

Abstract
Background and Purpose
Mild cognitive deficit in early Parkinson's disease (PD) has been widely studied. Here we have examined the effects of memantine in preventing memory deficit in experimental PD models and elucidated some of the underlying mechanisms.
Experimental Approaches
I.p. injection of 1-methyl-4- phenyl-1,2,3,6-tetrahydro pyridine (MPTP) in C57BL/6 mice was used to produce models of PD. We used behavioural tasks to test memory. In vitro, we used slices of hippocampus, with electrophysiological, Western blotting, real time PCR, elisa and immunochemical techniques.
Key Results
Following MPTP injection, long-term memory was impaired and these changes were prevented by pre-treatment with memantine. In hippocampal slices from MPTP treated mice, long-term potentiation (LTP) –induced by θ burst stimulation (10 bursts, 4 pulses) was decreased, while long-term depression (LTD) induced by low-frequency stimulation (1 Hz, 900 pulses) was enhanced, compared with control values. A single dose of memantine (i.p., 10 mg·kg−1) reversed the decreased LTP and the increased LTD in this PD model. Activity-dependent changes in tyrosine kinase receptor B (TrkB), ERK and brain-derived neurotrophic factor (BDNF) expression were decreased in slices from mice after MPTP treatment. These effects were reversed by pretreatment with memantine. Incubation of slices in vitro with 1-methyl-4-phenylpyridinium (MPP+) decreased depolarization-induced expression of BDNF. This effect was prevented by pretreatment of slices with memantine or with calpain inhibitor III, suggesting the involvement of an overactivated calcium signalling pathway.
Conclusions and Implications
Memantine should be useful in preventing loss of memory and hippocampal synaptic plasticity in PD models.
Tables of Links
| LIGANDS | |
|---|---|
| AP5 | MPP+ |
| BDNF | NBQX |
| DHPG, 3,5-dihydroxyphenylglycine | NMDA |
| Dopamine | |
| K252a | |
| Memantine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b,).
Introduction
Parkinson's disease (PD), characterized by both motor and non-motor dysfunctions is the second most common neurodegenerative disease after Alzheimer's disease (AD). Non-motor features of PD, especially cognitive impairment have increasingly attracted attention. In the early stages, PD patients frequently experience mild cognitive impairment, which is likely to progress into dementia in the later stages, that is PD with dementia (PDD). Dopamine replacement therapy with L-DOPA will ameliorate motor dysfunctions, although it is less effective in reversing cognitive impairment in PD (Kulisevsky et al., 2000; MacDonald et al., 2011; Miah et al., 2012; Poletti and Bonuccelli, 2013). From a consideration of the functional roles of memory in cognition, memory impairment in PD might not be simply caused by dopamine depletion and abnormality of the dopaminergic pathway or some other pathological mechanisms may contribute to the memory deficits in PD.
Neurotoxins, including 1-methyl-4- phenyl-1,2,3,6-tetrahydro pyridine (MPTP), 6-hydroxydopamine, paraquat and rotenone not only impair motor functions, but also affect cognitive networks (Bezard et al., 1998; Chiasserini et al., 2011; Martinez and Greenamyre, 2012). The hippocampus has recently been proposed to play a role in the pathophysiology of the non-motor symptoms of PD (Calabresi et al., 2013), particularly in memory deficit (Shohamy et al., 2009). Moreover, memory has been repeatedly reported to be decreased in neurotoxin-lesioned experimental animals (Costa et al., 2012; Moriguchi et al., 2012). In good agreement with the effects on impaired memory, hippocampal long-term potentiation (LTP), a cellular model for memory was also reduced in PD models (Zhu et al., 2011; Costa et al., 2012; Moriguchi et al., 2012). Although we and others demonstrated that depletion of hippocampal dopamine was likely to contribute to LTP deficit in PD models (Zhu et al., 2011; Costa et al., 2012; Moriguchi et al., 2012), the mechanisms are still unclear.
Brain-derived neurotrophic factor (BDNF) supports the survival of nigral dopaminergic neurons and expression of the BDNF protein and its mRNA are reduced in dopaminergic neurons in PD (Hyman et al., 1991; Erickson et al., 2001). In animal models, BDNF treatment reduced the loss of dopaminergic neurons in the substantia nigra in neurotoxin-induced lesions (Frim et al., 1994; Levivier et al., 1995). Thus, the use of BDNF in PD therapy has been recently considered (Nagahara and Tuszynski, 2011; Allen et al., 2013; He et al., 2013), but limited to the motor aspects of the disease. Interestingly, a functional role of BDNF in human memory is supported by the correlation between the Val66Met polymorphism, which decreased activity-dependent BDNF release, reduced hippocampal activation and impaired episodic memory (Mandelman and Grigorenko, 2012). Therefore, the hippocampal BDNF-tyrosine kinase receptor B (TrkB) signalling pathway in the hippocampus is likely to be modified in PD, which would contribute to the dysfunction of hippocampal LTP and memory.
Memantine is an non-competitive NMDA receptor antagonist with rapid blocking and unblocking kinetics. It is believed to block glutamate-related excitotoxicity and mitigate some of the symptoms of AD. In addition to the effects against AD symptoms, memantine also prevented dopaminergic neuron death and ameliorated PD symptoms (Aarsland et al., 2009; Emre et al., 2010). Whether memantine prevented memory deficits in experimental PD models and its mechanisms were not disclosed.
In our present study, we produced memory deficits through acute application of MPTP in C57BL/6 mice. Hippocampal LTP and memory were decreased after treatment with MPTP because of the inactivation of the BDNF-TrkB pathway. Importantly, memantine, given systemically, restored MPTP-impaired synaptic plasticity and memory through the BDNF-TrkB pathway. These results provide further advances in the characterization of memory deficits in PD and also provide experimental evidence for the possible clinical treatment of PDD.
Methods
MPTP models and drug treatment
All animal care and experimental procedures were approved by the Animal Care and Use Committee of Anhui University of Traditional Chinese Medicine. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 112 animals were used in the experiments described here.
Adult (8-week-old, male) C57BL/6 mice, obtained from the Animal Center of Chinese Academy of Sciences (CAS, Shanghai, China) were divided into five groups: saline control, MPTP group, high dose of memantine (10 mg·kg−1) plus MPTP, low dose of memantine (1 mg·kg−1) plus MPTP and the high dose of memantine alone. MPTP was administered by four i.p. injections, each at a dose of of 20 mg·kg−1 (in 100 μL), every 2 h, as previously described (Fig. 1A) (Zhu et al., 2011). Saline (control group) was administered in a similar manner. Memantine was injected i.p., 1 h before the MPTP regimen was started. The experimental assessments were carried out 7 days or 30 days after last MPTP or saline injection. A subchronic PD model was produced by injection of MPTP for 7 consecutive days (20 mg·kg·day−1, 100 μL; 140 mg·kg−1 cumulative) as described previously (Zhu et al., 2014; Fig. 1B). Memantine (10 mg·kg−1·day−1) or saline was given i.p. over 3 days before MPTP injection and further for another 7 days. One day after last injection, memory was tested.
Figure 1.
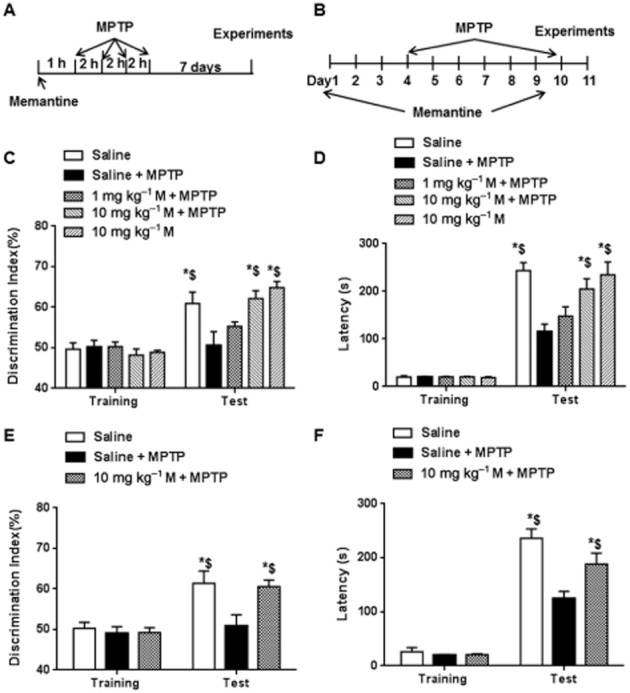
Memantine (M) reversed MPTP-induced impairment of long-term memory. (A) Scheme of the protocol for MPTP-induced acute Parkinson's disease model and memantine treatment. (B) Scheme of the protocol for MPTP-induced subchronic Parkinson's disease model and memantine treatment. (C) Novel object recognition was used to test the memory in acute MPTP model. During training session, preferences to the two objects were not different in each group. However, 24 h memory was significantly decreased in the MPTP group compared with the control. However, it was prevented by pretreatment with a high dose of memantine (10 mg·kg−1, i.p.). (D) One-trial passive avoidance task was used to test the memory. During training session, the latency to dark box was not different in different groups. In the MPTP group, 24 h memory was significantly decreased compared with control. However, it was prevented by high dose of memantine treatment. (E) Novel object recognition was used to test the memory in subchronic MPTP model. During training session, preferences to the two objects were not different in each group. In the MPTP group, 24 h memory was significantly decreased compared with the control and this decrease was prevented by the high dose of memantine, as consecutive treatment. (F) One-trial passive avoidance task was used to test the memory in subchronic MPTP model. During training session, the latency to dark box was not different in different groups. In the MPTP group, 24 h memory was significantly decreased compared with control and this decrease was reversed by the high dose of memantine, as consecutive administration. The data presented are means ± SEM (n = 5). *P < 0.05, compared with corresponding level at training session; $P < 0.05 compared with the value in MPTP group; two-way anova followed by Bonferroni test.
Hippocampal slice preparation
Brains were quickly removed and transferred to oxygenated, ice-cold cutting medium (in mM): 124 NaCl, 26 NaHCO3, 10 D-glucose, 3 KCl, 1.25 KH2PO4, 5 MgSO4, and 1.5 CaCl2. Transverse slices of hippocampus (350 μm thick) were prepared using a vibrotome (Leica, German) and transferred to an interface recording chamber and exposed to a warm, humidified atmosphere of 95%O2/5%CO2 and continuously perfused with oxygenated and preheated (33 ± 0.5°C) aCSF (in mM) [110 NaCl, 5 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.24 KH2PO4, 10 D-glucose, 27.4 NaHCO3] at a flow-rate of 1.6 ml·min−1.
Electrophysiological recordings
After 2 h incubation in recording chamber, a single glass pipette filled with 2 M NaCl was used to record field EPSPs (fEPSPs) elicited by stimulation of Schaffer collateral pathway with twisted nichrome wires (single bare wire diameter, 50 μm) placed in CA1 stratum radiatum. Responses were recorded through a differential amplifier (EXT-20F; npi electronic GmbH, Tamm, Germany) using 3 kHz low-pass and 0.1 Hz high-pass filters. Before each experiment, the input/output (I/O) relation was examined by varying stimulus intensity. Long term potentiation was induced by theta burst stimulation (TBS, 10 bursts of 4 pulses at 100 Hz delivered at 5 Hz). Long term depression (LTD) was induced by low frequency stimulation (1 Hz, 900 pulses). Bath application of DHPG, a mGluR5 agonist (100 μM, 10 min), was used to induce chemical LTD. Data were collected and digitized by Clampex, and the slope of the fEPSPs was analyzed. LTP level was normalized to the 30 min baseline.
Biochemical experiments
As the active metabolite of MPTP, MPP+ is a more effective neurotoxin in cultured cells or slices in vitro, we tested the effects of MPP+ on BDNF level after KCl-induced depolarization, as previously reported (Maharana et al., 2013). The drug treatments for hippocampal slices are shown in Fig.4A. Acute hippocampal slices including dentate gyrus were incubated in normal aCSF one hour for recovery. After recovery, the slices were treated with memantine alone (1 μM, 30 min), MPP+ alone (25 μM, 2 h) or MPP+ and memantine together. After treatment, drugs were washed out and the slices from different groups were depolarized by KCl (90 mM, 3 min), using KCl-aCSF. The composition of KCl-aCSF was as follows: 37.5 mM NaCl, 90 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 2 mM CaCl2, 1 mM MgCl2, 25 mM glucose. One hour after depolarization, the slices were collected in dry ice and kept at −80°C until use
BDNF elisa
Hippocampi were dissected, trimmed and homogenized in lysis buffer. Sample protein contents were measured using BCA protein assay kit. Volumes were adjusted to normalize protein content (μg·μL−1) and then aliquots were processed for elisa using the BDNF Emax Immunoassay System (Promega, Madison, WI, USA). BDNF levels were determined relative to a standard curve constructed from measures of BDNF protein standards (0–500 pg BDNF protein) that were assayed simultaneously with experimental samples. Data are presented as mean ± SEM pg BDNF/100 μg of sample protein content.
Western blotting
Whole hippocampus homogenates or collected hippocampal slices including dentate gyrus after in vitro drug treatments were obtained and lysed. Protein concentrations were measured using BCA protein assay kit (Thermo, Waltham, MA, USA). Equivalent amounts of proteins were processed for SDS-PAGE and Western blot. The primary antibodies used were BDNF (1:1000, Millipore, Darmstadt, Germany), Actin (1:10000, Millipore), phospho-Trk B (1:3000, Cell Signaling, Danvers, MA, USA), Trk B (1:3000, Cell signalling).
Real-time PCR
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, USA). Reverse transcription was carried out using random primer and Moloneymurine leukemia virus reverse transcriptase (Promega, Madison, USA). Real-time PCR was performed for the quantification of BDNF and TrkB with a quantitative thermal cycler (Mastercyclerep realplex, Eppendorf, Germany). Relative expression values were calculated as the ratio of target cDNA to β-actin; β-actin was used as the reference gene based on previous publications (Grunblatt et al., 2001; Domenger et al., 2012), which used experimental conditions similar to ours.
Immunohistochemistry
Ten minutes after TBS, hippocampal slices were fixed in 4% paraformaldehyde for 1 h and cryoprotected in 30% sucrose for 1 h and sectioned on a freezing microtome at 20 μm. Sections were blocked in 0.1 M PBS containing 10% goat serum and 0.4% Triton X-100, and then incubated with primary antibody mixture, including rabbit anti-p-TrkB (1:1000), mouse anti-PSD95 (1:500, MA1-045, Thermo), mouse anti-p-ERK (1:600) in 0.1 M PBS containing 5% goat serum and 0.4% Triton X-100 overnight at 4°C. Sections were washed 3 times (10 min each) in PBS and incubated in Alexa Fluor 594 goat anti-rabbit IgG (A-11037, Life Technologies) and Alexa Fluor 488 goat anti-mouse IgG (A-11001) for 2 h at room temperature. Mean fluorescence intensity in the area between stimulus and recording electrodes was analyzed. For puncta analysis, a Nikon C1 confocal laser-scanning microscope 100× objective was used. The localization of PSD95 with p-TrkB was analyzed by ImageJ software.
Novel object recognition
The novel object recognition task was conducted in an open field arena (40 cm × 50 cm open field surround by 50 cm high walls) with two different kinds of objects. Both objects were generally consistent in height and volume, but were different in shape and appearance. There was no object preference for all the animals before MPTP modelling and drug treatment. During habituation, animals were allowed to explore an empty arena for 5 min. Twenty-four h after habituation, animals were exposed to the familiar arena with two identical objects placed at an equal distance. The next day, mice were allowed to explore the open field for 5 min in the presence of one familiar object and a novel object to test long-term recognition memory. The discrimination index percentage was calculated as Tnew/(Tnew + Told) × 100%.
One-trial passive avoidance task
Briefly, the mice were put into a light compartment of a light–dark box. During habituation, mice were allowed to freely explore the box for 5 min with the sliding door between the light and dark compartments open. For conditioning, which was carried out 2 h after habituation, the mice were introduced into the light compartment, the sliding door was closed when both hindlimbs had entered into the dark box, and an electrical footshock was delivered via the floor grid in the dark compartment (0.2 mA, 3 s duration). The mice were left in the light–dark box for 5 min and then returned to their home cage. Tests were carried out 24 h after the conditioning by re-introducing the mice into the light compartment of the light–dark box. The latency time for mice to enter the dark compartment was measured (light–dark latency, with a 5 min cut-off). This procedure has been described previously (Jarvik and Kopp, 1967).
Data analyses
Data are presented as means ± SEM. All the statistical analyses were performed by one-way anova or two-way anova with GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, USA). Bonferroni correction for post hoc t-test was performed to compare the differences among groups when the first analysis revealed significant differences. P values less than 0.05 were considered statistically significant.
Materials
Recombinant BDNF (Millipore); NBQX (10 μM), AP5 (50 μM) (Tocris); calpain inhibitor III (10 μM, Calbiochem); Stock TrkB–Fc or control IgG–Fc (R & D Systems, Minneapolis, MN) was prepared in Tris-buffered saline containing 0.1% bovine serum albumin and diluted to working concentrations in artificial cerebrospinal fluid (aCSF); MPP+, MPTP (Sigma); memantine (Forest Laboratories, Jersey City, NJ; Purity: 99.3%); (RS)-3,5-DHPG (100 μM, Tocris); K252a (100 nM, Abcam); NMDA (25 μM, Tocris).
Results
Memantine prevents memory deficits in MPTP models
An acute PD model in mice was produced by i.p.injection of MPTP (Fig. 1A) (four times, 20 mg·kg−1 each time at 2 h interval) (Zhu et al., 2011). Seven days after MPTP treatment, TH-positive neurons in substantia nigra were reduced to 25% of the control level. High-frequency stimulation (three repeats of 100 Hz, 1 s, at 10 min intervals) induced LTP at Schaffer collateral-CA1 synapses was reduced in hippocampal slices from the mice with acute MPTP treatment. Here, we tested whether memantine prevented memory deficits in this MPTP-induced experimental PD model.
We then tested 24h memory 7 days after MPTP by applying the novel object recognition and one-trial passive avoidance tasks. As shown in Fig. 1C and D, treatment with MPTP inhibited the memory as shown by the decrease of discrimination index [interaction: F(4, 40) = 6.8, P < 0.05; row: F(1, 40) = 70.067, P < 0.05; column: F(4, 40) = 4.0, P < 0.05] and latency to dark box [interaction: F(4, 40) = 6.8, P < 0.05; row: F(1, 40) = 299.0, P < 0.05; column: F(4, 40) = 6.6, P < 0.05]. The decreased memory after MPTP was prevented by the high dose of memantine (10 mg·kg−1). Pretreatment with the low dose of memantine (1 mg·kg−1) did not significantly affect the loss of memory. Of note, high dose treatment (10 mg·kg−1) with memantine in control mice did not affect 24h memory. A subchronic PD model was used to confirm the protective effects of memantine on MPTP-induced memory loss (Fig. 1B). In this subchronic model, TH-positive neurons in the substantia nigra are reduced to 30% of the control level (Zhu et al., 2014) and we found 24h memory was decreased as shown by the novel object recognition and one-trial passive avoidance tests. Consistent with its effects in the acute PD model, memantine prevented memory loss in the subchronic model [interaction: F(2, 24) = 6.7, P < 0.05; row: F(1, 24) = 183, P < 0.05; column: F(2, 24) = 6.4, P < 0.05) (Fig. 1E and F). These data suggest that treatment with MPTP caused memory deficit, which was preserved by pretreatment with memantine.
Memantine reverses MPTP-mediated hippocampal synaptic plasticity in the acute MPTP model
A different LTP-inducing paradigm – θ burst stimulation (TBS, 10 bursts, 4 pulses) in hippocampal slices was used to confirm the effects of MPTP on LTP. In the slices obtained from control mice, TBS induced a consolidated LTP (Fig. 2A), whereas in slices from the MPTP mice, the amplitude of LTP was reduced. Pretreatment with a single dose of memantine (10 mg·kg−1) 1 h before the MPTP injections prevented the LTP decrease in this PD model. Summary data showed that compared with control, treatment with MPTP significantly decreased the LTP amplitude at 120 min after LTP induction (Fig. 2B), which was preserved by 10 mg·kg−1, but not by 1 mg·kg−1, memantine pretreatment [F(4, 19) = 18.20, P < 0.05]. In control mice, the high dose of memantine (10 mg·kg−1) did not increase the hippocampal LTP. Nevertheless, bath application of 1 μM memantine for 30 min, which rescued impaired LTP in an AD model (Martinez-Coria et al., 2010), did not rescue the decreased LTP in our MPTP model (Fig. 2C). To determine the stability of the LTP deficit in our acute MPTP model, we tested TBS-induced LTP in the slices obtained from mice 30 days after MPTP treatment. After memantine (10 mg·kg−1) pretreatment, the TBS-induced LTP decrease in MPTP model was also reversed (Fig. 2D). These data suggest that memantine prevents the hippocampal LTP deficit in the MPTP model.
Figure 2.
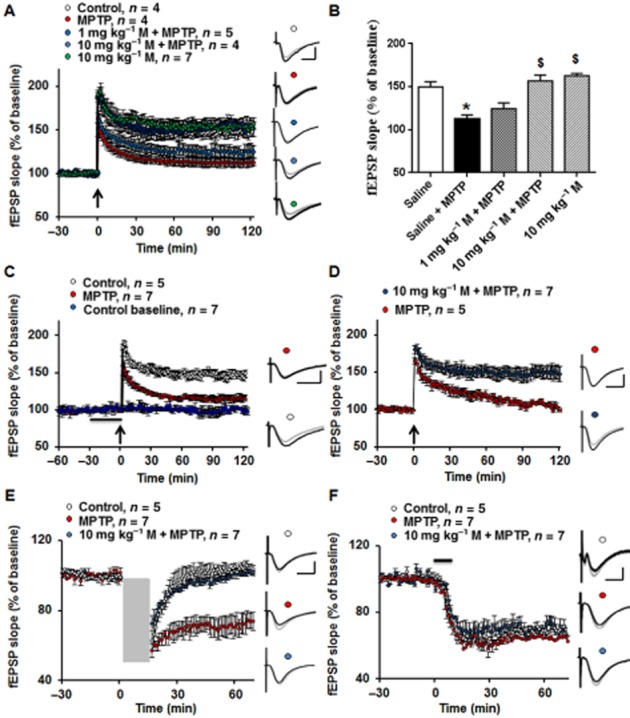
Memantine (M) pretreatment prevented the impairment of hippocampal synaptic plasticity in the MPTP model (A) Θ burst stimulation (TBS) induced LTP in acute hippocampal slices and this was decreased in the MPTP group, which was prevented by a high dose of memantine (10 mg·kg−1, i.p.). Black arrow indicates TBS. Inserts are analogue traces from baseline (grey) and 120 min after LTP induction (black). Scale bar: 1 mV/10 ms. (B)Summary data of the LTP values at 120 min after TBS. The data presented are means ± SEM (n = 5–10 slices from at least five animals). *P < 0.05 compared with control; $P < 0.05 compared with MPTP group; one-way anova followed by Bonferroni test. (C) Bath application of 1 μM memantine for 1 h did not ameliorate the decreased LTP in MPTP model. Inserts are analogue traces from baseline (grey) and 120 min after LTP induction (black). Scale bar: 1 mV per 10 ms. (D) Thirty days after MPTP treatment, TBS-induced LTP was still decreased compared with saline control group, which was rescued by memantine administration. Inserts are analogue traces from baseline (grey) and 120 min after LTP induction (black). Scale bar: 1 mV per 10 ms. (E) The low-frequency stimulation (LFS) induced LTD was enhanced in MPTP model compared with control. However, memantine administration reversed the abnormality of LTD in the MPTP model. Grey square indicates LFS. Inserts are analogue traces from baseline (grey) and 60 min after LTD induction (black). Scale bar: 1 mV per 10 ms. (F) The LTD induced by the mGluR5 agonist, DHPG, was not affected by MPTP treatment or memantine application. Black line indicates DHPG application. Inserts are analogue traces from baseline (grey) and 60 min after LTD induction (black). Scale bar: 1 mV per 10 ms.
Low-frequency stimulation and DHPG-induced LTD were proposed as models for memory consolidation or extinction (Collingridge et al., 2010). We further asked whether MPTP treatment or memantine application altered hippocampal LTD. Low-frequency stimulation induced LTD and this was enhanced in slices from mice 7 days after MPTP treatment, compared with control (Fig. 2E). However, pretreatment with 10 mg·kg−1 memantine fully prevented the enhanced LTD. The mGluR5 agonist, DHPG, also induced LTD (Fig. 2F) but neither MPTP nor memantine affected this chemically induced LTD. These data suggest that systemic application of memantine before MPTP treatment could protect against abnormalities of hippocampal synaptic plasticity involving activation of NMDA receptors.
Memantine reverses the decrease of hippocampal BDNF level in MPTP model
TBS-induced LTP in hippocampus requires activation of the BDNF/TrkB signalling pathway (Minichiello et al., 2002; Minichiello, 2009). As LTP was decreased after MPTP, we looked for changes in hippocampal BDNF content in the acute MPTP model, using elisa and Western blotting. The BDNF content of hippocampus homogenates assayed by ELISA, was significantly decreased in the MPTP group, compared with saline control, 7 days after MPTP treatment [F(4, 10) = 5.739, P < 0.05] (Fig. 3A). This decrease was prevented by the high dose of memantine. The low dose of memantine did not affect BDNF levels. Of note, the high dose of memantine did not affect BDNF expression in control mice. Similar results were obtained using Western blotting (Fig. 3B and C). There was a fall in hippocampal BDNF after MPTP, which was reversed by the high dose of memantine treatment [F(4, 10) = 3.232, P < 0.05]. The BDNF receptor, TrkB was also detected in the different experimental groups. Neither TrkB nor p-TrkB was affected by MPTP or memantine treatment (Fig. 3B and D). To ascertain the expression changes, BDNF and TrkB at mRNA level were also measured. Consistent with assays of the protein, MPTP treatement decreased expression of mRNA for BDNF and this decrease was reversed by memantine administration [F(4, 10) = 6.52, P < 0.05] (Fig. 3E). Levels of mRNA for TrkB were not affected by MPTP or memantine treatment (Fig. 3F). These results suggest that BDNF expression is reduced after treatment with MPTP and that memantine pretreatment prevented this decrease.
Figure 3.
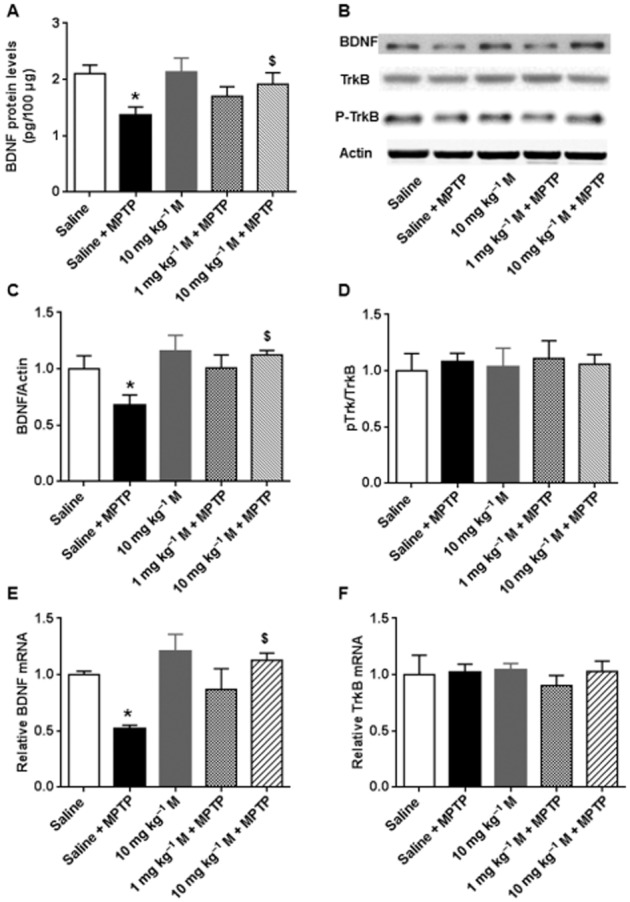
Memantine (M) prevented the MPTP-induced decrease of hippocampal BDNF. (A) The hippocampal BDNF contents in different groups were assayed by elisa. (B) Representative blots of BDNF, TrkB and p-Trk B in different groups. (C) Summary data of the BDNF expression in different groups. (D) Summary data of the p-Trk B/TrkB expression in different groups. Real-time PCR was used to detect BDNF (E) and TrkB (F). The data presented are means ± SEM (n = 3). *P < 0.05, compared with control; $P < 0.05, compared with MPTP group; one-way anova followed by Bonferroni test.
In vitro incubation with memantine reverses the effect of MPP+ on depolarization-mediated BDNF expression
In vitro experiments were carried out to assess the effects of MPP+, the active metabolite of MPTP on activity-dependent regulation of BDNF expression in the hippocampus. The drug treatment schemes are indicated in Fig. 4A. MPP+ or memantine incubation alone did not affect BDNF expression in control slices (Fig. 4B). KCl depolarization significantly increased BDNF expression in control slices and this stimulated increase was lower in slices incubated with MPP+. Pre-incubation with memantine (1 μM, 20 min before MPP+) blocked the MPP+-induced decrease of activity-dependent BDNF expression which was comparable with control levels. By contrast, post-treatment with memantine (immediately after MPP+ for 30 min) did not block the effect of MPP+ on KCl depolarization-induced BDNF expression. Importantly, memantine only inhibited the effect of MPP+ on hippocampal BDNF synthesis, but did not change BDNF synthesis in control slices [interaction: F(4, 40) = 10.2, P < 0.05; row: F(1, 40) = 158.0, P < 0.05; column: F(4, 40) = 8.8, P < 0.05].
Figure 4.
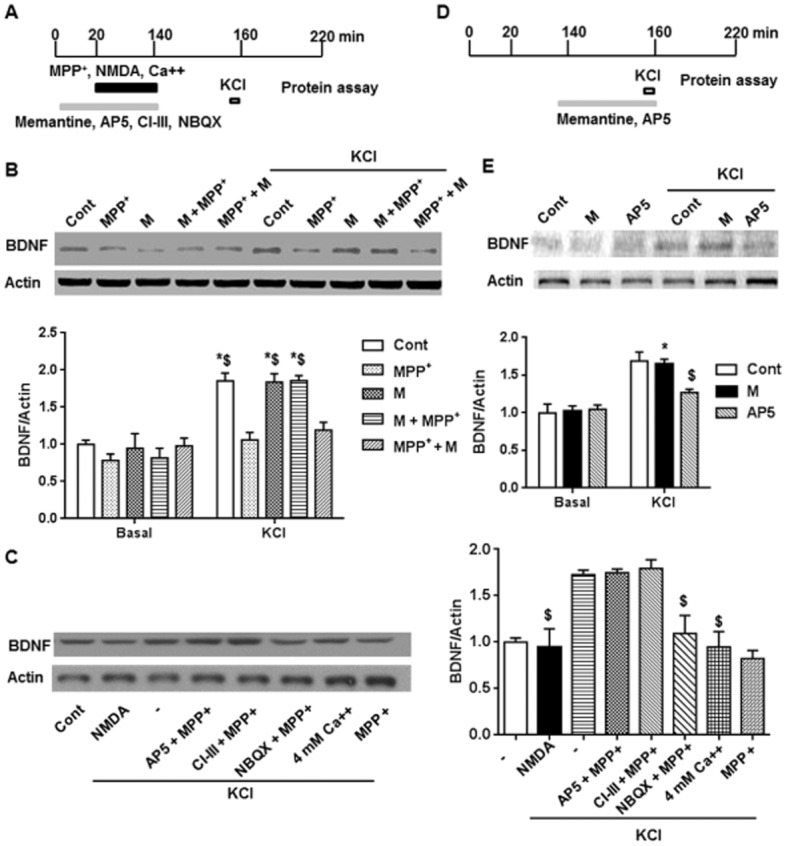
MPP+ incubation induced a decrease of BDNF expression after KCl depolarization and this was prevented by pre-application of memantine (M). (A) Scheme of the in vitro experimental procedure. The slices from control mice were incubated with memantine, NMDA receptor antagonist (AP5), calpain inhibitor (CI-III) or AMPA receptor inhibitor (NBQX) for 20 min, and then treated with MPP+, NMDA, or 4 mM Ca2+ for 2 h. After washing out the drug for about 20 min, the slices were depolarized by KCl for 3 min. One hour later, the slices were collected for protein assay. (B) Upper panel shows representative blots after different drug treatments. Lower panel shows summary data of BDNF expression after different drug treatments. (C) Left panel shows representative blots after different drug treatments. Right panel shows summary data of BDNF expression after different drug treatments. The data presented are means ± SEM (n = 5). $P < 0.05 compared with control group after KCl depolarization; two-way anova followed by Bonferroni test. (D) Direct effect of AP5 or memantine on KCl-induced BDNF expression was analysed as shown in the scheme. (E) Direct effect of AP5 or memantine on KCl-induced BDNF expression. Upper panel shows representative blots after different drug treatments. Lower panel shows summary data of BDNF expression after different drug treatments. The data presented are means ± SEM. Three slices from each animal and a total of three animals in each group were used. *P < 0.05 compared with corresponding basal level; $P < 0.05 compared with control group after KCl depolarization; two-way anova followed by Bonferroni test.
Next, we asked whether this decrease of depolarization-induced BDNF expression after MPP+ was a synaptic event. Incubation with the NMDA receptor antagonist (50 μM AP5), but not the AMPA receptor antagonist (10 μM NBQX) blocked the effect of MPP+ on depolarization-induced BDNF expression (Fig. 4C). In addition, a non-selective calpain inhibitor (calpain inhibitor III, 10 μM) also blocked the effect of MPP+ on depolarization-induced BDNF expression. Interestingly, incubation with artificial CSF containing a high concentration of calcium (4 mM) or 25 μM NMDA could mimic the effect of MPP+ on BDNF [F(6, 28) = 17.2, P < 0.05]. Furthermore, we tested the direct effects of memantine or AP5 on KCl-induced BDNF expression, i.e., in the absence of MPP+. Depolarization-induced BDNF expression was blocked by AP5, but not by memantine [interaction: F(2, 24) = 6.5, P < 0.05; row: F(1, 24) = 105.0, P < 0.05; column: F(2, 24) = 4.8, P < 0.05] (Fig. 4D and E). These data suggest that MPP+ probably over-activated a calcium signalling pathway in order to depress subsequent activity-dependent BDNF expression.
Memantine reverses inactivation of TrkB and ERK pathway during TBS in MPTP model
TrkB activation is an initial step for TBS-induced hippocampal LTP. As MPTP administration decreased BDNF level in the hippocampus, we looked for in situ p-TrkB expression after LTP induction using immunohistochemical methods (Fig. 5A). In slices from control mice, LTP induction increased p-TrkB expression significantly when compared with basal level (Fig. 5A and B) and this increase was blocked by treatment with MPTP. However, pretreatment with memantine before the MPTP treatment preserved the TrkB activation induced by LTP [interaction: F(2, 24) = 8.9, P < 0.05; row: F(1, 24) = 52.1, P < 0.05; column: F(2, 24) = 6.7, P < 0.05]. Using a high magnification to count p-TrkB-immunopositive synapses, similar results were obtained (Fig. 5C and D). Thus, these counts were increased after LTP induction, compared with baseline, in the control group but the increase was blocked by MPTP treatment and restored by memantine pre-treatment before MPTP [interaction: F(2, 24) = 8.98, P < 0.05; row: F(1, 24) = 221.0, P < 0.05; column: F(2, 24) = 6.4, P < 0.05].
Figure 5.
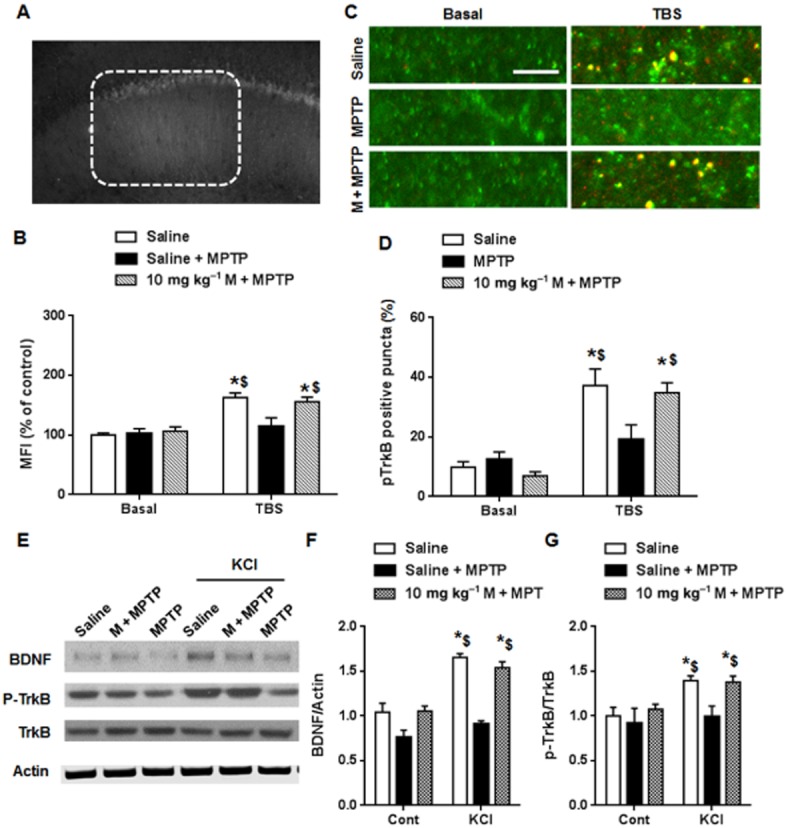
TrkB activation after LTP induction was abolished in the MPTP model, and was restored by memantine (M) pretreatment. (A) A representative image of p-TrkB after TBS. (B) Summary data of the mean fluorescence intensity (MFI) around stimulus electrode. The data presented are means ± SEM (n = 5). *P < 0.05 compared with corresponding basal level; $P < 0.05 compared with TBS in MPTP group; two-way anova followed by Bonferroni test. (C) Representative images of apical dendrite. Green: PSD95, Red: p-TrkB. (Scale bar: 5 μm). (D) p-TrkB positive synapses were significantly increased after LTP induction compared with baseline in control group. In MPTP group, p-TrkB positive synapses after TBS was not obviously increased. Memantine pretreatment reversed the defect caused by MPTP. (E) KCl depolarization was applied to activate BDNF expression and TrkB activation in hippocampal slices from different groups. Representative blots of BDNF and p-TrkB are shown. (F) Summary data of BDNF expression after different drug treatment. (G) Summary data of p-TrkB/TrkB after different drug treatment. The data presented are means ± SEM. In each group, three slices from each animal and a total of 5 animals were used. *P < 0.05 compared with corresponding basal level; $P < 0.05 compared with MPP+ group after KCl depolarization; two-way anova followed by Bonferroni test.
To confirm the effects on activity-dependent BDNF synthesis, hippocampal slices from control, and MPTP- and memantine+MPTP-treated groups were depolarized by KCl for 3 min. As shown in Fig. 5E and F, depolarization induced BDNF expression only in the control and memantine+MPTP-treated groups and not in the MPTP group [interaction: F(2, 24) = 8.2, P < 0.05; row: F(1, 24) = 149.0, P < 0.05; column: F(2, 24) = 6.8, P < 0.05] (). Consistent with these results, KCl-induced p-TrkB activation was not found in the MPTP group [interaction: F(2, 24) = 5.2, P < 0.05; row: F(1, 24) = 88.0, P < 0.05; column: F(2, 24) = 4.9, P < 0.05] (Fig. 5E and G) but was restored by memantine pretreatment. These data support the general conclusion that MPTP impaired activity-dependent BDNF expression in the hippocampus.
Activation of ERK is required for TBS-induced LTP and this event is thought to be downstream of the BDNF/TrkB signalling pathway (Wang et al., 2014). In the present experiments, we detected p-ERK expression using immunostaining, through analysing mean fluorescence intensity around the stimulus electrode in dendrites. The basal level of p-ERK was not altered in the different groups (Fig. 6A and B) and LTP induction activated ERK in slices from control mice, but not in MPTP-treated mice and pretreatment with memantine at the high dose restored ERK activation in MPTP-treated mice [interaction: F(2, 36) = 9.5, P < 0.05; row: F(1, 36) = 165.0, P < 0.05; column: F(2, 36) = 6.1, P < 0.05]. These data show that pretreatment with memantine protected the ERK pathway from the lack of activation induced by MPTP.
Figure 6.
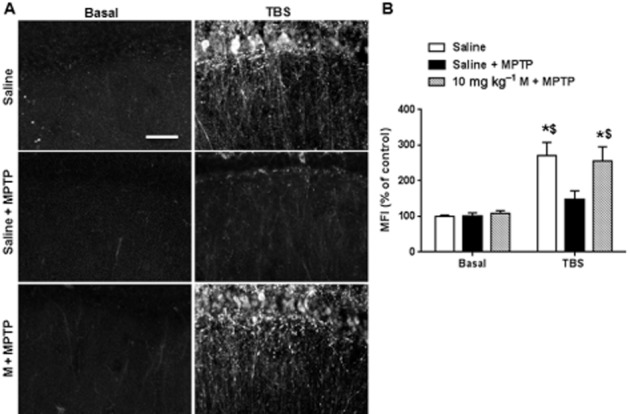
TBS-induced ERK activation during LTP was abolished in MPTP model, and was recovered by memantine (M) pretreatment. (A) The region of interest of p-ERK staining in different groups. (Scale bar: 20 μm) (B) Summary data of the mean fluorescence intensity (MFI) around stimulus electrode. The data presented are means ± SEM. In each group, three slices from each animal and total five animals were applied. *P < 0.05 compared with corresponding basal level; $P < 0.05 compared with TBS in MPTP group; two-way anova followed by Bonferroni test.
Memantine-rescued LTP was decreased by TrkB receptor blockers
We confirmed that pretreatment with memantine rescued LTP after exposure to MPTP model, through activating the BDNF-TrkB pathway, by using inhibitors of TrkB. The BDNF blocker (TrkB-FC) or control IgG–Fc was added in vitro to slices obtained from memantine+MPTP-treated mice. As shown in Fig. 7A, after incubation with TrkB-FC (2 μg·mL−1) for 1 h, the LTP amplitude was reduced, while the LTP was normal in IgG–Fc incubation group. Neither TrkB-FC nor corresponding IgG–Fc affected the baseline values (Fig. 7B). The non-specific TrkB inhibitor K252a also reduced LTP in memantine+MPTP-treated mice (Fig. 7C).
Figure 7.
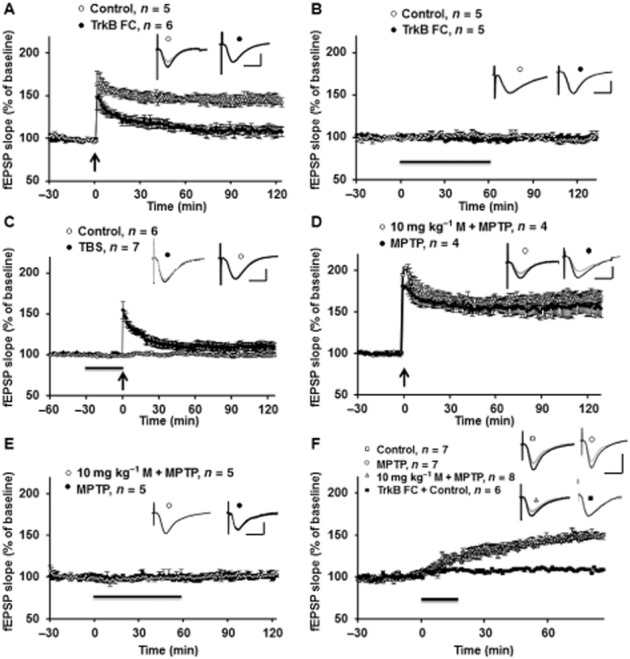
Memantine (M) prevented MPTP-induced decrease of hippocampal LTP through BDNF pathway. (A)The BDNF blocker, TrkB-FC (1 h incubation) blocked the memantine-induced reversal of LTP in the MPTP model. Inserts are analogue traces for baseline (grey) and 120 min after LTP induction (black). Scale bar: 1 mV per 10 ms. (B) TrkB-FC and Ig-FC did not affect basal responses. Black line indicates TrkB-FC and Ig-FC application. Inserts are analogue traces for baseline (grey) and 120 min after drug treatment (black). Scale bar: 1 mV per 10 ms. (C) The TrkB nonspecific inhibitor K252a also blocked the LTP in memantine + MPTP group, but did not affect baseline values. Black line indicates K252a application. Inserts are analogue traces from baseline (grey) and 90 min after LTP induction or drug treatment (black). (D) Incubation with a low concentration of BDNF (500 ng·mL−1) for 1 h rescues the impaired LTP in slices from the MPTP model. Inserts are analogue traces from baseline (grey) and 120 min after LTP induction (black). Scale bar: 1 mV per 10 ms. (E) Low concentration of BDNF did not affect the baseline values from the slices obtained from MPTP, and memantine treatment groups. Black line indicates BDNF application. Inserts are analogue traces from baseline (grey) and 120 min after drug treatment (black). Scale bar: 1 mV per 10 ms. (F) Incubation with a high concentration of BDNF (2 μg·mL−1) elicits comparable potentiation in control, MPTP, memantine and MPTP groups. Black line indicates BDNF application. Moreover, BDNF-induced chemical LTP was TrkB pathway dependent. Inserts are analogue traces from baseline (grey) and 120 min after drug treatment (black). Scale bar: 1 mV per 10 ms.
As memantine protected against the MPTP-induced decrease of BDNF and LTP, we tested the direct effect of exogenous BDNF on LTP in our MPTP model. As shown in Fig. 7D, incubation of hippocampal slices with BDNF (500 ng·mL−1; 30 min before TBS) rescued the decreased LTP in slices from the MPTP model. The final LTP level after BDNF treatment in the MPTP- and memantine-treated groups was comparable. It is notable that BDNF at 500 ng·mL−1 did not affect baseline synaptic transmission (Fig. 7E). A higher concentration of BDNF (2 μg·mL−1) was also tested. As shown in Fig. 7F, potentiation induced by exogenous BDNF, at this concentration, was similar in hippocampal slices obtained from control, MPTP- and memantine-treated groups. Moreover, BDNF-induced chemical LTP was TrkB pathway dependent (Fig. 7F, black square). These data demonstrated that the induction of LTP by BDNF was normal in the mice treated with MPTP.
Discussion
In this study, we present evidence for the inhibition of the BDNF-TrkB signalling pathway as a potential mechanism for MPTP-induced impairment of hippocampal synaptic plasticity and memory. Importantly, memantine effectively blocked the decrease in activity-dependent BDNF expression, induced by MPTP or MPP+ in acute hippocampal slices, thus restoring the BDNF-TrkB pathway, synaptic plasticity and memory abnormalities in the MPTP model.
MPTP impairs activity-dependent regulation of BDNF expression and hippocampal LTP
PD patients often suffer from depression, and patients suffering from depression exhibit specific reductions in hippocampal volume and hippocampal BDNF levels. In this study, we found that BDNF expression was reduced in the hippocampus after MPTP treatment, although Lesemann et al. (2012) have reported that MPTP injections for 3 consecutive days did not affect hippocampal BDNF expression in female transgenic mice. The reduced BDNF in the hippocampus contributes to impairment of synaptic plasticity and memory rather than cell death [the input–out response was not altered in the MPTP model (Zhu et al., 2011) ]. In the late stage of PD, BDNF abnormalities might lead to atrophy or cell death in the hippocampus, which has been widely reported in PD patients (Camicioli et al., 2003; Erickson et al., 2011). MPTP treatment could lead to decreased mRNA and protein levels of BDNF, not only in the substantia nigra as previously reported (Fumagalli et al., 2006), but also in the hippocampus, a region integrally involved in memory formation. The mechanisms underlying the reduced BDNF in the substantia nigra and hippocampus might be identical, but the functional phenotypes are different. The decrease of BDNF in dopaminergic neurons abolished neuroprotection, leading to cell death (Porritt et al., 2005). However, in hippocampal neurons, the lack of BDNF appeared only to decrease synaptic plasticity and memory storage. The important question of how endogenous BDNF cuodl be down-regulated in the brain of this PD model, is still not clear. One possible explanation is that the toxic oxidative product of MPTP, MPP+ accumulates in the mitochondria and causes oxidative stress, which is supposed to be a mechanism for the reduction of BDNF expression (Kapczinski et al., 2008). In addition, acute MPTP application is able to regulate hippocampal synaptic transmission and activity-dependent plasticity (Zhu et al., 2012).
Memantine reverses the abnormality of BDNF expression and plasticity in MPTP model
Memantine is efficient at ameliorating PDD or dementia with Lewy bodies (Aarsland et al., 2009). In this study, we found that the MPTP-induced decrease of BDNF expression was reversed by memantine pretreatment. In vitro experiments confirmed that MPP+, the active metabolite of MPTP abolished depolarization-induced BDNF synthesis, although MPP+ or memantine incubation alone did not affect BDNF expression. Pre-application but not post-application of memantine blocked the effect of MPP+ on BDNF expression. These data suggest that the hippocampus can be directly affected by MPTP or MPP+, even though the substantia nigra is particularly sensitive to MPTP. In addition, memantine only inhibited the effects of MPP+ on hippocampal BDNF and did not evoke BDNF expression in control slices. Besides memantine, AP5 and the calpain inhibitor also blocked the effects of MPP+ on depolarization-induced BDNF expression. Incubation with a high concentration of calcium or NMDA treatment for 2 h mimicked the effect of MPP+, which implied that an overactive calcium signalling pathway might underlie the MPP+ insult in the hippocampus. Although AP5 and the calpain inhibitor have similar protective effects to those of memantine against MPP+ insult, the time window for these drug applications is critical (as shown in Fig. 4A and D). AP5 not only blocked the effects of MPP+, but it also affected depolarization-induced BDNF expression.
The acute application of memantine can rescue Mg2+ reduction- or amyloid β oligomer-induced impairment of LTP (Frankiewicz and Parsons, 1999; Martinez-Coria et al., 2010) possibly through inhibiting the overactivated NMDA receptors, especially extrasynaptic NMDA receptors. In our study, bath application of memantine did not reverse the decrease of LTP in slices from the MPTP model, implying that activation of extrasynaptic NMDA receptors was not directly responsible for LTP impairment in the MPTP model, or at least extrasynaptic NMDA receptors were affected in the early phase after MPTP administration. Calcium entry through extrasynaptic NMDA receptors, triggered by bath glutamate exposure or hypoxic/ischemic conditions, activated a general and dominant cAMP response element-binding protein shut-off pathway and blocked induction of BDNF expression (Hardingham et al., 2002). Therefore, it is probable that MPTP/MPP+ activates extrasynaptic NMDA receptors and switches off BDNF expression system.
Using electrical stimulation under a protocol of TBS, mature BDNF can be synthesized rapidly and activates its specific receptor, TrkB receptor (Gartner and Staiger, 2002). In the present study, immunofluorescence and Western blot methods confirmed that p-TrkB levels were not affected 7 days after MPTP injection in CA1 region or whole hippocampal homogenate (Fig. 3D, 5B and E), although the BDNF level was reduced. This difference between expression of BDNF and p-TrkB is surprising. This discrepancy might be explained by the generation of a compensatory substance that can activate TrkB, 7 days after MPTP injection. A similar discrepancy was found in aged mice, where BDNF levels were reduced, but not those of p-TrkB (Martin et al., 2008). However, whether cellular cholesterol content is regulated to activate TrkB in the MPTP model still needs clarification. Although p-TrkB at the basal level was not affected following MPTP or memantine treatment, TBS-induced TrkB activation was inhibited in the MPTP model, and was rescued by memantine pretreatment. These results suggest that synaptic activation-related BDNF synthesis was impaired by MPTP treatment. To support that conclusion, KCl depolarization-induced BDNF expression was also impaired in the slices obtained from MPTP-treated mice.
The MAPK/ERK pathway is a critical signalling pathway for TBS-induced hippocampal LTP (Selcher et al., 2003; Wang et al., 2014). Activation of ERK can affect actin polymerization and gene expression, which are necessary for enlargement or formation of new spines (Huang et al., 2007). Here, we detected p-ERK under basal conditions and after LTP induction, with no difference in basal p-ERK levels with MPTP treatment or with memantine pretreatment. However, after LTP induction, p-ERK was decreased in the MPTP group. The decreased p-ERK was consistent with the decreased LTP in the MPTP group and memantine also rescued TBS-induced ERK activation in the MPTP model.
In this study, we have demonstrated that LTP impairment in the MPTP model was due to the reduction of hippocampal BDNF. Either protection of endogenous BDNF synthesis by memantine or application of exogenous BDNF restored the LTP deficit in the MPTP model. These data would suggest that compounds that can activate BDNF synthesis have the potential to protect memory in PD. In addition to the effects of memantine on LTP, MPTP-enhanced LTD was also reversed by memantine administration. Based on previous publications (Akaneya et al., 1996; Kumura et al., 2000; Aicardi et al., 2004), LTD did not benefit from endogenous BDNF release. Ikegaya et al. (2002) reported that the application of BDNF reduced LTD via the PLC-γ signalling pathway (Ikegaya et al., 2002). A recent study has shown that blocking of the BDNF-TrkB signalling pathway caused a switch from chemically induced LTP to LTD (Montalbano et al., 2013). However, in some other studies, pro-BDNF was suggested to promote NR2B-dependent LTD (Rosch et al., 2005; Woo et al., 2005), which was inconsistent with our recent study. In addition, a low level of BDNF and pro-BDNF in BDNF+/− mice affected the maintenance of LTD (Novkovic et al., 2014).
Memantine prevents against the impaired memory in the MPTP model
MPTP treatment caused motor dysfunction in mice, rats and primates (Fredriksson et al., 1997), but also affected memory (Moriguchi et al., 2012). However, the latencies for cognitive deficits are inconsistent in different MPTP models. In a subchronic MPTP model, memory was enhanced in the first 2 weeks, and then gradually decreased (Moriguchi et al., 2012). It is well known that MPTP has different effects in rats and mice (Tieu, 2011). Even in mice, the strains, age and sex should be taken into consideration regarding the behavioural results (Filipov et al., 2009). Here, using the novel object recognition and one-trial passive avoidance task, we obtained consistent results that in both acute and subchronic MPTP treatment regimens, the long-term memory was impaired. However, single or consecutive systemic administration with memantine could block the deleterious effects of MPTP.
Conclusion
In this study, we present a mechanism that potentially explains the memory deficits exhibited in PD. A deficiency in the activity-dependent regulation of BDNF expression in hippocampus might be the basis of memory loss and later dementia. Our data indicate that memantine would be a useful compound for preventing memory deficit in PD.
Acknowledgments
This research was supported by the Science Fund of Anhui University of Traditional Chinese Medicine (2011zr004A) and by the Natural Science Foundation of Anhui Province (1308085QH135).
Glossary
- DHPG
3,5-dihydroxyphenylglycine
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine
- PD
Parkinson's disease
- PDD
Parkinson's disease with dementia
- LTP
long-term potentiation
- LTD
long-term depression
Author contributions
G. Z. designed the study, did most of the experiments, analysed the data and wrote the paper; J. L., L. H., X. W. and X. H. did some of the experiments and revised the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Appendix S1 Supplemental experimental information.
References
- Aarsland D, Ballard C, Walker Z, Bostrom F, Alves G, Kossakowski K, et al. Memantine in patients with Parkinson's disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8:613–618. doi: 10.1016/S1474-4422(09)70146-2. [DOI] [PubMed] [Google Scholar]
- Aicardi G, Argilli E, Cappello S, Santi S, Riccio M, Thoenen H, et al. Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 2004;101:15788–15792. doi: 10.1073/pnas.0406960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaneya Y, Tsumoto T, Hatanaka H. Brain-derived neurotrophic factor blocks long-term depression in rat visual cortex. J Neurophysiol. 1996;76:4198–4201. doi: 10.1152/jn.1996.76.6.4198. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther. 2013;138:155–175. doi: 10.1016/j.pharmthera.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Bezard E, Imbert C, Gross CE. Experimental models of Parkinson's disease: from the static to the dynamic. Rev Neurosci. 1998;9:71–90. doi: 10.1515/revneuro.1998.9.2.71. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Castrioto A, Di Filippo M, Picconi B. New experimental and clinical links between the hippocampus and the dopaminergic system in Parkinson's disease. Lancet Neurol. 2013;12:811–821. doi: 10.1016/S1474-4422(13)70118-2. [DOI] [PubMed] [Google Scholar]
- Camicioli R, Moore MM, Kinney A, Corbridge E, Glassberg K, Kaye JA. Parkinson's disease is associated with hippocampal atrophy. Mov Disord. 2003;18:784–790. doi: 10.1002/mds.10444. [DOI] [PubMed] [Google Scholar]
- Chiasserini D, Tozzi A, de Iure A, Tantucci M, Susta F, Orvietani PL, et al. Mortalin inhibition in experimental Parkinson's disease. Mov Disord. 2011;26:1639–1647. doi: 10.1002/mds.23647. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Costa C, Sgobio C, Siliquini S, Tozzi A, Tantucci M, Ghiglieri V, et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson's disease. Brain. 2012;135(Pt 6):1884–1899. doi: 10.1093/brain/aws101. [DOI] [PubMed] [Google Scholar]
- Domenger D, Dea D, Theroux L, Moquin L, Gratton A, Poirier J. The MPTP neurotoxic lesion model of Parkinson's disease activates the apolipoprotein E cascade in the mouse brain. Exp Neurol. 2012;233:513–522. doi: 10.1016/j.expneurol.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Emre M, Tsolaki M, Bonuccelli U, Destee A, Tolosa E, Kutzelnigg A, et al. Memantine for patients with Parkinson's disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:969–977. doi: 10.1016/S1474-4422(10)70194-0. [DOI] [PubMed] [Google Scholar]
- Erickson JT, Brosenitsch TA, Katz DM. Brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor are required simultaneously for survival of dopaminergic primary sensory neurons in vivo. J Neurosci. 2001;21:581–589. doi: 10.1523/JNEUROSCI.21-02-00581.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. 2011;108:3017–3022. doi: 10.1073/pnas.1015950108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipov NM, Norwood AB, Sistrunk SC. Strain-specific sensitivity to MPTP of C57BL/6 and BALB/c mice is age dependent. Neuroreport. 2009;20:713–717. doi: 10.1097/WNR.0b013e32832aa95b. [DOI] [PubMed] [Google Scholar]
- Frankiewicz T, Parsons CG. Memantine restores long term potentiation impaired by tonic N-methyl-D-aspartate (NMDA) receptor activation following reduction of Mg2+ in hippocampal slices. Neuropharmacology. 1999;38:1253–1259. doi: 10.1016/s0028-3908(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Eriksson P, Archer T. MPTP-induced deficits in motor activity: neuroprotective effects of the spintrapping agent, alpha-phenyl-tert-butyl-nitrone (PBN) J Neural Transm. 1997;104:579–592. doi: 10.1007/BF01291877. [DOI] [PubMed] [Google Scholar]
- Frim DM, Uhler TA, Galpern WR, Beal MF, Breakefield XO, Isacson O. Implanted fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevent 1-methyl-4-phenylpyridinium toxicity to dopaminergic neurons in the rat. Proc Natl Acad Sci U S A. 1994;91:5104–5108. doi: 10.1073/pnas.91.11.5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Racagni G, Riva MA. Shedding light into the role of BDNF in the pharmacotherapy of Parkinson's disease. Pharmacogenomics J. 2006;6:95–104. doi: 10.1038/sj.tpj.6500360. [DOI] [PubMed] [Google Scholar]
- Gartner A, Staiger V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci U S A. 2002;99:6386–6391. doi: 10.1073/pnas.092129699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunblatt E, Mandel S, Maor G, Youdim MB. Gene expression analysis in N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice model of Parkinson's disease using cDNA microarray: effect of R-apomorphine. J Neurochem. 2001;78:1–12. doi: 10.1046/j.1471-4159.2001.00397.x. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- He YY, Zhang XY, Yung WH, Zhu JN, Wang JJ. Role of BDNF in central motor structures and motor diseases. Mol Neurobiol. 2013;48:783–793. doi: 10.1007/s12035-013-8466-y. [DOI] [PubMed] [Google Scholar]
- Huang F, Chotiner JK, Steward O. Actin polymerization and ERK phosphorylation are required for Arc/Arg3.1 mRNA targeting to activated synaptic sites on dendrites. J Neurosci. 2007;27:9054–9067. doi: 10.1523/JNEUROSCI.2410-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, et al. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. 1991;350:230–232. doi: 10.1038/350230a0. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Ishizaka Y, Matsuki N. BDNF attenuates hippocampal LTD via activation of phospholipase C: implications for a vertical shift in the frequency-response curve of synaptic plasticity. Eur J Neurosci. 2002;16:145–148. doi: 10.1046/j.1460-9568.2002.02051.x. [DOI] [PubMed] [Google Scholar]
- Jarvik ME, Kopp R. An improved one-trial passive avoidance learning situation. Psychol Rep. 1967;21:221–224. doi: 10.2466/pr0.1967.21.1.221. [DOI] [PubMed] [Google Scholar]
- Kapczinski F, Frey BN, Andreazza AC, Kauer-Sant'Anna M, Cunha AB, Post RM. Increased oxidative stress as a mechanism for decreased BDNF levels in acute manic episodes. Rev Bras Psiquiatr. 2008;30:243–245. doi: 10.1590/s1516-44462008000300011. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulisevsky J, Garcia-Sanchez C, Berthier ML, Barbanoj M, Pascual-Sedano B, Gironell A, et al. Chronic effects of dopaminergic replacement on cognitive function in Parkinson's disease: a two-year follow-up study of previously untreated patients. Mov Disord. 2000;15:613–626. doi: 10.1002/1531-8257(200007)15:4<613::aid-mds1005>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Kumura E, Kimura F, Taniguchi N, Tsumoto T. Brain-derived neurotrophic factor blocks long-term depression in solitary neurones cultured from rat visual cortex. J Physiol. 2000;524(Pt 1):195–204. doi: 10.1111/j.1469-7793.2000.t01-2-00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesemann A, Reinel C, Huhnchen P, Pilhatsch M, Hellweg R, Klaissle P, et al. MPTP-induced hippocampal effects on serotonin, dopamine, neurotrophins, adult neurogenesis and depression-like behavior are partially influenced by fluoxetine in adult mice. Brain Res. 2012;1457:51–69. doi: 10.1016/j.brainres.2012.03.046. [DOI] [PubMed] [Google Scholar]
- Levivier M, Przedborski S, Bencsics C, Kang UJ. Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson's disease. J Neurosci. 1995;15:7810–7820. doi: 10.1523/JNEUROSCI.15-12-07810.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald PA, MacDonald AA, Seergobin KN, Tamjeedi R, Ganjavi H, Provost JS, et al. The effect of dopamine therapy on ventral and dorsal striatum-mediated cognition in Parkinson's disease: support from functional MRI. Brain. 2011;134(Pt 5):1447–1463. doi: 10.1093/brain/awr075. [DOI] [PubMed] [Google Scholar]
- Maharana C, Sharma KP, Sharma SK. Feedback mechanism in depolarization-induced sustained activation of extracellular signal-regulated kinase in the hippocampus. Sci Rep. 2013;3:1103. doi: 10.1038/srep01103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelman SD, Grigorenko EL. BDNF Val66Met and cognition: all, none, or some? A meta-analysis of the genetic association. Genes Brain Behav. 2012;11:127–136. doi: 10.1111/j.1601-183X.2011.00738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MG, Perga S, Trovo L, Rasola A, Holm P, Rantamaki T, et al. Cholesterol loss enhances TrkB signaling in hippocampal neurons aging in vitro. Mol Biol Cell. 2008;19:2101–2112. doi: 10.1091/mbc.E07-09-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez TN, Greenamyre JT. Toxin models of mitochondrial dysfunction in Parkinson's disease. Antioxid Redox Signal. 2012;16:920–934. doi: 10.1089/ars.2011.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, et al. Memantine improves cognition and reduces Alzheimer's-like neuropathology in transgenic mice. Am J Pathol. 2010;176:870–880. doi: 10.2353/ajpath.2010.090452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miah IP, Olde Dubbelink KT, Stoffers D, Deijen JB, Berendse HW. Early-stage cognitive impairment in Parkinson's disease and the influence of dopamine replacement therapy. Eur J Neurol. 2012;19:510–516. doi: 10.1111/j.1468-1331.2011.03578.x. [DOI] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Montalbano A, Baj G, Papadia D, Tongiorgi E, Sciancalepore M. Blockade of BDNF signaling turns chemically-induced long-term potentiation into long-term depression. Hippocampus. 2013;23:879–889. doi: 10.1002/hipo.22144. [DOI] [PubMed] [Google Scholar]
- Moriguchi S, Yabuki Y, Fukunaga K. Reduced calcium/calmodulin-dependent protein kinase II activity in the hippocampus is associated with impaired cognitive function in MPTP-treated mice. J Neurochem. 2012;120:541–551. doi: 10.1111/j.1471-4159.2011.07608.x. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- Novkovic T, Mittmann T, Manahan-Vaughan D. BDNF contributes to the facilitation of hippocampal synaptic plasticity and learning enabled by environmental enrichment. Hippocampus. 2014;25:1–15. doi: 10.1002/hipo.22342. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poletti M, Bonuccelli U. Acute and chronic cognitive effects of levodopa and dopamine agonists on patients with Parkinson's disease: a review. Ther Adv Psychopharmacol. 2013;3:101–113. doi: 10.1177/2045125312470130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porritt MJ, Batchelor PE, Howells DW. Inhibiting BDNF expression by antisense oligonucleotide infusion causes loss of nigral dopaminergic neurons. Exp Neurol. 2005;192:226–234. doi: 10.1016/j.expneurol.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Rosch H, Schweigreiter R, Bonhoeffer T, Barde YA, Korte M. The neurotrophin receptor p75NTR modulates long-term depression and regulates the expression of AMPA receptor subunits in the hippocampus. Proc Natl Acad Sci U S A. 2005;102:7362–7367. doi: 10.1073/pnas.0502460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD. A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem. 2003;10:26–39. doi: 10.1101/lm.51103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohamy D, Myers CE, Hopkins RO, Sage J, Gluck MA. Distinct hippocampal and basal ganglia contributions to probabilistic learning and reversal. J Cogn Neurosci. 2009;21:1821–1833. doi: 10.1162/jocn.2009.21138. [DOI] [PubMed] [Google Scholar]
- Tieu K. A guide to neurotoxic animal models of Parkinson's disease. Cold Spring Harb Perspect Med. 2011;1:a009316. doi: 10.1101/cshperspect.a009316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu G, Briz V, Hsu YT, Bi X, Baudry M. A molecular brake controls the magnitude of long-term potentiation. Nat Commun. 2014;5:3051. doi: 10.1038/ncomms4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, et al. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci. 2005;8:1069–1077. doi: 10.1038/nn1510. [DOI] [PubMed] [Google Scholar]
- Zhu G, Chen Y, Huang Y, Li Q, Behnisch T. MPTP-meditated hippocampal dopamine deprivation modulates synaptic transmission and activity-dependent synaptic plasticity. Toxicol Appl Pharmacol. 2011;254:332–341. doi: 10.1016/j.taap.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Zhu G, Huang Y, Chen Y, Zhuang Y, Behnisch T. MPTP modulates hippocampal synaptic transmission and activity-dependent synaptic plasticity via dopamine receptors. J Neurochem. 2012;122:582–593. doi: 10.1111/j.1471-4159.2012.07815.x. [DOI] [PubMed] [Google Scholar]
- Zhu G, Wang X, Wu S, Li X, Li Q. Neuroprotective effects of puerarin on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induced Parkinson's disease model in mice. Phytother Res. 2014;28:179–186. doi: 10.1002/ptr.4975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplemental experimental information.