. Unlike the methods described earlier, radioactive tracer methods permitted the detection of small quantities in biological materials. For instance, Hadgraft et al. (1956) detected small quantities of radioactivity in the rat blood after the topical application of [131I]diiodofluorescein in five different ointment bases.
Development of topical products with systemic effects
The first quantitative report of clinically managing a systemic condition by topical application appears to be the work of Zondek, now some 70 years ago. He reported that chloroxylenol, an external disinfectant still present in antiseptic soaps and solutions today (Dettol®; Reckitt Benckiser, Slough, Berkshire, UK), could be effective in the treatment of urogenital infections when topically applied as a 30% lanolin ointment (Figure 1E) (Zondek, 1942a,b). Interestingly, the potential percutaneous absorption of the drugs now found in many of our current transdermal products has been demonstrated much earlier through inadvertent toxicity after topical exposure during manufacturing, consumer use of the products and in farming. For instance, nitroglycerin permeation across human skin, now used transdermally to prevent and to treat angina, first came to light in the early 1900s as a side effect –‘nitroglycerin head’ – a severe headache experienced by people working in the manufacture of explosives or otherwise handling nitroglycerin-containing materials (Laws, 1898; 1910; Evans, 1912). Experimentally, 1 and 10% alcoholic nitroglycerin solutions applied topically to the forearm of healthy humans led to prolonged systemic effects (i.e. headache, changes in BP and pulse rate), with volunteers eventually showing an acquired tolerance to headache effects after an average of 38 h (Crandall et al., 1931). However, it was not until 1948 that a nitroglycerin ointment was successfully applied to treat Raynaud's disease (Fox and Leslie, 1948; Lund, 1948). This work led to a 2% nitroglycerin ointment (Nitrol®; Kremers Urban Company, Seymour, IN, USA) being used to treat angina pectoris in the 1950s. Here, a wooden applicator was used to measure the dose of nitroglycerin applied to the chest (Davis and Wiesel, 1955). A clinical trial published in 1974 demonstrated a sustained prophylactic efficacy lasting for up to 5 h (Reichek et al., 1974). However, the ointment was messy and needed to be applied several times a day. Concerns remained about the exact amount of drug being applied each time (No authors listed, 1976). As another example, systemic adverse effects of nicotine, the transdermal smoking cessation drug, became apparent after topical contact associated with its use as a topical insecticide (Wilson, 1930; Faulkner, 1933; Lockhart, 1933). In addition, nicotine absorption was noted among workers harvesting tobacco leaves in the form of green tobacco sickness (Gehlbach et al., 1974; 1975,). The percutaneous absorption of oestrogens was discovered in the 1940s when men working in stilboestrol plants noticed an enlargement of their breasts (Scarff and Smith, 1942; Fitzsimons, 1944).
The development of adhesive transdermal delivery devices
Dale Wurster's contribution to the early understanding of transdermal delivery is seldom acknowledged (Roberts, 2013). Important components of that work, often associated with transdermal delivery, are the defined delivery system in dose, area, vehicle and device; the quantification of the time course of absorption into urine; and the application of pharmacokinetic principles to quantify the resulting drug delivery kinetics. In Wurster's first set of transdermal studies, his student Sherman Kramer glued a diffusion cell containing a defined dose of salicylate esters to the forearm of his human volunteers and then measured their systemic absorption by the excretion of salicylates in the urine. The extent of absorption could be modified by varying the diffusion area of the cell and by changing the level of skin hydration (Wurster and Kramer, 1961). The primitive diffusion cell designed (Figure 1G) and used in their study appears very much to be the forerunner of cells currently used in transdermal research and could even be considered a first prototype of today's commercial transdermal devices in that the in vivo diffusion cell permitted a precise, area-dependent dosing of a topically applied drug (Roberts, 2013). There are now a number of salicylate esters and other non-steroidal anti-inflammatory products on the market for local pain relief. Skin biopsies and microdialysis have been used to show their selective targeting of deeper tissues in preference to the systemic blood supply (Cross et al., 1998; Roberts and Cross, 1999). More recently, we have suggested that the dermal vasculature is a major conduit to deeper tissues for highly bound anti-inflammatory drugs based upon our analysis of the available microdialysis data (Dancik et al., 2012) and for corticosteroids by biopsy (Anissimov and Roberts, 2011).
Ten years after Kramer's studies, the first patent using a rate-controlling membrane to control the rate of transdermal delivery from a bandage for the continuous delivery through the skin of drugs into the systemic circulation was filled by the biochemist and entrepreneur Alejandro Zaffaroni (1923–2014) (Zaffaroni, 1971). In 1972, Beckett et al. compared the systemic absorption of ephedrine (and ephedrine analogues) through the skin to that achieved with p.o. administration. They fastened an ephedrine and ethanol solution spread over an adhesive, impervious occlusive tape to a male human subject (Beckett et al., 1972). The data obtained with this ‘transdermal patch’ were subsequently analysed by Riegelman (1974). It was concluded that the ‘patch’ delivery resulted in an absorption-limited terminal elimination phase (the pharmacokinetic phenomenon referred to as ‘flip-flop’ kinetics). Accordingly, patches were seen to offer the potential of maintaining sustained steady-state blood levels after topical application, with the levels being varied by manipulating the drug concentration and vehicle components in the patch and/or the area of skin exposed to the patch. The potency of the drug was noted as an important therapeutic determinant given that therapeutic blood levels would have to be achieved (Riegelman, 1974). The next step in this journey to a working transdermal system was to identify transdermal candidates. This step was taken in a pioneering work by Michaels et al. in 1975. Using diffusion cells fitted with human cadaver skin membranes, these researchers reported in vitro fluxes of a series of 10 drugs thought to have potential for the method (Michaels et al., 1975). Of the drugs studied, scopolamine, nitroglycerin, oestradiol and fentanyl have now been developed into marketed transdermal systems. We can now consider the history associated with the patch development of each of these drugs.
Scopolamine (hyoscine) patch for the treatment of motion sickness: the first transdermal patch to reach the market
Powder of Hyoscyamus (scopolamine's parent plant) was mentioned as an agent to be topically applied or taken orally for abdominal discomfort in the Papyrus Ebers. Scopolamine was first applied topically as an antiperspirant (MacMillan et al., 1964). In 1944, p.o. administration of 0.6 mg of scopolamine (hyoscine), tested with other drugs, was used to prevent seasickness in troops. A larger dose (1.2 mg) was shown to be more effective but was also associated with dry mouth (Holling et al., 1944). In 1947, dimenhydrinate (Dramamine®; Prestige Brands, Tarrytown, NY, USA), an antihistamine and anticholinergic drug, given experimentally to a woman to treat hives, led to the unexpected disappearance of the car sickness that she had suffered all her life. As a consequence, 100 mg of Dramamine was tested on 389 US soldiers suffering seasickness while sailing to Germany and found to be effective within 1 h in 372 of them (Gay and Carliner, 1949). Scopolamine was later used successfully to prevent airsickness in student navigators (Lilienthal, 1945; Smith, 1946b) but found to be only moderately effective in flexible gunnery students (Smith, 1946a). Unfortunately, scopolamine has a comparatively short elimination half-life of 4.5 h and is therefore expected to only have a short duration of action (Putcha et al., 1989).
The finding that scopolamine had a substantial flux through excised human skin (Michaels et al., 1975) led to a follow-up study in which the mechanism by which scopolamine penetrated the stratum corneum was studied in more depth (Chandrasekaran et al., 1976). This 1970s work culminated in the Alza Corporation developing a transdermal therapeutic system (TTS) for prevention and treatment of motion-induced nausea designed to provide controlled administration of scopolamine through the surface of the skin, such that the system governed drug input kinetics to the systemic circulation (Shaw et al., 1975; 1976,). Studies were performed to locate a highly permeable skin site. It was found that the transdermal patch with a Zaffaroni design applied behind the ear worked best. The patch had a drug reservoir and a microporous membrane that could control the delivery of scopolamine (Shaw and Urquhart, 1979). As a result of a redistribution of scopolamine into the contact adhesive lamina, an initial bolus (loading) dose of scopolamine was released upon application of the patch to the skin, enabling therapeutic scopolamine plasma levels to be achieved rapidly (Urquhart et al., 1977; Shaw and Urquhart, 1979). The device was first tested with Alza employees sailing in a large sailboat through a rough stretch of water close to the Golden Gate Bridge known as the ‘potato patch’. Employees wearing the placebo patch were sick, whereas most of those wearing the scopolamine patch did not (Hoffman, 2008). Controlled trials were then conducted as part of the programme for the American Spacelab missions; these demonstrated the efficacy of the transdermal scopolamine system (Graybriel et al., 1976; 1981,; Graybriel, 1979). In 1979, a 2.5 cm2-TTS (which is still one of the smallest patches on the market) programmed to deliver 1.5 mg of scopolamine over 3 days (Transderm Scōp®; Novartis Consumer Health, Parsippany, NJ, USA) was the first transdermal patch to reach the US market. Alza's scientists later conducted four double-blind clinical trials in healthy men and women with a history of motion sickness to evaluate the efficacy of transdermal scopolamine for the prevention of motion sickness at sea. Transdermal scopolamine not only provided significant protection against motion sickness compared with placebo and p.o. dimenhydrinate but was also associated with minimal side effects (Price et al., 1981).
Nitroglycerin for angina pectoris: from the ointment to the transdermal patches
Until the marketing of the transdermal scopolamine patch, a nitroglycerin ointment was the only transdermal product on the market. Whereas the nitroglycerin ointment led to more sustained serum levels than sublingual and p.o. sustained release capsule dose forms (Maier-Lenz et al., 1980), the plasma levels were dependent upon the surface area to which a given dose of ointment was applied (Sved et al., 1981). However, applying a precise dose to a stratified area is difficult. For example, the dosages of Nitro-Bid® (nitroglycerin ointment USP 2%; Fougera, Melville, NY, USA), used in clinical trials were determined using a ruler to define the length of ointment ribbon ejected from the ointment tube (Figure 1F) and ranged from 1.3 cm (1/2 in.; 7.5 mg) to 5.1 cm (2 in.; 30 mg), typically applied to 232 cm2 (36 in.2) of skin on the trunk of the body. An additional limitation of semi-solids is the need for frequent dosing, e.g. every 8 h for Nitro-Bid, to achieve the intended therapeutic effect, which is likely to lead to greater patient non-compliance than once daily dosing possible with patches. However, nitroglycerin volatilization appeared not to be an issue (Cossum and Roberts, 1981). In contrast, unintentional transfer through interpersonal contact was a problem, as evidenced by the report of spousal headache after intercourse with a partner who had rubbed a nitroglycerin patch on his penis to treat erectile dysfunction (Talley and Crawley, 1985).
In 1973, Alza Corporation filed an additional US patent based upon its topical rate-controlling membrane medicated adhesive bandage concept for the controlled systemic administration of vasodilators such as nitroglycerin. An embodiment of the patent was that the drug within the reservoir could be mixed with a transporting agent to assist drug delivery (Zaffaroni, 1973). At the beginning of the 1980s, Key Pharmaceuticals and Searle Laboratories disclosed two different nitroglycerin transdermal system designs: a water-soluble polymeric diffusion matrix containing nitroglycerin and a microsealed pad with a polymer matrix containing nitroglycerin within a hydrophobic solvent to enhance nitroglycerin transport and diffusion (Keith and Snipes, 1981a; Sanvordeker et al., 1982). Associated with these patents, three nitroglycerin transdermal patches varying in structure and dosages were introduced onto the US market in 1981 for the prevention and treatment of angina pectoris: Transderm-Nitro® (Ciba Pharmaceuticals Company), Nitro-Dur® (Key Pharmaceuticals) and Nitrodisc® (Searle Laboratories) (Dasta and Geraets, 1982). Since it had been learnt in clinical studies that nitroglycerin inactivated itself upon sustained delivery, each marketed patch was to be applied once daily with an approximately 12 h ‘rest period’ between wear times. A subsequent patent claimed that addition of ethanol as a permeation enhancer to a transdermal nitroglycerin system enabled nitroglycerin skin fluxes of at least 40 μg·cm−2·h−1 (preferably in the range of 50–150 μg·cm−2·h−1) greater than the prior art (Gale and Berggren, 1986). In the United States, Key Pharmaceuticals eventually developed a patch in which the drug was contained solely in the adhesive, the first successful commercial patch of this kind and this patch captured the greatest share of the nitroglycerin market. The patch was later marketed as Nitro-Dur II® and described in a US patent (Sablotsky et al., 1993).
Transdermal clonidine for the treatment of hypertension
Clonidine, approved by the US Food and Drug Administration (FDA) in 1984 for up to 1 week transdermal delivery to manage mild-to-moderate hypertension (Sica and Grubbs, 2005), was first applied to facial skin in the form of a shaving lotion, a soap or a cream for its pilomotor effect (Zeile et al., 1965), in which the stimulation of the arrector pili muscle of the skin causes goose bumps so that hairs are raised away from the skin. In the 1960s, the hypotensive effect of clonidine was discovered by accident when a solution of the drug was introduced into the nose of a woman suffering a cold to test the nasal decongestive properties of clonidine. Surprisingly, the woman then fell into a deep sleep until the next day. Controlled tests, run after she woke up, showed a significant drop in BP and heart rate (Stähle, 2000). Transdermal clonidine was developed to reduce drug side effects (mainly drowsiness and dry mouth) and to improve patient compliance (Shaw et al., 1983), which was estimated to be no more than 50% with p.o. hypertensive therapy (Haynes et al., 1978). In 1980, a US patent disclosed a transdermal patch for hypertension therapy. The system contained a gelled mineral oil–polyisobutene–clonidine reservoir and contact adhesive layer with a microporous membrane in-between that controlled the drug release rate (Chandrasekaran et al., 1980). In a subsequent patent, it was claimed that the drug release rate of a clonidine transdermal system could be modulated from 1.6 to 2.4 μg·cm−2·h−1 by modifying the polyisobutylene (PIB)/mineral oil ratios in the drug reservoir and in the contact adhesive with and without the presence of colloidal silicon dioxide (Enscore and Gale, 1985). First clinical trials showed that the clonidine transdermal patch was an effective alternative to p.o. administration in decreasing BP in healthy volunteers (Arndts and Arndts, 1984) and in patients with essential hypertension (Popli et al., 1983; Weber et al., 1984). However, clonidine patches have since been associated with a high rate of dermatological adverse reactions (e.g. allergic contract dermatitis), leading sometimes to treatment discontinuation (Boekhorst, 1983; Groth et al., 1983; Holdiness, 1989).
Transdermal oestradiol for female hormone replacement therapy
Cutaneous application of follicular hormone (follicle-stimulating hormone), oestrone, for amenorrhoea was introduced by Zondek (1938). In 1960, 2 g of an ointment containing both radiolabelled oestradiol-17β and progesterone was applied to human subjects. Between 16.5 and 44% of the radioactivity appeared in the urine within 72 h (Goldzieher and Baker, 1960). Oestradiol was first applied transdermally for post-menopausal replacement therapy as a hydroalcoholic gel (Oestrogel®; Benins-Iscovesco) (Holst et al., 1982; Holst, 1983). However, this dosage form was messy and dosage control was difficult. In 1983, a US patent disclosed a bandage to be applied to the skin for administration of oestradiol within a vehicle rich in ethanol, the latter used as a percutaneous absorption enhancer (Campbell and Chandrasekaran, 1983). A microporous polymer film membrane was used to maintain the fluxes of oestradiol and ethanol in the vicinity of 0.1 and 400 μg·cm−2·h−1 respectively. The sustained plasma levels of oestradiol obtained with the device overcame the key peak and trough profile limitation of the then marketed oestradiol ointment (Strecker et al., 1979). In 1984, the first transdermal oestradiol system reached the US market. Its application resulted in circulating oestradiol plasma levels (40–60 pg·mL−1) sufficient to meet the early follicular phase hormone levels (Good et al., 1985). A number of clinical trials demonstrated the efficacy of Alza's transdermal device in reducing hot flushes and showed the advantages of transdermal delivery as compared to conventional p.o. oestrogen treatment (i.e. reduction in daily dose required, limited effects on liver function) (Laufer et al., 1983; Powers et al., 1985). Eventually, patches with oestradiol exclusively in the adhesive were developed and these too assumed strong market positions. Today, an alternative approach is to use metered-dose applicators, exemplified by Elestrin® (oestradiol 0.06% in a hydroalcoholic gel base; Meda Pharmaceuticals, Somerset, NJ, USA) packed as 100 doses each of 0.87 g gel and Divigel® (Orion Corporation Pharm, Turku, Finland) packed as single use gel-filled sachets (0.25, 0.5 and 1.0 g gel-filled foil packets containing 0.25, 0.5 and 1 mg of oestradiol respectively).
Transdermal fentanyl for the treatment of pain
As pointed out by Watkinson (2012), the Alza fentanyl patch, marketed by Johnson & Johnson (J&J) as Duragesic®, has dominated the transdermal market with peak sales of greater than $2 billion in 2004. Michaels et al. (1975) showed its potential as a transdermal candidate by reporting maximum fluxes through human thigh skin of 0.8–3.8 μg·cm−2·h−1 (average, 2 μg·cm−2·h−1) at 30°C. A 1986 US patent, disclosing various transdermal system designs with different sizes (5–100 cm2) for the delivery of the free base of the narcotic fentanyl, observed that in vitro skin penetration rates of 0.5–10 μg·cm−2·h−1 could be maintained for at least 12 h and for up to 7 days (Gale et al., 1986). The system's in vivo delivery of fentanyl citrate and base (and sufentanil citrate and base) through the skin was demonstrated by applying 50 μg of the drug in water to the forearm skin of five volunteers (six volunteers for sufentanil) under an occlusive dressing, showing that about 20% of the absorbed dose was recovered in urine after 24 h (Sebel et al., 1987). The first clinical studies evaluating Alza's TTS-fentanyl patch, a standard Zaffaroni system with the drug in the pouch of a form-fill-seal design, were conducted in patients in the late 1980s (Duthie et al., 1988; Holley and van Steennis, 1988; Caplan et al., 1989). Further to their studies comparing permeation of fentanyl and sufentanil across human skin in vitro, the relationship to their physicochemical properties and their suitability for transdermal delivery (Roy and Flynn, 1989; 1990,), Roy et al. (1996) showed that optimum flux of fentanyl through human skin from various adhesive patches was achieved when its thermodynamic activity in the patch was maximal. The Alza patch ran into difficulties in 2006 when its patent expired and it was found that fentanyl could leak out of the patch reservoir (Watkinson, 2012). However, while the US FDA approved the Mylan fentanyl matrix [drug-in-adhesive (DIA)] patch, described in a US patent (Miller et al., 2009), in January 2005 and another from Lavipharm in August 2006, J&J had sales of more than $1.2 billion in 2006 and $900 million in 2009, mainly due to J&J's assertive marketing and patent protection (Watkinson, 2012). Interestingly, although Noven received approval for a new generic patch in 2009, its initial application in September 2005 failed because its patch contained much more fentanyl than that in Duragesic. Ultimately, these matrix designs, together with Activis (2007), Watson (2007) and Teva (2008), dominated the market (Watkinson, 2012).
Nicotine patches for smoking cessation aid: first transdermal blockbuster
Nicotine was first used in a transdermal form as a smoking reduction and cessation aid in 1984. One study showed significant levels of nicotine in the saliva between 30 and 90 min after the topical application of 9 mg of nicotine base in a 30% aqueous solution to the volar forearm of a volunteer; there was also an increase in both the pulse and the systolic BP (Rose et al., 1984). A follow-up study showed a reduced craving in 10 cigarette smokers after application of 8 mg of nicotine base in a 30% aqueous solution in a polyethylene patch in comparison to an inactive placebo solution (Rose et al., 1985). The first German patches containing nicotine proved to be successful in suppressing the urge to smoke in clinical trials in Münster/Germany in 1989 (Buchkremer et al., 1989). One of the first US patents dealing with transdermal delivery of nicotine claimed an occlusive transdermal pad to be attached to the skin with a reservoir liquid nicotine base (Etscorn, 1986). In this invention, the delivery of nicotine from the device was controlled with the use of a microporous membrane. Its duration of delivery was on the order of 30–45 min, thus requiring the application of several patches over the course of a day to maintain nicotine plasma levels. A subsequent patent disclosed a monolithic patch with a polyurethane matrix layer that contained between 5 and 50% nicotine. This system was to deliver nicotine through human skin over at least 24 h (Baker and Kochinke, 1989). A later US patent suggested that the concentration of nicotine in the patch reservoir should preferably be at a thermodynamic activity of less than 0.50 (Osborne et al., 1991). Between the end of 1991 and early 1992, four nicotine patches with different designs, all obviously approved by the US FDA, reached the US market within a few months. These were Ciba-Geigy/Lohmann Therapie-Systeme (LTS): Habitrol® (matrix); Lederle/Elan: Prostep® (matrix); Marion Merrell Dow/Alza: Nicoderm® (reservoir/membrane); and Warner-Lambert/Cygnus: Nicotrol® (DIA). Collectively, they became a huge commercial success with total sales approaching US $1 billion during their year of introduction. Over a million smokers gave up smoking with the help of nicotine patches (Prausnitz et al., 2004). Although transdermal patches had been on the market for around 10 years, it was the arrival of nicotine patches that led to them being widely accepted.
Transdermal testosterone for hypogonadism
Testosterone was initially applied as a cream in order to treat male hypogonadism (Jacobs et al., 1975; Klugo and Cerny, 1978; Ben-Galim et al., 1980). However, skin-to-skin transfer of testosterone gel from parents to their young children or from male to their female sexual partners was reported, resulting in precocious puberty or pronounced virilization (Delanoe et al., 1984; Kunz et al., 2004; Busse and Maibach, 2011). The first TTS for administration of testosterone was developed and tested in nine healthy normal men and seven hypogonadal patients (Bals-Pratsch et al., 1986). The first systems were developed by Alza Corporation and designed to be applied to the highly permeable scrotal tissue (Testoderm® TTS) (Campbell and Eckenhoff, 1987; Korenman et al., 1987; Campbell et al., 1988; 1989a,). However, Ahmed et al. (1988) reported high serum dihydrotestosterone levels after scrotal application and expressed concern about the possible detrimental effects on the prostate. Moreover, the site of application was inconvenient for patients who had to clip their scrotal hair to enable these patches to adhere adequately (Nieschlag, 2006). The next-generation testosterone patch (Androderm®; Watson Laboratories, Inc., Salt Lake City, UT, USA) was therefore designed for application to non-scrotal skin (i.e. the back or the chest) to overcome these difficulties. The naturally low skin penetration rate of testosterone was overcome by raising its concentration to just below saturation and including ethanol or comparable solvent as a skin penetration enhancer (Ebert et al., 1992; Meikle et al., 1992).
Not all transdermal candidates result in successful, marketed products
In vitro and in vivo skin permeation studies showed that ephedrine might be a likely candidate for administration by way of the transdermal route (Beckett et al., 1972; Michaels et al., 1975). It was thought that the drug could be incorporated in a polymeric transdermal patch for its decongestant effect (Keith and Snipes, 1981d) and for potential anti-asthmatic therapy (Bhalla and Toddywala, 1988). Subsequent in vitro drug release studies from a polymeric matrix patch and in vivo absorption studies in nine healthy volunteers looked promising (Jain et al., 1990). Inventions describing matrix patches containing phenylephrine and phenylpropanolamine were also reported (Keith and Snipes, 1981b,c,). A phenylpropanolamine transdermal patch was investigated in a pilot study with three subjects and showed effective plasma levels for appetite suppression (Devane et al., 1991). However, none of these transdermal patches reached the market. Nevertheless, the lay press has also reported the use of ephedrine patches as an aid to weigh loss (Real Pharma, 2014). However, since 2004, ephedra-containing dietary supplements have been banned by the FDA due to serious toxicities (FDA, 2004).
Despite encouraging results in healthy volunteers, neither a transdermal timolol ointment (Vlasses et al., 1985) nor a transdermal timolol patch (Kubota et al., 1993) has received clinical and therefore regulatory acceptance. Captopril, an angiotensin-converting enzyme inhibitor, has also been incorporated into transdermal patches and tested in vivo in animal models. However, its physicochemical properties are not favourable for transdermal delivery and the drug is associated with severe skin irritation (Helal and Lane, 2014).
Avoidance of first-pass metabolism and transdermal blood level profile
Administration of therapeutic agents across the skin enables drugs to avoid p.o. first-pass chemical or enzymatic degradation in the gastrointestinal tract or liver. Transdermal delivery is therefore of particular interest for molecules with limited systemic (p.o.) bioavailabilities and short half-lives, providing that the molecule can also be shown not to have a high skin first-pass effect. Examples of molecules with a high skin first pass that are used in topical and transdermal products include testosterone (∼60%, in vitro mouse skin) (Kao and Hall, 1987); methyl salicylate (>90%, in vivo human volunteers) (Cross et al., 1998); nitroglycerin (∼20%, in vivo rhesus monkeys) (Wester and Maibach, 1983) and others (Dancik et al., 2010). The zero-order (constant rate of delivery) kinetics of transdermal delivery has been one of the cornerstones in the development of transdermal systems for the treatment, for instance, of neurodegenerative disorders (Poewe et al., 2007; Lefèvre et al., 2008).
Design of patches based upon engineering and pharmacokinetics principles
Reservoir and rate-controlling membrane
The variability in dosing and possible transfer of the active to others with ointment and cream transdermal systems has emphasized the need to have controlled, occluded and safer delivery systems. This has been a major driver in the development of the more sophisticated TTSs that are commonly known as ‘transdermal patches’. The first of these systems was a combination of a reservoir containing the active and a rate-controlling membrane pioneered in the early 1970s by the entrepreneur Alejandro Zaffaroni through his company Alza. His first commercialized TTS was a scopolamine TTS. Alza championed the view that the co-existence of a reservoir and a rate-limiting membrane in their system was a key requirement to minimize variability in skin permeability within and between individuals and subsequent drug blood levels. A key premise was that the device, and not the skin, controlled drug input into the bloodstream (Shaw and Theeuwes, 1985). In turn, the precisely controlled delivery into the systemic circulation through intact skin not only attained an adequate therapeutic effect (i.e. to prevent motion sickness) but also minimized undesired CNS adverse events such as drowsiness and confusion (Shaw and Urquhart, 1979). A patent filled in August 1971 (US Patent 3,797,494) described a patch using this concept, which was quite revolutionary in comparison to previously existing transdermal systems (Zaffaroni, 1974). The reservoir/membrane patch design is illustrated in Figure 1H. In this type of patch design (also known as form-fill-seal design), the drug is contained in a compartment and is usually present in the form of a liquid (i.e. solution or suspension) or a gel. This liquid or gel reservoir is separated from a continuous adhesive layer by a permeable membrane that controls the release of the active from the device. Figure 2A and B shows, for the reservoir patch, the process of form-filling-sealing and coating-drying respectively.
Figure 2.
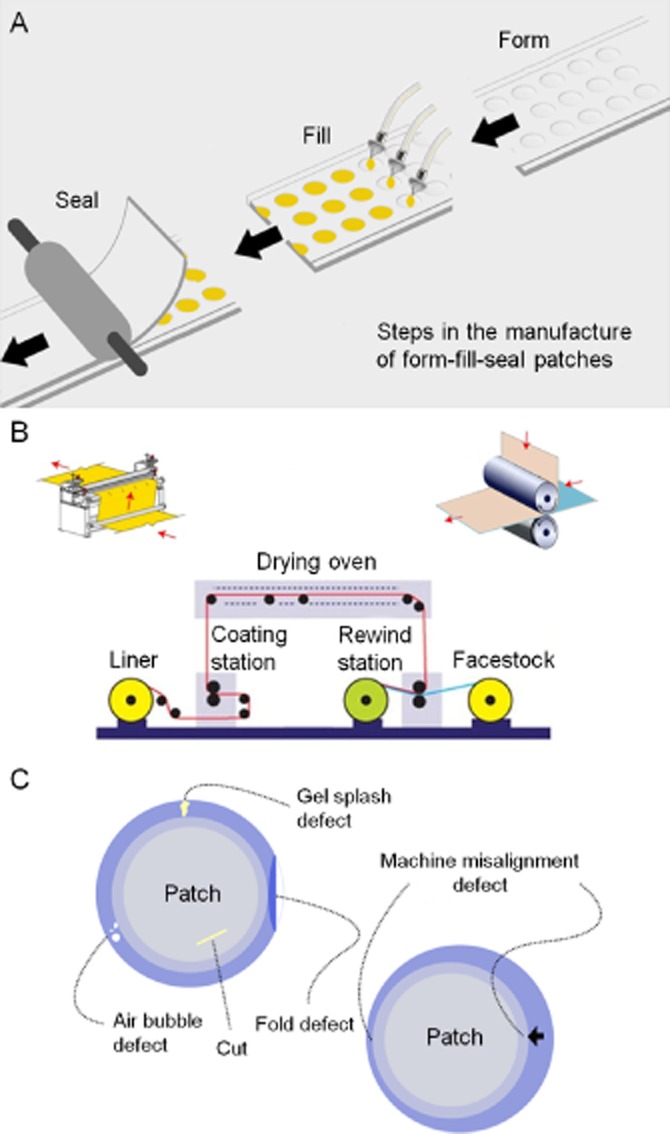
Manufacturing process for and potential failures of reservoir patches: (A) form-filling and sealing process; (B) coating and drying process; and (C) potential problems arising during patch reservoir manufacturing process.
An unplanned benefit in this initial patch design is that the drug in the reservoir equilibrates with the adhesive layer so that upon application to the skin, the drug in the adhesive acts as a priming dose of drug that when released can saturate skin binding sites. The advantage of a reservoir/membrane-type patch is that it provides a constant release rate of drug from the system (zero-order kinetics). However, this design also has the disadvantage of requiring a larger patch to achieve its delivery goal as the membrane rate control is increased. One should also mention that the membrane function only applies to the dynamic in vivo phase. During storage, drug in a patch will diffuse into and saturate all the membranes of the system as well as the in-line adhesive layer, in this way possibly resulting in overly high initial delivery rates. This phenomenon is a general disadvantage for high-solubility molecules that need some kind of flux moderation.
A major limitation in this system is potential for leakage from its sealed liquid reservoir that could arise from an aberration in the manufacturing of the patch. Uncontrolled drug release from the reservoir and potentially drug overdosing (a dose-dumping effect) could arise, for instance, from an accidental rupture of a backing membrane (Govil, 1988; Peterson et al., 1997). Indeed, recalled lots of the form-fill-seal type of fentanyl patches were apparently associated with this problem and similar problems in the early 2000s. Figure 2C shows some examples of issues that may arise with this patch design. In addition, the use of reservoir solution can also lead to other difficulties. As an example, a design fault in the Estraderm® device, patented by Alza in 1984 (US Patent 4,460,372) (Campbell and Chandrasekaran, 1984) led to an unexpected drug delivery profile despite the presence of a rate-controlling membrane (Paoletti et al., 2001). In a system with a ‘rate-controlling’ membrane, the putative membrane will affect the overall flux of both the drug and the enhancer. Early on, Alza created an oestradiol patch intended to yield a constant flux of oestradiol over 4 days, in which the reservoir contained oestradiol in an ethanolic solution. However, an unexpected oestradiol plasma concentration–time profile was found when the transdermal system was applied to human skin. On day 2, there were higher than expected blood levels, most probably as a result of the back diffusion of moisture from the skin into the patch reservoir reducing the solubility of oestradiol in the reservoir and greatly increasing its thermodynamic activity leading ultimately to the formation of a supersaturated solution and marked skin penetration. However, on day 3, the blood levels significantly fell as the thermodynamic activity of oestradiol in the reservoir solution was reduced by the formation of oestradiol hemihydrates and their crystallizing out of solution.
A key concept Alza advocated to protect their patent was that ‘… each TTS under development or in clinical testing, incorporates a rate-controlling membrane …’ (Shaw et al., 1975). They argued that ‘the microporous membrane is chosen to ensure that the delivery rate of scopolamine to the skin surface is much less than the rate at which even the most impermeable skin can absorb the drug. Hence, the system, and not the skin, controls the entry of drug into the systemic circulation. This means that differences in skin permeability among different subjects will be negated; all will receive scopolamine into the circulation at the same rate, predetermined by the system's delivery characteristics’ (Shaw and Chandrasekaran, 1978). In support of these assertions, Shaw and Theeuwes (1985) estimated the coefficient of variability in net transdermal flux from a patch through the skin as 25% (=SD.100/mean). This value was based upon an intrinsic variability in the transdermal flux of nitroglycerin through human skin in vivo being 46% (based upon the variability in the nitroglycerin lost from a transdermal ointment applied to 12 volunteers for 24 h) and an almost equal resistance to the skin being imposed by the patch in controlling the transdermal flux of nitroglycerin (in vitro flux from Transderm-Nitro patch on the skin accounts for 45% of the total resistance when applied to the skin).
However, more important than what is lost from the site of application, as used in these calculations, is the actual systemic plasma nitroglycerin concentration arising from the transdermal products – as these are more reflective of the likely pharmacodynamic effects for the products. The data reported by McAllister et al. (1986) for the nitroglycerin concentrations in plasma for 24 male subjects receiving a single application of Transderm-Nitro 50 mg, 1 in. of Nitro-Bid 2% ointment and two other products show a very different nitroglycerin plasma concentration–time profile for the Nitro-Bid ointment versus the other products that show similar profiles. Importantly, the variability in the extent of absorption, as defined by SD.100/mean for AUC0–24 (pg·h·mL−1), is comparable: 77.5% for Nitro-Bid ointment and 52% for the Transderm-Nitro patch. An additional source for the higher Nitro-Bid variability is the variation in dose per area applied (Sved et al., 1981). The variability in plasma nitroglycerin concentrations of transdermal systems lacking a rate-limiting membrane (Nitrodisc, 43%; Nitro-Dur, 55%) is also similar to that for Transderm-Nitro (McAllister et al., 1986), suggesting that this membrane is not essential for controlled transdermal delivery. In reality, pharmacokinetic differences mainly define the variations in plasma concentrations and systemic effects for patches, as can be seen by nitroglycerin patch doses for angina pectoris being normally titrated to give a decrease of 10 mmHg in systolic BP (Thadani et al., 1986). The variability in maximum-tolerated doses of nitroglycerin after i.v. infusion, which normally determines the infusion rate in practice, is 64% (Zimrin et al., 1988).
A key technology advancement implemented to enable efficacious delivery of certain drugs is the inclusion of a skin penetration enhancer. As an example, in the US Patent 4,588,580 filed by Alza in 1984 for the patch, later named Duragesic, the analgesic fentanyl was formulated in a gel matrix using ethanol as a vehicle to both maximize its thermodynamic activity and enhance skin penetration as well as enable its membrane barrier to partly control the release of fentanyl into the skin (Gale et al., 1986; Santus and Baker, 1993). In practice, many adjuvants are included in transdermal formulations to either: (i) increase drug diffusivity in the skin; (ii) increase drug solubility in the skin; and/or (iii) increase the degree of drug saturation in the formulation (Moser et al., 2001). Typical adjuvants in patches include ethanol, oleic acid, oleyl oleate, dipropylene glycol and triacetin (Govil et al., 1993; Lane, 2013). The most important consideration is the maximal delivery rate through the skin. This is evident in the delivery area for the Mylan matrix fentanyl patches, which came onto the market in the early 2000s, being only slightly smaller than Duragesic patch. In 2011, as a consequence of leakage problems, J&J introduced a matrix patch, in which fentanyl existed in an essentially saturated state in the adhesive.
Matrix patches
Several of Alza's early competitors – Key Pharmaceuticals, Theratech, Cygnus, Noven and LTS – used the matrix concept for nitroglycerin, oestradiol and testosterone to overcome the intellectual property challenges associated with Alza's technology in the 1980s. Collectively and at times individually, these matrix designs became the dominant products within the transdermal market (Figure 3). This market position was achieved because they were not only generally thinner and more flexible and so more comfortable and adhering, but they were also less expensive to manufacture. The matrix design overcame both the Alza intellectual property ownership in the liquid reservoir/rate-controlling membrane design and most of the limitations detailed herein associated with that design.
Figure 3.
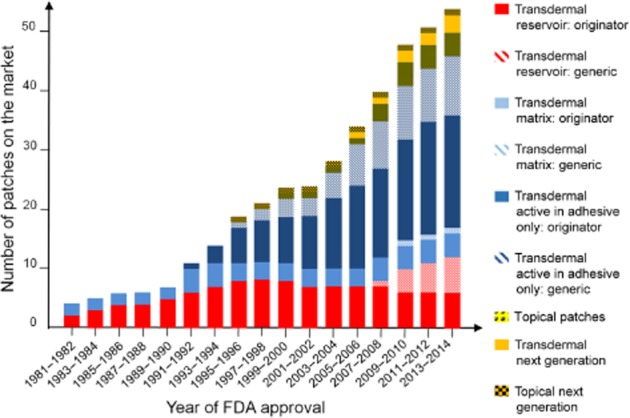
Evolution of commercial topical and transdermal patches – transdermal reservoir: originator, generic; transdermal matrix: originator, generic; transdermal active in adhesive only: originator, generic; topical patches; transdermal next generation; topical next generation.
In general, all patches that do not contain a liquid reservoir may be regarded as matrix patches and these can be applied to the skin by either gluing the backing to the skin adjacent to the matrix or an adhesive on the matrix to the skin (Figure 1I and J). Patches in which drug is mainly incorporated in a polymeric or viscous adhesive (DIA), and discussed later, are also matrix patches. In principle, when a drug is suspended in an internal polymer matrix, in the pouch of a form-fill-seal system or in the adhesive of a patch without a distinct internal reservoir, the delivery can be steady (zero-order), depending upon just how any such system is designed. Mylan's fentanyl patch has its drug suspended in the adhesive (approximately 75% is suspended at the outset of patch wear) and it delivers at a constant rate over a multiple day course because as the drug is released from the patch and absorbed, suspended drug dissolves back in the adhesive and compensates for that which is released. The thermodynamic activity of fentanyl is therefore virtually constant over the whole time the Mylan patch is worn.
Active in adhesive patches
The original design of matrix patches was that the matrix was an alternative to the internal reservoir in the reservoir/rate-limiting membrane patch. Later patches, the DIA patches, simply incorporated the drug entirely in the pressure-sensitive adhesive (PSA). This design, which, in principle, is also a matrix patch, constitutes the simplest, state-of-the-art transdermal patch design. The drug is directly included in the adhesive polymer that not only fulfils its adhesion function but also holds the drug and controls its delivery rate (Peterson et al., 1997; Tan and Pfister, 1999). A US patent filled in August 1981 (US Patent 4,409,206) described a transdermal release system in which the active (e.g. clonidine, haloperidol, nitroglycerin or dihydroergotamine) was directly incorporated into a skin-compatible polyacrylate adhesive but not in a large amount (0–30% by weight) (Stricker, 1983). A transdermal tape, where nitroglycerin (25–45% by weight) was incorporated into an acrylic adhesive polymer, was later disclosed in a US patent in 1988 (Wick, 1988). In 1993, a US patent describing a DIA design for delivery of fentanyl was disclosed (Cleary and Roy, 1993). It has been suggested that the concept of a DIA patch came from the concept of the bubble jet printer where the ink was printed on the surface of some appropriate materials. It was realized that the DIA could be loaded onto the patch backing in the same way (G.W. Cleary, pers. comm. to M. S. Roberts, 8th World Congress on Clinical Pharmacology and Therapeutics, Brisbane, 1–6 August 2004). The DIA patch design is illustrated in Figure 1K.
However, while the DIA patch appears easier to make than its reservoir/rate-controlling membrane and traditional matrix patch counterparts, the formulation of such a patch is rather challenging (Padula et al., 2007). A key outcome from the DIA design are lighter, thinner and more flexible patches that are more comfortable to wear, have better conformity with skin surface variations and a significant improvement in patient acceptability (Hougham et al., 1989; Wick et al., 1989; Lake and Pinnock, 2000). In 1996, Roy et al. evaluated the physicochemical properties of adhesives used in the design of DIA transdermal patches (Roy et al., 1996). The effect of various adhesive formulations on transdermal delivery of fentanyl was investigated. Various PSAs (acrylate, silicone-2675, silicone-2920 and PIB) were characterized with respect to fentanyl's solubility, partition coefficient and diffusion coefficient. The fentanyl release profiles from these adhesives and the in vitro flux through human cadaver membranes were also evaluated. The silicone-2920 with 2% drug loading, characterized by low drug solubility, a low partition coefficient and a high diffusion coefficient, provided the highest skin flux. Thus, this adhesive appeared to be a promising candidate to design a transdermal patch for the delivery of fentanyl at a therapeutic rate. Interestingly, even though the acrylate adhesive exhibited a relatively higher release rate in water in these studies, its skin flux was considerably lower compared with the silicone-2675 and PIB adhesive formulations. This was seemingly because the acrylate adhesive was a good solvent for fentanyl and the systems in which this adhesive was used were of lower thermodynamic activity relative to the other adhesives.
However, a major disadvantage associated with these patches is that, if the drug is completely in solution, the rate of drug release from the device is dependent upon the drug concentration in the adhesive (first-order kinetics), thus bringing about a decrease in the release rate with wear time (Levin and Maibach, 2008). Hence, a constant rate of delivery could only be achieved if 80% of the amount of drug remained in the patch when the patch was spent and removed or if the drug was in suspension. The early nitroglycerin matrix patches were based upon a high residual content of drug in the patch. Alternatively, like the membrane control for the reservoir patch, the matrix could also provide some resistance to the penetration of drug into the skin, leading to a lower required drug content in the patch. Guy and Hadgraft (1992) estimated that the percentage control exerted by various nitroglycerin patches to the overall penetration of nitroglycerin through the skin was as follows: Transderm-Nitro, 45%: Nitro-Dur II, 13%; Minitran® (3M Drug Delivery Systems, Northridge, CA, USA), 28%; and Deponit® (UCB Pharma, Slough, Berkshire, UK), 87%.
In conclusion, the design of all transdermal patches is characterized by a multi-layered structure with most frequently three or four basic elements: an impermeable backing film, a preparation containing the drug(s) together with the excipient(s), an adhesive responsible for skin adhesion and a protective release liner that is peeled off before applying the patch to the skin. Transdermal patch systems used by the pharmaceutical industry today are mainly reservoir/controlled-release membrane and DIA patches, with the latter becoming the standard in practice (Hopp, 2002).
Drug candidates for transdermal delivery
Not all drugs are suitable for patch delivery. The only drugs that can be used are those that can penetrate the skin, that are sufficiently potent to be active and that meet a clinical need. To date, nearly two dozen molecules have been approved by the regulatory authorities for transdermal administration and have reached the market. The overriding commercial need for any new product is, as Watkinson (2012) puts it, the ‘meeting of unmet medical needs’ at ‘a reasonable cost’.
In principle, the maximal skin penetration flux for a drug is determined by the product of its solubility in the stratum corneum and its diffusivity in the stratum corneum (Kasting et al., 1987; Roberts, 2013). In turn, solubility can be related to melting point (MP), and drug–stratum corneum interactions and diffusivity can be related to molecular weight (MW) or molar volume (Roberts and Cross, 2002). While molecular size can dominate other variables when a wide variety of drugs are used to study percutaneous penetration (Magnusson et al., 2004), the drugs used in topical and transdermal patches have a limited size range. Table 1 shows the properties of the current drugs in transdermal patches. Recently, Wiedersberg and Guy (2014) used some of these properties, a combination of MW and drug–solvent interaction parameters [such as aqueous solubility (Saq) and log octanol–water partition coefficient (log P)], to first estimate the delivery rate of drugs through human skin. They then defined the predicted to actual flux ratios for all marketed drugs. As the average ratio is 5.8 times that expected of 1.0, with a percent coefficient of variation (=SD.100/mean) of 129, the precise prediction of the skin penetration rate for drugs in patches is not straightforward. Wiedersberg and Guy (2014) suggested that higher than expected ratios may arise when penetration enhancers were present in patches, whereas lower ratios arise when the drug concentrations in patches were below saturation. Figure 4 shows a plot of the various drugs now marketed in patches on the Berner–Cooper nomogram (Kydonieus et al., 1999), widely used by the pharmaceutical industry to predict potential candidate drugs for use in transdermal patches. The equation underpinning this nomogram assumes a two-pathway (polar and lipid) model for drug transport through the stratum corneum (Berner and Cooper, 1987). It is apparent from Figure 4 that this nomogram lacks precision in its prediction of the skin penetration rate for the various sized drugs used in patches.
Table 1.
Physicochemical, pharmacokinetic and safety data for currently marketed transdermal drugs
| Drug | MWa (Da) | MP (°C)a Unionized | log Pb | Saq (mg·mL−1)a Unionized (25°C) | Cl (L·h−1) (70 kg) | t1/2 (h) | Oral F (%) | Target plasma level (ng·mL−1) | Estimated Jskin required (μg·h−1) | In vivo Jskin (μg·cm−2·h−1)a″ | Equivalent maximum hourly dose (μg·h−1)a‴ | Safety margin = max dose per h/in vivo Jskin |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buprenorphine | 468 | 209 | 3.8 | 0.047, 0.008h (32°C) | 77 i.v.a′, 55 i.m.b′ | 3 i.v.a′, 28 s.l.a′, 19 bca′, 26 t.d.b′ | 51 s.l.a′, 28 bca′ | >0.1c′ | 7.7 | 0.8 | 70.8 | ∼100 |
| Clonidine | 230 | 130 | 2.7 | 0.17, 13.58i | 15d′ | 8–13 i.v.d′, 12–16 i.v.e′, 20 t.d.e′ | 95d′ | 0.2–2d′ | 3–30 | 1.2 | 12.5 | ∼10 |
| Oestradiol | 272 | 173–179 | 4.2 | 0.003, 0.003j (30°C), 0.0015 1k (25°C) | 600–800f′ | ∼1 p.o.g′ | 5h′ | 0.04–0.06i′ | 24–48 | 0.2b″, 0.17c″, 0.14d″, 0.12e″, 0.42f″, 0.18g″, 0.63h″, 0.23i″, 0.09j″ | 4.2 | ∼20 |
| Ethinyl oestradiol | 296 | 141–146 | 4.3 | 0.039, 0.0092k (25°C) | 70 p.o.j′ | 7.7 p.o.j′, 17 t.d.k′ | 55h′ | 0.025–0.075l′ | 1.75–5.25 | 0.07 | 0.8 | ∼10 |
| Fentanyl | 337 | 83–84 | 3.9 | 0.15, 0.2j (30°C), 0.2l (25°C) | 27–75m′ | 3–12 i.v.m′, 20–27 t.d.m′ | 50 o.t.n′ | 1–3o′ | 27–225 | 2.4 | 100 | ∼40 |
| Granisetron | 312 | 152–154c | 2.6 | 0.017 | 33–76p′ | 4–6 i.v.p′, 36 t.d.p′ | 60q′ | 3.9 (t.d. mean Cmax)p′ | 129–296 | 2.5 | 129 | ∼50 |
| Levonorgestrel | 312 | 235–237 | 3.8 | 0.017 | 5.7 p.o.j′ | 19.3 p.o.j′, 28 t.d.r′ | 94h′ | 0.17 (t.d. Css)r′ | 1 | 0.03 | – | – |
| Methylphenidate | 233 | 74–75, liquid | 2.1 | 1.8 | 12(d); 21(l) (children 30 kg)s′ | 1.5–5 p.o. (children)s′ | 22(d); 5(l)s′ | 5–15t′ | 60–315 | 88 | 1250 | ∼15 |
| Nicotine | 162 | −79, liquid | 1.1 | 62, 1085i (30°C) | 77u′ | 2 i.v.u′ | 20–45v′ | 5–30w′ | 385–2310 | 40k″, 31l″, 69m″, 29n″ | 875 | ∼20 |
| Nitroglycerin (glyceryl trinitrate) | 227 | 13, liquid | 1 | 0.66, 1.3j (30°C) | 216–3270x′ | 0.03–0.05 p.o.x′ | <1y′ | 0.02–0.4z′ | 4.32–1308 | 20o″, 30p″ | 833 | ∼30 |
| Norelgestromin | 327 | 110–130 | 3.67 (pred) | 0.0043 | – | 28 t.d.k′ | – | 0.6–1.2l′ | – | 0.31 | 6.25 | ∼20 |
| Norethindrone acetate (norethisterone acetate) | 341 | 161–162 | 3.2 | 0.0065 | 20.6aa′ | 34.8 p.o.aa′, 6–8 t.d.ab′ | 60h′ | 0.5–0.8ab′ | 10.3–16.5 | 0.65 | 10.4 | ∼20 |
| Oxybutynin | 358 | 56–58d | 4.3 | 0.0093 | 10–64ac′ | 2 i.v.ad′, 7–8 t.d.ad′ | 6ad′ | 0.5–3ae′ | 5–192 | 4.2 | 162.5 | ∼40 |
| Rivastigmine | 250 | Oil at 25°Ce | 2.3 | 25 | 108af′ | 1.3–2 p.o.ag′, 3.4 t.d.ag′ | 36af′ | 2.5–20 (t.d. mean Cmax)ah′ | 270–2160 | 39 | 396 | ∼10 |
| Rotigotine | 316 | 75–77f | 4.7 | 0.017 | 600ai′ | 7 t.d.ai′ | – | 0.4–2aj′ | 240–1200 | 8.3 | 250 | ∼30 |
| Scopolamine (hyoscine) | 303 | 55, liquid | 0.8 | 1.8, 75j (30°C) | 65–121ak′ | 1–5 p.o.ak′ | 4–27ak′ | >0.05al′ | 3.25–6.05 | 5.6 | 210b‴ | ∼40 |
| Selegiline (deprenyl) | 187 | Liquid at 25°Cg | 2.7 | 0.73 | 84am′ | 9–15 p.o.an′, 15–25 t.d.am′ | 4an′ | 2ao′ | 168 | 12.5 | 500 | ∼40 |
| Testosterone | 288 | 155 | 3.6 | 0.02, 0.02i (25°C) | 41ap′ | 0.17–1.7aq′ | 7ar′ | 3–10.5aq′ | 123–430.5 | 13.9 | 417 | ∼30 |
bc, buccal; Cl, total body clearance; d, dextro isomer; F, oral bioavailability; i.m., intramuscular; i.v., intravenous; Jskin, skin flux; l, levo isomer; log P, log octanol–water partition coefficient; MP, melting point of the unionized form; MW, molecular weight; o.t., transmuscosal; p.o., per oral; Saq, aqueous solubility of the unionized form; s.l., sublingual; t1/2, elimination half-life; t.d., transdermal.
aSci Finder Scholar, 2014. bChambers Fox, 2014. cGafni et al., 2008. dTang et al., 2008. eChiang et al., 2009. fKrivonos and Weisman, 2013. gGovil and Weimann, 2006. hRoy et al., 1994. iMagnusson et al., 2004. jMichaels et al., 1975. kShareef et al., 2006. lRoy and Flynn, 1988.
a′Kuhlman et al., 1996. b′Butrans® PI. c′Sittl et al., 2003. d′Lowenthal et al., 1988. e′Catrapres-TTS® PI. f′Cleary, 1993. g′Estraderm® PI. h′Fotherby, 1996. i′Good et al., 1985. j′Kanarkowski et al., 1988. k′Ortho Evra® PI. l′Abrams et al., 2002. m′Duragesic® PI. n′Actiq® PI. o′Duthie et al., 1988. p′Sancuso® PI. q′Kytril® PI. r′Climara Pro® PI. s′Ritalin LA® PI. t′Greenhill et al., 2001. u′Benowitz et al., 1982. v′Hukkanen et al., 2005. w′Bannon et al., 1994. x′Bogaert, 1987. y′Bauer and Seifert, 2005. z′Noonan and Benet, 1986.
aa′Singh et al., 1979. ab′Combipatch® PI. ac′Douchamps et al., 1988. ad′Oxytrol® PI. ae′Lee et al., 1995. af′Exelon® capsules PI. ag′Lefèvre et al., 2008. ah′Frank et al., 2014. ai′Neupro® PI. aj′Lauterbach et al., 2002. ak′Guay, 2003. al′Nachum et al., 2006. am′Emsam® PI. an′Azzaro et al., 2007. ao′Kolli et al., 2010. ap′Horton et al., 1965. aq′Axiron® PI. ar′Tauber et al., 1986.
a″In vivo Jskin is calculated by dividing the labelled dose rate by the patch size (active area). b″Estraderm. c″Climara®. d″Vivelle®. e″Alora®. f″Vivelle-Dot®. g″Menostar®.
h″Minivelle®. i″Combipatch®. j″Climara Pro® . k″Nicoderm CQ®. l″Nicorette®. m″Nicorette® Invisi patch®. n″Habitrol®. o″Nitro-Dur®. p″Minitran®.
Figure 4.
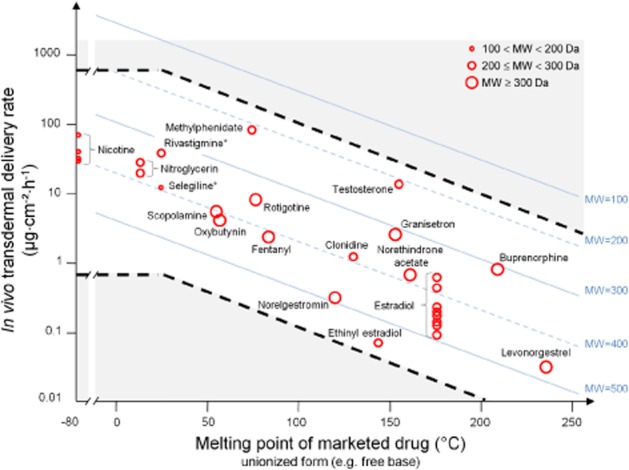
Transdermal delivery rate for currently marketed drugs in patches (log scale) (with symbol size being used to show the actual variation in molecular weight: 100 < MW < 200 Da; 200 ≤ MW < 300 Da; MW ≥ 300 Da) plotted against the active drug melting point (where unknown melting point given by an asterisk is represented as liquid at 25°C) and overlaid on the Berner–Cooper nomogram for a drug with a log P of 5 (Kydonieus et al., 1999). Also shown, as dashed black lines, are the estimated upper and lower boundary lines for marketed drug delivery rate from patches as defined by the rates for small (MW = 100), polar (log P = 1) and large (MW = 500), lipophilic (log P = 5) solutes respectively. [The dashed black lines are calculated from the expression: log maximum delivery rate (μg·cm−2·h−1) = 1.6 + log MW − 0.0086 MW − 0.01 (MP − 25) − 0.219 log P and is based on a regression of maximum transdermal flux (in nmol, equation 7) versus MP, MW and log P for the combined data set of Magnusson et al. (2004) (Milewski and Stinchcomb, 2012). The level region in this plot recognises that 25°C is an approximate lower skin surface temperature for patches applied to human skin in vivo and at which all drugs with MP < 25°C will be liquid.]
An alternative approach to predicting individual skin penetration fluxes for candidate drugs to be used in patches is to define the physicochemical boundaries within which all candidates in the patch systems should fall. As shown in Figure 4, most, but not all, of the marketed drugs used in patches are above the lower Berner–Cooper boundary of MW = 500, log P = 5 and MP < 250°C. All currently marketed drugs in the patch data fall within boundaries derived using a single pathway model similar to that used by Wiedersberg and Guy (2014) and a larger data set (Magnusson et al., 2004; Milewski and Stinchcomb, 2012) (Figure 4). It is evident from Table 1 that a candidate drug for transdermal patches should normally be moderately lipophilic (log P range from 1 to 5), have a low molecular weight (MW < 500 Da) and a low melting point (MP < 250°C). Implicitly, an upper skin limit is also defined by the risk of local skin reactions.
The second requirement of drugs in a patch is that they are sufficiently potent to be active. This generally means that they have therapeutically attainable plasma concentrations, Css (Table 1), that are defined by the rate of delivery of a drug from a patch through the skin, R0, divided by the systemic clearance, Cl (i.e. 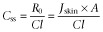 , noting also that: R0 = Jskin × A, where Jskin is the per unit area transdermal drug flux and A is the area of application) (Roberts and Walters, 1998). Indeed, this plasma concentration and the transdermal delivery rate (Figure 4) define the patch area required for therapeutic effect as we now illustrate with a fentanyl patch. Fentanyl, a moderate MW, low melting point and moderate high lipophilicity (MW = 337 Da, MP = 83°C and log P = 3.9) solute, has an average systemic blood plasma clearance in humans of ∼50 L·h−1 and a therapeutic blood level of ∼2 ng·mL−1. Accordingly, assuming a complete skin bioavailability and a maximum flux of 0.8–3.8 μg·cm−2·h−1 (Michaels et al., 1975) through excised human skin, the desired skin flux requires a patch of 25–125 cm2. In reality, the choice of an appropriate skin site and the presence of a skin penetration enhancer can lead to a higher fentanyl skin flux of 5–10 μg·cm−2·h−1, requiring the use of a patch of 10–20 cm2 (Cleary, 1993). Accordingly, fentanyl is now widely used in transdermal delivery to manage post-operative pain. Similarly, a 50 cm2 nitroglycerin patch meets its target therapeutic concentration of 1 ng·mL−1 and requires a transdermal flux of 20 μg·cm−2·h−1 (Naik et al., 2000).
, noting also that: R0 = Jskin × A, where Jskin is the per unit area transdermal drug flux and A is the area of application) (Roberts and Walters, 1998). Indeed, this plasma concentration and the transdermal delivery rate (Figure 4) define the patch area required for therapeutic effect as we now illustrate with a fentanyl patch. Fentanyl, a moderate MW, low melting point and moderate high lipophilicity (MW = 337 Da, MP = 83°C and log P = 3.9) solute, has an average systemic blood plasma clearance in humans of ∼50 L·h−1 and a therapeutic blood level of ∼2 ng·mL−1. Accordingly, assuming a complete skin bioavailability and a maximum flux of 0.8–3.8 μg·cm−2·h−1 (Michaels et al., 1975) through excised human skin, the desired skin flux requires a patch of 25–125 cm2. In reality, the choice of an appropriate skin site and the presence of a skin penetration enhancer can lead to a higher fentanyl skin flux of 5–10 μg·cm−2·h−1, requiring the use of a patch of 10–20 cm2 (Cleary, 1993). Accordingly, fentanyl is now widely used in transdermal delivery to manage post-operative pain. Similarly, a 50 cm2 nitroglycerin patch meets its target therapeutic concentration of 1 ng·mL−1 and requires a transdermal flux of 20 μg·cm−2·h−1 (Naik et al., 2000).
The third driver for transdermal patch systems is a cost-effective safety advantage they may provide over other dosage forms for specific drugs. As discussed earlier, patches have less variability than arbitrarily applied solutions, creams and ointments. Also shown in Table 1 is the estimated maximum hourly systemic exposure based upon the maximum systemic daily dose given by Watkinson (2012). The ratio of this value divided by the in vivo patch flux gives a safety ratio for a given transdermal patch and is generally 10–100. An exception based upon Watkinson's data appears to be scopolamine (hyoscine). However, in practice, up to 5 mg (0.65 mg each 8 h) can be given to adults over 24 h (Drugs, 2014). As Dorne and Renwick (2005) pointed out, there should be at least a 10-fold safety factor to allow for human variability. Drugs such as oestradiol, nitroglycerin, oxybutynin, scopolamine, selegiline and testosterone may be unsuitable for p.o. delivery because of a high p.o. first-pass effect or a low intrinsic water solubility with that of oestradiol, norelgestromin, norethindrone acetate and oxybutynin being less than 10 mg·L−1 (Table 1). Further, the controlled release that avoids fluctuating blood levels (Figure 5) and the convenience offered by patches make them an ideal delivery system for drugs with short elimination half-lives (Table 1). As Wiedersberg and Guy (2014) pointed out, only i.v. infusion and transdermal patches allow systemic delivery to be stopped at any time, the latter by simply removing the patch.
Figure 5.
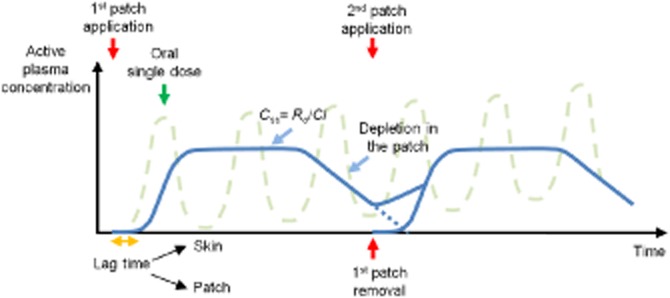
Typical active plasma concentration profile after patch application showing the lag-time, reaching and achieving steady-state, depletion and patch removal as well as the corresponding profile for repeated p.o. dosing of the same active.
An example of a drug that would be unwise to formulate as a patch is paracetamol (MW = 151 Da, MP = 169°C, log P = 0.46), with a clearance of about 15 L·h−1 (McNeil, 2002), a therapeutic analgesic concentration of 3–5 μg·mL−1 (Bacon et al., 2002) and an estimated human skin penetration flux of 0.94 μg·cm−2·h−1 (based upon the derived expression in Figure 4). Accordingly, a 6 m2 paracetamol patch would be needed to be effective. Given that paracetamol is well absorbed and is readily available in various p.o. dosage forms, such a patch is unlikely to be commercially viable. Naik et al. (2000) showed that formulating an aspirin patch for use as anti-inflammatory was equally impractical as an area of 22 m2 would be required based upon a 150 μg·mL−1 therapeutic concentration and a skin penetration flux of 20 μg·cm−2·h−1. However, the dose for its antithrombotic effect is about an order of magnitude lower than that of its anti-inflammatory actions. McAdam et al. (1996) showed that repeated application of a 50 cm2 aspirin patch, containing 120 mg of aspirin and limonene as a permeation enhancer, released 33 mg of aspirin daily and led to a 90% suppression of platelet-produced thromboxane B2 serum levels at day 21 in nine male volunteers.
Table 2 summarizes the approximately 20–25 drugs or drug combinations that are now available as transdermal products and have appeared since the approval of the first transdermal patch for treatment of motion sickness more than 30 years ago. Most of these drugs are for prescription use only, with many being available as generic patches following patent expirations.
Table 2.
Commercially available transdermal patches approved by the US FDA
| Drug (Trade name, year of FDA approval) | Type | Indication | Patch design | Dose and size of patch – Delivery rate | Site of application | Duration of application |
|---|---|---|---|---|---|---|
| Buprenorphine (Butrans®, 2010) | Therapeutic | Chronic pain | DIA | 5 mg in 20.25 (6.25)a cm2 – 5 μg·h−1 7.5 mg in 33.65 (7.5) cm2 – 7.5 μg·h−1 10 mg in 30.60 (12.5) cm2 – 10 μg·h−1 15 mg in 42.48 (18.75) cm2 – 15 μg·h−1 20 mg in 51.84 (25) cm2 – 20 μg·h−1 |
Upper outer arm, upper chest, upper back or the side of the chest | 7 days |
| Clonidine (Catapres-TTS®, 1984) | Therapeutic | Hypertension | Reservoir/Membrane | 2.5 mg in 3.5 cm2 – 0.1 mg·day−1 5.0 mg in 7.0 cm2 – 0.2 mg·day−1 7.5 mg in 10.5 cm2 – 0.3 mg·day−1 |
Upper outer arm or upper chest | 7 days |
| Oestradiol (Estraderm®, 1986) | Therapeutic | Female HRT | Reservoir/Membrane | 4 mg in 18 (10) cm2 – 0.05 mg·day−1 8 mg in 31 (20) cm2 – 0.10 mg·day−1 |
Trunk of the body including the buttocks and abdomen | 3–4 days |
| Oestradiol (Climara®, 1994) | Therapeutic | Female HRT | DIA | 2 mg in 6.5 cm2 – 0.025 mg·day−1 2.85 mg in 9.375 cm2 – 0.0375 mg·day−1 3.8 mg in 12.5 cm2 – 0.05 mg·day−1 4.55 mg in 15 cm2 – 0.06 mg·day−1 5.7 mg in 18.75 cm2 – 0.075 mg·day−1 7.6 mg in 25 cm2 – 0.1 mg·day−1 |
Lower abdomen or upper quadrant of the buttock | 7 days |
| Oestradiol (Vivelle®, 1994) | Therapeutic | Female HRT | DIA | 4.33 mg in 14.5 cm2 – 0.05 mg·day−1 8.66 mg in 29.0 cm2 – 0.1 mg·day−1 |
Trunk of the body including abdomen and buttocks | 3–4 days |
| Oestradiol (Alora®, 1996) | Therapeutic | Female HRT | DIA | 0.77 mg in 9 cm2 – 0.025 mg·day−1 1.5 mg in 18 cm2 – 0.05 mg·day−1 2.3 mg in 27 cm2 – 0.075 mg·day−1 3.1 mg in 36 cm2 – 0.1 mg·day−1 |
Lower abdomen, upper quadrant of the buttock or outer aspect of the hip | 3–4 days |
| Oestradiol (Vivelle-Dot®, 1999) | Therapeutic | Female HRT | DIA | 0.39 mg in 2.5 cm2 – 0.025 mg·day−1 0.585 mg in 3.75 cm2 – 0.0375 mg·day−1 0.78 mg in 5.0 cm2 – 0.05 mg·day−1 1.17 mg in 7.5 cm2 – 0.075 mg·day−1 1.56 mg in 10.0 cm2 – 0.1 mg·day−1 |
Lower abdomen | 3–4 days |
| Oestradiol (Menostar®, 2004) | Therapeutic | Female HRT | DIA | 1 mg in 3.25 cm2 – 0.014 mg·day−1 | Lower abdomen | 7 days |
| Oestradiol (Minivelle®, 2012) | Therapeutic | Female HRT | DIA | 0.62 mg in 2.48 cm2 – 0.0375 mg·day−1 0.83 mg in 3.30 cm2 – 0.05 mg·day−1 1.24 mg in 4.95 cm2 – 0.075 mg·day−1 1.65 mg in 6.6 cm2 – 0.1 mg·day−1 |
Lower abdomen or buttocks | 3–4 days |
| Oestradiol (E)/Norethindrone (NT) (Combipatch®, 1998) | Therapeutic | Female HRT | DIA | 0.62 mg E/2.7 mg NT in 9 cm2 − 0.05/0.14 mg E/NT per day 0.51 mg E/4.8 mg NT in 16 cm2 − 0.05/0.25 mg E/NT per day |
Lower abdomen | 3–4 days |
| Ethinyl oestradiol (EE)/Norelgestromin (NL) (Ortho Evra®, 2001) | Therapeutic | Female contraception | DIA | 0.75 mg EE/6.00 mg NL in 20 cm2 − 0.035/0.15 mg EE/NL per day | Buttock, abdomen, upper outer arm or upper torso | 7 days |
| Oestradiol (E)/Levonorgestrel (L) (Climara Pro®, 2003) | Therapeutic | Female HRT | DIA | 4.40 mg E/1.39 mg L in 22 cm2 – 0.045/0.015 mg E/L per day | Lower abdomen | 7 days |
| Fentanyl (Duragesic®, 1990) | Therapeutic | Chronic pain | DIAb | 2.1 mg in 5.25 cm2 – 12.5 μg·h−1 4.2 mg in 10.5 cm2 – 25 μg·h−1 8.4 mg in 21 cm2 – 50 μg·h−1 12.6 mg in 31.5 cm2 – 75 μg·h−1 16.8 mg in 42 cm2 – 100 μg·h−1 |
Chest, back, flank or upper arm | 72 h |
| Granisetron (Sancuso®, 2008) | Therapeutic | Chemotherapy-induced nausea and vomiting | DIA | 34.3 mg in 52 cm2 – 3.1 mg per 24 h | Upper outer arm | Up to 7 days |
| Methylphenidate (Daytrana®, 2006) | Therapeutic | ADHD | DIA | 27.5 mg in 12.5 cm2 – 1.1 mg·h−1 41.3 mg in 18.75 cm2 – 1.6 mg·h−1 55 mg in 25 cm2 – 2.2 mg·h−1 82.5 mg in 37.5 cm2 – 3.3 mg·h−1 |
Hip area, avoiding the waistline | Up to 9 h in a day |
| Nitroglycerin (Nitro-Dur®, 1995) | Therapeutic | Angina pectoris | DIA | 20 mg in 5 cm2 – 0.1 mg·h−1 40 mg in 10 cm2 – 0.2 mg·h−1 60 mg in 15 cm2 – 0.3 mg·h−1 80 mg in 20 cm2 – 0.4 mg·h−1 120 mg in 30 cm2 – 0.6 mg·h−1 160 mg in 40 cm2 – 0.8 mg·h−1 |
Chest, shoulder, upper arm or back (hairless area) | 12–14 h |
| Nitroglycerin (Minitran®, 1996) | Therapeutic | Angina pectoris | DIA | 9 mg in 3.3 cm2 – 0.1 mg·h−1 18 mg in 6.7 cm2 – 0.2 mg·h−1 36 mg in 13.3 cm2 – 0.4 mg·h−1 54 mg in 20.0 cm2 – 0.6 mg·h−1 |
Chest, shoulder, upper arm or back (hairless area) | 12–14 h |
| Oxybutynin (Oxytrol®, 2003) | Therapeutic | Overactive bladder | DIA | 36 mg in 39 cm2 – 3.9 mg·day−1 | Abdomen, buttocks or hip | 3–4 days |
| Rivastigmine (Exelon®, 2007) | Therapeutic | Alzheimer's and Parkinson's disease | Matrix | 9 mg in 5 cm2 – 4.6 mg per 24 h 18 mg in 10 cm2 – 9.5 mg per 24 h 27 mg in 15 cm2 – 13.3 mg per 24 h |
Upper/lower back, upper arm or chest | 24 h |
| Rotigotinec (Neupro®, 2007) | Therapeutic | Parkinson's disease Restless legs syndrome |
DIA | 2.25 mg in 5 cm2 – 1 mg per 24 h (*) 4.5 mg in 10 cm2 – 2 mg per 24 h 6.75 mg in 15 cm2 – 3 mg per 24 h (*) 9 mg in 20 cm2 – 4 mg per 24 h 13.5 mg in 30 cm2 – 6 mg per 24 h 18 mg in 40 cm2 – 8 mg per 24 h (*) |
Abdomen, thigh, hip, flank, shoulder or upper arm | 24 h |
| Scopolamine (Transderm Scōp®,1981) | Therapeutic | Motion sickness | Reservoir/Membrane | 1.5 mg in 2.5 cm2 – 1.0 mg per 3 days | Behind one ear | 72 h |
| Selegiline (Emsam®, 2006) | Therapeutic | Major depressive disorder | DIA | 20 mg in 20 cm2 – 6 mg per 24 h 30 mg in 30 cm2 – 9 mg per 24 h 40 mg in 40 cm2 – 12 mg per 24 h |
Upper chest or back, upper thigh or the outer surface of the upper arm | 24 h |
| Testosteroned (Androderm®, 1995) | Therapeutic | Hypogonadism | Reservoir/Membrane | 9.7 mg in 32 cm2 (6) – 2 mg·day−1 (#) 12.2 mg in 37 cm2 (7.5) – 2.5 mg·day−1 19.5 mg in 39 cm2 – (12) 4 mg·day−1 (#) 24.3 mg in 44 cm2 – (15) 5 mg·day−1 |
Back, abdomen, thighs or upper arm | 24 h |
| Nicotine (Nicoderm CQ®, 1991)e | OTC | Smoking cessation | Reservoir/Membrane | 36 mg in 7 cm2 – 7 mg per 24 h 75 mg in 15 cm2 – 14 mg per 24 h 114 mg in 22 cm2 – 21 mg per 24 h |
Anywhere on the body, avoiding joints | 24 h |
| Nicotine (Nicorette®)f | OTC | Smoking cessation | Matrix | 8.3 mg in 10 cm2 – 5 mg per 16 h 16.6 mg in 20 cm2 – 10 mg per 16 h 24.9 mg in 30 cm2 – 15 mg per 16 h |
To an area on the upper body or upper outer arm that is non-hairy, intact, non-irritated, clean and dry | 16 h |
| Nicotine (Nicorette® Invisipatch®)f | OTC | Smoking cessation | Matrix | 15.75 mg in 9 cm2 – 10 mg per 16 h 23.62 mg in 13.5 cm2 – 15 mg per 16 h 39.7 mg in 22.5 cm2 – 25 mg per 16 h |
A clean, intact, dry and hairless skin of the thigh, arm or chest | 16 h |
| Nicotine (Habitrol®, 1990)g | OTC | Smoking cessation | Matrix | 17.5 mg in 10 cm2 – 7 mg per 24 h 35 mg in 20 cm2 – 14 mg per 24 h 52.5 mg in 30 cm2 – 21 mg per 24 h |
Upper body or the outer part of the arm | 24 h |
| Sumatriptan (Zecuity®, 2013) | Active | Migraine | Iontophoretic system | 36 mg in 7 cm2 – 6.5 mg per 4 h | Upper arm or tight | 4 h |
| Capsaicin (Qutenza®, 2009) | Topical | Neuropathic pain | DIA | 179 mg in 280 cm2 – No information on delivery rate | The most painful areas, excluding face and scalp | Single 60 min application of up to four patches |
| Diclofenac epolamine (Flector®, 2007) | Topical | Topical treatment acute pain | DIA | 180 mg in 140 cm2 No information on delivery rate |
The most painful area | 12 h |
| Lidocaine (Lidoderm®, 1999) | Topical | Post-herpetic neuralgia pain | DIA | 700 mg in 140 cm2 21 mg·12 h−1 |
The most painful area, avoiding the contact with the eyes | Up to three patches only once for up to 12 h within a 24 h period |
| Lidocaine (L)/Tetracaine (T) (Synera®, 2005) | Topical | Local dermal analgesia | Eutectic mixture – CHADD® technology | 70 mg L per 70 mg T in 50 cm2 – 1.7/1.6 mg L/T per 30 min | Site of venipuncture, i.v. cannulation or superficial dermatological procedure | 20–30 min |
| Menthol (M)/Methyl salicylate (MS) (Salonpas®, 2008) | Topical | Muscles and joints pain | DIA | 3% M/10% MS in 70 cm2 | The affected area | Up to 8–12 h |
| Oestradiol (Evamist®, 2007)h | Therapeutic | Menopausal symptoms | Cutaneous solution | 1.53 mg per spray (90 μL) | The inside of the forearm between the elbow and the wrist | One spray once daily (starting dose) |
| Testosterone (Axiron®, 2010)h | Therapeutic | Hypogonadism | Cutaneous solution | 30 mg per pump actuation | The axilla (armpit) | 2 pump actions once daily (starting dose) |
a(x) Size of patch reported corresponds to the active surface except for Butrans, Estraderm and Androderm patches where both active and overall surface are reported. bPrior to July 2009, a reservoir/membrane patch design was on the market. Following numerous reports of deaths and life-threatening side effects due to a serious design defect of the reservoir patch (risk of drug leakage from the patches), the company moved to a DIA patch design. cIn 2008, the product has been withdrawn from the US market due to the formation of rotigotine crystals in the patches and in 2012 Neupro was re-approved by the FDA with three new strengths (*). dIn 2011, the two patch strengths available on the market were discontinued and replaced by two new smaller size and lower-dose patches (#) but not as a result of any safety or efficacy concerns. eNicoderm CQ in the United States, NiQuitin® in the UK and Nicabate® in Australia. fNicorette is not FDA approved and available in the UK. gHabitrol in the United States and Canada, Nicotinell® in the UK. hEvamist and Axiron are cutaneous solutions using the Patchless Patch® delivery method developed by Acrux Ltd. Data source: FDA (2014) and products' PI.
ADHD, attention deficit hyperactivity disorder; CHADD, controlled heat-aided drug delivery; HRT, hormone replacement therapy.
These include [generic name, reference trade name, generic trade name(s)]: clonidine, Catapres-TTS® (Boehringer Ingelheim, Ingelheim am Rhein, Germany), Clonidine Transdermal System [Aveva (Miramar, FL, USA), Barr Pharm Labs Div Teva (Montvale, NJ, USA), Mylan Technologies (Albans City, VT, USA) and Watson Labs (Dublin, Ireland)]; oestradiol, Climara® (Bayer Healthcare, Montville, NJ, USA), Estradiol Transdermal System (Mylan Technologies); ethinyl oestradiol/norelgestromin, Ortho-Evra® (Janssen Pharms, Titusville, NJ, USA), Xulane® (Mylan Technologies); fentanyl, Duragesic (Janssen Pharms), Fentanyl Transdermal System [Aveva, Lavipharm Labs (Hightstown, NJ, USA), Mallinckrodt (Hazelwood, MO, USA), Mylan Technologies, Par Pharm (Woodcliff Lake, NJ, USA) and Watson Labs]; nitroglycerin, Nitro-Dur® (Merck, Whitehouse Station, NJ, USA), Nitroglycerin Transdermal System [Hercon Pharm (Emigsville, PA, USA), Kremers Urban Pharms (Princeton, NJ, USA) and Mylan Technologies]; oxybutynin, Oxytrol® (Watson Labs), Oxybutynin Transdermal System (Barr Pharm Labs Div Teva). The corresponding transdermal patches for Japan were first developed by the Nitto Denko Corporation in the 1970s and include isosorbide dinitrate (Frandol® Tape-S) for angina pectoris, tulobuterol (Hokunalin® Tape) for asthma (Tamura et al., 2012) and bisoprolol patch (Bisono® Tape) for treating hypertension (Nitto, 2013). Table 2 also lists examples of patches applied to the skin for topical effects. The main active agents used are capsaicin, various diclofenac ion pairs and lidocaine.
In general, the bioequivalence of patch formulations of the same drug can be undertaken using either ex vivo human epidermal penetration studies or by assessment of the plasma drug concentration profiles. These are not always equivalent as shown by the similar skin penetration profiles for the nicotine products, Nicoderm and Habitrol (Ho and Chien, 1993), but significantly different nicotine plasma concentrations after 5 days multiple dosing (Cmax, Tmax, P < 0.001; AUC, P < 0.05) (Gupta et al., 1995). The higher dose of nicotine delivered from the Nicoderm patch, particularly during the first 8 h after application, was attributed to the presence of nicotine in Nicoderm adhesive layer acting as a priming dose (Gupta et al., 1995). In these studies, 21 mg of nicotine was applied to the upper back for 24 h. Later patch designs were for 16 and 21 h so that patients were not exposed to nicotine during their sleep. Fant et al. (2000) conducted a crossover study of three nicotine transdermal patches (a 15 mg per 16 h patch (Nicorette®, Maidenhead, UK) and two brands 21 mg per 24 h patches [Nicoderm (NiQuitin®; GlaxoSmithKline Consumer Healthcare, Brentford, UK)] and Habitrol (Nicotinell®; Novartis Consumer Health, Horsham, UK) and showed significant differences in the pharmacokinetic profiles between the two 21 mg patches and the 15 mg patch (AUC0–24 h and Cmax; P < 0.05). This study showed an unexpected peak-like delivery of nicotine from the reservoir/matrix patch arising from nicotine equilibrating in the adhesive layer during the storage of the patch. DeVeaugh-Geiss et al. (2010) also showed significant differences in the single-dose pharmacokinetic profiles of two nicotine transdermal patches, the Nicoderm (NiQuitin) 21 mg per 24 h patch and a newly UK available, Nicorette 25 mg per 16 h patch. A limitation in these studies was the lack of any apparent clinical efficacy or adverse profile comparisons.
A number of comparative bioequivalence studies have also been conducted with nitroglycerin (discussed earlier) and with fentanyl. Sathyan et al. (2005) suggested that the Duragesic DIA and reservoir fentanyl patches were bioequivalent, based upon single and multiple dose randomized controlled trials. However, Fiset et al. (1995) attributed an observed greater variability in absorption rate and fentanyl concentration for the matrix transdermal fentanyl patch developed by Cygnus compared with Alza's reservoir fentanyl patch to the absence of rate-controlling membrane. More recently, Marier et al. (2007) showed bioequivalence between a novel matrix formulation of fentanyl with a rate-controlling membrane (developed by Nycomed and known as Matrifen® in Europe) to the original reservoir Duragesic formulation in an open-label, randomized, fully replicated, four-way crossover study in healthy male subjects over a 72 h single patch application.
Variability, safety and regulatory issues for patches
Site of application
It has been well established that human skin penetration fluxes are highly dependent upon the site of application (Feldmann and Maibach, 1967; Scheuplein and Blank, 1971; Roberts et al., 1982; Roberts and Walters, 1998). However, some parts of the body (trunk and upper arm) appear to have similar fluxes, enabling patches to be interchangeably placed at those sites and to achieve similar plasma concentrations. For instance, MacGregor et al. (1985) showed that plasma concentrations obtained after the application of a 3.5 cm2 clonidine patch (Catapres-TTS) on chest and arm were not significantly different over the recommended wear time. Schenkel et al. (1986) also showed that Estraderm could be applied to different sites of the trunk and to the upper arm without significant differences in the oestradiol uptake. Gorsline et al. (1992) later showed that bioequivalent (AUC0–t, AUC0–∞ and Tmax) plasma were achieved irrespective of the application site on the upper body (upper back, upper outer arm, upper chest) from Nicoderm 14 mg per 24 h. Yu et al. (1997) showed that a testosterone transdermal system (D-Trans testosterone gel system®) could be applied interchangeably to the skin of the upper buttocks, upper arms or upper back, giving similar drug plasma concentrations at three different skin sites (AUC0–27, Cmax parameters not significantly different). Further, the plasma concentrations of norelgestromin and ethinyl oestradiol after application of the contraceptive patch Ortho Evra® remained within the reference ranges during the wear-period after application on abdomen, buttock, arm and torso (Abrams et al., 2002). However, Lefèvre et al. (2007) showed a higher plasma exposure of rivastigmine (AUC0–∞ and AUC0–last) after the application of Exelon® 8.5 mg per 24 h patch to the upper back, chest or upper arm rather than on the thigh and abdomen. Similarly, Taggart et al. (2000) showed that the extent of drug absorption (AUC0–168 and AUC0–last) from an oestradiol patch (Climara 0.1 mg per 24 h) application on buttock was significantly higher than when applied to the abdomen. However, the observed plasma drug concentrations for both sites were consistent with physiological oestradiol levels required for the relief of menopausal symptoms (Taggart et al., 2000). Finally, the systemic exposure of nicotine from Nicorette 15 mg per 16 h applied to the upper arm was higher compared with the abdomen but equivalent to the back (Sobue et al., 2005). Practically, transdermal systems should not be applied to the waistline as tight clothing may rub or remove the patch.
Safety
As discussed earlier, the safety ratio for the systemic percutaneous absorption of drugs presently marketed in patches relative to the maximum dose for that drug is usually at least 10 or more (Table 1). However, these safety ratios mainly relate to adult skin. Liebelt and Shannon (1993) pointed out that many commonly used over-the-counter (OTC) topical medications, including those containing methyl salicylate, camphor, topical imidazolines and benzocaine, can cause serious toxicity in children when ingested in small doses. Further, whereas the barrier function in full-term infants is fully developed, that in premature infants is incomplete (Fluhr et al., 2010; Delgado-Charro and Guy, 2014). Accordingly, transdermal administration has been used to deliver theophylline and caffeine in the premature infant, for whom dosing by conventional routes of administration can be difficult (Barrett and Rutter, 1994). However, this impaired skin barrier function in neonates also puts them more at risk (Kalia et al., 1998; Delgado-Charro and Guy, 2014) so that any unplanned percutaneous absorption in neonates is potentially hazardous (Rutter, 1987).
Transdermal patches have an additional drawback relative to other dosage forms and that is the potential for their ingredients, including both the active drug and the excipients, to induce adverse skin reactions, especially when the dosage form has prolonged contact with the skin for a long period of time. There are typically two types of skin reactions with patches: irritant contact dermatitis, which is the most common adverse effect associated with transdermal patch systems, and allergic contact dermatitis, which is infrequent (Ale et al., 2009). Most of the cutaneous adverse reactions reported in the literature with transdermal drug delivery systems have been induced by the drug itself, whereas the components of the patch (e.g. adhesive materials and chemical enhancers) have caused skin side effects to a lesser extent. Although generally mild and transient, these reactions can result in the discontinuation of the treatment by the patients (Murphy and Carmichael, 2000; Singh and Maibach, 2002). On the contrary, even the clonidine patch, with a noticeable degree of sensitization (Hogan and Maibach, 1990), is still well accepted and performs well in many patients.
Fentanyl patches have been a continual source for safety concerns. Duragesic was the first fentanyl patch to reach the market in 1990 and was characterized by a drug reservoir containing fentanyl and ethanol combined within a gel (Prodduturi et al., 2009).
Manufacturing defects (i.e. seal and membrane defects) with the possibility of dangerous drug leakage during use have led to patches being recalled in 2004 and 2008; as such leakage may expose patients to a potentially fatal overdose. The Duragesic leakage problem was addressed by a redesign of this patch to a DIA design in 2009 (Prodduturi et al., 2010). However, Oliveira et al. (2012) concluded that the possibility of fentanyl intoxication from the reservoir leakage of a commercially available fentanyl transdermal patch was unlikely to be toxic.
Fentanyl may also lead to patient issues as a result of the illicit use of fentanyl from these patches or after swallowing fentanyl patches. The US FDA issued Public Health Advisories in 2005 and 2007 to raise public awareness of the safe use of fentanyl patches and the dangers of accidental exposure (FDA, 2005; 2007,) after receiving reports of death and life-threatening side effects in patients using brand name Duragesic and the generic product due to an inappropriate use (e.g. multiple patch application) (Edinboro et al., 1997). Table 3 describes the initial amount of fentanyl on supply and the anticipated residual amount of fentanyl in a patch at the end of an application period. Of particular concern is the risk of fatal exposure for young children who have swallowed or left fentanyl patches on their skin (Teske et al., 2007). As a consequence, the US FDA has reinforced education of patients and caregivers for a proper disposal of fentanyl patches after the reports of 26 cases of paediatric accidental exposure to fentanyl over the past 15 years, including 10 deaths and 12 hospitalizations (FDA, 2012a,c,). The illicit use of fentanyl by recreational users is also of concern as fentanyl is 100 times more potent than morphine (Arvanitis and Satonik, 2002; Lilleng et al., 2004). Recreational users have extracted fentanyl from patches for subsequent injection (Firestone et al., 2009) and placed the patches into their mouth so that fentanyl can be absorbed through buccal mucosa (Nelson and Schwaner, 2009).
Table 3.
Drug utilization rate and residual amount of drug after use of recently approved, fentanyl and nicotine transdermal patches
| Drug (Trade name, year of FDA approval) | Patch design | Dose and size of patch – Delivery rate | Drug utilization rate (%)a | Residual amount of drug in the patch (mg)b | Patch area activity (%·cm−2)c |
|---|---|---|---|---|---|
| Methylphenidate (Daytrana®, 2006) | DIA | 27.5 mg in 12.5 cm2 – 1.1 mg·h−1 | 36 | 17.6 | 2.9 |
| 41.3 mg in 18.75 cm2 – 1.6 mg·h−1 | 34.9 | 26.9 | 1.9 | ||
| 55 mg in 25 cm2 – 2.2 mg·h−1 | 36 | 35.2 | 1.4 | ||
| 82.5 mg in 37.5 cm2 – 3.3 mg·h−1 | 36 | 52.8 | 1 | ||
| Selegiline (Emsam®, 2006) | DIA | 20 mg in 20 cm2 – 6 mg per 24 h | 30 | 14 | 1.5 |
| 30 mg in 30 cm2 – 9 mg per 24 h | 30 | 21 | 1 | ||
| 40 mg in 40 cm2 – 12 mg per 24 h | 30 | 28 | 0.75 | ||
| Rivastigmine (Exelon®, 2007) | Matrix | 9 mg in 5 cm2 – 4.6 mg per 24 h | 51.1 | 4.4 | 10.2 |
| 18 mg in 10 cm2 – 9.5 mg per 24 h | 52.8 | 8.5 | 5.3 | ||
| 27 mg in 15 cm2 – 13.3 mg per 24 h | 49.3 | 13.7 | 3.3 | ||
| Rotigotine (Neupro®, 2007) | DIA | 2.25 mg in 5 cm2 – 1 mg per 24 h | 44.4 | 1.25 | 8.9 |
| 4.5 mg in 10 cm2 – 2 mg per 24 h | 44.4 | 2.5 | 4.4 | ||
| 6.75 mg in 15 cm2 – 3 mg per 24 h | 44.4 | 3.75 | 3 | ||
| 9 mg in 20 cm2 – 4 mg per 24 h | 44.4 | 5 | 2.2 | ||
| 13.5 mg in 30 cm2 – 6 mg per 24 h | 44.4 | 7.5 | 1.5 | ||
| 18 mg in 40 cm2 – 8 mg per 24 h | 44.4 | 10 | 1.1 | ||
| Granisetron (Sancuso®, 2008) | DIA | 34.3 mg in 52 cm2 – 3.1 mg per 24 h | 63.3 | 12.6 | 1.2 |
| Buprenorphine (Butrans®, 2010) | DIA | 5 mg in 6.25 cm2 – 5 μg·h−1 | 16.8 | 4.26 | 2.7 |
| 7.5 mg in 7.5 cm2 – 7.5 μg·h−1 | 16.8 | 6.24 | 2.2 | ||
| 10 mg in 12.5 cm2 – 10 μg·h−1 | 16.8 | 8.32 | 1.3 | ||
| 15 mg in 18.75 cm2 – 15 μg·h−1 | 16.8 | 12.48 | 0.9 | ||
| 20 mg in 25 cm2 – 20 μg·h−1 | 16.8 | 16.64 | 0.7 | ||
| Oestradiol (Minivelle®, 2012) | DIA | 0.62 mg in 2.48 cm2 – 0.0375 mg·day−1 | 21.17 | 0.49 | 8.5 |
| 0.83 mg in 3.30 cm2 – 0.05 mg·day−1 | 21.1 | 0.66 | 6.4 | ||
| 1.24 mg in 4.95 cm2 – 0.075 mg·day−1 | 21.17 | 0.98 | 4.3 | ||
| 1.65 mg in 6.6 cm2 – 0.1 mg·day−1 | 21.21 | 1.3 | 3.2 | ||
| Fentanyl (Duragesic®, 1990) discontinued | Reservoir/Membrane | 1.25 mg in 5 cm2 – 12.5 μg·h−1 | 72 | 0.35 | 14.4 |
| 2.5 mg in 10 cm2 – 25 μg·h−1 | 72 | 0.7 | 7.2 | ||
| 5 mg in 20 cm2 – 50 μg·h−1 | 72 | 1.4 | 3.6 | ||
| 7.5 mg in 30 cm2 – 75 μg·h−1 | 72 | 2.1 | 2.4 | ||
| 10 mg in 40 cm2 – 100 μg·h−1 | 72 | 2.8 | 1.8 | ||
| Fentanyl (Mylan FTS, 2005) | DIA | 1.28 mg in 3.13 cm2 – 12.5 μg·h−1 | 70.3 | 0.38 | 22.5 |
| 2.55 mg in 6.25 cm2 – 25 μg·h−1 | 70.6 | 0.75 | 11.3 | ||
| 5.10 mg in 12.5 cm2 – 50 μg·h−1 | 70.6 | 1.5 | 5.6 | ||
| 7.65 mg in 18.75 cm2 – 75 μg·h−1 | 70.6 | 2.25 | 3.8 | ||
| 10.20 mg in 25 cm2 – 100 μg·h−1 | 70.6 | 3 | 2.8 | ||
| Fentanyl (Lavipharm Labs FTS, 2006) | DIA | 1.375 mg in 5 cm2 – 12 μg·h−1 | 62.8 | 0.51 | 12.6 |
| 2.75 mg in 10 cm2 – 25 μg·h−1 | 65.4 | 0.95 | 6.5 | ||
| 5.5 mg in 20 cm2 – 50 μg·h−1 | 65.4 | 1.9 | 3.3 | ||
| 8.25 mg in 30 cm2 – 75 μg·h−1 | 65.4 | 2.85 | 2.2 | ||
| 11.0 mg in 40 cm2 – 100 μg·h−1 | 65.4 | 3.8 | 1.6 | ||
| Fentanyl (Par Pharm FTS, 2007) | Reservoir/Membrane | 2.5 mg in 10 cm2 – 25 μg·h−1 | 72 | 0.7 | 7.2 |
| 5 mg in 20 cm2 – 50 μg·h−1 | 72 | 1.4 | 3.6 | ||
| 7.5 mg in 30 cm2 – 75 μg·h−1 | 72 | 2.1 | 2.4 | ||
| 10 mg in 40 cm2 – 100 μg·h−1 | 72 | 2.8 | 1.8 | ||
| Fentanyl (Watson FTS, 2007) | Reservoir/Membrane | 2.5 mg in 10 cm2 – 25 μg·h−1 | 72 | 0.7 | 7.2 |
| 5 mg in 20 cm2 – 50 μg·h−1 | 72 | 1.4 | 3.6 | ||
| 7.5 mg in 30 cm2 – 75 μg·h−1 | 72 | 2.1 | 2.4 | ||
| 10 mg in 40 cm2 – 100 μg·h−1 | 72 | 2.8 | 1.8 | ||
| Fentanyl (Aveva FTS, 2008) | DIA | 2.76 mg in 10.7 cm2 – 25 μg·h−1 | 65.2 | 0.96 | 6.1 |
| 5.52 mg in 21.4 cm2 – 50 μg·h−1 | 65.2 | 1.92 | 3 | ||
| 8.28 mg in 32.1 cm2 – 75 μg·h−1 | 65.2 | 2.88 | 2 | ||
| 11.04 mg in 42.8 cm2 – 100 μg·h−1 | 65.2 | 3.84 | 1.5 | ||
| Fentanyl (Duragesic®, 2009) | DIA | 2.1 mg in 5.25 cm2 – 12.5 μg·h−1 | 42.9 | 1.2 | 8.2 |
| 4.2 mg in 10.5 cm2 – 25 μg·h−1 | 42.9 | 2.4 | 4.1 | ||
| 8.4 mg in 21 cm2 – 50 μg·h−1 | 42.9 | 4.8 | 2 | ||
| 12.6 mg in 31.5 cm2 – 75 μg·h−1 | 42.9 | 7.2 | 1.4 | ||
| 16.8 mg in 42 cm2 – 100 μg·h | 42.9 | 9.6 | 1 | ||
| Fentanyl (Noven TTS, 2009) discontinued | DIA | 2.55 mg in 19 cm2 – 25 μg·h−1 | 70.6 | 0.75 | 3.7 |
| 5.10 mg in 38 cm2 – 50 μg·h−1 | 70.6 | 1.5 | 1.9 | ||
| 7.65 mg in 57 cm2 – 75 μg·h−1 | 70.6 | 2.25 | 1.2 | ||
| 10.20 mg in 76 cm2 – 100 μg·h−1 | 70.6 | 3 | 0.9 | ||
| Fentanyl (Mallinckrodt FTS, 2011) | Reservoir/Membrane | 2.75 mg in 7.8 cm2 – 25 μg·h−1 | 65.5 | 0.95 | 8.4 |
| 5.50 mg in 15.6 cm2 – 50 μg·h−1 | 65.5 | 1.9 | 4.2 | ||
| 8.25 mg in 23.4 cm2 – 75 μg·h−1 | 65.5 | 2.85 | 2.8 | ||
| 11.0 mg in 31.2 cm2 – 100 μg·h−1 | 65.5 | 3.8 | 2.1 | ||
| Nicotine (Nicoderm CQ®, 1991) | Reservoir/Matrix | 36 mg in 7 cm2 – 7 mg per 24 h | 19.4 | 29 | 2.8 |
| 75 mg in 15 cm2 – 14 mg per 24 h | 18.7 | 61 | 1.2 | ||
| 114 mg in 22 cm2 – 21 mg per 24 h | 18.4 | 93 | 0.8 | ||
| Nicotine (Habitrol®, 1991) | Matrix | 17.5 mg in 10 cm2 – 7 mg per 24 h | 40 | 10.5 | 4 |
| 35 mg in 20 cm2 – 14 mg per 24 h | 40 | 21 | 2 | ||
| 52.5 mg in 30 cm2 – 21 mg per 24 h | 40 | 31.5 | 1.3 | ||
| Nicotine (Prostep®, 1992) discontinued | Matrix | 15 mg in 24 (3.5) cm2 – 11 mg per 24 h | 73.3 | 4 | 20.9 |
| 30 mg in 32 (7) cm2 – 22 mg per 24 h | 73.3 | 8 | 10.5 | ||
| Nicotine (Nicotrol®, 1992) discontinued | DIA | 8.3 mg in 10 cm2 – 5 mg per 16 h | 60.2 | 3.3 | 6 |
| 16.6 mg in 20 cm2 – 10 mg per 16 h | 60.2 | 6.6 | 3 | ||
| 24.9 mg in 30 cm2 – 15 mg per 16 h | 60.2 | 9.9 | 2 |
For instance, the rivastigmine transdermal patch (Exelon) dosage strength 4.6 mg per 24 h, application time 24 h, patch size (active surface) 5 cm2, overall amount of drug substance incorporated into the patch 9 mg. Drug utilization = 4.6 mg; drug utilization rate = 4.6/9 ≈ 50%; residual amount = 9 − 4.6 = 4.4 mg; patch area activity = 50/5 ≈ 10%·cm−2. When the patch has to be applied twice weekly (every 3–4 days), t = 3.5 days is considered for calculation.
aDrug utilization rate (%) = (delivery rate × duration of application)/drug content. bResidual amount (mg) = drug content − drug utilization. cPatch area activity (%·cm−2) = drug utilization rate/patch size − ‘it is a measure of the formulation's intrinsic capability to release drug substance from the patch in vivo and as such a surrogate measurement of thermodynamic activity’ (EMEA, 2012).
The US FDA, in a Drug Safety Communication, has recently alerted the public that certain OTC topical muscle and joint pain relievers may cause burns (FDA, 2012b), especially for OTC topical patches containing menthol as the single active ingredient at 3% or more and methyl salicylate combinations above 10%. Concerns have also been reported for capsaicin, which normally leads to local warmth or coolness but no burns.
The presence of metals (e.g. aluminium) in the backing layer of certain transdermal patches such as Catapres-TTS, Habitrol, Nicotine CQ®, Neupro® and Transderm Scōp can pose safety concerns for patients undergoing an MRI scan (Ball and Smith, 2008; Durand et al., 2012). Skin burns have been reported at the patch site in several patients wearing an aluminized transdermal system during these types of procedures (Hong et al., 2010). Consequently, safe practice recommendations have been issued and the temporary removal of the transdermal system before such procedures may be the safest approach (FDA, 2009a; Kanal et al., 2013). Nowadays, most patches contain no conducting metal surfaces.
The prescribing information (PI) of recently approved transdermal patches, such as Butrans®, Exelon and Neupro, warns patients to avoid exposing the application site and surrounding area to direct external heat sources (e.g. heating pads, electric blankets, sunbathing, heat or tanning lamps, saunas, hot tubs or hot baths and heated water beds) while wearing the patch. In theory, fever could also result in an increase in plasma drug concentration due to temperature-dependent increases in drug release from the transdermal patch. In an open, randomized crossover study with 12 healthy smokers, Vanakoski et al. (1996) showed that a sauna significantly increased the amount of nicotine absorbed (P < 0.01) and transiently increased plasma drug concentration (Cmax and AUC0–1 significantly higher in the sauna session, P < 0.01) from nicotine transdermal patches (Nicorette) without adverse symptoms. Fentanyl overdoses have been described in case reports in which a fentanyl patch was covered by a warming blanket (Frolich et al., 2001) or a heating pad (Rose et al., 1993).
Regulatory
Three types of studies are normally used to evaluate a finished transdermal patch product: product quality tests, in vitro drug product performance tests and in vivo drug product performance test. The product quality attributes typically include description (visual examination of the patch), identification, assay (content of drug product), impurities, dosage form uniformity, residual solvent levels, cold flow property (adhesive migration out of the edge of the patch during storage or when the patch is applied to the patient), polymorphism and microbial limits. Other quality attributes may be product-specific such as water content (for hydroalcoholic reservoir patches), particle size (when the drug substance is suspended in the patch), crystal formation test (when a patch contains dissolved drug substance) and leak test (for liquid reservoir patch) (Van Buskirk et al., 2012; USP, 2014a).
Crystallization is a particular problem that may arise from supersaturated systems that are thermodynamically unstable and where drug may potentially crystallize out during storage. Crystallization was first observed with scopolamine patches in the late 1980s when the previously liquid base showed up instantly as crystalline hydrates (Campbell et al., 1989b). Later, more stable but less soluble and permeable polymorphic semi-hydrate oestradiol crystals could be generated in the presence of ambient humidity for any marketed oestradiol patch (Horstmann et al., 1998; Muller and Horstmann, 1999). The formation of ‘snowflake’ crystals in rotigotine transdermal patches led to the withdrawal of the product from some markets, underlining the severe impact that crystallization can have on a patch formulation (Chaudhuri, 2008; Waters, 2013). Low MW surfactants (e.g. Cremophor®), co-polymers of methacrylic (e.g. Eudragit®) (Kotiyan and Vavia, 2001; Cilurzo et al., 2005) and polyvinylpyrrolidone (Jain and Banga, 2012) are now often included in patches as crystallization inhibitors.
In vitro drug product performance usually involves three tests: in vitro drug release, in vitro skin permeation studies and in vitro adhesive tests. In vitro drug release tests evaluate the rate and the extent of release of drug from a transdermal patch as described in both European Pharmacopoeia (Ph Eur) and USP, including the paddle over disk method (USP Apparatus 5/Ph Eur 2.9.4.1), the rotating cylinder method (USP Appartus 6/Ph Eur 2.9.4.3) and the reciprocating holder method (USP Apparatus 7) (USP, 2014b; Ph Eur, 2015). The Organization for Economic Cooperation and Development and the European Medicine Agency (EMEA) provides guidance documents on the performance of in vitro permeation studies to evaluate the rate of transport (Organization for Economic Cooperation and Development, 2004; EMEA, 2012). Four tests are generally used to evaluate in vitro adhesive properties: the liner release test (force required to remove the liner from the adhesive prior to application of the patch, to determine the feasibility of removal by the patient), the probe tack test (ability of the adhesive to adhere to the surface with minimal contact pressure), the peel adhesion test (force required to peel away an adhesive after it has been attached to the substrate) and the shear test (static or dynamic) (the internal or cohesive strength of the adhesive) (Venkatraman and Gale, 1998; Mausar, 2011; Banerjee et al., 2014). Stainless steel remains the preferred substrate used for in vitro testing as it represents an acceptable alternative to human skin, which usually poses ethical issues, restricted availability (Cilurzo et al., 2012) and high variability. An ideal PSA used as part of a transdermal patch, (i) allows easy removal of the (properly selected) protective liner of the patch before use; (ii) has an initial affinity for human skin; (iii) adheres properly to human skin upon application; (iv) remains in place on the skin surface during the whole labelled wear-period; and (v) permits easy and clean removal of the patch after the period of use (Mausar, 2011; Van Buskirk et al., 2012).
In vivo drug product performance pharmacokinetic and in vivo adhesive performances are usually conducted in parallel. Clinical studies should determine the pharmacokinetic parameters – Cmax, Tmax, AUC0–∞ and AUC0–last (EMEA, 2012) – and the percentage of the patch area that remains attached to the skin throughout the proposed period of use should be assessed with an expectation of a mean adherence greater than 90% (Minghetti et al., 2004; EMEA, 2012). In principle, the most probable pharmacokinetic parameters for a new active in a patch can be estimated from a predicted delivery rate of the drug from patches as defined in Figure 4 and the drug pharmacokinetics in vivo. However, as shown in the recent correspondence on attempts to estimate steady-state transdermal patch structure–activity relationships based upon observed drug plasma concentrations, care is required in (i) the choice of physicochemical values, such as aqueous solubility, in calculations, regression models; (ii) identification of the role of rate-controlling membranes and/or enhancer effects, prediction of clearance and dose duration; and (iii) last but not least, consistency of units (Maibach and Farahmand, 2009a, b; Kissel and Bunge, 2010).
Another key regulatory aspect is the amount of unused drug left in the patch when it is removed from the skin, as defined by the FDA's guidance in August 2011 on Residual Drug in Transdermal and Related Drug Delivery Systems (FDA, 2011). The drug utilization rate and residual amount of drug after use in various marketed patches, in addition to fentanyl discussed earlier, are summarized in Table 3. Transdermal patches retain up to 95% of the initial total amount of drug after the intended wearing period (e.g. oestradiol patches). Alza's nicotine (membrane/reservoir) patch delivers only 18% of the nicotine contained, whereas LTS's construction delivers 40% and the PIB formula of Cygnus even reached 60%.
Future prospects of transdermal patches and transdermal drug delivery systems
In 2013, four drugs (oestradiol, fentanyl, nicotine and testosterone) accounted for around 50% of all transdermal clinical trials (463) listed on ClinicalTrials.gov (Watkinson, 2013). Of all the drugs contained in marketed transdermal patches, rotigotine is the only active compound that was originally developed to be administered via the transdermal route (McAfee et al., 2014). We began this review with a discussion of the original solution and semi-solid products for topical and transdermal delivery. Watkinson (2012) pointed out that there are at least nine non-occlusive passive transdermal products, including the 1988 approved Nitro-Bid nitroglycerin ointment (Fougera) delivering about 7.5 mg per dose and contrasting with the 0.2% nitroglycerin ointment used for anal fissures, a range of oestradiol products (Estrasorb®, Estragel®, Elestrin, Divigel and Evamist®, approved in 2003, 2004, 2006, 2007 and 2007, respectively) and oxybutynin (Gelnique®, approved in 2009). In addition, the bulk of the $2.15 billion testosterone market at the end of 2013 were cutaneous solutions (including gels), consisting of Androgel® ∼ 66% (approved in 2000), Axiron® ∼ 12.6% (approved in 2010), Testim® gel ∼12.6% (approved in 2002) and Fortesta® gel ∼5.6% (approved in 2010) with the patch, Androderm, at ∼3.2% (Acrux Ltd, 2014). Importantly, two testosterone replacement gels, Androgel and Testim, now carry FDA's strongest black-box warning for secondary exposure in children to application sites, left over gel and unwashed linen (FDA, 2009b). In this context, it is of note that two systems, developed by Acrux Ltd., use a ‘no-touch’ metered-dose pump technology: Evamist (oestradiol) (Figure 1L) and Axiron (testosterone) (Perumal et al., 2013).
Today, there is a move towards ‘active’ transdermal delivery systems that use non- and minimally invasive technologies, such as iontophoresis, microneedles, electroporation and sonophoresis, to enhance drug delivery across the skin as well as challenging drug candidates, such as actives that have a low penetration flux and low potency (Naik et al., 2000; Gratieri et al., 2013). The development of active patches has however been associated with much false hope with initial commercial success being hampered by commercial, technical and consumer issues (Watkinson, 2012). This history is probably best illustrated by the mixed success so far in achieving painless local anaesthesia with lidocaine. One of the first FDA-approved topical (local) iontophoretic patch system, Iontocaine® from Iomed (Salt Lake City, UT, USA), was approved in 1995 and discontinued in 2005. This was followed by ultrasound Sonoprep® and iontophoretic LidoSite® both approved in 2004 but discontinued in 2007 and 2008, respectively, and then by the i.d. powder injector Zingo® that was approved in 2008, withdrawn in 2008 and re-launched in September 2014 (Marathon Pharmaceuticals News, 2014). The failed iontophoretic GlucoWatch Biographer® is the only non-invasive glucose monitor to have been approved by the FDA (Wiedersberg and Guy, 2014). The only success story appears to be Synera® (Zars Pharma, now Nuvo Research), a heat-activated topical lidocaine/tetracaine patch, approved in 2005 and still on the market (Synera, 2014). Transdermal systems also face challenges as illustrated by the transdermal iontophoretic patch, Ionsys®, approved in 2006 for the systemic delivery of fentanyl for fast relief of post-operative pain. Ionsys was initially suspended by the EMEA in November 2008 due to patch corrosion, which could potentially lead to self-activation of the system and a potential overdose (Watkinson, 2012; Li et al., 2013). Its safety features are now being revamped by Incline Therapeutics ($43 million Series A funding) to be launched in the United States in 2014–2016 (The Medicines Company, 2012; Watkinson, 2012). Much hope therefore rests with Zecuity® (NuPathe, now Teva) (Figure 1M), which uses iontophoresis to actively deliver sumatriptan through the skin to manage the migraine-related nausea and vomiting that can limit p.o. dosing (Goldstein et al., 2012; Smith et al., 2012).
The most recent ‘hype’ for a drug delivery system is the use of microneedles with the main focus being on single-dose vaccine delivery (Quinn et al., 2014). For instance, the Nanopatch® (Figure 1N) required a second-order lower dose of antigen to be delivered to the skin to achieve antibody responses comparable to conventional i.m. injection (Fernando et al., 2010). The use of microneedles for long-term treatment has also been recently investigated for the treatment of opiate and alcohol dependence with naltrexone, an opioid antagonist (Wermeling et al., 2008). A parathyroid hormone (1–34)-coated microneedle patch, developed by Zosano Pharma (formerly, Macroflux® Alza Corporation) for the treatment of osteoporosis, has been shown to be efficacious in a Phase II clinical trial (Daddona et al., 2011). A key question asked by Wiedersberg and Guy (2014), concluding a review on these technologies, is: ‘where is the obvious unmet medical need that microneedles (or indeed any of the poration approaches) can address better, more reliably and safer than a conventional needle-and syringe?’.
Finally, transdermal delivery systems, particularly transdermal patches, are increasingly being used in the paediatric population. A range of transdermal patches (i.e. about 10 drugs) have been used in children and some have been specifically developed for paediatric use, as illustrated by the methylphenidate patch for the treatment of attention deficit hyperactivity disorder. However, while transdermal delivery can be regarded as a convenient non-invasive method of drug delivery for term infants and older children requiring smaller doses than adults, formulation challenges remain for premature neonates with an immature skin barrier (Delgado-Charro and Guy, 2014).
Conclusions
Topical delivery systems have been used for various ailments and as cosmetics since the arrival of man. Over time, there has been a definition of suitable drug candidates for transdermal delivery and the associated development of technologies, both passive and active, that has led to delivery enhancement, precision in drug dosing and a better meeting of individual needs. A focus in the further development of drugs in transdermal patches and associated delivery forms remains the finding of sufficiently potent drugs that can penetrate the skin with an appropriate transdermal technology. A key challenge is to meet clinical and cosmetic needs, which cannot be appropriately met in a cost-effective manner through other routes of delivery.
Acknowledgments
Two authors (M. N. P. and M. S. R.) thank the National Heath & Medical Research Council of Australia for their support. We also thank Professor Françoise Falson and Professor Hamid Moghimi for their suggestions on the early history of topical products, Dr Lorraine Mackenzie for proofreading the final revised manuscript as well as Professor Gordon Flynn for reviewing the historical section of this review and making Figure 2 available for us, for which he retains copyright. We also thank Mr Ben Miller and all reviewers for the helpful comments.
Glossary
- DIA
drug-in-adhesive
- EMEA
European Medicine Agency
- FDA
Food and Drug Administration
- J&J
Johnson & Johnson
- LTS
Lohmann Therapie-Systeme
- OTC
over-the-counter
- Ph Eur
European Pharmacopoeia
- PI
prescribing information
- PIB
polyisobutylene
- PSA
pressure-sensitive adhesive
- TTS
transdermal therapeutic system
- USP
United States Pharmacopoeia
Conflict of interest
The authors disclose no potential conflicts of interest.
References
- Abrams LS, Skee DM, Natarajan J, Wong FA, Anderson GD. Pharmacokinetics of a contraceptive patch (Evra/Ortho Evra) containing norelgestromin and ethinyloestradiol at four application sites. Br J Clin Pharmacol. 2002;53:141–146. doi: 10.1046/j.0306-5251.2001.01532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acrux Ltd. 2014. Data estimated from the ‘US testosterone therapy market histogram’. Annual Report 2014 [Online]. Available at: http://www.acrux.com.au/IRM/Company/ShowPage.aspx/PDFs/1381-10000000/2014AnnualReport (accessed 10/28/2014)
- Ahmed SR, Boucher AE, Manni A, Santen RJ, Bartholomew M. Transdermal testosterone therapy in the treatment of male hypogonadism. J Clin Endocrinol Metab. 1988;66:546–551. doi: 10.1210/jcem-66-3-546. [DOI] [PubMed] [Google Scholar]
- Aiache JM. Historique des emplâtres. Bull Tech Gattefossé. 1984;77:9–17. [Google Scholar]
- Ale I, Lachapelle J-M, Maibach HI. Skin tolerability associated with transdermal drug delivery systems: an overview. Adv Ther. 2009;26:920–935. doi: 10.1007/s12325-009-0075-9. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1458. [Google Scholar]
- Anissimov YG, Roberts MS. Modelling dermal drug distribution after topical application in human. Pharm Res. 2011;28:2119–2129. doi: 10.1007/s11095-011-0437-2. [DOI] [PubMed] [Google Scholar]
- Arndts D, Arndts K. Pharmacokinetics and pharmacodynamics of transdermally administered clonidine. Eur J Clin Pharmacol. 1984;26:79–85. doi: 10.1007/BF00546713. [DOI] [PubMed] [Google Scholar]
- Arvanitis ML, Satonik RC. Transdermal fentanyl abuse and misuse. Am J Emerg Med. 2002;20:58–59. doi: 10.1053/ajem.2002.29562. [DOI] [PubMed] [Google Scholar]
- Azzaro AJ, Ziemniak J, Kemper E, Campbell BJ, VanDenBerg C. Pharmacokinetics and absolute bioavailability of selegiline following treatment of healthy subjects with the selegiline transdermal system (6 mg/24 h): a comparison with oral selegiline capsules. J Clin Pharmacol. 2007;47:1256–1267. doi: 10.1177/0091270007304779. [DOI] [PubMed] [Google Scholar]
- Bacon TH, Hole JG, North M, Burnett I. Analgesic efficacy of sustained release paracetamol in patients with osteoarthritis of the knee. Br J Clin Pharmacol. 2002;53:629–636. doi: 10.1046/j.1365-2125.2002.01603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RW, Kochinke F. 1989. Novel transdermal nicotine patch. US Patent 4,839,174, Pharmetrix Corporation.
- Ball AM, Smith KM. Optimizing transdermal drug therapy. Am J Health Syst Pharm. 2008;65:1337–1346. doi: 10.2146/ajhp070554. [DOI] [PubMed] [Google Scholar]
- Bals-Pratsch M, Knuth UA, Yoon YD, Nieschlag E. Transdermal testosterone substitution therapy for male hypogonadism. Lancet. 1986;2:943–946. doi: 10.1016/s0140-6736(86)90600-8. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Chattopadhyay P, Ghosh A, Datta P, Veer V. Aspect of adhesives in transdermal drug delivery systems. Int J Adhes Adhes. 2014;50:70–84. [Google Scholar]
- Bannon YB, Corish J, Corrigan OI, Geoghegan EJ, Masterson JG. 1994. Method for the treatment of withdrawal symptoms associated with smoking cessation and preparations for use in said method. US Patent 5,298,257, Elan Transdermal Limited.
- Barrett DA, Rutter N. Transdermal delivery and the premature neonate. Crit Rev Ther Drug Carrier Syst. 1994;11:1–30. [PubMed] [Google Scholar]
- Bauer JA, Seifert J. Sublingual transmucosal delivery of organic nitrates: treatment of angina pectoris. In: Ghosh TK, Pfister WR, editors. Drug Delivery to the Oral Cavity Molecules to Market. London: Taylor & Francis Group; 2005. pp. 111–124. [Google Scholar]
- Beckett A, Gorrod J, Taylor D. Comparison of oral and percutaneous routes in man for the systemic administration of ‘ephedrines’. J Pharm Pharmacol. 1972;24:65P–70P. [PubMed] [Google Scholar]
- Ben-Galim E, Hillman RE, Weldon W. Topically applied testosterone and phallic growth. Its effects in male children with hypopituitarism and microphallus. Am J Dis Child. 1980;134:296–298. doi: 10.1001/archpedi.1980.02130150050013. [DOI] [PubMed] [Google Scholar]
- Bender GA, Thom RA. Great Moments in Medicine and Pharmacy: a History of Medicine and Pharmacy in Pictures. Detroit, MI: Northwood Institute Press; 1966. [Google Scholar]
- Benowitz NL, Jacob P, 3rd, Jones RT, Rosenberg J. Interindividual variability in the metabolism and cardiovascular effects of nicotine in man. J Pharmacol Exp Ther. 1982;221:368–372. [PubMed] [Google Scholar]
- Berner B, Cooper ER. Models of skin permeability. In: Kydonieus AF, Berner B, editors. Transdermal Delivery of Drugs. Boca Raton, FL: CRC Press; 1987. pp. 41–56. [Google Scholar]
- Bhalla HL, Toddywala RD. Transdermal films of ephedrine. Drug Dev Ind Pharm. 1988;14:119–131. [Google Scholar]
- Boekhorst JC. Allergic contact dermatitis with transdermal clonidine. Lancet. 1983;2:1031–1032. doi: 10.1016/s0140-6736(83)91020-6. [DOI] [PubMed] [Google Scholar]
- Bogaert MG. Clinical pharmacokinetics of glyceryl trinitrate following the use of systemic and topical preparations. Clin Pharmacokinet. 1987;12:1–11. doi: 10.2165/00003088-198712010-00001. [DOI] [PubMed] [Google Scholar]
- Brown EW, Scott WO. The absorption of methyl salicylate by the human skin. J Pharmacol Exp Ther. 1934;50:32–50. [Google Scholar]
- Bryan CP. The Papyrus Ebers. Chicago, IL: Ares; 1930. [Google Scholar]
- Buchkremer G, Bents H, Horstmann M, Opitz K, Tölle R. Combination of behavioral smoking cessation with transdermal nicotine substitution. Addict Behav. 1989;14:229–238. doi: 10.1016/0306-4603(89)90054-3. [DOI] [PubMed] [Google Scholar]
- Busse KL, Maibach HI. Transdermal estradiol and testosterone transfer in man: existence, models, and strategies for prevention. Skin Pharmacol Physiol. 2011;24:57–66. doi: 10.1159/000321444. [DOI] [PubMed] [Google Scholar]
- Campbell PS, Chandrasekaran SK. 1983. Dosage for coadministering drug and percutaneous absorption enhancer. US Patent 4,379,454, Alza Corporation.
- Campbell PS, Chandrasekaran SK. 1984. Percutaneous absorption enhancer dispenser for use in coadministering drug and percutaneous absorption enhancer. US Patent 4,460,372, Alza Corporation.
- Campbell PS, Eckenhoff JB. 1987. Transdermal therapeutic system having improved delivery characteristics. US Patent 4,704,282, Alza Corporation.
- Campbell PS, Eckenhoff JB, Place VA. 1988. Transdermal drug delivery device. US Patent 4,725,439, Alza Corporation.
- Campbell PS, Eckenhoff JB, Place VA. 1989a. Transdermal drug delivery device. US Patent 4,867,982, Alza Corporation.
- Campbell PS, Enscore DJ, Gale R, Kaufman A. 1989b. Method for preventing the formation of a crystalline hydrate in a dispersion of a liquid in a monaqueous matrix. US Patent 4,832,953, Alza Corporation.
- Caplan RA, Ready LB, Oden RV, Matsen FA, 3rd, Nessly ML, Olsson GL. Transdermal fentanyl for postoperative pain management. A double-blind placebo study. JAMA. 1989;261:1036–1039. [PubMed] [Google Scholar]
- Castle T. Lexicon Pharmaceuticum: Or a Pharmaceutical Dictionary, Comprehending the Pharmacopoeias of London, Edinburgh, and Dublin, with a Variety of Other Use. 2nd edn. London: E. Cox & Son; 1828. [Google Scholar]
- Chambers Fox S. Remington Education: Pharmaceutics. 1st edn. London: Pharmaceutical Press; 2014. [Google Scholar]
- Chandrasekaran SK, Michaels AS, Campbell PS, Shaw JE. Scopolamine permeation through human skin in vitro. AIChE J. 1976;22:828–832. [Google Scholar]
- Chandrasekaran SK, Darda S, Michaels AS, Cleary GW. 1980. Therapeutic system for administering clonidine transdermally. US Patent 4,201,211, Alza Corporation & Boehringer Ingelheim GmbH.
- Chaudhuri KR. Crystallisation within transdermal rotigotine patch: is there cause for concern? Expert Opin Drug Deliv. 2008;5:1169–1171. doi: 10.1517/17425240802500870. [DOI] [PubMed] [Google Scholar]
- Chiang C-C, Lin T-S, Chen R. 2009. Compositions and methods for the transdermal delivery of pharmaceutical compounds. US Patent Application 2009/0297591 A1, Orient Pharma Co., Ltd.
- Chien YW. Development of transdermal drug delivery systems. Drug Dev Ind Pharm. 1987;13:589–651. [Google Scholar]
- Cilurzo F, Minghetti P, Casiraghi A, Tosi L, Pagani S, Montanari L. Polymethacrylates as crystallization inhibitors in monolayer transdermal patches containing ibuprofen. Eur J Pharm Biopharm. 2005;60:61–66. doi: 10.1016/j.ejpb.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Cilurzo F, Gennari CGM, Minghetti P. Adhesive properties: a critical issue in transdermal patch development. Expert Opin Drug Deliv. 2012;9:33–45. doi: 10.1517/17425247.2012.637107. [DOI] [PubMed] [Google Scholar]
- Cleary GW. Transdermal delivery systems: a medical rationale. In: Shah VP, Maibach HI, editors. Topical Drug Bioavailability, Bioequivalence, and Penetration. New York: Plenum Press; 1993. pp. 17–68. [Google Scholar]
- Cleary GW, Roy SD. 1993. Laminated composite for transdermal administration of fentanyl. US Patent 5,186,939, Cygnus Therapeutic Systems.
- Cole HN, Gammel JA, Rauschkolb JE, Schreiber N, Sollmann T. Excretion of mercury after intramuscular injection of mercuric bromide, inunction and rectal suppositories. Arch Derm Syphilol. 1926;14:683–692. [Google Scholar]
- Cole HN, Schreiber N, Sollmann T. Mercurial ointments in the treatment of syphilis: their absorption as measured by studies on excretion. Arch Derm Syphilol. 1930;21:372–393. [Google Scholar]
- Coleman JW. Nitric oxide in immunity and inflammation. Int Immunopharmacol. 2001;1:1397–1406. doi: 10.1016/s1567-5769(01)00086-8. [DOI] [PubMed] [Google Scholar]
- Cossum PA, Roberts MS. Stability of nitroglycerin ointment. J Pharm Sci. 1981;70:832–833. doi: 10.1002/jps.2600700740. [DOI] [PubMed] [Google Scholar]
- Coxe JR. The American Dispensatory, Containing the Natural, Chemical, Pharmaceutical and Medical History of the Different Substances Employed in Medicine, Together with the Operations of Pharmacy. 8th edn. Philadelphia, PA: Carey & Lea; 1830. [Google Scholar]
- Crandall LA, Leake CD, Loevenhart AS, Muehlberger CW. Acquired tolerance to and cross tolerance between the nitrous and nitric acid esters and sodium nitrite in man. J Pharmacol Exp Ther. 1931;41:103–119. [Google Scholar]
- Cross SE, Anderson C, Roberts MS. Topical penetration of commercial salicylate esters and salts using human isolated skin and clinical microdialysis studies. Br J Clin Pharmacol. 1998;46:29–35. doi: 10.1046/j.1365-2125.1998.00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daddona PE, Matriano JA, Mandema J, Maa Y-F. Parathyroid hormone (1–34)-coated microneedle patch system: clinical pharmacokinetics and pharmacodynamics for treatment of osteoporosis. Pharm Res. 2011;28:159–165. doi: 10.1007/s11095-010-0192-9. [DOI] [PubMed] [Google Scholar]
- Dancik Y, Thörling C, Krishnan G, Roberts M. Cutaneous metabolism and active transport in transdermal drug delivery. In: Monteiro-Riviere N, editor. Toxicology of the Skin: Target Organ Toxicology Series. Boca Raton, FL: Taylor & Francis Group; 2010. pp. 69–82. [Google Scholar]
- Dancik Y, Anissimov YG, Jepps OG, Roberts MS. Convective transport of highly plasma protein bound drugs facilitates direct penetration into deep tissues after topical application. Br J Clin Pharmacol. 2012;73:564–578. doi: 10.1111/j.1365-2125.2011.04128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasta JF, Geraets DR. Topical nitroglycerin: a new twist to an old standby. Am Pharm. 1982;NS22:29–35. [PubMed] [Google Scholar]
- Davis JA, Wiesel BH. The treatment of angina pectoris with a nitroglycerin ointment. Am J Med Sci. 1955;230:259–263. doi: 10.1097/00000441-195509000-00004. [DOI] [PubMed] [Google Scholar]
- Delanoe D, Fougeyrollas B, Meyer L, Thonneau P. Androgenization of female partners of men on medroxyprogesterone acetate/percutaneous testosterone contraception. Lancet. 1984;1:276. doi: 10.1016/s0140-6736(84)90144-2. [DOI] [PubMed] [Google Scholar]
- Delgado-Charro MB, Guy RH. Effective use of transdermal drug delivery in children. Adv Drug Deliv Rev. 2014;73:63–82. doi: 10.1016/j.addr.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Devane J, Mulligan S, Foynes M, Martin M. In vivo pharmacokinetic characteristics of a transdermal phenylpropanolamine (PPA) preparation. Eur J Drug Metab Pharmacokinet. 1991;3:297–299. [PubMed] [Google Scholar]
- DeVeaugh-Geiss AM, Chen LH, Kotler ML, Ramsay LR, Durcan MJ. Pharmacokinetic comparison of two nicotine transdermal systems, a 21-mg/24-hour patch and a 25-mg/16-hour patch: a randomized, open-label, single-dose, two-way crossover study in adult smokers. Clin Ther. 2010;32:1140–1148. doi: 10.1016/j.clinthera.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Dorne JL, Renwick AG. The refinement of uncertainty/safety factors in risk assessment by the incorporation of data on toxicokinetic variability in humans. Toxicol Sci. 2005;86:20–26. doi: 10.1093/toxsci/kfi160. [DOI] [PubMed] [Google Scholar]
- Douchamps J, Derenne F, Stockis A, Gangji D, Juvent M, Herchuelz A. The pharmacokinetics of oxybutynin in man. Eur J Clin Pharmacol. 1988;35:515–520. doi: 10.1007/BF00558247. [DOI] [PubMed] [Google Scholar]
- 2014. Drugs Scopolamine dosage [Online]. Available at: http://www.drugs.com/dosage/scopolamine.html (accessed 10/14/2014)
- Durand C, Alhammad A, Willett KC. Practical considerations for optimal transdermal drug delivery. Am J Health Syst Pharm. 2012;69:116–124. doi: 10.2146/ajhp110158. [DOI] [PubMed] [Google Scholar]
- Duthie DJ, Rowbotham DJ, Wyld R, Henderson PD, Nimmo WS. Plasma fentanyl concentrations during transdermal delivery of fentanyl to surgical patients. Br J Anaesth. 1988;60:614–618. doi: 10.1093/bja/60.6.614. [DOI] [PubMed] [Google Scholar]
- Ebbell B. The Papyrus Ebers: the Greatest Egyptian Medical Document. Copenhagen: Levin & Munksgaard; 1937. [Google Scholar]
- Ebert CD, Patel D, Heiber W. 1992. Method and device for transdermally administering testosterone across nonscrotal skin at therapeutically effective levels. US Patent 5,152,997, Theratech, Inc.
- Edinboro LE, Poklis A, Trautman D, Lowry S, Backer R, Harvey CM. Fatal fentanyl intoxication following excessive transdermal application. J Forensic Sci. 1997;42:741–743. [PubMed] [Google Scholar]
- EMEA. 2012. Guidelines on quality of transdermal patches.
- Enscore DJ, Gale RM. 1985. Matrix composition for transdermal therapeutic system. US Patent 4,559,222, Alza Corporation.
- Etscorn FT. 1986. Transcutaneous application of nicotine. US Patent 4,597,961.
- Evans ES. A case of nitroglycerin poisoning. JAMA. 1912;58:550. [Google Scholar]
- Fant RV, Henningfield JE, Shiffman S, Strahs KR, Reitberg DP. A pharmacokinetic crossover study to compare the absorption characteristics of three transdermal nicotine patches. Pharmacol Biochem Behav. 2000;67:479–482. doi: 10.1016/s0091-3057(00)00399-3. [DOI] [PubMed] [Google Scholar]
- Faulkner JM. Nicotine poisoning by absorption through the skin. JAMA. 1933;100:1164–1665. [Google Scholar]
- FDA. 2004. FDA acts to remove ephedra-containing dietary supplements from market.
- FDA. 2005. Safety warnings regarding use of fentanyl transdermal (skin) patches.
- FDA. 2007. FDA Public Health Advisory: important information for the safe use of fentanyl transdermal system (patch)
- FDA. 2009a. Public Health Advisory: risk of burns during MRI scans from transdermal drug patches with metallic backings.
- FDA. 2009b. Topical testosterone gel products (marketed as AndroGel 1% and Testim 1%): secondary exposure of children to topical testosterone products.
- FDA. 2011. Guidance for industry: residual drug in transdermal and related drug delivery systems.
- FDA. 2012a. Fentanyl patch can be deadly to children.
- FDA. 2012b. FDA Drug Safety Communication: rare cases of serious burns with the use of over-the-counter topical muscle and joint pain relievers.
- FDA. 2012c. FDA reminds the public about the potential for life-threatening harm from accidental exposure to fentanyl transdermal systems (‘patches’)
- FDA. 2014. Orange Book. Annual Edition, 34th edn. U. S. Food and Drug Administration: Silver Spring, MD. Available at: http://www.fda.gov/Drugs/InformationOnDrugs/ucm129662.htm (accessed 2/23/2015)
- Feldmann RJ, Maibach HI. Regional variation in percutaneous penetration of 14C cortisol in man. J Invest Dermatol. 1967;48:181–183. doi: 10.1038/jid.1967.29. [DOI] [PubMed] [Google Scholar]
- Fernando GJ, Chen X, Prow TW, Crichton ML, Fairmaid EJ, Roberts MS, et al. Potent immunity to low doses of influenza vaccine by probabilistic guided micro-targeted skin delivery in a mouse model. PLoS ONE. 2010;5:e10266. doi: 10.1371/journal.pone.0010266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestone M, Goldman B, Fischer B. Fentanyl use among street drug users in Toronto, Canada: behavioural dynamics and public health implications. Int J Drug Policy. 2009;20:90–92. doi: 10.1016/j.drugpo.2008.02.016. [DOI] [PubMed] [Google Scholar]
- Fiset P, Cohane C, Browne S, Brand SC, Shafer SL. Biopharmaceutics of a new transdermal fentanyl device. Anesthesiology. 1995;83:459–469. doi: 10.1097/00000542-199509000-00004. [DOI] [PubMed] [Google Scholar]
- Fitzsimons MP. Gynaecomastia in stilboestrol worker. Br J Ind Med. 1944;1:235–237. [Google Scholar]
- Fleischer R. 1877. Untersuchgen uber das resporptions-vermogen der menschlichen haut. Habilitationsschrift: 81.
- Fluhr JW, Darlenski R, Taieb A, Hachem JP, Baudouin C, Msika P, et al. Functional skin adaptation in infancy – almost complete but not fully competent. Exp Dermatol. 2010;19:483–492. doi: 10.1111/j.1600-0625.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- Forbes RJ. Studies in Ancient Technology. III. Leiden: EJ Brill; 1955. [Google Scholar]
- Fotherby K. Bioavailability of orally administered sex steroids used in oral contraception and hormone replacement therapy. Contraception. 1996;54:59–69. doi: 10.1016/0010-7824(96)00136-9. [DOI] [PubMed] [Google Scholar]
- Fox MJ, Leslie CL. Treatment of Raynaud's diseases with nitroglycerine. Wis Med J. 1948;47:855–858. [PubMed] [Google Scholar]
- Frank T, Wall B, Platt B, Lane R. 2014. Transdermal therapeutic system. US Patent Application 2014/0134230 A1, Novartis AG.
- Frolich M, Giannotti A, Modell JH. Opioid overdose in a patient using a fentanyl patch during treatment with a warming blanket. Anesth Analg. 2001;93:647–648. doi: 10.1097/00000539-200109000-00023. [DOI] [PubMed] [Google Scholar]
- Gafni Y, Weisman A, Adin I. 2008. Crystalline granisetron base and production process therefor. US Patent Application 2008/0242696 A1, Chemagis Ltd.
- Gale RM, Berggren RG. 1986. Transdermal delivery system for delivering nitroglycerin at high transdermal fluxes. US Patent 4,615,699, Alza Corporation.
- Gale RM, Goetz V, Lee ES, Taskovich LT, Yum SI. 1986. Transdermal administration of fentanyl and device thereof. US Patent 4,588,580, Alza Corporation.
- Gay LN, Carliner PE. The prevention and treatment of motion sickness. I. Seasickness. Science. 1949;109:359. doi: 10.1126/science.109.2832.359. [DOI] [PubMed] [Google Scholar]
- Gehlbach SH, Williams WA, Perry LD, Woodall JS. Green-tobacco sickness. An illness of tobacco harvesters. JAMA. 1974;229:1180–1883. [PubMed] [Google Scholar]
- Gehlbach SH, Perry LD, Williams WA, Freeman JI, Langone JJ, Peta LV, et al. Nicotine absorption by workers harvesting green tobacco. Lancet. 1975;1:478–480. [PubMed] [Google Scholar]
- Geller MJ. Ancient Babylonian Medicine. 1st edn. Malden, MA: Wiley-Blackwell; 2010. [Google Scholar]
- Gemmell D, Morrison J. The release of medicinal substances from topical applications and their passage through the skin. J Pharm Pharmacol. 1957;9:641–656. doi: 10.1111/j.2042-7158.1957.tb12320.x. [DOI] [PubMed] [Google Scholar]
- Goldstein J, Smith TR, Pugach N, Griesser J, Sebree T, Pierce M. A sumatriptan iontophoretic transdermal system for the acute treatment of migraine. Headache. 2012;52:1402–1410. doi: 10.1111/j.1526-4610.2012.02198.x. [DOI] [PubMed] [Google Scholar]
- Goldzieher JW, Baker RE. The percutaneous absorption of estradiol-17β and progesterone. J Invest Dermatol. 1960;35:215–218. [PubMed] [Google Scholar]
- Good WR, Powers MS, Campbell P, Schenkel L. A new transdermal delivery system for estradiol. J Control Release. 1985;2:89–97. [Google Scholar]
- Gorsline J, Okerholm RA, Rolf CN, Moos CD, Hwang SS. Comparison of plasma nicotine concentrations after application of Nicorderm (nicotine transdermal system) to different skin sites. J Clin Pharmacol. 1992;32:576–581. doi: 10.1177/009127009203200615. [DOI] [PubMed] [Google Scholar]
- Govil SK. Transdermal drug delivery devices. In: Tyle P, editor. Drug Delivery Devices: Fundamentals and Applications. New York: Marcel Dekker; 1988. pp. 385–419. [Google Scholar]
- Govil SK, Weimann LJ. 2006. Adhesive mixture for transdermal delivery of highly plasticizing drugs. US Patent 7,150,881 B2, Mylan Technologies, Inc.
- Govil SK, Rudnic EM, Sterner DG. 1993. Transdermal nitroglycerin patch with penetration enhancers. US Patent 5,262,165, Schering Corporation.
- Gratieri T, Alberti I, Lapteva M, Kalia YN. Next generation intra- and transdermal therapeutic systems: using non- and minimally-invasive technologies to increase drug delivery into and across the skin. Eur J Pharm Sci. 2013;18:609–622. doi: 10.1016/j.ejps.2013.03.019. [DOI] [PubMed] [Google Scholar]
- Graybriel A. Prevention and treatment of space sickness in shuttle-orbiter missions. Aviat Space Environ Med. 1979;50:171–176. [PubMed] [Google Scholar]
- Graybriel A, Kneption J, Shaw J. Prevention of experimental motion sickness by scopolamine absorbed through the skin. Aviat Space Environ Med. 1976;47:1096–1100. [PubMed] [Google Scholar]
- Graybriel A, Cramer DB, Wood CD. Experimental motion sickness: efficacy of transdermal scopolamine plus ephedrine. Aviat Space Environ Med. 1981;52:337–339. [PubMed] [Google Scholar]
- Greenhill LL, Perel JM, Rudolph G, Feldman B, Curran S, Puig-Antich J, et al. Correlations between motor persistence and plasma levels in methylphenidate-treated boys with ADHD. Int J Neuropsychopharmacol. 2001;4:207–215. doi: 10.1017/S1461145701002413. [DOI] [PubMed] [Google Scholar]
- Groth H, Vetter H, Knusel J, Boerlin HJ, Walger P, Baumgart P, et al. Clonidine through the skin in the treatment of essential hypertension: is it practical? J Hypertens Suppl. 1983;1:120–122. [PubMed] [Google Scholar]
- Guay DR. Clinical pharmacokinetics of drugs used to treat urge incontinence. Clin Pharmacokinet. 2003;42:1243–1285. doi: 10.2165/00003088-200342140-00004. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Okerholm RA, Eller M, Wei G, Rolf CN, Gorsline J. Comparison of the pharmacokinetics of two nicotine transdermal systems: nicoderm and habitrol. J Clin Pharmacol. 1995;35:493–498. doi: 10.1002/j.1552-4604.1995.tb04093.x. [DOI] [PubMed] [Google Scholar]
- Guy RH, Hadgraft J. Rate control in transdermal delivery? Int J Pharm. 1992;82:R1–R6. [Google Scholar]
- Hadgraft JW, Somers GF. A method for studying percutaneous absorption in the rat. J Pharm Pharmacol. 1954;6:944–949. doi: 10.1111/j.2042-7158.1954.tb11029.x. [DOI] [PubMed] [Google Scholar]
- Hadgraft JW, Somers GF, Williams HS. Percutaneous absorption using diiodofluorescein 131I. J Pharm Pharmacol. 1956;8:1027–1033. doi: 10.1111/j.2042-7158.1956.tb12232.x. [DOI] [PubMed] [Google Scholar]
- Hallmann. Proceedings of the Third Central European Conference of Young Egyptologists. Pułtusk: The Pułtusk Academy of Humanities; 2009. [Google Scholar]
- Harrison JB. The effects of a belladonna plaster. Br Med J. 1872;1:520–521. doi: 10.1136/bmj.1.594.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes RB, Sackett DL, Taylor DW. Practical management of low compliance with antihypertensive therapy: a guide for the busy practitioner. Clin Invest Med. 1978;1:175–180. [PubMed] [Google Scholar]
- Helal F, Lane ME. Transdermal delivery of angiotensin converting enzyme inhibitors. Eur J Pharm Biopharm. 2014;88:1–7. doi: 10.1016/j.ejpb.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Henshilwood CS, d'Errico F, van Niekerk KL, Coquinot Y, Jacobs Z, Lauritzen SE, et al. A 100 000-year-old ochre-processing workshop at Blombos Cave, South Africa. Science. 2011;334:219–222. doi: 10.1126/science.1211535. [DOI] [PubMed] [Google Scholar]
- Ho H, Chien YW. Kinetic evaluation of transdermal nicotine delivery systems. Drug Dev Ind Pharm. 1993;19:295–313. [Google Scholar]
- Hoffman AS. The origins and evolution of ‘controlled’ drug delivery systems. J Control Release. 2008;132:153–163. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Hogan DJ, Maibach HI. Adverse dermatologic reactions to transdermal drug delivery systems. J Am Acad Dermatol. 1990;22:811–814. doi: 10.1016/0190-9622(90)70113-v. [DOI] [PubMed] [Google Scholar]
- Holdiness MR. A review of contact dermatitis associated with transdermal therapeutic systems. Contact Dermatitis. 1989;20:3–9. doi: 10.1111/j.1600-0536.1989.tb03087.x. [DOI] [PubMed] [Google Scholar]
- Holley FO, van Steennis C. Postoperative analgesia with fentanyl: pharmacokinetics and pharmacodynamics of constant-rate i.v. and transdermal delivery. Br J Anaesth. 1988;60:608–613. doi: 10.1093/bja/60.6.608. [DOI] [PubMed] [Google Scholar]
- Holling HE, McArdle B, Trotter WR. Prevention of seasickness by drugs. Lancet. 1944;146:126–129. [Google Scholar]
- Holst J. Percutaneous estrogen therapy. Endometrical response and metabolic effects. Acta Obstet Gynecol Scand. 1983;115:676–678. [PubMed] [Google Scholar]
- Holst J, Hofer PA, Cajander S, von Schoultz B. Percutaneous estrogen therapy – a semi-quantitative histologic study of the abdominal skin. Acta Obstet Gynecol Scand. 1982;61:515–516. doi: 10.3109/00016348209156605. [DOI] [PubMed] [Google Scholar]
- Hong I, Gabay M, Lodolce A. Safety concerns involving transdermal patches and magnetic resonance imaging (MRI) Hosp Pharm. 2010;45:771–778. [Google Scholar]
- Hopp MS. Developing custom adhesive systems for transdermal drug delivery products. Pharm Tech. 2002;26:30–36. [Google Scholar]
- Horstmann M, Kursawe M, Dzekan H. 1998. Transdermal therapeutic system comprising the active substance 17-β-estradiol (anhydrous). US Patent 5,827,245, LTS Lohmann Therapie-Systeme GmbH & Co.
- Horton R, Shinsako J, Forsham PH. Testosterone production and metabolic clearance rates with volumes of distribution in normal adult men and women. Acta Endocrinol (Copenh) 1965;48:446–458. doi: 10.1530/acta.0.0480446. [DOI] [PubMed] [Google Scholar]
- Hougham AJ, Hawkinson RW, Crowley JK, Wilson RR, Glode JE, Hilty RW, et al. Improved skin adherence and patient acceptance in a new transdermal nitroglycerin delivery system. Clin Ther. 1989;11:23–31. [PubMed] [Google Scholar]
- Hukkanen J, Jacob P, 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- Jacobs SC, Kaplan GW, Gittles RF. Topical testosterone therapy for penile growth. Urology. 1975;6:708–710. doi: 10.1016/0090-4295(75)90801-8. [DOI] [PubMed] [Google Scholar]
- Jain P, Banga AK. Induction and inhibition of crystallization in drug-in-adhesive-type transdermal patches. Pharm Res. 2012;30:562–571. doi: 10.1007/s11095-012-0901-7. [DOI] [PubMed] [Google Scholar]
- Jain SK, Vyas SP, Dixit VK. Effective and controlled transdermal delivery of ephedrine. J Control Release. 1990;12:257–263. [Google Scholar]
- Johnstone RT. Occupational Medicine and Industrial Hygiene. St Louis, MO: CV Mosby; 1948. [Google Scholar]
- Kalia YN, Nonato LB, Lunch CH, Guy RH. Development of skin barrier function in premature infants. J Invest Dermatol. 1998;111:320–326. doi: 10.1046/j.1523-1747.1998.00289.x. [DOI] [PubMed] [Google Scholar]
- Kanal E, Barkovich AJ, Bell C, Borgstede JP, Bradley WG, Jr, Froelich JW, et al. ACR guidance document on MR safe practices: 2013. J Magn Reson Imaging. 2013;37:501–530. doi: 10.1002/jmri.24011. [DOI] [PubMed] [Google Scholar]
- Kanarkowski R, Tornatore KM, D'Ambrosio R, Gardner MJ, Jusko WJ. Pharmacokinetics of single and multiple doses of ethinyl estradiol and levonorgestrel in relation to smoking. Clin Pharmacol Ther. 1988;43:23–31. doi: 10.1038/clpt.1988.7. [DOI] [PubMed] [Google Scholar]
- Kao J, Hall J. Skin absorption and cutaneous first pass metabolism of topical steroids: in vitro studies with mouse skin in organ culture. J Pharmacol Exp Ther. 1987;241:482–487. [PubMed] [Google Scholar]
- Kasting GB, Smith RL, Cooper ER. Effect of lipid solubility and molecular size on percutaneous absorption. In: Shroot B, Schaefer H, editors. Pharmacology and the Skin. Basel: Karger; 1987. pp. 138–153. [Google Scholar]
- Keith AD, Snipes W. 1981a. Polymeric diffusion matrix containing a vasodilatator. US Patent 4,291,015, Key Pharmaceuticals, Inc.
- Keith AD, Snipes W. 1981b. Polymeric diffusion matrix containing phenylephrine. US Patent 4,294,820, Key Pharmaceuticals, Inc.
- Keith AD, Snipes W. 1981c. Polymeric diffusion matrix containing phenylpropanolamine. US Patent 4,289,749, Key Pharmaceuticals, Inc.
- Keith AD, Snipes W. 1981d. Polymeric diffusion matrix containing ephedrine. US Patent 4,292,301, Key Pharmaceuticals, Inc.
- Kissel JC, Bunge AL. Response to Farahmand and Maibach's corrigenda. Int J Pharm. 2010;398:254–256. doi: 10.1016/j.ijpharm.2010.06.024. [DOI] [PubMed] [Google Scholar]
- Klugo RC, Cerny JC. Response of micropenis to topical testosterone and gonadotropin. J Urol. 1978;119:667–668. doi: 10.1016/s0022-5347(17)57584-9. [DOI] [PubMed] [Google Scholar]
- Kolli CS, Chadha G, Xiao J, Parsons DL, Babu RJ. Transdermal iontophoretic delivery of selegiline hydrochloride, in vitro. J Drug Target. 2010;18:657–664. doi: 10.3109/10611860903494237. [DOI] [PubMed] [Google Scholar]
- Korenman SG, Viosca S, Garza D, Guralnik M, Place V, Campbell P, et al. Androgen therapy of hypogonadal men with transscrotal testosterone systems. Am J Med. 1987;83:471–478. doi: 10.1016/0002-9343(87)90757-1. [DOI] [PubMed] [Google Scholar]
- Kotiyan PN, Vavia PR. Eudragits: role as crystallization inhibitors in drug-in-adhesive transdermal systems of estradiol. Eur J Pharm Biopharm. 2001;52:173–180. doi: 10.1016/s0939-6411(01)00174-6. [DOI] [PubMed] [Google Scholar]
- Kramer SN. The Sumerians: Their History, Culture, and Character. Chicago and London: The University of Chicago Press; 1963. [Google Scholar]
- Kremers E. Kremers and Urdang's History of Pharmacy. Philadelphia, PA: JB Lippincott Co; 1976. [Google Scholar]
- Krivonos S, Weisman A. 2013. Crystalline rotigotine base and production process therefor. US Patent 8,344,165 B2, Chemagis Ltd.
- Kubota K, Yamada T, Kikuchi K, Koyama E, Ishizaki T. Pharmacokinetics and β-blocking effects of transdermal timolol. Eur J Clin Pharmacol. 1993;44:493–495. doi: 10.1007/BF00315551. [DOI] [PubMed] [Google Scholar]
- Kuhlman JJ, Jr, Lalani S, Magluilo J, Jr, Levine B, Darwin WD. Human pharmacokinetics of intravenous, sublingual, and buccal buprenorphine. J Anal Toxicol. 1996;20:369–378. doi: 10.1093/jat/20.6.369. [DOI] [PubMed] [Google Scholar]
- Kunz GJ, Klein KO, Clemons RD, Gottschalk ME, Jones KL. Virilization of young children after topical androgen use by their parents. Pediatrics. 2004;114:282–284. doi: 10.1542/peds.114.1.282. [DOI] [PubMed] [Google Scholar]
- Kydonieus AF, Wille JJ, Murphy GF. Fundamental concepts in transdermal delivery of drugs. In: Kydonieus AF, Wille JJ, editors. Biochemical Modulation of Skin Reactions: Transdermals, Topicals, Cosmetics. Boca Raton, FL: CRC Press; 1999. pp. 1–14. [Google Scholar]
- Lake Y, Pinnock S. Improved patient acceptability with a transdermal drug-in-adhesive oestradiol patch. Aust N Z J Obstet Gynaecol. 2000;40:313–316. doi: 10.1111/j.1479-828x.2000.tb03341.x. [DOI] [PubMed] [Google Scholar]
- Lane ME. Skin penetration enhancers. Int J Pharm. 2013;447:12–21. doi: 10.1016/j.ijpharm.2013.02.040. [DOI] [PubMed] [Google Scholar]
- Laufer LR, DeFazio JL, Lu JK, Meldrum DR, Eggena P, Sambhi MP, et al. Estrogen replacement therapy by transdermal estradiol administration. Am J Obstet Gynecol. 1983;146:533–540. doi: 10.1016/0002-9378(83)90796-2. [DOI] [PubMed] [Google Scholar]
- Lauterbach T, Schacht DW, Wolff H-M, Muller W. 2002. Transdermal therapeutic system for Parkinson's disease inducing high plasma levels of rotigotine. Eur Patent 1256339 A1, Schwarz Pharma AG.
- Laws GC. The effects of nitroglycerin upon those who manufacture it. JAMA. 1898;31:793–794. [Google Scholar]
- Laws GE. Nitroglycerin head. JAMA. 1910;54:793. [Google Scholar]
- LaWall CH. Four Thousand Years of Pharmacy: an Outline History of Pharmacy and the Allied Sciences. Philadelphia, PA and London: JB Lippincott Co; 1927. [Google Scholar]
- Lee ES, Nedberge DE, Yum SI. 1995. Transdermal administration of oxybutynin. US Patent 5,411,740, Alza Corporation.
- Lefèvre G, Sedek G, Huang H-LA, Saltzman M, Rosenberg M, Kiese B, et al. Pharmacokinetics of a rivastigmine transdermal patch formulation in healthy volunteers: relative effects of body site application. J Clin Pharmacol. 2007;47:471–478. doi: 10.1177/0091270006297748. [DOI] [PubMed] [Google Scholar]
- Lefèvre G, Sedek G, Jhee SS, Leibowitz MT, Huang HL, Enz A, et al. Pharmacokinetics and pharmacodynamics of the novel daily rivastigmine transdermal patch compared with twice-daily capsules in Alzheimer's disease patients. Clin Pharmacol Ther. 2008;83:106–114. doi: 10.1038/sj.clpt.6100242. [DOI] [PubMed] [Google Scholar]
- Levin C, Maibach HI. Transdermal drug delivery systems: an overview. In: Zhai H, Wilhelm K-P, Maibach HI, editors. Marzulli and Maibach's Dermatotoxicology. 7th edn. Boca Raton, FL: CRC Press; 2008. pp. 101–106. [Google Scholar]
- Li H, Yu Y, Faraji Dana S, Li B, Lee C-Y, Kang L. Novel engineered systems for oral, mucosal and transdermal drug delivery. J Drug Target. 2013;21:611–629. doi: 10.3109/1061186X.2013.805335. [DOI] [PubMed] [Google Scholar]
- Liebelt EL, Shannon MW. Small doses, big problems: a selected review of highly toxic common medications. Pediatr Emerg Care. 1993;9:292–297. [PubMed] [Google Scholar]
- Lilienthal JL. The effect of hyoscine on airsickness. J Aviat Med. 1945;16:59–68. [Google Scholar]
- Lilleng PK, Mehlum LI, Bachs L, Morild I. Deaths after intravenous misuse of transdermal fentanyl. J Forensic Sci. 2004;49:1364–1366. [PubMed] [Google Scholar]
- Lockhart LP. Nicotine poisoning. Br Med J. 1933;1:246–247. [Google Scholar]
- Lowenthal DT, Matzek KM, MacGregor TR. Clinical pharmacokinetics of clonidine. Clin Pharmacokinet. 1988;14:287–310. doi: 10.2165/00003088-198814050-00002. [DOI] [PubMed] [Google Scholar]
- Lucas A, Harris JR. Ancient Egyptian Materials and Industries. 4th edn. London: Edward Arnold; 1962. [Google Scholar]
- Lund F. Percutaneous nitroglycerin treatment in cases of peripheral circulatory disorders, especially Raynaud's disease. Acta Med Scand. 1948;131:196–206. doi: 10.1111/j.0954-6820.1948.tb12036.x. [DOI] [PubMed] [Google Scholar]
- MacGregor TR, Matzek KM, Keirns JJ, Vanwayjen RGA, Vandenende A, Vantol RGL. Pharmacokinetics of transdermally delivered clonidine. Clin Pharmacol Ther. 1985;38:278–284. doi: 10.1038/clpt.1985.171. [DOI] [PubMed] [Google Scholar]
- Macht DI. The absorption of drugs and poisons through the skin and mucous membranes. JAMA. 1938;110:409–414. [Google Scholar]
- MacMillan FS, Reller HH, Synder FH. The antiperspirant action of topically applied anticholinergics. J Invest Dermatol. 1964;43:363–377. doi: 10.1038/jid.1964.167. [DOI] [PubMed] [Google Scholar]
- Magner LN. A History of Medicine. 2nd edn. Boca Raton, FL: Taylor & Francis Group; 2005. [Google Scholar]
- Magnusson BM, Anissimov YG, Cross SE, Roberts MS. Molecular size as the main determinant of solute maximum flux across the skin. J Invest Dermatol. 2004;122:993–999. doi: 10.1111/j.0022-202X.2004.22413.x. [DOI] [PubMed] [Google Scholar]
- Maibach HI, Farahmand S. Transdermal drug pharmacokinetics in man: interindividual variability and partial prediction. Int J Pharm. 2009a;367:1–15. doi: 10.1016/j.ijpharm.2008.11.020. [DOI] [PubMed] [Google Scholar]
- Maibach HI, Farahmand S. Estimating skin permeability from physicochemical characteristics of drugs: a comparison between conventional models and an in vivo-based approach. Int J Pharm. 2009b;375:41–47. doi: 10.1016/j.ijpharm.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Maier-Lenz H, Ringwelski L, Windorfer A. Pharmacokinetics and relative bioavailability of a nitroglycerin ointment formulation. Arzneimittelforschung. 1980;30:320–324. [PubMed] [Google Scholar]
- Malkinson FD, Rothman S. Percutaneous absorption. In: Jadassohn J, editor. Handbuch der Haut und Geschlecht Skrauberten, Normale, und Pathologische Physiologic der Haut. Berlin: Springer; 1963. pp. 90–156. [Google Scholar]
- Marathon Pharmaceuticals News. 2014. Marathon pharmaceuticals launches ZiNGO® (lidocaine Hydrochloride monohydrate) powder intradermal injection system, topical local [Online]. Available at: http://marathonpharma.com/marathon-pharmaceuticals-launches-zingo-lidocaine-hydrochloride-monohydrate-powder-intradermal-injection-systemtopical-local-anesthetic-to-manage-venous-access-pain/ (accessed 11/4/2014)
- Marier JF, Lor M, Morin J, Roux L, Di Marco M, Morelli G, et al. Comparative bioequivalence study between a novel matrix transdermal delivery system of fentanyl and a commercially available reservoir formulation. Br J Clin Pharmacol. 2007;63:121–124. doi: 10.1111/j.1365-2125.2006.02758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Bouyer G, Toga M, Lebreton R, Stolley P, Lockhart J. Outbreak of accidental hexachlorophene poisoning in France. Lancet. 1982;319:91–95. doi: 10.1016/s0140-6736(82)90225-2. [DOI] [PubMed] [Google Scholar]
- Mausar JT. Testing and evaluating the performance of pressure sensitive adhesives (PSAs) for TDDS patches. TransDermal. 2011;5:15–22. [Google Scholar]
- McAdam B, Keimowitz RM, Maher M, Fitzgerald DJ. Transdermal modification of platelet function: an aspirin patch system results in marked suppression of platelet cyclooxygenase. J Pharmacol Exp Ther. 1996;277:559–564. [PubMed] [Google Scholar]
- McAfee DA, Hadgraft J, Lane ME. Rotigotine: the first new chemical entity for transdermal drug delivery. Eur J Pharm Biopharm. 2014;88:586–593. doi: 10.1016/j.ejpb.2014.08.007. [DOI] [PubMed] [Google Scholar]
- McAllister A, Mosberg H, Settlage JA, Steiner JA. Plasma levels of nitroglycerin generated by three nitroglycerin patch preparations, Nitradisc, Transiderm-Nitro and Nitro-Dur and one ointment formulation, Nitrobid. Br J Clin Pharmacol. 1986;21:365–369. doi: 10.1111/j.1365-2125.1986.tb05208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil Consumer & Specialty Pharmaceuticals. 2002. McNeil Consumer & Specialty Pharmaceuticals Report 2002 [Online]. Available at: http://www.fda.gov/ohrms/dockets/ac/02/briefing/3882B1_13_McNeil-Acetaminophen.pdf (accessed 11/30/2014)
- Meikle AW, Mazer NA, Moellmer JF, Stringhman JD, Tolman KG, Sanders SW, et al. Enhanced transdermal delivery of testosterone across nonscrotal skin produces physiological concentrations of testosterone and its metabolites in hypogonadal men. J Clin Endocrinol Metab. 1992;74:623–628. doi: 10.1210/jcem.74.3.1740497. [DOI] [PubMed] [Google Scholar]
- Mellick GD, Roberts MS. Structure-hepatic disposition relationships for phenolic compounds. Toxicol Appl Pharmacol. 1999;158:50–60. doi: 10.1006/taap.1999.8682. [DOI] [PubMed] [Google Scholar]
- Michaels AS, Chandrasekaran SK, Shaw JE. Drug permeation through human skin: theory and in vitro experimental measurement. AIChE J. 1975;21:985–996. [Google Scholar]
- Milewski M, Stinchcomb AL. Estimation of maximum transdermal flux of nonionized xenobiotics from basic physicochemical determinants. Mol Pharm. 2012;9:2111–2120. doi: 10.1021/mp300146m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KJ, Govil SK, Bhatia KS. 2009. Fentanyl suspension-based silicone adhesive formulations and devices for transdermal delivery of fentanyl. US Patent 7,556,823 B2, Mylan Pharmaceuticals, Inc.
- Minghetti P, Cilurzo F, Casiraghi A. Measuring adhesive performance in transdermal delivery systems. Am J Drug Deliv. 2004;2:193–206. [Google Scholar]
- Moghimi HR, Shafizade A, Kamlinejad M. Drug Delivery Systems in Iranian Traditional Pharmacy. Tehran: Traditional Medicine and Materia Medica Research Center, SBMU; 2011. [Google Scholar]
- Moore CL, Lamar JK, Beck N. Cutaneous absorption of sex hormones. JAMA. 1938;111:11–14. [Google Scholar]
- Morgan WF. Poisoning by the external application of belladonna. Br Med J. 1866;2:621. [Google Scholar]
- Moser K, Kriwet K, Naik A, Kalia YN, Guy RH. Passive skin penetration enhancement and its quantification in vitro. Eur J Pharm Biopharm. 2001;52:103–112. doi: 10.1016/s0939-6411(01)00166-7. [DOI] [PubMed] [Google Scholar]
- Muehlberger CW. Shoe dye poisoning. JAMA. 1925;84:1987–1991. [Google Scholar]
- Muller W, Horstmann M. 1999. Estradiol-TTS having water-binding additives. US Patent 5,902,602, LTS Lohmann Therapie-Systeme GmbH.
- Murphy M, Carmichael AJ. Transdermal drug delivery systems and skin sensitivity reactions. Incidence and management. Am J Clin Dermatol. 2000;1:361–368. doi: 10.2165/00128071-200001060-00004. [DOI] [PubMed] [Google Scholar]
- Nachum Z, Shupak A, Gordon CR. Transdermal scopolamine for prevention of motion sickness: clinical pharmacokinetics and therapeutic applications. Clin Pharmacokinet. 2006;45:543–566. doi: 10.2165/00003088-200645060-00001. [DOI] [PubMed] [Google Scholar]
- Nadkarni MV, Meyers DB, Carney RG, Zopf LC. Clinical studies in percutaneous absorption. AMA Arch Derm Syphilol. 1951;64:294–300. doi: 10.1001/archderm.1951.01570090041005. [DOI] [PubMed] [Google Scholar]
- Naik A, Kalia YN, Guy RH. Transdermal drug delivery: overcoming the skin's barrier function. Pharm Sci Technolo Today. 2000;3:318–326. doi: 10.1016/s1461-5347(00)00295-9. [DOI] [PubMed] [Google Scholar]
- Nelson L, Schwaner R. Transdermal fentanyl: pharmacology and toxicology. J Med Toxicol. 2009;5:230–241. doi: 10.1007/BF03178274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieschlag E. Testosterone treatment comes of age: new options for hypogonadal men. Clin Endocrinol (Oxf) 2006;65:275–281. doi: 10.1111/j.1365-2265.2006.02618.x. [DOI] [PubMed] [Google Scholar]
- Nitto. 2013. Nitto press release [Online]. Available at: http://www.nitto.com/press/2013/0628.jsp (accessed 4/6/2014)
- No authors listed. Nitroglycerin ointment. Lancet. 1976;308:1287. [PubMed] [Google Scholar]
- Noonan PK, Benet LZ. The bioavailability of oral nitroglycerin. J Pharm Sci. 1986;75:241–243. doi: 10.1002/jps.2600750306. [DOI] [PubMed] [Google Scholar]
- Nyiri W, Jannitti M. About the fate of free iodine upon application to the unbroken animal skin: an experimental study. J Pharmacol Exp Ther. 1932;45:85–107. [Google Scholar]
- Oliveira G, Hadgraft J, Lane ME. Toxicological implications of the delivery of fentanyl from gel extracted from a commercial transdermal reservoir patch. Toxicol in Vitro. 2012;26:645–648. doi: 10.1016/j.tiv.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Organization for Economic Cooperation and Development. 2004. OECD guidelines for testing chemicals – test no. 428: skin absorption: in vitro method.
- Osborne JL, Nelson M, Enscore DJ, Yum SI, Gale RM. 1991. Subsaturated nicotine transdermal therapeutic system. US Patent 5,004,610, Alza Corporation.
- Padula C, Nicoli S, Aversa V, Colombo P, Falson F, Pirot F, et al. Bioadhesive film for dermal and transdermal drug delivery. Eur J Dermatol. 2007;17:309–312. doi: 10.1684/ejd.2007.0205. [DOI] [PubMed] [Google Scholar]
- Paoletti AM, Pilia I, Nannipieri F, Bigini C, Melis GB. Comparison of pharmacokinetic profiles of a 17β-estradiol gel 0.6 mg/g (Gelestra) with a transdermal delivery system (Estraderm TTS 50) in postmenopausal women at steady state. Maturitas. 2001;40:203–209. doi: 10.1016/s0378-5122(01)00239-0. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira J. The Elements of Materia Medica; Comprehending the Natural History, Preparation, Properties, Composition, Effects, and Uses of Medicines. London: Longman, Orme, Brown, Green, and Longmans; 1839. [Google Scholar]
- Perumal O, Murthy SN, Kalia YN. Turning theory into practice: the development of modern transdermal drug delivery systems and future trends. Skin Pharmacol Physiol. 2013;26:331–342. doi: 10.1159/000351815. [DOI] [PubMed] [Google Scholar]
- Peterson TA, Wick SM, Ko C. Design, development manufacturing and testing of transdermal drug delivery systems. In: Ghosh TK, Pfister WR, Yum SI, editors. Transdermal and Topical Drug Delivery Systems. Buffalo Grove, IL: Interpharm Press; 1997. pp. 249–297. [Google Scholar]
- Pfister WR. Transdermal and dermal therapeutic systems: current status. In: Ghosh TK, Pfister WR, Yum SI, editors. Transdermal and Topical Drug Delivery Systems. Buffalo Grove, IL: Interpharm Press; 1997. pp. 249–297. [Google Scholar]
- Ph Eur. 2015. 2.9.4. Dissolution test for transdermal patches. Ph Eur 8th edn.
- Poewe WH, Rascol O, Quinn N, Tolosa E, Oertel WH, Martignoni E, et al. Efficacy of pramipexole and transdermal rotigotine in advanced Parkinson's disease: a double-blind, double-dummy, randomised controlled trial. Lancet Neurol. 2007;6:513–520. doi: 10.1016/S1474-4422(07)70108-4. [DOI] [PubMed] [Google Scholar]
- Popli S, Stroka G, Ing TS, Daugirdas JT, Norusis MJ, Hano JE, et al. Transdermal clonidine for hypertensive patients. Clin Ther. 1983;5:624–628. [PubMed] [Google Scholar]
- Powers MS, Schenkel L, Darley PE, Good WR, Balestra JC, Place VA. Pharmacokinetics and pharmacodynamics of transdermal dosage forms of 17β-estradiol: comparison with conventional oral estrogens used for hormone replacement. Am J Obstet Gynecol. 1985;152:1099–1106. doi: 10.1016/0002-9378(85)90569-1. [DOI] [PubMed] [Google Scholar]
- Prausnitz MR, Mitragotri S, Langer R. Current status and future potential of transdermal drug delivery. Nat Rev Drug Discov. 2004;3:115–124. doi: 10.1038/nrd1304. [DOI] [PubMed] [Google Scholar]
- Price NM, Schmitt LG, McGuire J, Shaw JE, Trobough G. Transdermal scopolamine in the prevention of motion sickness at sea. Clin Pharmacol Ther. 1981;29:414–419. doi: 10.1038/clpt.1981.57. [DOI] [PubMed] [Google Scholar]
- Prodduturi S, Smith GJ, Wokovich AM, Doub WH, Westenberger BJ, Buhse L. Reservoir based fentanyl transdermal drug delivery systems: effect of patch age on drug release and skin permeation. Pharm Res. 2009;26:1344–1352. doi: 10.1007/s11095-009-9843-0. [DOI] [PubMed] [Google Scholar]
- Prodduturi S, Sadrieh N, Wokovich AM, Doub WH, Westenberger BJ, Buhse L. Transdermal delivery of fentanyl from matrix and reservoir systems: effect of heat and compromised skin. J Pharm Sci. 2010;99:2357–2366. doi: 10.1002/jps.22004. [DOI] [PubMed] [Google Scholar]
- Putcha L, Cintron NM, Tsui J, Vanderploeg JM, Kramer WG. Pharmacokinetics and oral bioavailability of scopolamine in normal subjects. Pharm Res. 1989;6:481–485. doi: 10.1023/a:1015916423156. [DOI] [PubMed] [Google Scholar]
- Quinn HL, Kearney MC, Courtenay AJ, McCrudden MT, Donnelly RF. The role of microneedles for drug and vaccine delivery. Expert Opin Drug Deliv. 2014;11:1769–1780. doi: 10.1517/17425247.2014.938635. [DOI] [PubMed] [Google Scholar]
- Real Pharma. 2014. Fat & weight loss patch [Online]. Available at: http://www.realpharma.com/generic/fat_patch.htm (accessed 3/30/2014)
- Reichek N, Goldstein RE, Redwood DR, Epstein SE. Sustained effects of nitroglycerin ointment in patients with angina pectoris. Circulation. 1974;50:348–352. doi: 10.1161/01.cir.50.2.348. [DOI] [PubMed] [Google Scholar]
- Riegelman S. Pharmacokinetics. Pharmacokinetic factors affecting epidermal penetration and percutaneous adsorption. Clin Pharmacol Ther. 1974;16:873–883. doi: 10.1002/cpt1974165part2873. [DOI] [PubMed] [Google Scholar]
- Roberts MS. Solute-vehicle-skin interactions in percutaneous absorption: the principles and the people. Skin Pharmacol Physiol. 2013;26:356–370. doi: 10.1159/000353647. [DOI] [PubMed] [Google Scholar]
- Roberts MS, Cross SE. Percutaneous absorption of topically applied NSAIDS and other compounds: role of solute properties, skin physiology and delivery systems. Inflammopharmacology. 1999;7:339–350. doi: 10.1007/s10787-999-0028-6. [DOI] [PubMed] [Google Scholar]
- Roberts MS, Cross SE. Skin transport. In: Walters KA, editor. Dermatological and Transdermal Formulations. New York: Marcel Dekker; 2002. pp. 97–215. [Google Scholar]
- Roberts MS, Walters KA. Human skin morphology and dermal absorption. In: Roberts MS, Walters KA, editors. Dermal Absorption and Toxicity Assessment. 2nd edn. New York: Informa Healthcare; 1998. pp. 1–15. [Google Scholar]
- Roberts MS, Anderson RA, Swarbrick J. Permeability of human epidermis to phenolic compounds. J Pharm Pharmacol. 1977;29:677–683. doi: 10.1111/j.2042-7158.1977.tb11434.x. [DOI] [PubMed] [Google Scholar]
- Roberts MS, Favretto WA, Meyer A, Reckmann M, Wongseelashote T. Topical bioavailability of methyl salicylate. Aust N Z J Med. 1982;12:303–305. doi: 10.1111/j.1445-5994.1982.tb02485.x. [DOI] [PubMed] [Google Scholar]
- Rose JE, Jarvik ME, Rose KD. Transdermal administration of nicotine. Drug Alcohol Depend. 1984;13:209–213. doi: 10.1016/0376-8716(84)90061-9. [DOI] [PubMed] [Google Scholar]
- Rose JE, Herskovic JE, Trilling Y, Jarvik ME. Transdermal nicotine reduces cigarette craving and nicotine preference. Clin Pharmacol Ther. 1985;38:450–456. doi: 10.1038/clpt.1985.203. [DOI] [PubMed] [Google Scholar]
- Rose PG, Macfee MS, Boswell MV. Fentanyl transdermal system overdose secondary to cutaneous hyperthermia. Anesth Analg. 1993;77:390–391. doi: 10.1213/00000539-199308000-00029. [DOI] [PubMed] [Google Scholar]
- Roy SD, Flynn GL. Solubility and related physicochemical properties of narcotic analgesics. Pharm Res. 1988;5:580–586. doi: 10.1023/a:1015994030251. [DOI] [PubMed] [Google Scholar]
- Roy SD, Flynn GL. Transdermal delivery of narcotic analgesics: comparative permeabilities of narcotic analgesics through human cadaver skin. Pharm Res. 1989;6:825–832. doi: 10.1023/a:1015944018555. [DOI] [PubMed] [Google Scholar]
- Roy SD, Flynn GL. Transdermal delivery of narcotic analgesics: pH, anatomical, and subject influences on cutaneous permeability of fentanyl and sufentanil. Pharm Res. 1990;7:842–847. doi: 10.1023/a:1015912932416. [DOI] [PubMed] [Google Scholar]
- Roy SD, Roos E, Sharma K. Transdermal delivery of buprenorphine through cadaver skin. J Pharm Sci. 1994;83:126–130. doi: 10.1002/jps.2600830204. [DOI] [PubMed] [Google Scholar]
- Roy SD, Gutierrez M, Flynn GL, Cleary GW. Controlled transdermal delivery of fentanyl: characterizations of pressure-sensitive adhesives for matrix patch design. J Pharm Sci. 1996;85:491–495. doi: 10.1021/js950415w. [DOI] [PubMed] [Google Scholar]
- Rutter N. Percutaneous drug absorption in the newborn: hazards and uses. Clin Perinatol. 1987;14:911–930. [PubMed] [Google Scholar]
- Sablotsky S, Questel JM, Thompson JA. 1993. Adhesive transdermal dosage. US Patent 5,186,938, Key Pharmaceuticals, Inc.
- Santus GC, Baker RW. Transdermal enhancer patent literature. J Control Release. 1993;25:1–20. [Google Scholar]
- Sanvordeker DR, Cooney JG, Wester RC. 1982. Transdermal nitroglycerin pad. US Patent 4,336,243, G. F Searle & Co.
- Sathyan G, Guo C, Sivakumar K, Gidwani S, Gupta S. Evaluation of the bioequivalence of two transdermal fentanyl systems following single and repeat applications. Curr Med Res Opin. 2005;21:1961–1968. doi: 10.1185/030079905X65259. [DOI] [PubMed] [Google Scholar]
- Scarff RW, Smith CP. Proliferative and other lesions of the male breast. With notes on 2 cases of proliferative mastitis in stilbœstrol workers. Br J Surg. 1942;29:393–396. [Google Scholar]
- Schenkel L, Barlier D, Riera M, Barner A. Transdermal absorption of estradiol from different body sites is comparable. J Control Release. 1986;4:195–201. [Google Scholar]
- Scheuplein RJ, Blank IH. Permeability of the skin. Physiol Rev. 1971;51:702–747. doi: 10.1152/physrev.1971.51.4.702. [DOI] [PubMed] [Google Scholar]
- Schwenkenbecker A. Das absorptions verniogen der haut. Arch Anat Physiol. 1904;28:121–165. [Google Scholar]
- Sci Finder Scholar. 2014. Substance identifier [Online]. Available at: https://scifinder.cas.org/ (accessed 2/23/2015)
- Sebel PS, Barrett CW, Kirk CJ, Heykants J. Transdermal absorption of fentanyl and sufentanil in man. Eur J Clin Pharmacol. 1987;32:529–531. doi: 10.1007/BF00637682. [DOI] [PubMed] [Google Scholar]
- Shareef A, Angove MJ, Wells JD, Johnson BB. Sorption of bisphenol A, 17α-ethynylestradiol and estrone to mineral surfaces. J Colloid Interface Sci. 2006;297:62–69. doi: 10.1016/j.jcis.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Shaw JE, Chandrasekaran SK. Controlled topical delivery of drugs for systemic action. Drug Metab Rev. 1978;8:223–233. doi: 10.3109/03602537808993785. [DOI] [PubMed] [Google Scholar]
- Shaw JE, Theeuwes F. Transdermal dosage forms. In: Prescott LF, Nimmo WS, editors. Rate Control in Drug Therapy. New York: Churchill Livingstone; 1985. pp. 65–70. [Google Scholar]
- Shaw JE, Urquhart J. Programmed, systemic drug delivery by the transdermal route. Trends Pharmacol Sci. 1979;1:208–211. [Google Scholar]
- Shaw JE, Chandrasekaran SK, Taskovich L. Use of percutaneous absorption for systemic administration of drugs. Pharm J. 1975;215:32–328. [Google Scholar]
- Shaw JE, Chandrasekaran SK, Campbell P. Percutaneous absorption: controlled drug delivery for topical or systemic therapy. J Invest Dermatol. 1976;67:677–678. [Google Scholar]
- Shaw JE, Enscore D, Chu L. Clonidine rate-controlled system: technology and kinetics. In: Weber MA, Mathias CJ, editors. Mild Hypertension. Darmstadt: Steinkopff Verlag; 1983. pp. 134–140. [Google Scholar]
- Sica DA, Grubbs R. Transdermal clonidine: therapeutic considerations. J Clin Hypertens. 2005;7:558–562. doi: 10.1111/j.1524-6175.2005.04133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Uniyal JP, Jha P, Murugesan K, Takkar D, Hingorani V, et al. Pharmacokinetics of norethindrone acetate in women. Am J Obstet Gynecol. 1979;135:409–414. doi: 10.1016/0002-9378(79)90715-4. [DOI] [PubMed] [Google Scholar]
- Singh J, Maibach HI. Transdermal delivery and cutaneous reactions. In: Walters KA, editor. Dermatological and transdermal formulations. New York: Marcel Dekker; 2002. pp. 570–589. [Google Scholar]
- Sittl R, Griessinger N, Likar R. Analgesic efficacy and tolerability of transdermal buprenorphine in patients with inadequately controlled chronic pain related to cancer and other disorders: a multicenter, randomized, double-blind, placebo-controlled trial. Clin Ther. 2003;25:150–168. doi: 10.1016/s0149-2918(03)90019-1. [DOI] [PubMed] [Google Scholar]
- Smith PK. Use of hyoscine hydrobromide for the prevention or airsickness in flexible gunnery students. J Aviat Med. 1946a;17:346–347. [PubMed] [Google Scholar]
- Smith PK. The effectiveness of some motion sickness remedies in preventing airsickness in air force navigation students. J Aviat Med. 1946b;17:343–345. [PubMed] [Google Scholar]
- Smith TR, Goldstein J, Singer R, Pugach N, Silberstein S, Pierce MW. Twelve-month tolerability and efficacy study of NP101, the sumatriptan iontophoretic transdermal system. Headache. 2012;52:612–624. doi: 10.1111/j.1526-4610.2012.02094.x. [DOI] [PubMed] [Google Scholar]
- Sobue S, Sekiguchi K, Kikkawa H, Irie S. Effect of application sites and multiple doses on nicotine pharmacokinetics in healthy male Japanese smokers following application of the transdermal nicotine patch. J Clin Pharmacol. 2005;45:1391–1399. doi: 10.1177/0091270005282632. [DOI] [PubMed] [Google Scholar]
- Stähle H. A historical perspective: development of clonidine. Best Pract Res Clin Anaesthesiol. 2000;14:237–246. [Google Scholar]
- Strecker JR, Lauritzen C, Goessens L. Plasma concentrations of unconjugated and conjugated estrogens and gonadotrophins following application of various estrogen preparations after oophorectomy and in the menopause. Maturitas. 1979;1:183–190. doi: 10.1016/0378-5122(79)90007-0. [DOI] [PubMed] [Google Scholar]
- Stricker H. 1983. Transdermal release system for pharmaceutical preparation. US Patent 4,409,206, Boehringer Ingelheim GmbH.
- Sved S, McLean WM, McGilveray IJ. Influence of the method of application on pharmacokinetics of nitroglycerin from ointment in humans. J Pharm Sci. 1981;70:1368–1369. doi: 10.1002/jps.2600701220. [DOI] [PubMed] [Google Scholar]
- Synera. 2014. Prescribing information [Online]. Available at: http://www.synera.com/ (accessed 10/15/2014)
- Taggart W, Dandekar K, Ellman H, Notelovitz M. The effect of site of application on the transcutaneous absorption of 17-β estradiol from a transdermal delivery system (Climara) Menopause. 2000;7:364–369. doi: 10.1097/00042192-200007050-00010. [DOI] [PubMed] [Google Scholar]
- Talley JD, Crawley IS. Transdermal nitrate, penile erection, and spousal headache. Ann Intern Med. 1985;103:804. doi: 10.7326/0003-4819-103-5-804_1. [DOI] [PubMed] [Google Scholar]
- Tamura G, Ichinose M, Fukuchi Y, Miyamoto T. Transdermal tulobuterol patch, a long-acting β2-agonist. Allergol Int. 2012;61:219–229. doi: 10.2332/allergolint.11-RA-0358. [DOI] [PubMed] [Google Scholar]
- Tan HS, Pfister WR. Pressure-sensitive adhesives for transdermal drug delivery systems. Pharm Sci Technolo Today. 1999;2:60–69. doi: 10.1016/s1461-5347(99)00119-4. [DOI] [PubMed] [Google Scholar]
- Tang J, Deverich JM, Miller JM, Beste RD. 2008. Amorphous drug transdermal systems, manufacturing methods, and stabilization. US Patent Application 2008/0226698 A1, Mylan Technologies, Inc.
- Tapsoba I, Arbault S, Walter P, Amatore C. Finding out Egyptian gods' secret using analytical chemistry: biomedical properties of Egyptian black makeup revealed by amperometry at single cells. Anal Chem. 2010;82:457–460. doi: 10.1021/ac902348g. [DOI] [PubMed] [Google Scholar]
- Tauber U, Schroder K, Dusterberg B, Matthes H. Absolute bioavailability of testosterone after oral administration of testosterone-undecanoate and testosterone. Eur J Drug Metab Pharmacokinet. 1986;11:145–149. doi: 10.1007/BF03189840. [DOI] [PubMed] [Google Scholar]
- Teske J, Weller JP, Larsch K, Troger HD, Karst M. Fatal outcome in a child after ingestion of a transdermal fentanyl patch. Int J Legal Med. 2007;121:147–151. doi: 10.1007/s00414-006-0137-3. [DOI] [PubMed] [Google Scholar]
- Thadani U, Hamilton SF, Olson E, Anderson J, Voyles W, Prasad R, et al. Transdermal nitroglycerin patches in angina pectoris. Dose titration, duration of effect, and rapid tolerance. Ann Intern Med. 1986;105:485–492. doi: 10.7326/0003-4819-105-4-485. [DOI] [PubMed] [Google Scholar]
- The Lancet annotations. Poisoning by cutaneous absorption of aniline. Lancet. 1902;159:463–464. [Google Scholar]
- The Medicines Company. 2012. News release [Online]. Available at: http://ir.themedicinescompany.com/phoenix.zhtml?c=122204&p=irol-newsArticle&ID=1766469&highlight= (accessed: 3/26/2014)
- USP. 2014a. General Chapter <3> Topical and transdermal drug products – product quality tests. First Supplement to USP 37-NF 32.
- USP. 2014b. General Chapter <724> Drug release. Second Supplement to USP 37-NF 32.
- Urquhart J, Kumar S, Chandrasekaran SK, Shaw JE. 1977. Bandage for transdermally administering scopolamine to prevent nausea. US Patent 4,031,894, Alza Corporation.
- Van Buskirk GA, Arsulowicz D, Basu P, Block L, Cai B, Cleary GW, et al. Passive transdermal systems whitepaper incorporating current chemistry, manufacturing and controls (CMC) development principles. AAPS PharmSciTech. 2012;13:218–230. doi: 10.1208/s12249-011-9740-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanakoski J, Seppala T, Sievi E, Lunell E. Exposure to high ambient temperature increases absorption and plasma concentrations of transdermal nicotine. Clin Pharmacol Ther. 1996;60:308–315. doi: 10.1016/S0009-9236(96)90057-0. [DOI] [PubMed] [Google Scholar]
- Venkatraman S, Gale R. Skin adhesives and skin adhesion 1. Transdermal drug delivery systems. Biomaterials. 1998;19:1119–1136. doi: 10.1016/s0142-9612(98)00020-9. [DOI] [PubMed] [Google Scholar]
- Vlasses PH, Ribeiro LG, Rotmensch HH, Bondi JV, Loper AE, Hichens M, et al. Initial evaluation of transdermal timolol: serum concentrations and β-blockade. J Cardiovasc Pharmacol. 1985;7:245–250. doi: 10.1097/00005344-198503000-00006. [DOI] [PubMed] [Google Scholar]
- Waters C. The development of the rotigotine transdermal patch: a historical perspective. Neurol Clin. 2013;31:S37–S50. doi: 10.1016/j.ncl.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Watkinson AC. Transdermal and topical drug delivery today. In: Benson HA, Watkinson AC, editors. Topical and Transdermal Drug Delivery – Principles and Practice. Hoboken, NJ: John Wiley & Sons Inc; 2012. pp. 357–366. [Google Scholar]
- Watkinson AC. A commentary on transdermal drug delivery systems in clinical trials. J Pharm Sci. 2013;102:3082–3088. doi: 10.1002/jps.23490. [DOI] [PubMed] [Google Scholar]
- Weber MA, Drayer JIM, Brewer DD, Lipson JL. Transdermal continuous antihypertensive therapy. Lancet. 1984;1:9–11. doi: 10.1016/s0140-6736(84)90180-6. [DOI] [PubMed] [Google Scholar]
- Wermeling DP, Banks SL, Hudson DA, Gill HS, Gupta J, Prausnitz MR, et al. Microneedles permit transdermal delivery of a skin-impermeant medication to humans. Proc Natl Acad Sci U S A. 2008;105:2058–2063. doi: 10.1073/pnas.0710355105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wester RC, Maibach HI. Cutaneous pharmacokinetics: 10 steps to percutaneous absorption. Drug Metab Rev. 1983;14:169–205. doi: 10.3109/03602538308991388. [DOI] [PubMed] [Google Scholar]
- White R. Poisoning from aniline black on shoes. Lancet. 1909;173:349. [Google Scholar]
- Wick KA, Wick SM, Hawkinson RW, Holtzman JL. Adhesion-to-skin performance of a new transdermal nitroglycerin adhesive patch. Clin Ther. 1989;11:417–424. [PubMed] [Google Scholar]
- Wick SM. 1988. Transdermal nitroglycerin delivery system. US Patent 4,751,087, Riker Laboratories, Inc.
- Wiedersberg S, Guy RH. Transdermal drug delivery: 30+ years of war and still fighting. J Control Release. 2014;190:150–156. doi: 10.1016/j.jconrel.2014.05.022. [DOI] [PubMed] [Google Scholar]
- Wild RB. Part I. On the official ointments, with special reference to the substabces used as bases. Br Med J. 1911;2:161–162. doi: 10.1136/bmj.2.2638.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild RB, Roberts I. The absorption of mercurials from ointments applied to the skin. Br Med J. 1926;1:1076–1079. doi: 10.1136/bmj.1.3416.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wile UJ, Elliott JA. Mode of absorption of mercury in the inunction treatment of syphilis: preliminary report. JAMA. 1917;68:1024–1028. [Google Scholar]
- Wilson JB. Nicotine poisoning by absorption through the skin. Br Med J. 1930;2:601–602. [Google Scholar]
- Wurster DE, Kramer SF. Investigation of some factors influencing percutaneous absorption. J Pharm Sci. 1961;50:288–293. doi: 10.1002/jps.2600500403. [DOI] [PubMed] [Google Scholar]
- Yu ZL, Gupta SK, Hwang SS, Cook DM, Duckett MJ, Atkinson LE. Transdermal testosterone administration in hypogonadal men: comparison of pharmacokinetics at different sites of application and at the first and fifth days of application. J Clin Pharmacol. 1997;37:1129–1138. doi: 10.1002/j.1552-4604.1997.tb04297.x. [DOI] [PubMed] [Google Scholar]
- Zaffaroni A. 1971. Bandage for administering drugs. US Patent 3,598,122, Alza Corporation.
- Zaffaroni A. 1973. Bandage for controlled release of vasodilatators. US Patent 3,742,951, Alza Corporation.
- Zaffaroni A. 1974. Bandage for the administration of drug by controlled metering through microporous materials. US Patent 3,797,494, Alza Corporation.
- Zeile K, Hauptmann K-H, Stahle H. 1965. Shaving composition and method of using same. US Patent 3,190,802, Boehringen Ingelheim GmbH.
- Zimrin D, Reichek N, Bogin KT, Aurigemma G, Douglas P, Berko B, et al. Antianginal effects of intravenous nitroglycerin over 24 hours. Circulation. 1988;77:1376–1384. doi: 10.1161/01.cir.77.6.1376. [DOI] [PubMed] [Google Scholar]
- Zondek B. Cutaneous application of follicular hormone. Lancet. 1938;231:1107–1110. [Google Scholar]
- Zondek B. Chemotherapeutical use of halogenized phenols as external disinfectants. Nature. 1942a;149:334–335. [Google Scholar]
- Zondek B. The excretion of halogenated phenols and their use in the treatment of urogenital infections. J Urol. 1942b;48:747–758. [Google Scholar]
- Zondek B, Shapiro B, Hestrin S. Application of the Millon reaction to the determination of chlorophenols in body fluids and tissues. Biochem J. 1943;37:589–591. doi: 10.1042/bj0370589. [DOI] [PMC free article] [PubMed] [Google Scholar]