

Abstract
Background and Purpose
The catalytic topoisomerase II inhibitor dexrazoxane has been associated not only with improved cancer patient survival but also with secondary malignancies and reduced tumour response.
Experimental Approach
We investigated the DNA damage response and the role of the activating transcription factor 3 (ATF3) accumulation in tumour cells exposed to dexrazoxane.
Key Results
Dexrazoxane exposure induced topoisomerase IIα (TOP2A)-dependent cell death, γ-H2AX accumulation and increased tail moment in neutral comet assays. Dexrazoxane induced DNA damage responses, shown by enhanced levels of γ-H2AX/53BP1 foci, ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3-related), Chk1 and Chk2 phosphorylation, and by p53 accumulation. Dexrazoxane-induced γ-H2AX accumulation was dependent on ATM. ATF3 protein was induced by dexrazoxane in a concentration- and time-dependent manner, which was abolished in TOP2A-depleted cells and in cells pre-incubated with ATM inhibitor. Knockdown of ATF3 gene expression by siRNA triggered apoptosis in control cells and diminished the p53 protein level in both control and dexrazoxane -treated cells. This was accompanied by increased γ-H2AX accumulation. ATF3 knockdown also delayed the repair of dexrazoxane -induced DNA double-strand breaks.
Conclusions and Implications
As with other TOP2A poisons, dexrazoxane induced DNA double-strand breaks followed by activation of the DNA damage response. The DNA damage-triggered ATF3 controlled p53 accumulation and generation of double-strand breaks and is proposed to serve as a switch between DNA damage and cell death following dexrazoxane treatment. These findings suggest a mechanistic explanation for the diverse clinical observations associated with dexrazoxane.
Tables of Links
| TARGETS | |
|---|---|
| Enzymes | |
| ATM kinase | |
| ATR kinase | |
| Caspase 3 | |
| Caspase 7 | |
| Chk1, checkpoint kinase1 | |
| Chk2, checkpoint kinase2 | |
| DNA-PK, DNA-dependent protein kinase | |
| JNK | |
| Topoisomerase IIα |
| LIGANDS | |
|---|---|
| Cyclophosphamide | |
| Dexrazoxane | |
| Doxorubicin | |
| Fluorouracil | |
| KU55933 | |
| NU7026 | |
| SB203580 | |
| SP600125 | |
| Suramin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
The irreversible inhibition (‘poisoning’) of topoisomerase IIα (TOP2A) represents one of the most successful oncological strategies. This strategy takes advantage of the essential role of TOP2A in proliferating cells in resolving DNA supercoiling and/or intra- and intermolecular knots resulting from DNA replication, transcription, chromosomal recombination and segregation. TOP2A generates transient DNA double-strand breaks (DSB), which allow for the passage of another nucleic acid segment and are followed by DSB re-ligation. TOP2A ‘poisons’, such as doxorubicin, turn transient DSB into permanent ones. The level of the resulting DSB is considered to be a key determinant of tumour cell apoptosis and thereby of the therapeutic response. Correspondingly, the response of cancer cells to doxorubicin correlates with the expression level of TOP2A (Burgess et al., 2008), although the application of TOP2A levels as a therapeutic predictor has been unsuccessful (Bonnefoi, 2011).
Some other drugs are thought to kill cancer cells via inhibition of TOP2A catalytic activity rather than by ‘poisoning’ and DSB formation. Depending upon the affected step of the TOP2A activity cycle, these so-called catalytic inhibitors are thought to prevent binding between DNA and TOP2A (aclarubicin and suramin), to stabilize non-covalent DNA–TOP2A complexes (merbarone and bisdioxopiperazines) or to inhibit ATP binding (novobiocin) (Larsen et al., 2003). The clinical applications of catalytic inhibitors are much less common, but more diverse, in comparison to TOP2 poisons. While aclarubicin and the bisdioxopiperazine MST-16 serve as antineoplastic drugs, low doses of suramin and novobiocin can be used to enhance the efficacy of other cytotoxic agents (Larsen et al., 2003).
The most versatile effects and applications have been described for the bisdioxopiperazine dexrazoxane (ICRF-187). Dexrazoxane was originally described as an anti-tumour agent (Creighton et al., 1969), consistent with increased median survival time of patients with therapy-responsive advanced breast cancer (Swain et al., 1997a) and with data from a variety of pre-clinical animal cancer models (Hasinoff et al., 1998). Notwithstanding these effects, dexrazoxane is currently used to prevent congestive heart failure, which develops in a fraction of patients treated with anthracyclines, and to treat tissue damage resulting from accidental anthracycline extravasation. For congestive heart failure prevention, dexrazoxane is the only approved drug. Despite the demonstrated 80% reduction of this side effect (van Dalen et al., 2011a), the use of dexrazoxane as a cardioprotectant is limited. This is, in part, due to the unclear cardioprotective mechanism, which may combine iron chelation with the depletion of the other TOP2 isozyme, topoisomerase IIβ (TOP2B), in cardiomyocytes (Lyu et al., 2007). Even more important are the persisting concerns of interference with anti-tumour efficacy and of induction of secondary malignancies. These concerns originate from two reports of, respectively, lower breast cancer response rate to the fluorouracil, doxorubicin and cyclophosphamide regimen in combination with dexrazoxane (Swain et al., 1997b), and of dexrazoxane -induced secondary malignancies in children treated for Hodgkin's disease (Tebbi et al., 2007). Importantly, meta-analyses of anti-tumour efficacy and of secondary malignancies after dexrazoxane do not support these individual observations (van Dalen et al., 2011b).
The controversy about dexrazoxane may be resolved through a better understanding of its effects on cancer cells. Despite its designation as catalytic inhibitors, bisdioxopiperazines leave TOP2A trapped on DNA, which might interfere with DNA metabolism in a manner analogous to TOP2A poisons (Nitiss, 2009). Accordingly, bisdioxopiperazines have been proposed to generate either cleavable complexes unverifiable by standard procedures (Huang et al., 2001) or a novel form of DNA lesion (van Hille and Hill, 1998; Jensen et al., 2000; Snyder, 2003). Previously, we reported that dexrazoxane increased the level of the DSB marker γ-H2AX in the fibrosarcoma-derived tumour cell line HTETOP. This was dependent upon the presence of TOP2A and associated with cell apoptosis (Yan et al., 2009). The data suggest the formation of TOP2A-mediated DSB by dexrazoxane. We reasoned that in this case dexrazoxane may activate the DNA damage response (DDR). This hypothesis is addressed in the present study.
Little is known about the cellular response to dexrazoxane-induced DNA damage. Genome-wide RNA microarray analysis using relatively stringent criteria revealed up-regulation of only one gene, the activating transcription factor 3 (ATF3) (Yan et al., 2009), in dexrazoxane-treated HTETOP cells. ATF3 is a member of the activation transcription factor/cAMP responsive element-binding (ATF/CREB) protein family of basic-leucine zipper (b-Zip)-type transcription factors. The level of ATF3 is low in quiescent cells but can be rapidly induced by diverse stimuli, including genotoxic stressors. In vitro studies support cytostatic and pro-apoptotic, but also proliferative and anti-apoptotic effects of ATF3 (Nobori et al., 2002; Janz et al., 2006; Huang et al., 2008; Turchi et al., 2008). Remarkably, ATF3 was the only gene significantly induced by dexrazoxane exposure (Yan et al., 2009). Therefore, in the present study, we also investigated the mechanism and cellular effects of dexrazoxane -induced ATF3 accumulation.
Methods
Cell culture and chemicals
HTETOP cells were derived from the human fibrosarcoma cell line HT1080 through the deletion of both endogenous TOP2A alleles and the insertion of a tetracycline-repressible TOP2A transgene (Carpenter and Porter, 2004). The expression level of TOP2A in HTETOP cells can be reduced by >95% 24 h after the addition of tetracycline (1 μg·mL–1). HTETOP, HT1080, NYH and DLD-1 cells were cultured as previously described (Wessel et al., 1999; Yan et al., 2009; Yamada et al., 2013). ATM mutant GM05849 (ATM mt) and the wild-type GM637 (ATM wt) cell lines have been described before (Eich et al., 2013). Cells were exposed to drugs with indicated concentrations for various time periods, or to a single dose of 10 Gy of ionizing radiation (IR).Ddexrazoxane was purchased from Chiron (Amsterdam, the Netherlands). KU55933, SB203580 and SP600125 were obtained from Calbiochem (Darmstadt, Germany). NU7026 and VE-821 were purchased from Selleckchem (Munich, Germany). ICRF-161 was kindly provided by Dr Annemette Vinding Thougaard (TopoTarget A/S, Denmark).
Cell viability assay
Cell viability in response to dexrazoxane exposure was assessed using Cell Titer-Glo (Promega, Mannheim, Germany) according to the instructions of the manufacturer. This assay is based upon the measurement of ATP content, which is proportional to the number of living cells. HTETOP cells were cultured in 96-well plates, tetracycline at 1 μg·mL–1 was added 24 h before dexrazoxane. Cell viability was measured 24 h following dexrazoxane treatment by a luminometer and was expressed relative to the survival of cells without dexrazoxane (=100%).
Transfections
Transfections with small interfering RNA (siRNA) or plasmid DNA were performed 24 h before drug treatment using the jetPRIME™ transfection reagent (Polyplus Transfection SA, Illkirch, France), according to the specifications of the manufacturer. siRNA oligonucleotides targeting the sequence of ATF3 mRNA GAGGCGACGAGAAAGAAAT (ATF3-1) or GAAGAAGGAGAAGACGGAG (ATF3-2) (Janz et al., 2006) are capable of knocking down both the full-length and the shorter isoforms of ATF3 (ATF3 ΔZip), the latter lacks the leucine zipper protein-dimerization motif (Chen et al., 1994).
TaqMan assay
Total RNA was isolated from cells using TriFast (PeqLab, Erlangen, Germany). One μg of total RNA was reverse-transcribed to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Darmstadt, Germany). One μL of the resulting 20 μL of cDNA solution was mixed with a TaqMan Universal PCR Master Mix (Applied Biosystems) and ATF3 Gene Expression Assay (Hs00231069_m1; Applied Biosystems), followed by real-time PCR with a BioRad iCycler (Bio-Rad, Hercules, CA, USA). 18S rRNA (Hs99999901_s1; Applied Biosystems) was used as the internal control. The results were calculated using the ΔΔCt method.
Western blot
Western blots were performed as previously described (Yan et al., 2009; Berdelle et al., 2011). About 20–50 μg of protein was loaded onto each lane, with the exception of ATM and ATR detection, where equal portions of cell dish lysates were loaded. Briefly, proteins lysed in sample buffer were separated by SDS-PAGE and subsequently transferred to PVDF membrane by semi-dry blotting. For large proteins (ATR and ATM), LDS-PAGE was performed on 4.5%:6% gels (acrylamide : bisacrylamide, 59:1) followed by blotting onto nitrocellulose membranes. The primary antibodies used were as follows: anti-ATF3 (1:500; Santa Cruz Biotechnology, Heidelberg, Germany), anti-p53 (1:5,000; Dianova, Hamburg, Germany), anti-γ-H2AX (phospho-Ser139) (1:400; Abcam, Berlin, Germany), anti-TOP2A (1:2000; Stressgen, Hamburg, Germany), anti-GAPDH (1:20 000; Santa Cruz Biotechnology), anti-β-tubulin (1:10 000; Sigma-Aldrich, Munich, Germany), anti-pATM (Ser1981) (1:750; Millipore, Darmstadt, Germany), anti-ATM (1:1000; Cell Signaling, Frankfurt, Germany), anti-pATR (Ser428) (1:1000; Cell Signaling), anti-ATR (1: 1000; Cell Signaling), anti-pChk1 (Ser317) (1:1000; Cell Signaling) and anti-pChk2 (Thr68) (1:1000; Cell Signaling).
Apoptosis measurement by FACS
Early stage of apoptosis was detected using Annexin V staining followed by FACS analysis as previously described (Yan et al., 2009). Briefly, cells were gently harvested using Accutase (PAA, Cölbe, Germany) and pelleted by centrifugation at 400× g for 5 min. After washing with PBS, the cell pellets were resuspended in binding buffer and stained with Annexin V-FITC and To-Pro-3. FACS analysis was performed within 1 h.
Caspase 3/7 activity assay
Caspase 3/7 activity was measured with the Caspase-Glo 3/7 Assay kit (Promega), according to the instructions of the manufacturer. HTETOP cells were seeded in 96-well plates, one day before dexrazoxane administration. After specified incubation periods, the caspase 3/7 assay reagent was added to each well followed by 1 h of incubation at room temperature. Luminescence was detected in a plate-reading luminometer. The luminescence intensity was expressed as relative light units.
γ-H2AX and 53BP1 immunofluorescence staining
HTETOP cells grown on coverslips were fixed with ice-cold methanol/acetone (v/v = 7:3) at −20°C for 10 min followed by three times washing with PBS. After blocking with PBS containing 10% goat serum and 0.3% Triton X-100 at room temperature for 1 h, cells were incubated with a mixture of monoclonal anti-γ-H2AX (1:1000; Millipore) and polyclonal anti-53BP1 (1:500; Millipore) antibodies at 4°C overnight. After washing with PBS, the cells were incubated with Alexa Fluor 488-conjugated goat anti-mouse (1:300; Invitrogen, Darmstadt, Germany) and DyLight 549-conjugated goat anti-rabbit (1:600; Jackson ImmunoResearch Laboratories, Dianova, Hamburg, Germany) antibodies at room temperature for 1 h. Finally, the nuclei were stained with 1 μM To-Pro-3 for 15 min and the slides were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). Fluorescence images were recorded with a laser scanning microscope (LSM 710) and fluorescent intensities were quantified with the ZEN Software from Carl Zeiss (Jena, Germany). Each value represents the average fluorescence of at least 50 nuclei. When only γ-H2AX foci were determined, microscopic images were recorded using Zeiss Axio Imager M1 (Carl Zeiss) supplied with the Metafer4 Software (MetaSystems, Altlussheim, Germany), as previously described (Nikolova et al., 2014).
Single-cell gel electrophoresis (comet assay)
Following drug treatment, HTETOP cells were trypsinized and washed with ice-cold PBS. Cell lysis and electrophoresis were performed as previously described (Berdelle et al., 2011). About 1 mg·mL−1 formamidopyrimidine DNA glycosylase was added to the agarose gel on the microscope slides, followed by 45 min of incubation at 37°C. Electrophoresis (23 V) was carried out at 4°C for 15 min in buffer containing 90 mM Tris, 90 mM boric acid and 2 mM EDTA (pH 7.5). Following staining with 50 μg·mL−1 propidium iodide, the slides were evaluated with a fluorescence microscope and the Komet 4.0.2 software (Kinetic Imaging Ltd., Liverpool, UK). Data were expressed as tail moment, which represents the percentage of DNA in the tail (tail DNA intensity/cell DNA intensity) multiplied by the length between the centre of the head and of the tail. This assessment method accounts for nucleus size differences such as the TOP2A inhibition-driven endopolyploidy caused by dexrazoxane.
Statistical analysis
All experiments were repeated at least three times and the results were expressed as mean ± SE. Data were analysed using Student's t-test, one-way or two-way anova. Differences with P < 0.05 were considered statistically significant.
Results
Dexrazoxane induces TOP2A-mediated DSB
Cell viability in response to dexrazoxane was evaluated in TOP2A expressing and non-expressing HTETOP cells. Higher percentages of viable cells were observed at 100 μM and 1 mM dexrazoxane in TOP2A non-expressing cells compared with TOP2A expressing ones (Figure 1A). In the following experiments, dexrazoxane was used at 100 μM, which is in the range of concentrations seen in patients (Hochster et al., 1992; Sparano et al., 1999). Treatment of HTETOP cells with dexrazoxane (100 μM, 24 h) resulted in an accumulation of the DSB marker γ-H2AX, which was abolished by TOP2A depletion with tetracycline (Yan et al., 2009), indicating that TOP2A inhibition was the primary source of dexrazoxane-induced γ-H2AX. The kinetics of dexrazoxane-induced DSB formation was determined by γ-H2AX foci staining and compared with that of the classical TOP2A poison doxorubicin. Enhanced foci formation was observed as early as 1 h after either drug (Figure 1B). Foci induced by dexrazoxane lasted longer than those evoked by doxorubicin, suggesting distinct kinetics of cleavable complex formation. Additionally, we investigated whether dexrazoxane-induced DSB formation is a general phenomenon in tumour cells using NYH cells derived from human small cell lung cancer, LDL-1 cells from colorectal adenocarcinoma and fibrosarcoma-derived HT1080, the parental cell line of HTETOP. All cell lines exhibited increased foci formation following dexrazoxane exposure (Figure 1C).
Figure 1.

Dexrazoxane induces DSB in tumour cells. (A) HTETOP cells expressing (+TOP2A) or depleted of TOP2A (−TOP2A) by tetracycline (TET) pre-treatment (1 μg·mL–1 for 24 h) were treated with dexrazoxane (DRZ) at the indicated concentrations. Cell viability was determined 24 h following dexrazoxane exposure. *P < 0.05, **P < 0.01, significantly different from +TOP2A. (B) TOP2A-expressing HTETOP cells were treated with 100 μM dexrazoxane or 1 μM doxorubicin (DOX) for specified time periods. γ-H2AX foci were detected by immunofluorescent staining. (C) γ-H2AX foci following 24 h treatment with 100 μM dexrazoxane in HT1080, NYH and DLD-1 cells were determined by immunofluorescent staining. *P < 0.05, **P < 0.01, significantly different as indicated. (D) Neutral comet assay of TOP2A-expressing HTETOP cells performed after 24 h of treatment with 100 μM dexrazoxane. Con: untreated controls, dexrazoxane : dexrazoxane -treated cells. n = 3. *P < 0.05, significantly different as indicated. (E) Immunofluorescent staining of 53BP1 and γ-H2AX in TOP2A-expressing HTETOP cells treated with 100 μM dexrazoxane for 24 h. Quantitative data are mean values from three experiments. *P < 0.05, **P < 0.01, significantly different as indicated.
However, γ-H2AX is an indirect DSB marker and its induction may also reflect inhibition of DNA synthesis rather than generation of free DSB (Kinner et al., 2008). Therefore, we characterized the effects of dexrazoxane using the neutral comet assay, which allows for direct DSB detection in single cells. Dexrazoxane (100 μM, 24 h) enlarged the size of nuclei of dexrazoxane-treated cells, probably due to endopolyploidy caused by TOP2A inhibition (Hasinoff et al., 2001) or to G2/M cell cycle arrest (Supporting Information Fig. S1). Dexrazoxane significantly increased the tail moment as compared to that of untreated cells (Figure 1D). The formation of DSB by dexrazoxane treatment was further verified by γ-H2AX and 53BP1 immunohistochemistry. Dexrazoxane treatment led to the formation of γ-H2AX and 53BP1 foci, many of which co-localized (Figure 1E). Both the increased tail moment and the γ-H2AX/53BP1 co-localization were consistent with dexrazoxan -induced and TOP2A-mediated γ-H2AX being true DSB (FitzGerald et al., 2009).
Dexrazoxane activates the DDR
We next investigated if dexrazoxane-induced DSB result in DDR activation. Firstly, we measured the phosphorylation of DDR transducers and kinases Chk1 and Chk2 at serine 317 and threonine 68 respectively. Both kinases were largely phosphorylated in response to 100 μM dexrazoxane 4 h following the drug exposure (Figure 2A). We then investigated the phosphorylation status of ATR and ATM, the respective activating kinases of Chk1 and Chk2. The phosphorylation of ATR at Ser428 and of ATM at Ser1981, which act as the markers for enzyme activation following DNA damage (Sahu et al., 2009), reached peak values 4 h following dexrazoxane exposure (Figure 2B). As expected, the accumulation of the dexrazoxane-induced DDR mediator γ-H2AX was nearly abolished by the ATM inhibitor KU55933 (Figure 4B) and absent in ATM mutant cells (Figure 4C). Consistent with enhanced ATR phosphorylation (Figure 2B), its specific inhibitor VE-821 successfully prevented the downstream target Chk1 phosphorylation (Figure 2C), indicating ATR contribution to Chk1 activation following dexrazoxane. We also assessed the status of the DDR (Chk1 and Chk2) target p53 and the resultant apoptosis. Distinct from the kinetics of ATM or ATR activation, total p53 protein level increased first after 8 h of dexrazoxane exposure (Figure 2D). The apoptosis marker caspase 3/7 reached the highest value 24 h after dexrazoxane treatment (Figure 2E), which was accompanied by a tripled rate of apoptosis (Figure 5C). Taken together, we obtained evidence consistent with the activation of DDR sensors (ATM, ATR), mediators (53BP1, γ-H2AX), transducers (Chk1, Chk2) and effectors (p53) in response to dexrazoxane.
Figure 2.
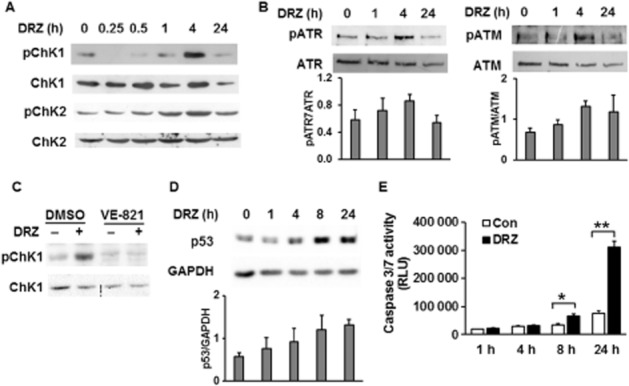
Dexrazoxane activates DDR. Phosphorylations of Chk1 at Ser317 (pChk1) and Chk2 at Thr68 (pChk2) (A), and of ATR at Ser428 (pATR) and ATM at Ser1981 (pATM) (B) were detected by Western blot. Total Chk1, Chk2, ATM or ATR was used as loading controls. TOP2A-expressing HTETOP cells were exposed to 100 μM dexrazoxane (DRZ) for the indicated time periods. (C) HTETOP cells were exposed to ATR inhibitor VE-821 (10 μM for 30 min), followed by 24 h treatment with 100 μM dexrazoxane. pChk1 (Ser317) and total Chk1 protein levels were detected by Western blot. (D) Total p53 protein level determined by Western blot in TOP2A-expressing HTETOP cells incubated with 100 μM dexrazoxane for the indicated time periods. Quantitative data are mean values from three experiments. (E) Caspase 3/7 activity in TOP2A-expressing HTETOP cells following 100 μM dexrazoxane for the indicated time periods. n = 5. *P < 0.05, **P < 0.01, significantly different as indicated. All Western blots represent at least three independent experiments. RLU, relative light units.
Figure 4.
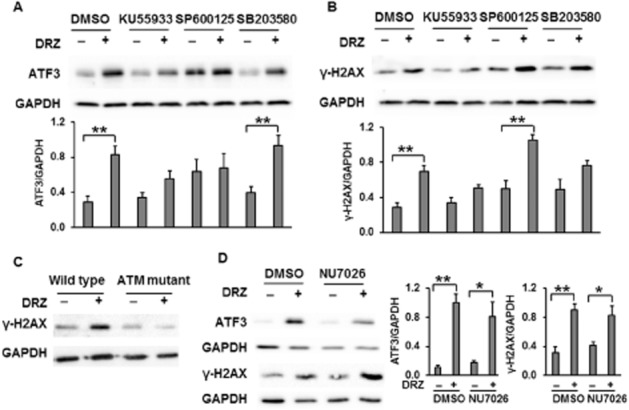
The effects of ATM, JNK, p38 and DNA-PK on the induction of ATF3 and γ-H2AX by dexrazoxane. Protein levels of ATF3 (A) and γ-H2AX (B) following dexrazoxane (DRZ; 100 μM, 24 h) in the presence or absence of ATM inhibitor KU55933 (20 μM), JNK inhibitor SP600125 (20 μM) or p38 inhibitor SB203580 (20 μM) were assessed by Western blot in TOP2A-expressing HTETOP cells. The inhibitors were administered 30 min prior to dexrazoxane treatment. Quantitative data are mean values from three experiments. **P < 0.01, significantly different as indicated. (C) γ-H2AX Western blot was performed in dexrazoxane-treated (100 μM for 24 h) ATM mutant (GM05849) and wild-type (GM637) cells. (D) HTETOP cells were first incubated with DNA-PK inhibitor NU7026 (10 μM) for 30 min then with dexrazoxane (100 μM) for 4 h. Western blots were used to evaluate ATF3 and γ-H2AX protein levels. *P < 0.05, **P < 0.01, significantly different as indicated.
Figure 5.
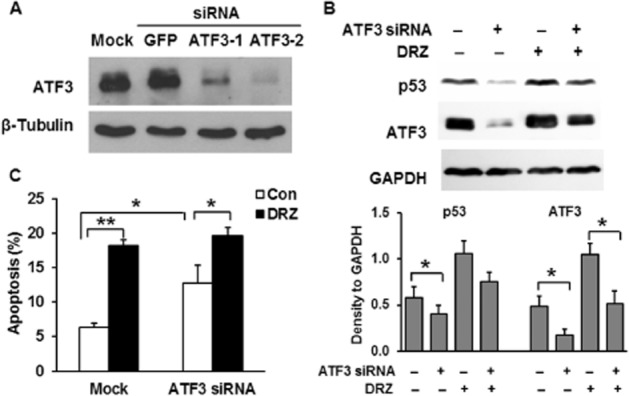
The effect of ATF3 on p53 induction and apoptosis. (A) ATF3 expression 48 h after the transfection with siRNA targeting GFP or two different loci of ATF3 mRNA assessed by Western blot. (B) p53 and ATF3 protein levels assessed by Western blot in TOP2A-expressing HTETOP cells incubated for 24 h with 100 μM dexrazoxane (DRZ) beginning at 24 h after ATF3-2 siRNA transfection. Quantitative data are mean values from three experiments. *P < 0.05, significantly different as indicated. (C) FACS analysis of Annexin V staining in cells pre-treated with or without ATF3-2 siRNA for 24 h followed by exposure to 100 μM dexrazoxane for another 24 h. n = 5, *P < 0.05, **P < 0.01, significantly different as indicated.
Dexrazoxane triggers TOP2A-dependent ATF3 induction
ATF3 was the only gene up-regulated genome-wide by dexrazoxane in HTETOP cells (Yan et al., 2009). Using an ATF3-specific antibody, we first investigated if this induction also affected ATF3 protein level. Dexrazoxane exposure resulted in the up-regulation of AFT3 protein in a concentration-dependent (Figure 3A) and incubation time-dependent manner (Figure 3B). The accumulation of ATF3 was TOP2A-dependent, as it was nearly absent in cells pre-incubated with tetracycline (-TOP2A). Consistent with our previous study, prolonged (24 h, Figure 3B) exposure to high dexrazoxane concentrations (100 and 500 μM, Figure 3A) reduced the TOP2A protein level, which was accompanied by the repression of TOP2A mRNA (Yan et al., 2009). No ATF3 induction was observed following the incubation with the dexrazoxane analogue ICRF-161 (Figure 3C), which chelates iron but does not bind to TOP2 (Martin et al., 2009). These results together demonstrate that dexrazoxane-induced ATF3 up-regulation was mediated by TOP2A inhibition and was unrelated to iron chelation. As the expression of TOP2A from the TOP2A transgene in HTETOP cells is cell cycle-independent (Carpenter and Porter, 2004), we investigated if dexrazoxane induces ATF3 also in cells with a physiological TOP2A expression. This was confirmed on the mRNA level in the parental HTETOP cell line HT1080 (Figure 3D).
Figure 3.
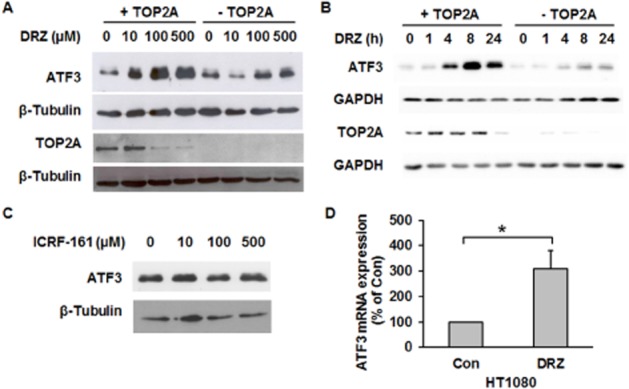
Dexrazoxane, but not ICRF-161, induces TOP2A-dependent ATF3 overexpression. HTETOP cells expressing (+TOP2A) or depleted of TOP2A (−TOP2A) by tetracycline pre-treatment were treated with increasing concentrations of dexrazoxane (DRZ) for 24 h (A) or with 100 μM dexrazoxane for the indicated time periods (B). ATF3 and TOP2A protein levels were analysed by Western blot. (C) ATF3 protein expression determined by Western blot in TOP2A-expressing HTETOP cells following 24 h of ICRF-161 treatment with indicated concentrations. (D) ATF3 mRNA expression assessed by TaqMan in HT1080 cells incubated with 100 μM dexrazoxane for 24 h. n = 3, *P < 0.05, significantly different as indicated.
ATF3 induction by dexrazoxane is mediated by ATM
Several protein kinases including ATM, JNK and JNK have been implicated both in the cellular response to DSB and in ATF3 regulation. ATF3 induction by ionizing radiation (IR) has been linked to ATM, JNK and p38 MAP kinases (Kool et al., 2003). We investigated the induction of ATF3 by dexrazoxane in the presence of the ATM inhibitor KU55933 (Hickson et al., 2004), the p38 inhibitor SB203580 (Eyers et al., 1999) and the JNK inhibitor SP600125 (Bennett et al., 2001). ATF3 induction by dexrazoxane was attenuated in the presence of the ATM inhibitor KU55933, whereas the p38 inhibitor SB203580 had no effect (Figure 4A). Although ATF3 was not up-regulated by dexrazoxane in the presence of the JNK inhibitor SP600125, SP600125 alone significantly increased the ATF3 basal level (Figure 4A). JNK and p38 inhibitors slightly increased the level of γ-H2AX, but they did not affect the dexrazoxane-induced γ-H2AX accumulation (Figure 4B). As DNA-dependent protein kinase (DNA-PK) is also involved in γ-H2AX phosphorylation following various DNA-damaging stressors, its specific inhibitor NU7026 was applied to investigate its role in dexrazoxane -induced DSB and ATF3 formation. As shown in Figure 4D, NU7026 did not affect ATF3 or γ-H2AX protein level in untreated or dexrazoxane-treated cells. The above results suggest that ATM is involved both in dexrazoxane-induced DSB formation and in the associated ATF3 accumulation.
The effects of ATF3 on p53 accumulation and apoptosis
To assess the role of ATF3 in the induction of p53 and subsequent apoptosis, its expression level was manipulated by siRNA-mediated knockdown. Of the two siRNA molecules tested, ATF3-2 better suppressed ATF3 expression (Figure 5A) and it was therefore used in all further experiments. The specificity of the ATF3-2 depletion was verified by transfecting HTETOP cells with a GFP-targeting siRNA, which had no effect on ATF3 protein level (Figure 5A). dexrazoxane -induced apoptosis in HTETOP cells has been suggested to be p53-dependent, as it was compromised by a p53 inhibitor (Yan et al., 2009). In agreement, p53 protein accumulated in HTETOP cells following dexrazoxane treatment (Figures 2D and 5B). We then investigated whether p53 accumulation resulted from ATF3 induction. Although ATF3 was still induced by dexrazoxane in the presence of ATF3 siRNA, knockdown of ATF3 mitigated p53 accumulation in both untreated and dexrazoxane -treated cells (Figure 5B), suggesting the involvement of ATF3 in p53 induction. In agreement with previous data (Yan et al., 2009), dexrazoxane increased the rate of apoptosis in HTETOP cells (Figure 5C). However, the percentage of cells undergoing apoptosis in response to dexrazoxane was unaffected by ATF3 depletion. ATF3 siRNA alone significantly increased apoptosis in cells unexposed to dexrazoxane (Figure 5C), suggesting that ATF3 has an anti-apoptotic effect in untreated HTETOP cells.
ATF3 knockdown facilitates DSB formation following dexrazoxane exposure
We have shown that ATF3 was capable of up-regulating p53 protein, which was involved in dexrazoxane-triggered apoptosis (Yan et al., 2009), indicating a pro-apoptotic potential of ATF3. However, ATF3 knockdown did not attenuate dexrazoxane-induced apoptosis and even promoted apoptosis in untreated cells (Figure 5C), suggesting that an alternative signalling pathway of ATF3 may counter dexrazoxane-induced and p53-mediated apoptosis. We subsequently investigated if ATF3 has an effect on dexrazoxane-initiated DNA damage. ATF3 knockdown increased γ-H2AX level (Figure 6A) in both untreated and dexrazoxane-treated cells. As this suggested ATF3 involvement in various aspects of DSB metabolism, we investigated DSB levels during a recovery phase following 24 h of incubation with dexrazoxane. Compared with dexrazoxane-untreated cells (‘Con’ in the left part of Figure 6B), cells exposed to dexrazoxane exhibited detectable levels of DSB immediately after dexrazoxane removal (‘0 h’ in the left part of Figure 6B), which declined during the following 24 h. Consistent with Figure 6A, ATF3 siRNA alone increased γ-H2AX (‘Con’ in the right part of Figure 6B). Compared with cells without ATF3 siRNA (‘0 h’ in the left part of Figure 6B), cells depleted of ATF3 exhibited much higher levels of DSB immediately after dexrazoxane removal (‘0 h’ in the right part of Figure 6B), which persisted over the entire post-dexrazoxane exposure period investigated. Similar results were observed when the specific DSB marker γ-H2AX/53BP1 foci was determined (Figure 6C). Immediately after dexrazoxane removal, ATF3 siRNA-treated cells showed higher number of foci than cells without ATF3 (‘0 h’ in ATF3 siRNA vs. ‘0 h’ in SCR siRNA). We than investigated the role of ATF3 in IR-triggered DSB generation (Figure 6D). Similar to Figure 6A and B, ATF3 siRNA alone increased γ-H2AX protein level in untreated (‘Φ’ in ATF3 siRNA vs. ‘Φ’ in SCR siRNA) and IR-exposed cells [0.5 and 1 h after IR exposure as compared to SCR siRNA (Figure 6D)]. These results support an involvement of ATF3 in the processing of both spontaneous and induced DSB, irrespective of the inducing agent.
Figure 6.
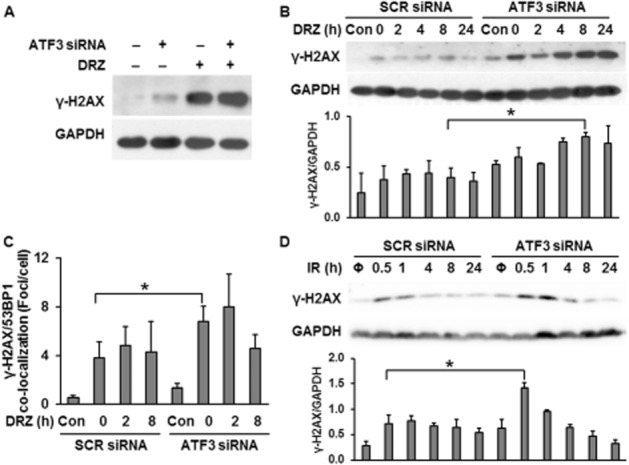
The effect of ATF3 on DSB formation in TOP2A-expressing HTETOP cells in response to dexrazoxane (DRZ) or IR. (A) γ-H2AX protein levels were assessed by Western blot in TOP2A-expressing HTETOP cells. ATF3 siRNA was transfected 24 h before 100 μM dexrazoxane exposure for 24 h. (B) γ-H2AX levels in scrambled (SCR) or ATF3-2 siRNA-transfected HTETOP cells treated 24 later with 100 μM dexrazoxane for another 24 h. Cells were subsequently washed with PBS, fed with fresh medium and γ-H2AX levels were assessed at the indicated time points after washing. *P < 0.05, significantly different as indicated. (C) The quantification of γ-H2AX/53BP1 foci determined by immunofluorescent staining. The treatment was the same as in (B). n = 3, *P < 0.05, significantly different as indicated. (D) γ-H2AX levels in SCR or ATF3-2 siRNA-transfected cells exposed 24 h later to 10 Gy of IR (Φ: without IR exposure). γ-H2AX levels were determined at the indicated time points after IR exposure. *P < 0.05, significantly different as indicated. All histograms represent mean values from three independent experiments.
Discussion and conclusions
Catalytic TOP2 inhibitors are thought to exert cytotoxic effects predominantly via inhibiting ATPase activity, forming a closed clamp and blocking the turnover of the enzyme. As bisdioxopiperazines are specific for TOP2, they are the most commonly used catalytic TOP2 inhibitors in human cells. The question of whether bisdioxopiperazines are pure catalytic inhibitors has been raised following observations that bisdioxopiperazine ICRF-193 is able to trap TOP2 covalent complexes undetectable by standard procedures (Huang et al., 2001). Using a genetic model of conditional TOP2A expression, we demonstrated a correlation between TOP2-mediated DSB marker γ-H2AX and apoptosis in response to dexrazoxane (Yan et al., 2009). However, a limitation of γ-H2AX is that it detects both free DSB and blocked replication forks (Kinner et al., 2008). In the present study, we demonstrate that TOP2A inhibition by dexrazoxane induces true DSB. This is followed by activation of the DDR and by apoptosis. dexrazoxane also triggers ATF3 gene expression, which affects the level of p53 accumulation as well as dexrazoxan -induced DSB.
Dexrazoxane-induced DSB were demonstrated by γ-H2AX protein level, by the formation of γ-H2AX foci co-localization with 53BP1 and by the tail moment in the neutral comet assay. These data are consistent with the formation of dexrazoxane–TOP2A cleavable complexes demonstrated by others (Sehested et al., 1998; Wessel et al., 1999; Lyu et al., 2007). DSB triggered by TOP2A poisons have been shown to activate the DDR, beginning with the activation of ATR and/or ATM, which signals downstream to Chk1, Chk2 and p53. p53 induces transcriptional activation of pro-apoptotic factors such as FAS, PUMA and BAX. As dexrazoxane induced true DSB, we investigated if it also activated DDR. This was confirmed by increased γ-H2AX/53BP1 co-localization as well as by the phosphorylation of ATR, ATM, Chk1 and Chk2, which was accompanied by p53 accumulation and apoptosis. The time-course of these events was consistent with γ-H2AX induction being followed by DDR activation, ATF3, as well as p53 accumulation and ultimately apoptosis (Figure 7). Besides the time-course, the dependency of dexrazoxane-induced apoptosis on p53 in HTETOP cells is supported by its block by a p53 inhibitor (Yan et al., 2009). Therefore, in addition to blocking TOP2 catalytic activity and similar to TOP2 poisons, dexrazoxane treatment clearly results in DSB formation, which is followed by DDR activation and ultimately by cell death from p53-dependent apoptosis.
Figure 7.
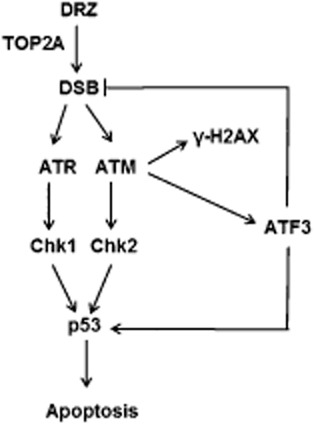
HTETOP cell response to dexrazoxane (DRZ), including DSB induction, DDR activation, ATF3 accumulation and apoptosis.
Accumulation of p53 following dexrazoxane treatment was dependent upon ATF3 (Yan et al., 2009). ATF3 acts as an adaptive response gene that participates in cellular processes in response to extra- and/or intracellular changes. The induction of ATF3 by dexrazoxane was TOP2A-dependent, as it was absent both after TOP2A depletion and upon treatment with a dexrazoxane analogue incapable of TOP2 inhibition. The induction of ATF3 also involved the protein kinase ATM, as it was blocked by a specific ATM inhibitor and in ATM mutant cells. ATM acts as a primary transducer in response to DSB and phosphorylates key factors in DNA damage response pathways. Among others, it is suggested to interact with and phosphorylate ATF2, which is capable of increasing ATF3 expression via promoter activation (Lee et al., 2010). The JNK inhibitor SP600125 alone significantly increased the ATF3 level in untreated, but not in dexrazoxane-treated, cells (Figure 4A). This is consistent with the SP600125-induced ATF3 expression in human colon cancer HCT116 cells, via an unknown mechanism (Hackl et al., 2010). Taken together, dexrazoxane-driven accumulation of ATF3 was most likely to be mediated by TOP2A-generated DSB detected by ATM.
ATF3 has been suggested to be pro-apoptotic (Huang et al., 2008; Kashiwakura et al., 2008) and to function as tumour suppressor, as its down-regulation promotes tumour growth and metastasis (Hackl et al., 2010). On the contrary, ATF3 overexpression has been found to enhance the tumorigenic potential (Wu et al., 2010) and contribute to the malignant growth of tumour cells. Despite many efforts trying to clarify the oncogenic properties of ATF3, ATF3 cannot be defined exclusively as an oncogene or tumour repressor and this assessment is consistent with the various effects of ATF3 perturbation observed in HTETOP cells. ATF3 was clearly anti-apoptotic in untreated cells, as its knockdown by siRNA caused significant apoptosis. In contrast, down-regulation of ATF3 could not affect dexrazoxan-induced apoptosis, despite ATF3-mediated p53 accumulation, although the latter one is required for dexrazoxane-induced apoptosis (Yan et al., 2009).
How can we explain the apparent contradiction between the dependency of dexrazoxane-induced p53 accumulation on ATF3 and the disparate effects of these proteins on apoptosis? One possibility is that induction of ATF3 facilitates apoptosis by up-regulating p53, but detecting this effect requires more than simple manipulation of the ATF3 level. Alternatively, this contradiction may reflect the involvement of ATF3 in the processing of dexrazoxane-induced DSB, which may counter the pro-apoptotic effect of p53 stabilized by ATF3 (Figure 7). ATF3 knockdown led to increased γ-H2AX protein accumulation in dexrazoxane-treated cells, which may be the consequence of ongoing apoptosis rather than DSB. However, the specific marker of true DSB, γ-H2AX/53BP1 foci, was also enhanced by ATF3 siRNA in dexrazoxane-treated cells. The involvement of ATF3 in DSB processing would be consistent with the capability of its transcriptional activator ATF2 to enhance DNA repair (Hayakawa et al., 2003). Consistent with our data, the repression of ATF3 after UV-mediated genotoxic stress impaired the DNA repair process mediated through ATF3 transcriptional target PCNA-associated factor KIAA0101/p15PAF (Turchi et al., 2009). The mechanisms by which ATF3 protects against dexrazoxane-induced DNA damage need to be further investigated. p15PAF elevation was not observed in dexrazoxane-treated HTETOP cells from the microarray experiments (data not shown), indicating that this protein may not be involved in the protective effect of ATF3, or that such an effect is p15PAF transcription independent. Taken together, the DNA damage-triggered ATF3 induction controls the level of p53 accumulation and may as well as regulate DSB repair. This is consistent with ATF3 serving as a switch between DNA damage repair and cell death following dexrazoxane treatment.
The apparent roles of ATF3 in apoptosis and DNA damage process are reminiscent of the puzzling heterogeneity of cancer-related clinical endpoints of dexrazoxane treatments. Dexrazoxane remains underused due to the reported reduction of the anti-tumour effect of anthracyclines (Swain et al., 1997b) and to its association with secondary malignancies (Tebbi et al., 2007). On the contrary, dexrazoxane has increased survival both in patients (Swain et al., 1997a) and in pre-clinical animal cancer models when combined with TOP2 poisons (Hasinoff et al., 1998; Hofland et al., 2005). The latter findings are in agreement with dexrazoxane's capacity to induce DSB, DDR and apoptosis, which resemble and may enhance the anti-tumour effects of anthracyclines. They are also consistent with meta-analyses demonstrating undiminished anti-tumour efficacy of anthracyclines when combined with dexrazoxane (van Dalen et al., 2011b). Finally, based upon our observation that dexrazoxane is clearly genotoxic, secondary malignancies could arise from dexrazoxane-induced DNA damage in cells with diminished capacity for apoptosis and/or DNA repair. The application of dexrazoxane as a cardioprotectant may require a consideration of these variables in the context of individual patients and tumour entities. These complex relationships in dividing, that is, TOP2A-expressing cells, contrast the apparently more straightforward mechanism of cardioprotection conferred by dexrazoxane. The predominant cardiac TOP2 isozyme TOP2B undergoes rapid proteosomal degradation following exposure to dexrazoxane, thereby preventing doxorubicin-induced and TOP2B-mediated DNA damage (Lyu et al., 2007).
Acknowledgments
This project was supported by the Deutsche Forschungsgemeinschaft (DFG) grant (WO505/3-1).
Glossary
- ATF3
activating transcription factor 3
- ATM
ataxia telangiectasia mutated
- ATR
ATM and Rad3-related
- Chk
checkpoint kinase
- DDR
DNA damage response
- DNA-PK
DNA-dependent protein kinase
- DSB
double-strand breaks
- IR
ionizing radiation
- siRNA
small interfering RNA
- TOP2A
topoisomerase IIα
- TOP2B
topoisomerase IIβ
Author Contributions
S. D., B. K. and L. W. participated in research design. S. D., T. Y., T. N., D. F., A. N. and U. G.-A. conducted the experiments. T. N. contributed new reagents or analytical tools. S. D. and T. Y. performed data analysis. S. D., B. K. and L. W. wrote or contributed to the writing of the manuscript.
Conflicts of interest
No conflict of interest is declared.
Supporting Information
Figure S1 Dexrazoxane exposure led to cell cycle arrest at G2/M. HTETOP cell was exposed to 100 μM dexrazoxane (DRZ) or DMSO for indicated time periods. DNA was stained with PI and cell cycle distribution was examined by FACS analysis. The PI intensity was plotted against cell numbers.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdelle N, Nikolova T, Quiros S, Efferth T, Kaina B. Artesunate induces oxidative DNA damage, sustained DNA double-strand breaks, and the ATM/ATR damage response in cancer cells. Mol Cancer Ther. 2011;10:2224–2233. doi: 10.1158/1535-7163.MCT-11-0534. [DOI] [PubMed] [Google Scholar]
- Bonnefoi HR. Anthracyclines, HER2, and TOP2A: the verdict. Lancet Oncol. 2011;12:1084–1085. doi: 10.1016/S1470-2045(11)70241-8. [DOI] [PubMed] [Google Scholar]
- Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, et al. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A. 2008;105:9053–9058. doi: 10.1073/pnas.0803513105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AJ, Porter AC. Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol Biol Cell. 2004;15:5700–5711. doi: 10.1091/mbc.E04-08-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BP, Liang G, Whelan J, Hai T. ATF3 and ATF3 delta Zip. Transcriptional repression versus activation by alternatively spliced isoforms. J Biol Chem. 1994;269:15819–15826. [PubMed] [Google Scholar]
- Creighton AM, Hellmann K, Whitecross S. Antitumour activity in a series of bisdiketopiperazines. Nature. 1969;222:384–385. doi: 10.1038/222384a0. [DOI] [PubMed] [Google Scholar]
- van Dalen EC, Caron HN, Dickinson HO, Kremer LC. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev. 2011a;(6) doi: 10.1002/14651858.CD003917.pub4. CD003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dalen EC, van den Berg H, Raphael MF, Caron HN, Kremer LC. Should anthracyclines and dexrazoxane be used for children with cancer? Lancet Oncol. 2011b;12:12–13. doi: 10.1016/S1470-2045(10)70301-6. [DOI] [PubMed] [Google Scholar]
- Eich M, Roos WP, Nikolova T, Kaina B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol Cancer Ther. 2013;12:2529–2540. doi: 10.1158/1535-7163.MCT-13-0136. [DOI] [PubMed] [Google Scholar]
- Eyers PA, van den Ijssel P, Quinlan RA, Goedert M, Cohen P. Use of a drug-resistant mutant of stress-activated protein kinase 2a/p38 to validate the in vivo specificity of SB 203580. FEBS Lett. 1999;451:191–196. doi: 10.1016/s0014-5793(99)00552-9. [DOI] [PubMed] [Google Scholar]
- FitzGerald JE, Grenon M, Lowndes NF. 53BP1: function and mechanisms of focal recruitment. Biochem Soc Trans. 2009;37:897–904. doi: 10.1042/BST0370897. [DOI] [PubMed] [Google Scholar]
- Hackl C, Lang SA, Moser C, Mori A, Fichtner-Feigl S, Hellerbrand C, et al. Activating transcription factor-3 (ATF3) functions as a tumor suppressor in colon cancer and is up-regulated upon heat-shock protein 90 (Hsp90) inhibition. BMC Cancer. 2010;10:668. doi: 10.1186/1471-2407-10-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasinoff BB, Hellmann K, Herman EH, Ferrans VJ. Chemical, biological and clinical aspects of dexrazoxane and other bisdioxopiperazines. Curr Med Chem. 1998;5:1–28. [PubMed] [Google Scholar]
- Hasinoff BB, Abram ME, Barnabe N, Khelifa T, Allan WP, Yalowich JC. The catalytic DNA topoisomerase II inhibitor dexrazoxane (ICRF-187) induces differentiation and apoptosis in human leukemia K562 cells. Mol Pharmacol. 2001;59:453–461. doi: 10.1124/mol.59.3.453. [DOI] [PubMed] [Google Scholar]
- Hayakawa J, Depatie C, Ohmichi M, Mercola D. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J Biol Chem. 2003;278:20582–20592. doi: 10.1074/jbc.M210992200. [DOI] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- van Hille B, Hill BT. Yeast cells expressing differential levels of human or yeast DNA topoisomerase II: a potent tool for identification and characterization of topoisomerase II-targeting antitumour agents. Cancer Chemother Pharmacol. 1998;42:345–356. doi: 10.1007/s002800050828. [DOI] [PubMed] [Google Scholar]
- Hochster H, Liebes L, Wadler S, Oratz R, Wernz JC, Meyers M, et al. Pharmacokinetics of the cardioprotector ADR-529 (ICRF-187) in escalating doses combined with fixed-dose doxorubicin. J Natl Cancer Inst. 1992;84:1725–1730. doi: 10.1093/jnci/84.22.1725. [DOI] [PubMed] [Google Scholar]
- Hofland KF, Thougaard AV, Dejligbjerg M, Jensen LH, Kristjansen PE, Rengtved P, et al. Combining etoposide and dexrazoxane synergizes with radiotherapy and improves survival in mice with central nervous system tumors. Clin Cancer Res. 2005;11:6722–6729. doi: 10.1158/1078-0432.CCR-05-0698. [DOI] [PubMed] [Google Scholar]
- Huang KC, Gao H, Yamasaki EF, Grabowski DR, Liu S, Shen LL, et al. Topoisomerase II poisoning by ICRF-193. J Biol Chem. 2001;276:44488–44494. doi: 10.1074/jbc.M104383200. [DOI] [PubMed] [Google Scholar]
- Huang X, Li X, Guo B. KLF6 induces apoptosis in prostate cancer cells through up-regulation of ATF3. J Biol Chem. 2008;283:29795–29801. doi: 10.1074/jbc.M802515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz M, Hummel M, Truss M, Wollert-Wulf B, Mathas S, Johrens K, et al. Classical Hodgkin lymphoma is characterized by high constitutive expression of activating transcription factor 3 (ATF3), which promotes viability of Hodgkin/Reed-Sternberg cells. Blood. 2006;107:2536–2539. doi: 10.1182/blood-2005-07-2694. [DOI] [PubMed] [Google Scholar]
- Jensen LH, Nitiss KC, Rose A, Dong J, Zhou J, Hu T, et al. A novel mechanism of cell killing by anti-topoisomerase II bisdioxopiperazines. J Biol Chem. 2000;275:2137–2146. doi: 10.1074/jbc.275.3.2137. [DOI] [PubMed] [Google Scholar]
- Kashiwakura Y, Ochiai K, Watanabe M, Abarzua F, Sakaguchi M, Takaoka M, et al. Down-regulation of inhibition of differentiation-1 via activation of activating transcription factor 3 and Smad regulates REIC/Dickkopf-3-induced apoptosis. Cancer Res. 2008;68:8333–8341. doi: 10.1158/0008-5472.CAN-08-0080. [DOI] [PubMed] [Google Scholar]
- Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool J, Hamdi M, Cornelissen-Steijger P, van der Eb AJ, Terleth C, van Dam H. Induction of ATF3 by ionizing radiation is mediated via a signaling pathway that includes ATM, Nibrin1, stress-induced MAPkinases and ATF-2. Oncogene. 2003;22:4235–4242. doi: 10.1038/sj.onc.1206611. [DOI] [PubMed] [Google Scholar]
- Larsen AK, Escargueil AE, Skladanowski A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther. 2003;99:167–181. doi: 10.1016/s0163-7258(03)00058-5. [DOI] [PubMed] [Google Scholar]
- Lee SH, Bahn JH, Whitlock NC, Baek SJ. Activating transcription factor 2 (ATF2) controls tolfenamic acid-induced ATF3 expression via MAP kinase pathways. Oncogene. 2010;29:5182–5192. doi: 10.1038/onc.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–8846. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- Martin E, Thougaard AV, Grauslund M, Jensen PB, Bjorkling F, Hasinoff BB, et al. Evaluation of the topoisomerase II-inactive bisdioxopiperazine ICRF-161 as a protectant against doxorubicin-induced cardiomyopathy. Toxicology. 2009;255:72–79. doi: 10.1016/j.tox.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Nikolova T, Dvorak M, Jung F, Adam I, Kramer E, Gerhold-Ay A, et al. The γH2AX assay for genotoxic and nongenotoxic agents: comparison of H2AX phosphorylation with cell death response. Toxicol Sci. 2014;140:103–117. doi: 10.1093/toxsci/kfu066. [DOI] [PubMed] [Google Scholar]
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobori K, Ito H, Tamamori-Adachi M, Adachi S, Ono Y, Kawauchi J, et al. ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: a novel cardioprotective role of ATF3. J Mol Cell Cardiol. 2002;34:1387–1397. doi: 10.1006/jmcc.2002.2091. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu RP, Batra S, Srivastava SK. Activation of ATM/Chk1 by curcumin causes cell cycle arrest and apoptosis in human pancreatic cancer cells. Br J Cancer. 2009;100:1425–1433. doi: 10.1038/sj.bjc.6605039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehested M, Wessel I, Jensen LH, Holm B, Oliveri RS, Kenwrick S, et al. Chinese hamster ovary cells resistant to the topoisomerase II catalytic inhibitor ICRF-159: a Tyr49Phe mutation confers high-level resistance to bisdioxopiperazines. Cancer Res. 1998;58:1460–1468. [PubMed] [Google Scholar]
- Snyder RD. Evidence from studies with intact mammalian cells that merbarone and bis(dioxopiperazine)s are topoisomerase II poisons. Drug Chem Toxicol. 2003;26:15–22. doi: 10.1081/dct-120017554. [DOI] [PubMed] [Google Scholar]
- Sparano JA, Speyer J, Gradishar WJ, Liebes L, Sridhara R, Mendoza S, et al. Phase I trial of escalating doses of paclitaxel plus doxorubicin and dexrazoxane in patients with advanced breast cancer. J Clin Oncol. 1999;17:880–886. doi: 10.1200/JCO.1999.17.3.880. [DOI] [PubMed] [Google Scholar]
- Swain SM, Whaley FS, Gerber MC, Ewer MS, Bianchine JR, Gams RA. Delayed administration of dexrazoxane provides cardioprotection for patients with advanced breast cancer treated with doxorubicin-containing therapy. J Clin Oncol. 1997a;15:1333–1340. doi: 10.1200/JCO.1997.15.4.1333. [DOI] [PubMed] [Google Scholar]
- Swain SM, Whaley FS, Gerber MC, Weisberg S, York M, Spicer D, et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol. 1997b;15:1318–1332. doi: 10.1200/JCO.1997.15.4.1318. [DOI] [PubMed] [Google Scholar]
- Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J Clin Oncol. 2007;25:493–500. doi: 10.1200/JCO.2005.02.3879. [DOI] [PubMed] [Google Scholar]
- Turchi L, Aberdam E, Mazure N, Pouyssegur J, Deckert M, Kitajima S, et al. Hif-2alpha mediates UV-induced apoptosis through a novel ATF3-dependent death pathway. Cell Death Differ. 2008;15:1472–1480. doi: 10.1038/cdd.2008.74. [DOI] [PubMed] [Google Scholar]
- Turchi L, Fareh M, Aberdam E, Kitajima S, Simpson F, Wicking C, et al. ATF3 and p15PAF are novel gatekeepers of genomic integrity upon UV stress. Cell Death Differ. 2009;16:728–737. doi: 10.1038/cdd.2009.2. [DOI] [PubMed] [Google Scholar]
- Wessel I, Jensen LH, Jensen PB, Falck J, Rose A, Roerth M, et al. Human small cell lung cancer NYH cells selected for resistance to the bisdioxopiperazine topoisomerase II catalytic inhibitor ICRF-187 demonstrate a functional R162Q mutation in the Walker A consensus ATP binding domain of the alpha isoform. Cancer Res. 1999;59:3442–3450. [PubMed] [Google Scholar]
- Wu X, Nguyen BC, Dziunycz P, Chang S, Brooks Y, Lefort K, et al. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature. 2010;465:368–372. doi: 10.1038/nature08996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada N, Noguchi S, Mori T, Naoe T, Maruo K, Akao Y. Tumor-suppressive microRNA-145 targets catenin delta-1 to regulate Wnt/beta-catenin signaling in human colon cancer cells. Cancer Lett. 2013;335:332–342. doi: 10.1016/j.canlet.2013.02.060. [DOI] [PubMed] [Google Scholar]
- Yan T, Deng S, Metzger A, Godtel-Armbrust U, Porter AC, Wojnowski L. Topoisomerase II{alpha}-dependent and -independent apoptotic effects of dexrazoxane and doxorubicin. Mol Cancer Ther. 2009;8:1075–1085. doi: 10.1158/1535-7163.MCT-09-0139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Dexrazoxane exposure led to cell cycle arrest at G2/M. HTETOP cell was exposed to 100 μM dexrazoxane (DRZ) or DMSO for indicated time periods. DNA was stained with PI and cell cycle distribution was examined by FACS analysis. The PI intensity was plotted against cell numbers.