

Abstract
Background and Purpose
The Ca2+ sensitizer pimobendan is a unique inotropic agent that improves cardiac contractility with less of an increase in oxygen consumption and potentially fewer adverse effects on myocardial remodelling and arrhythmia, compared with traditional inotropes. However, clinical trials report contradictory effects of pimobendan in patients with heart failure (HF). We provide mechanistic experimental evidence of the efficacy of pimobendan using a novel mouse model of progressive HF.
Experimental Approach
A knock-in mouse model of human genetic dilated cardiomyopathy, which shows a clear transition from compensatory to end-stage HF at a fixed time during growth, was used to evaluate the efficacy of pimobendan and explore the underlying molecular and cellular mechanisms.
Key Results
Pimobendan prevented myocardial remodelling in compensated HF and significantly extended life span in both compensated and end-stage HF, but dose-dependently increased sudden death in end-stage HF. In cardiomyocytes isolated from end-stage HF mice, pimobendan induced triggered activity probably because of early or delayed afterdepolarizations. The L-type Ca2+ channel blocker verapamil decreased the incidence of triggered activity, suggesting that this was from over-elevated cytoplasmic Ca2+ through increased Ca2+ entry by PDE3 inhibition under diminished sarcoplasmic reticulum Ca2+ reuptake and increased Ca2+ leakage from sarcoplasmic reticulum in end-stage HF.
Conclusions and Implications
Pimobendan was beneficial regardless of HF stage, but increased sudden cardiac death in end-stage HF with extensive remodelling of Ca2+ handling. Reduction of cytoplasmic Ca2+ elevated by PDE3 inhibition might decrease this risk of sudden cardiac death.
Tables of Links
| LIGANDS |
|---|
| Candesartan |
| Cilostazol |
| Metoprolol |
| Verapamil |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guideto PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c,,).
Introduction
Symptomatic heart failure (HF) is an end-stage condition of cardiovascular diseases, occurring after the progressive breakdown of the homeostatic mechanisms of the body. Complete recovery remains an elusive goal even after recent advances in medical technology. One major cause of HF is dilated cardiomyopathy (DCM), which develops as a compensatory response to a variety of insults to the myocardium. These insults include coronary artery disease, alcoholism, thyroid disease, diabetes mellitus, viral infections and genetic mutations.
Mutations in more than 30 genes account for approximately 25–30% of all DCM cases (Burkett and Hershberger, 2005; Morimoto, 2008) and cause congenital myocardial systolic dysfunction, often leading to premature death from HF or sudden cardiac death. We previously created a knock-in mouse model of genetic DCM to explore the molecular mechanisms of its pathogenesis and potential therapies (Du et al., 2007; Zhan et al., 2009; Wang et al., 2010; Li et al., 2012). In this model, using a gene targeting technique (Du et al., 2007), a causative deletion mutation (ΔK210) in cardiac troponin T (cTnT) found in humans has been incorporated into the mouse endogenous cTnT gene. This mutation decreases cardiac myofilament sensitivity to cytoplasmic Ca2+ by causing a malfunction in the troponin–tropomyosin system, which regulates cardiomyocyte contraction/relaxation in a Ca2+-dependent manner (Morimoto et al., 2002). These knock-in mice develop DCM as a compensatory response to impaired myocardial contractility caused by decreased myofilament Ca2+ sensitivity and die in distinct ways depending on the genetic background before reaching the age of 2 months (Du et al., 2007; Li et al., 2012). C57Bl6/J background DCM mice develop a stable compensated HF, but suffer mostly from sudden cardiac death because of lethal arrhythmia before reaching 2 months. This resembles a human family with a high incidence of sudden cardiac death before 30 years of age (Kamisago et al., 2000). Conversely, BALB/c background DCM mice die mostly from congestive HF before reaching 2 months, resembling a human family with a high incidence of congestive HF death before 20 years of age (Kamisago et al., 2000).
Current standard therapy for HF is a combination of cardioprotective agents, including β-adrenoceptor antagonists, vasodilators (ACE inhibitors or angiotensin AT1 receptor blockers) and diuretics. These can retard the progression of myocardial remodelling in compensatory HF by reducing oxygen consumption in the heart (CIBIS-II, 1999; MERIT-HF, 1999; Packer et al., 2001; McAlister et al., 2009). However, patients with end-stage HF, who are refractory to standard therapy, require specialized interventions, including experimental surgery or drugs, heart transplantation, and chronic positive inotropes.
Positive inotropic agents, such as PDE inhibitors and β-adrenoceptor agonists, improve cardiac contractility through increasing cellular cAMP. This is followed by an elevation of systolic intracellular Ca2+ via phosphorylation of Ca2+-handling proteins with PKA. These drugs are considered beneficial for the acute treatment of patients with severe myocardial systolic dysfunction. However, long-term treatment with these drugs exacerbates HF by facilitating maladaptive myocardial remodelling because of increased oxygen consumption in Ca2+ handling and increases the risk of arrhythmic death from Ca2+ overload in cardiomyocytes (Amsallem et al., 2005; Endoh, 2013).
Pimobendan is a unique positive inotrope that directly increases cardiac myofilament sensitivity to cytoplasmic Ca2+ by making the troponin–tropomyosin system respond to lower Ca2+ by enhancing the Ca2+ binding affinity of cardiac troponin C (cTnC). This makes it possible to improve cardiac contractility with less of an increase in oxygen consumption compared with traditional positive inotropes although it also increases cellular cAMP through PDE3 inhibition (Fujino et al., 1988; Scheld et al., 1989; Solaro et al., 1989; Ovaska and Taskinen, 1991; Endoh, 2013). The PICO trial (Pimobendan in Congestive Heart Failure trial) in Europe reported that pimobendan improved exercise duration (bicycle ergometry), but showed a trend towards increased mortality, although not of statistical significance, largely because of an increased incidence of sudden cardiac death in HF patients (Lubsen et al., 1996). However, the EPOCH study (Effects of Pimobendan on Chronic Heart Failure study) in Japan reported that long-term treatment with pimobendan resulted in a significantly decreased incidence of adverse cardiac events and improved physical activity based on the Specific Activity Scale questionnaire in HF patients (EPOCH Study, 2002). As a result, pimobendan is currently approved only in Japan for patients with HF refractory to standard therapy (Murai et al., 2013). These clinical studies examined the effects of pimobendan on patients with mild to moderate HF (New York Heart Association classes II–III), and no evidence has been provided for the efficacy of pimobendan in severe end-stage HF.
We previously reported on studies using C57Bl/6 background DCM mice that cardioprotective drugs widely used for HF as standard therapy (i.e., the β-adrenoceptor antagonist metoprolol and the AT1 receptor antagonist candesartan) have beneficial effects on compensated HF (Zhan et al., 2009; Odagiri et al., 2014). The beneficial effect of pimobendan has also been reported in the compensated HF model (Du et al., 2007). As there is no reported systematic study investigating the efficacy of pimobendan for the treatment of decompensated end-stage HF despite its promising nature, the present study examined the therapeutic effects of pimobendan on the BALB/cJ background DCM mouse model of refractory end-stage HF.
Methods
Animal models
All animal care and experimental procedures complied with the Guidelines for Animal Experiments of the Faculty of Medicine, Kyushu University, and the Law (No. 105) and Notification (No. 6) of the Japanese Government and were approved by the Committee of Ethics on Animal Experiments of the Faculty of Medicine, Kyushu University. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 137 animals were used in the experiments described here.
Knock-in mice on the genetic background of BALB/cJ, in which three base-pairs coding for K210 in cTnT was deleted from the endogenous Tnnt2 gene, were created by backcrossing a knock-in mouse with the same mutation on the genetic background of C57Bl/6J (Du et al., 2007) to BALB/cJ line for at least 12 generations. Homozygous mutant mice and wild-type (WT) mice were obtained by crossing heterozygous mutant mice. Homozygous mutant mice and WT mice of nearly equally mixed gender from the same colony were used as DCM and healthy models respectively. The mice were housed in a temperature- and humidity-controlled (22.9–23.2°C and 30–50%, respectively) room on a 12 h light/dark cycle. They were provided with a normal chow diet and water ad libitum.
Measurements of body temperature and locomotor activity
Body (rectal) temperature was measured once daily at 09:00 h under conscious conditions using TH-5 Thermalert Monitoring Thermometer (Physitemp, Clifton, NJ, USA). Locomotor activity was measured using ACTIMO system (Shinfactory, Fukuoka, Japan).
Echocardiography
Transthoracic echocardiography (M-mode) was measured under conscious conditions using a 14 MHz linear array probe with a diagnostic ultrasound system (Nemio SSA-550A, Toshiba Medical Systems, Ohtawara, Tochigi, Japan).
Histochemistry
The hearts excised from mice after killing by cervical dislocation were fixed in a 10% formalin neutral buffered solution as described previously (Du et al., 2007) and cut transversely at the mid-ventricular level, embedded in paraffin, sectioned at 5 μm and stained with Masson trichrome. The extent of the fibrosis in left ventricular (LV) myocardium was quantified using the ImageJ (National Institutes of Health, Bethesda, MD, USA).
Myosin isoform contents
Myosin heavy chain (MyHC) isoforms in the LV myocardium were separated on SDS–PAGE and relative β-isoform expression (percentage of total MyHC) was determined as described previously (Du et al., 2007).
Western blot analysis
Ventricles were dissected from the heart excised from mice after being killed by cervical dislocation and homogenized in Laemmli's sample buffer as described previously (Du et al., 2007). To determine the phosphorylation levels of cardiac troponin I (cTnI), isolated cardiomyocytes in a thermostatically controlled chamber containing continuously oxygenated Krebs–Henseleit solution (in mM: 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 2.5 CaCl2, 0.5 EDTA-Na2, 10 HEPES, 11 D-glucose) with 0.1 μM noradrenaline and 0, 3 or 10 μM pimobendan were electrically stimulated at 1 Hz for 10 min at 37°C. Cardiomyocytes were then added into Laemmli's sample buffer. LV homogenate and cardiomyocyte samples were subjected to Western blot analysis as described previously (Nakaura et al., 1999), using an anti-sarcoplasmic/endoplasmic reticulum Ca2+ transport ATPase 2a (SERCA2a) monoclonal antibody (ab2861) (Abcam, Tokyo, Japan); an anti-Na+/Ca2+ exchanger 1 (NCX1) monoclonal antibody (ab6495) (Abcam); an anti-calsequestrin (CSQ) polyclonal antibody (ab3516) (Abcam,); an anti-GAPDH monoclonal antibody (ab9484) (Abcam); an anti-phospholamban (PLB) monoclonal antibody (A010-14) (Badrilla, Leeds, UK); an anti-PLB phospho Ser16 polyclonal antibody (A010-12) (Badrilla); an anti-PLB phospho Thr17 polyclonal antibody (A010-13) (Badrilla); an anti-ryanodine receptor 2 (RyR2) polyclonal antibody (Chugun et al., 2003); an anti-RyR2 phospho Ser2808 polyclonal antibody (A010-30) (Badrilla); an anti-RyR2 phospho Ser2814 polyclonal antibody (A010-31) (Badrilla); an anti-cTnI monoclonal antibody (8E10) (HyTest, Turku, Finland); and anti-cTnI phospho Ser22/23 monoclonal antibody (ab8291) (Abcam).
Drug administration
Pimobendan and UD-CG 212 CL were supplied by Nippon Boehringer Ingelheim Co., Ltd. (Tokyo, Japan). Pimobendan suspended in 0.25% methylcellulose solution was administered p.o. once daily at 09:00 h in two doses, 10 (low dose) and 100 (high dose) mg·kg−1, to DCM mice.
Blood plasma concentrations of pimobendan and its metabolite UD-CG 212 CL
Blood plasma concentrations of pimobendan and UD-CG 212 CL were quantified using an HPLC (Alliance 2695 Separations Module, Waters, Milford, MA, USA). Pimobendan was administered p.o. 10 or 100 mg·kg−1 to 8-week-old BALB/cJ WT male fasted mice (three mice per group), and about 40 μL of blood was taken from the caudal vein under anaesthesia with 1.5% isoflurane before and after pimobendan administration. Plasma was separated by centrifugation at 2300× g for 15 min at room temperature and stored at −20°C for 30 min. Three volumes of cold ethanol (−20°C) and 1/100 volume of 3 M NaOAc (pH 4.9) were added to plasma and stored at −20°C for 3 h. The samples were then centrifuged at 16 500× g for 30 min at −10°C, and the supernatant was weighed and evaporated to dryness for 2 h. Evaporated samples were reconstituted with 40 μL of methanol and 40 μL of a mobile phase of acetonitrile/methanol/ammonium acetate (0.6% w/v) (1:3:4 v/v/v). Pimobendan and UD-CG 212 CL were separated on a reverse-phase column (Inertsil ODS-2, 4.6 × 150 mm, GL Sscience, Tokyo, Japan) at a flow rate of 1.3 mL·min−1 and monitored at a wavelength of 330 nm.
Isolation of cardiomyocytes and simultaneous measurements of sarcomere shortening and Ca2+ transients (CaT)
Cardiomyocytes were isolated from LVs by collagenase treatment as described previously (Du et al., 2007), with 5 μM ACh being added to all solutions to reduce the cell excitability during preparation. Isolated cardiomyocytes suspended in Krebs–Henseleit solution containing 5 μM ACh were plated onto laminin-coated glass coverslips and incubated for 15 min at 37°C with 5% CO2. The cardiomyocyte-attached coverslips were then kept in oxygenated Krebs–Henseleit solution containing 0.1 μM noradrenaline at 37°C until measurements.
The cardiomyocyte-attached coverslip was mounted on a thermostatically controlled chamber with a capacity of 1 mL, which was located on the stage of an inverted microscope and perfused with oxygenated Krebs–Henseleit solution containing 0.1 μM noradrenaline at 37°C at a flow rate of 2.8 mL·min−1. Cardiomyocytes were loaded with fura-2 AM (1 μM) for 10 min at 37°C in oxygenated Krebs–Henseleit solution containing 0.1 μM noradrenaline. Contraction was evoked by bipolar electrical field stimulation of 5 ms duration with a voltage about 30% above the threshold at 1 Hz via platinum wire electrodes. CaT and sarcomere shortening in a single cardiomyocyte were simultaneously monitored through a 20× objective lens using a fluorescence and contractility recording system (MyoCam and Photo-Multiplier System with Galvo-Driven HyperSwitch Dual Excitation Light Source, IonOptix, Milton, MA, USA). Effects of pimobendan, cilostazol (Sigma-Aldrich, St. Louis, MO, USA) and verapamil (Research Biochemical International, Natick, MA, USA) on isolated cardiomyocytes were determined by dissolving these agents into the perfusate with final concentration of DMSO being 0.01 and 0.1% in the cases of pimobendan and cilostazol respectively. The peak amplitude of CaT induced by rapid application of 10 mM caffeine after steady-state electrical stimulation at 1 Hz was used as an index of sarcoplasmic reticulum (SR) Ca2+ content. Note that noradrenaline was added to the Krebs–Henseleit solution during these experiments because the intracellular cAMP level should be increased to detect the effect of the inhibition of PDE3 by pimobendan (Bohm et al., 1991).
Data analysis
Data are shown as the mean ± SEM. Differences between two mean values were analysed with unpaired or paired t-test. If the data did not conform to a normal distribution, mean values were analysed with Welch's correction. Mean values for more than three groups were compared by one-way anova, followed by a post hoc Dunnett's or Tukey's multiple comparison test. Survival data utilized the standard Kaplan–Meier analysis. P < 0.05 was considered to be statistically significant.
Results
Baseline characteristics of mice with compensated or end-stage HF
BALB/c background knock-in mice with the ΔK210 mutation in cTnT develop symptomatic HF gradually after reaching the age of 4 weeks, as shown by a progressive decrease in locomotor activity as well as body temperature, an index of HF severity (Ahmed et al., 2010), and usually die before reaching 8 weeks (Supporting Information Figure S1). Therefore, DCM mice in the present study were allocated to two groups: (i) the compensated HF group of 4-week-old mice, with an ejection fraction (EF) > 45% and body temperature >36°C; and (ii) the end-stage HF group of 6- to 9-week-old mice, with an EF <30% and body temperature <35°C.
Body weight significantly decreased in the end-stage HF group, but not in the compensated HF group, compared with age-matched, healthy WT controls (Table 1). LV internal dimension in end-diastole were markedly enlarged in both groups, with significant increases in heart weight normalized to tibia length by 97% in the compensated HF group and 125% in the end-stage HF group, compared with age-matched WT controls (Table 1, Figure 1A). Lung weight normalized to tibia length, indicative of congestive HF, did not increase in the compensated HF group, but significantly increased (by 102%) in the end-stage HF group (Table 1). Myocardial fibrosis was found in both groups with a greater extent in the end-stage HF group (Figure 1A and B). The expression of β-cardiac MyHC (β-MyHC), known to be up-regulated in HF (Miyata et al., 2000), also increased in both groups with a greater extent in the end-stage HF group (Figure 1C).
Table 1.
Echocardiography, and heart and lung weight in DCM mice
| WT | Compensated HF | WT | End-stage HF | |
|---|---|---|---|---|
| Age (days old) | 28 | 28 | 56 | 55 ± 4 |
| Echocardiography | ||||
| Number of mice | 5 | 5 | 5 | 5 |
| HR, bpm | 635 ± 32 | 579 ± 39 | 677 ± 14 | 399 ± 75* |
| IVST, mm | 0.92 ± 0.02 | 0.58 ± 0.02** | 0.96 ± 0.12 | 0.42 ± 0.04**†† |
| LVESD, mm | 1.14 ± 0.07 | 3.94 ± 0.34** | 1.06 ± 0.10 | 6.14 ± 0.23**†† |
| LVEDD, mm | 2.10 ± 0.19 | 5.10 ± 0.39** | 2.36 ± 0.11 | 6.74 ± 0.27**†† |
| LVPWT, mm | 0.70 ± 0.10 | 0.60 ± 0.03 | 0.70 ± 0.03 | 0.44 ± 0.02**†† |
| FS, % | 68 ± 2 | 22 ± 1** | 70 ± 1 | 9 ± 1**†† |
| EF, % | 96 ± 1 | 51 ± 2** | 97 ± 0.4 | 23 ± 1**†† |
| Heart and lung weight | ||||
| BT, °C | 37.2 ± 0.15 | 36.6 ± 0.18* | 37.1 ± 0.30 | 34.4 ± 0.2**†† |
| BW, g | 17.7 ± 1.6 | 12.9 ± 1.4 | 23.3 ± 1.2 | 14.8 ± 1.2** |
| TL, mm | 17.0 ± 0.5 | 16.7 ± 0.6 | 18.6 ± 0.4 | 18.3 ± 0.7 |
| HW, mg | 114.5 ± 4.0 | 218.9 ± 23.5* | 134.0 ± 5.0 | 308.2 ± 35.7** |
| HW/TL, mg·mm−1 | 6.2 ± 0.1 | 12.2 ± 1.7* | 6.3 ± 0.2 | 14.2 ± 1.5** |
| LW/TL, mg·mm−1 | 6.9 ± 0.4 | 8.2 ± 0.7 | 6.3 ± 0.2 | 12.7 ± 0.7**†† |
bpm, beats per min; BT, body temperature; BW, body weight; EF, ejection fraction; FS, fractional shortening; HR, heart rate; HW, heart weight; IVST, intraventricular septal wall thickness; LVEDD, LV end-diastolic dimension; LVESD, LV end-systolic dimension; LVPWT, LV posterior wall thickness; LW, lung weight; TL, tibia length. *P < 0.05. **P < 0.01 versus age-matched WT (unpaired t-test). †P < 0.05. †† P < 0.01 versus compensated HF (unpaired t-test).
Figure 1.
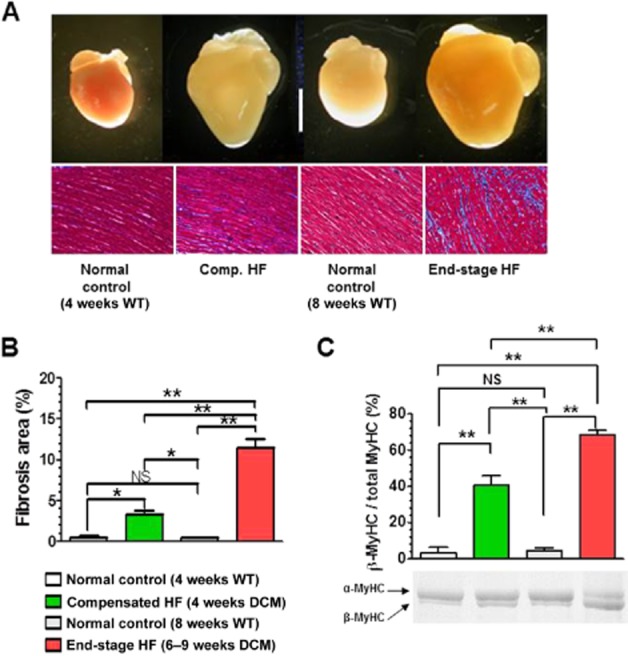
Morphological and histological changes in the heart of DCM mice during progression of HF. (A) Gross morphology of the heart (top images) (scale bar, 5 mm) and histology of the LV (bottom images). Connective tissues were stained blue with Masson trichrome. (B) Quantitative analysis of the fibrosis area in the LV (n = 3 mice per group). (C) Protein expression level of MyHC isoforms in the LV (n = 5 mice per group). *P < 0.05; **P < 0.01; significantly different as indicated; one-way anova and post hoc Tukey's multiple comparison test.
Figure 2 shows expression levels of Ca2+ handling proteins in cardiomyocytes. SERCA2a, which plays a critical role in cardiomyocyte relaxation through reuptake of cytoplasmic Ca2+ into SR, was markedly down-regulated in the end-stage HF group; with no significant changes in the compensated HF group (Figure 2A and B). In contrast, NCX1, which also plays an important role in cardiomyocyte relaxation through extrusion of cytoplasmic Ca2+ into the extracellular space, showed a 20-fold up-regulation in the end-stage HF group, with no significant changes observed in the compensated HF group (Figure 2A and C). CSQ, which plays an important role in Ca2+ storage and release as a Ca2+ sensor in SR (Terentyev et al., 2003; Kubalova et al., 2004), was up-regulated over twofold in the end-stage HF group owing to a marked increase in CSQ1 isoform; with no significant changes observed in the compensated HF group (Figure 2A and D). PLB, which inhibits SERCA2a in its unphosphorylated form, was down-regulated in the end-stage HF group; with no significant changes observed in the compensatory HF group (Figure 2E and F). The fraction of PLB phosphorylated at Ser16 (PKA site), but not at Thr17 [Ca2+/calmodulin-dependent PK II (CaMKII) site], in total PLB greatly decreased in both stages. RyR2, which plays an important role in Ca2+ release into the cytoplasm as an SR Ca2+ release channel, was down-regulated in both stages (Figure 2E and F). The fractions of RyR2 phosphorylated at Ser2808 (PKA site) and Ser2804 (CaMKII site) in total RyR2 were greatly increased in both stages. In human failing myocardium, PKA phosphorylation has been reported to be decreased in PLB, but increased in RyR2, suggesting the presence of local signalling causing distinct phosphorylation levels of PKA substrates in cardiomyocytes of HF patients (Schwinger et al., 1999; Marx et al., 2000). The present data support this concept and suggest that this local signalling becomes more evident as HF progresses into the decompensated stage.
Figure 2.

Up- and down-regulation of expression and phosphorylation of Ca2+ handling proteins in LV myocardium of DCM mice during progression of HF. (A) Representative Western blots of SERCA2a, NCX1, CSQ1 and CSQ2. (B) Relative expression levels of SERCA2a. (C) Relative expression levels of the NCX1. (D) Relative expression levels of CSQ1, CSQ2 and total CSQ. (E) Representative Western blots of PLB and RyR2. (F) Relative expression and phosphorylation levels of PLB and RyR2. *P < 0.05; **P < 0.01; significantly different as indicated; unpaired t-test. n = 5 mice per group except for the end-stage HF group in (F), where n = 4 mice.
Therapeutic effects of pimobendan on DCM mice in different stages of HF
Pimobendan dose-dependently improved life expectancy in the compensated HF group, with a greater effect at a high dose of 100 mg·kg−1·day−1 (Figure 3A). Pimobendan also improved life expectancy in the end-stage HF group, but to a lower extent at the high dose, compared with a low dose of 10 mg·kg−1·day−1 (Figure 3A). Maximum blood plasma concentrations of pimobendan were 1.6 and 18 μM at the doses of 10 and 100 mg·kg−1 respectively (Supporting Information Figure S2). DCM mice died from HF only during low-dose pimobendan treatment in both stages (Table 2). In contrast, mice died from sudden cardiac death only during high-dose treatment, with a much higher incidence in the end-stage HF group. High-dose pimobendan increased the body weight of DCM mice in the compensated HF group to the level of healthy WT mice while not changing the body weight in the end-stage HF group; whereas low-dose pimobendan had no effect in either stage (Figure 3B). High-dose pimobendan also prevented the decrease of body temperature in the compensated HF group and returned temperature levels to normal in the end-stage HF group; whereas low-dose pimobendan was not able to prevent the decrease of body temperature in either stage (Figure 3B). Additionally, high-, but not low-dose pimobendan prevented the progressive decrease in locomotor activity in the compensated HF group, and markedly improved locomotor activity, as well as dyspnoea, in end-stage HF (Supporting Information Figure S3). Pimobendan treatment did not improve LV systolic function in either stages, but dose-dependently prevented the progressive decrease in EF in the compensated stage of HF (Figure 3C, Supporting Information Table S1). Pimobendan treatment also prevented the progressive increase in heart and lung weights (Figure 4A), and markedly decreased myocardial fibrosis to levels lower than those before treatment in both stages of HF (Figure 4B and C). Consistent with these therapeutic effects of pimobendan, down-regulation of SERCA2a and up-regulation of NCX1 and β-MyHC were dose-dependently prevented in the compensated HF group by pimobendan treatment, but were not reversed in the end-stage HF group (Figure 5).
Figure 3.
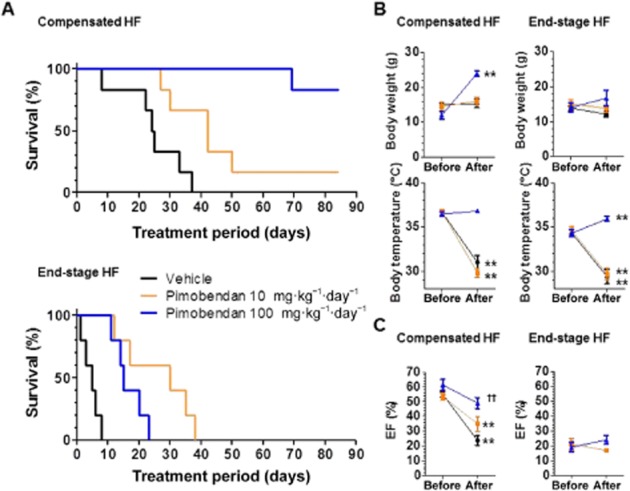
Effects of pimobendan treatment on survival, body weight and temperature, and LVEF of mice with DCM in different stages of HF. (A) Effects of pimobendan treatment on survival. Pimobendan significantly extended the life span of DCM in both stages, with more beneficial effects being observed at a high dose (100 mg·kg−1·day−1) in compensated HF, but at a low dose (10 mg·kg−1·day−1) in the end-stage HF (log–rank test). (B) Effects of pimobendan treatment on body weight and temperature. Data were collected at the start of pimobendan treatment and 1 day before death during pimobendan treatment or at the end of treatment. **P < 0.01 versus the value before treatment (paired t-test.); n = 5–6 mice per group. (C) Effects of pimobendan treatment on LVEF of DCM mice. The EF before treatment represents the data collected from echocardiography at the start of pimobendan treatment, and the EF after treatment represents the last data before death collected from echocardiography every 2 weeks during pimobendan treatment. Note that DCM mice in end-stage HF treated with vehicle all died within 2 weeks after treatment, making it impossible to collect any data after treatment. *P < 0.05; **P < 0.01; versus the value before treatment; paired t-test. ††P < 0.01; versus vehicle treatment; one-way anova and post hoc Dunnett's comparison test); n = 5–6 mice per group.
Table 2.
Incidence of HF death and sudden cardiac death during pimobendan treatment of DCM mice
| Pimobendan (mg·kg-1·day−1) | ||||||
|---|---|---|---|---|---|---|
| Compensated HF | End-stage HF | |||||
| 0 (vehicle) | 10 | 100 | 0 (vehicle) | 10 | 100 | |
| Number of mice | 6 | 6 | 6 | 5 | 5 | 5 |
| Maximum treatment period (days) | 37 | 84 | 84 | 8 | 38 | 23 |
| Number of all-cause deaths | 6 | 5 | 1 | 5 | 5 | 5 |
| Number of HF deaths | 6 | 5 | 0 | 5 | 5 | 0 |
| Number of sudden cardiac deaths | 0 | 0 | 1 | 0 | 0 | 5 |
Deaths after progressive decreases in body weight and temperature were considered to be HF death, while deaths despite preservation or recovery of body weight and temperature were considered to be sudden cardiac death.
Figure 4.
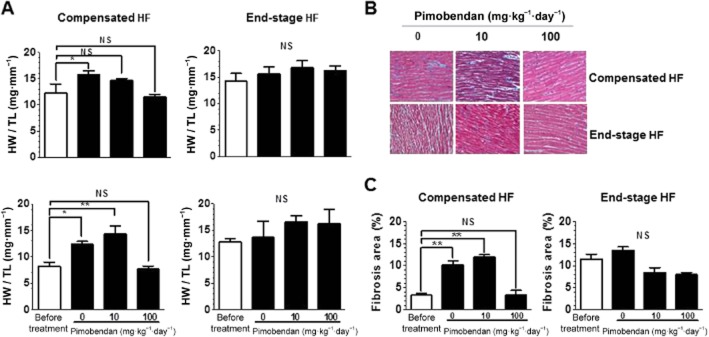
Effects of pimobendan treatment on heart and lung weight and myocardial fibrosis of DCM mice in different stages of HF. (A) Effects of pimobendan treatment on heart and lung weight (n = 5–6 mice per group). (B) Histology of the LV. Connective tissues were stained blue with Masson trichrome. (C) Quantitative analysis of the fibrosis area in the LV (n = 3 mice per group). *P < 0.05; **P < 0.01; versus the value before treatment; one-way anova and post hoc Dunnett's comparison test).
Figure 5.
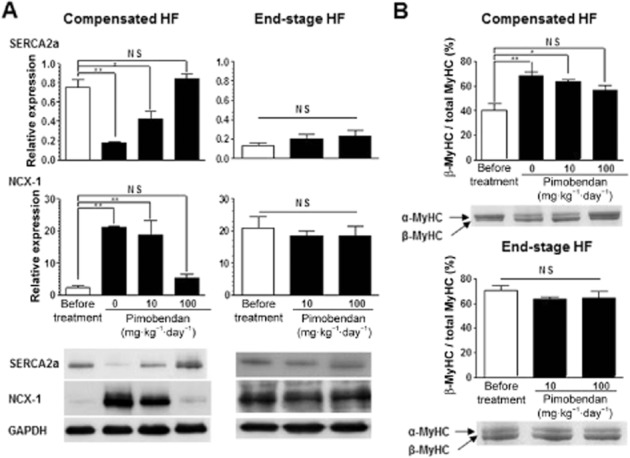
Effects of pimobendan treatment on up- and down-regulation of Ca2+ handling proteins and MyHC isoform transition in LV myocardium of DCM mice during progression of HF. (A) Effects of pimobendan treatment on the expression levels of SERCA2a and NCX1 (n = 3–5 mice per group). (B) Effects of pimobendan treatment on the relative expression level of β-MyHC isoform (n = 3–5 mice per group). *P < 0.05; **P < 0.01 versus the value before treatment; one-way anova and post hoc Dunnett's comparison test.
These results indicate that pimobendan is beneficial in the treatment of DCM mice in both the compensated and end-stage HF groups. However, the treatment with pimobendan at a high dose may increase the risk of lethal arrhythmia, especially with end-stage HF, despite dramatically improving HF symptoms. This counters the effect of high-dose pimobendan to improve life expectancy when compared with low-dose treatment.
Effects of pimobendan on CaT and contraction in single cardiomyocytes isolated from DCM mice in different stages of HF
To explore the cellular mechanisms underlying the high incidence of sudden death in end-stage HF during high-dose pimobendan treatment, effects of pimobendan were examined on contraction and CaT in single cardiomyocytes. When compared with age-matched, healthy WT mice, resting sarcomere length (SL) decreased in cardiomyocytes isolated from HF mice, with a greater decrease seen in the end-stage HF group, because of increased resting cytoplasmic Ca2+ levels (Figure 6A). Peak amplitudes of SL shortening and CaT also decreased in cardiomyocytes isolated from HF mice, with a greater decrease seen in the end-stage HF group (Figure 6B). Consistent with this, SR Ca2+ content decreased significantly in the end-stage HF group (Figure 6C), probably because of down-regulation of SERCA2a with decreased phosphorylation of PLB and increased phosphorylation of RyR2 (Figure 2).
Figure 6.

Pathophysiological changes in cardiomyocytes of DCM mice during progression of HF. (A) Representative SL shortening and CaT in a single cardiomyocyte after electrical stimulation. Normal control, 8-week-old WT. (B) Resting levels and peak amplitudes of SL shortening and CaT. Data were obtained by analysing 24–33 cardiomyocytes isolated (n = 5 hearts per group). (C) SR Ca2+ contents. Data were obtained by analysing five cardiomyocytes isolated from one heart in each group. *P < 0.05; **P < 0.01; significantly different as indicated; one-way anova and post hoc Tukey's multiple comparison.
Pimobendan exerted an inotropic action without affecting CaT (i.e., Ca2+ sensitization) on cardiomyocytes isolated from healthy WT mice at 3 μM (Supporting Information Figure S4). However, pimobendan further enhanced SL shortening with concomitant increases in resting Ca2+ levels and CaT at 10 μM, probably because of PDE3 inhibition as well as Ca2+ sensitization of cardiac myofilaments. The Ca2+ sensitizing action of pimobendan at 3 and 10 μM was evident from a delayed onset of relaxation, which should be caused by an enhanced Ca2+ binding to cTnC (i.e., a decrease in the off-rate of Ca2+) (Supporting Information Figure S4). Pimobendan had similar effects on cardiomyocytes isolated from compensated HF mice, with slight increases in resting Ca2+ levels and CaT even at 3 μM (Figure 7A, Supporting Information Figure S5). Pimobendan further increased in resting Ca2+ levels and CaT in cardiomyocytes isolated from end-stage HF mice, and frequently induced spontaneous contraction at 10 μM with a concomitant increase in cytoplasmic Ca2+ immediately after regular contraction evoked by electrical stimulation, which was probably induced by triggered activity because of early or delayed afterdepolarizations (EADs/DADs) (Figure 7A, Supporting Information Figure S6). It should be noted that sarcomere shortening is evoked by an even much higher Ca2+ concentration than estimated from the fura-2 ratio of the peak CaT, because the local cytosolic Ca2+ in the vicinity of RyR2, and thus cTnC in the thin filaments increases much more rapidly than in other cytoplasmic regions immediately after electrical stimulation. On the other hand, the fura-2 ratio before electrical stimulation should reflect the cytosolic Ca2+ level in the vicinity of cTnC. It is, therefore, not surprising that the resting SL in the presence of 10 μM pimobendan is much longer than the peak SL in the absence of pimobendan even at a comparable level of fura-2 ratio. Spontaneous contraction with a concomitant increase in cytoplasmic Ca2+ induced by triggered activity was also frequently induced by a specific PDE3 inhibitor (cilostazol) in cardiomyocytes isolated from end-stage HF mice (Figure 7B and E). Pimobendan markedly increased the phosphorylation of cTnI at Ser22/23 (PKA sites) at 10 μM in cardiomyocytes isolated from WT and end-stage HF mice, strongly suggesting that the triggered activity is caused by PDE3 inhibition and subsequent PKA activation (Figure 7C). The voltage-dependent L-type Ca2+ channel blocker verapamil decreased the incidence of triggered activity-induced spontaneous contraction induced by 10 μM pimobendan with concomitant decreases in resting Ca2+ levels and CaT in cardiomyocytes isolated from end-stage HF mice (Figure 7D and E).
Figure 7.

Effects of pimobendan on contraction and CaT in single cardiomyocytes isolated from DCM mice in different stages of HF. (A) Representative SL shortening and CaT in a single cardiomyocytes isolated from DCM mice in compensated or end-stage HF, which are perfused with Krebs–Henseleit solution containing 0, 3 and 10 μM pimobendan. (B) Effects of cilostazol (30 μM) on SL shortening and CaT in single cardiomyocytes isolated from end-stage HF DCM mice. (C) Effects of pimobendan on phosphorylation of cTnI at Ser22/23 in isolated cardiomyocytes (n = 3 hearts per group). **P < 0.01; significantly different as indicated; one-way anova and post hoc Tukey's multiple comparison. (D) Effects of verapamil (3 μM) on SL shortening and CaT in a single cardiomyocytes isolated from end-stage HF DCM mice, which is perfused with Krebs–Henseleit solution containing 10 μM pimobendan. (E) Percentage of cardiomyocytes showing triggered activity in investigated cardiomyocytes of end-stage HF DCM mice.
These results suggest that, in cardiomyocytes of end-stage HF mice, pimobendan may induce triggered activity through EADs/DADs via markedly up-regulated electrogenic NCX1 activation (Sipido et al., 2007), by overly elevating cytoplasmic Ca2+ thorough PDE3 inhibition under diminished SR Ca2+ reuptake by SERCA2a and increased Ca2+ leakage from SR through RyR2.
Discussion and conclusions
The present study used a DCM mouse model with ΔK210 mutation in cTnT on the BALB/c genetic background, which was created by backcrossing a knock-in mouse with the same mutation on the different genetic background of C57Bl/6 to BALB/c line. The BALB/c background DCM mice is phenotypically quite distinct from the original C57Bl/6 background DCM mice, which develop a stable compensated HF, but suffer mostly from sudden cardiac death before reaching 2 months without progressing into the decompensated stage characterized by dyspnoea, pulmonary congestion and decreased locomotor activity (Du et al., 2007; Li et al., 2012) (Supporting Information Figure S7). Until around 4 weeks old, the BALB/c background DCM mice do not show any HF symptoms despite developing a significantly enlarged heart and are therefore thought to be in the compensated stage of HF. However, they gradually develop symptoms of HF, as well as severe myocardial fibrosis, after this period and die from severe end-stage HF by around 8 weeks old. The present study using this unique mouse model for human inherited DCM, which shows a clear transition from compensated to end-stage HF at a fixed time during the growth period, provided important insights into the therapeutic efficacy of pimobendan in patients with end-stage HF, as well as compensated HF. Pimobendan was profoundly beneficial in preventing the progression of HF and increased the life span at the compensated stage. Pimobendan also improved HF symptoms and increased the life span at the end-stage. However, a high dose of pimobendan increased the risk of sudden death and had a lower effect on life expectancy than at a lower dose.
Cardiomyocyte contraction is evoked by a large release of Ca2+ into the cytoplasm from SR through the SR Ca2+ release channel RyR2, an event triggered by a small amount of Ca2+ influx into the cytoplasm through voltage-dependent L-type Ca2+ channels across the sarcolemma. Increased Ca2+ in the cytoplasm activates contractile proteins, and is rapidly removed from the cytoplasm into SR through SERCA2a and the extracellular space through sarcolemmal NCX1, leading to relaxation. The amplitude of CaT in cardiomyocytes is known to be significantly reduced in end-stage HF because of a decrease in the Ca2+ content stored in SR from altered expression and phosphorylation levels of Ca2+ handling proteins (Beuckelmann et al., 1992; Piacentino et al., 2003; Bers, 2006). Ca2+ reuptake into SR through SERCA2a has been shown to be impaired in human end-stage HF because of down-regulation of SERCA2a (Arai et al., 1994; Dash et al., 2001; DiPaola et al., 2001) and diminished phosphorylation of PLB resulting in an increased inhibition of SERCA2a activity (Schwinger et al., 1999; Houser et al., 2000). However, Ca2+ extrusion from the cell during diastole and Ca2+ entry into the cell during systole have been shown to be enhanced in human end-stage HF through up-regulation of NCX1 (Studer et al., 1994; Flesch et al., 1996; Reinecke et al., 1996). Ca2+ has been shown to leak from the SR through the RyR2 during diastole in human end-stage HF because of hyperphosphorylation of RyR2 at Ser2808 (Marx et al., 2000) and Ser2814 (Fischer et al., 2013).
These remodelling events in Ca2+ handling in human end-stage HF, together with up-regulation of β-MyHC isoform with lower ATPase activity, are likely to occur as an intrinsic cardioprotective mechanism to reduce oxygen consumption keeping the reduction in cardiomyocyte contractility as low as possible. Thus, pimobendan, an inotropic agent that could improve cardiac contractility with less of an increase in oxygen consumption compared with traditional inotropes, seems potentially beneficial in end-stage HF. However, the present study suggests that pimobendan may induce triggered activity because of EADs/DADs via markedly up-regulated electrogenic NCX1. The NCX1 could be activated by the over-elevated cytoplasmic Ca2+ through increased Ca2+ entry via L-type Ca2+ channel because of PDE3 inhibition under diminished Ca2+ reuptake into SR and increased Ca2+ leakage from SR in end-stage HF. The present study demonstrates that the voltage-dependent L-type Ca2+ channel blocker verapamil can decrease the incidence of triggered activity induced by pimobendan, by antagonizing the elevation of cytoplasmic Ca2+ through PDE3 inhibition. Further studies are warranted to explore whether the combined use of Ca2+ channel blockers and pimobendan could decrease the incidence of sudden cardiac death in DCM mice with end-stage HF.
The maximum blood plasma concentration of pimobendan after single dose p.o. administration of a standard tablet to healthy adult men is about 25 nM according to the package insert. However, pimobendan has been reported to exert a positive inotropic effect on papillary muscle fibres isolated from HF patients at considerably higher concentrations (EC50 = 0.4–26.0 μM) (Bohm et al., 1991). The present study also shows that the inotropic action of pimobendan through Ca2+ sensitization is exerted on mouse isolated cardiomyocytes at micromolar concentrations. These findings suggest that the current clinical dose is too low to obtain an optimal blood plasma concentration in order to exert the inotropic action of pimobendan through Ca2+ sensitization. Pimobendan and its demethylated metabolite UD-CG 212 CL may increase the risk of sudden cardiac death because of Ca2+ overload through PDE3 inhibition at high doses in end-stage HF. However, the Ca2+ overload through PDE3 inhibition could be reduced, at least to some extent, by the myofilament Ca2+ sensitization, which decreases the cytoplasmic Ca2+ level through enhanced Ca2+ binding to cTnC. UD-CG 212 CL has an approximately 10 times higher IC50 value for PDE3 inhibition than pimobendan (Bohm et al., 1991). The present study indicates that the maximum plasma concentration of UD-CG 212 CL is less than 5% of that of pimobendan in mice treated with pimobendan at the doses of 10–100 mg·kg−1, suggesting that contribution of UD-CG 212 CL to PDE3 inhibition is no more than 50% of that of pimobendan at these doses, at least, in mice (Supporting Information Figure S2). Currently available ‘Ca2+ sensitizers’ have a more or less inhibitory action on PDE3 that might cause adverse outcomes through Ca2+ overload. However, a specific Ca2+ sensitizer, if one exists, might have a risk of causing diastolic dysfunction as in the case of inherited hypertrophic cardiomyopathy caused by increased myofilament Ca2+ sensitivity (Morimoto, 2008). The lusitropic effect exerted by PKA phosphorylation of cTnI via PDE3 inhibition may play a potential beneficial role in antagonizing diastolic dysfunction caused by myofilament Ca2+ sensitization.
As myocardial remodelling in knock-in mice with ΔK210 mutation in cTnT is gradually produced to compensate for the decreased myocardial contractility because of a low myofilament Ca2+ sensitivity intrinsically conferred by the mutation, it may not be surprising that pimobendan can prevent the progression of myocardial remodelling during the compensated stage of HF. However, pimobendan has been demonstrated to delay the onset of clinical signs and extend the survival of Doberman Pinscher dogs with asymptomatic DCM, which is caused by several gene mutations unlikely to affect the myofilament Ca2+ sensitivity (Summerfield et al., 2012). This study strongly suggests that pimobendan can exert beneficial effects on compensated HF caused by various insults unrelated to myofilament Ca2+ sensitivity. Also noteworthy is that pathophysiological changes in the myocardium of DCM mice with end-stage HF are very similar to those observed in human end-stage HF, even if they should be caused by quite different aetiologies. This suggests that the decreased myofilament Ca2+ sensitivity plays a promoting role in myocardial remodelling as a root cause that induces myocardial systolic dysfunction, just as do other aetiological insults. The inotropic action through myofilament Ca2+ sensitization could be more pronounced and thus expected to be even more beneficial in end-stage HF caused by insults that do not change the myofilament Ca2+ sensitivity. Further studies are warranted to explore the potential benefits of pimobendan for human end-stage HF, as well as for compensated HF.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan to S. M. (No. 25670130, 23300145); and Grants-in-Aid for Graduate Students at Kyushu University to M. N. We would like to thank Ms Hayakawa and Ms Takami for their technical assistance at the Research Support Centre, Research Centre for Human Disease Modelling, Kyushu University Graduate School of Medical Sciences.
Glossary
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CaT
Ca2+ transients
- CSQ
calsequestrin
- cTnC
cardiac troponin C
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- DCM
dilated cardiomyopathy
- EADs/DADs
early or delayed afterdepolarizations
- EF
ejection fraction
- HF
heart failure
- LV
left ventricle
- MyHC
myosin heavy chain
- NCX1
Na+/Ca2+ exchanger 1
- PLB
phospholamban
- RyR2
ryanodine receptor 2
- SERCA2a
sarcoplasmic/endoplasmic reticulum Ca2+ transport ATPase 2a
- SL
sarcomere length
- SR
sarcoplasmic reticulum
- WT
wild-type
Author contributions
M. N. and S. M. performed entire experiments and wrote the paper. S. M. designed and supervised the experiments. T. M. carried out the Western blot analyses of PLB and RyR2. N. K. provided antibodies of PLB and RyR2. L. L. and Y. W. generated the mouse model by backcrossing. M. A., T. Y. and F. T.-Y. provided technical and material support. T. S. contributed to data interpretation and critically reviewed the paper.
Conflicts of interest
None.
Supporting Information
Figure S1 Time course of changes in locomotor activity and body temperature in WT and DCM mice on the genetic background of BALB/c. (A) Locomotor activity in WT (n = 3) and DCM mice (n = 5). (B) Body temperature in DCM mice (n = 6). **P < 0.01 versus 4 weeks old (one-way anova and post hoc Dunnett's comparison test).
{kind=link}
Figure S2 Blood plasma concentrations of pimobendan and UD-CG 212 CL in 8-week-old WT male mice after low-dose (10 mg·kg−1) and high-dose (100 mg·kg−1) p.o. administration of pimobendan.
{kind=link}
Figure S3 Effects of pimobendan treatment on locomotor activity in DCM mice in different stages of HF.
{kind=link}
Figure S4 Effects of pimobendan on SL shortening and CaT in single cardiomyocytes isolated from WT mice.
{kind=link}
Figure S5 Effects of pimobendan on relaxation in single cardiomvocytes isolated from compensated and end-stage HF mice.
{kind=link}
Figure S6 Continuous record of SL shortening and CaT in single cardiomyocytes isolated from DCM mice with end-stage HF, which are electrically stimulated at 1 Hz in the absence and presence of 10 μM pimobendan.
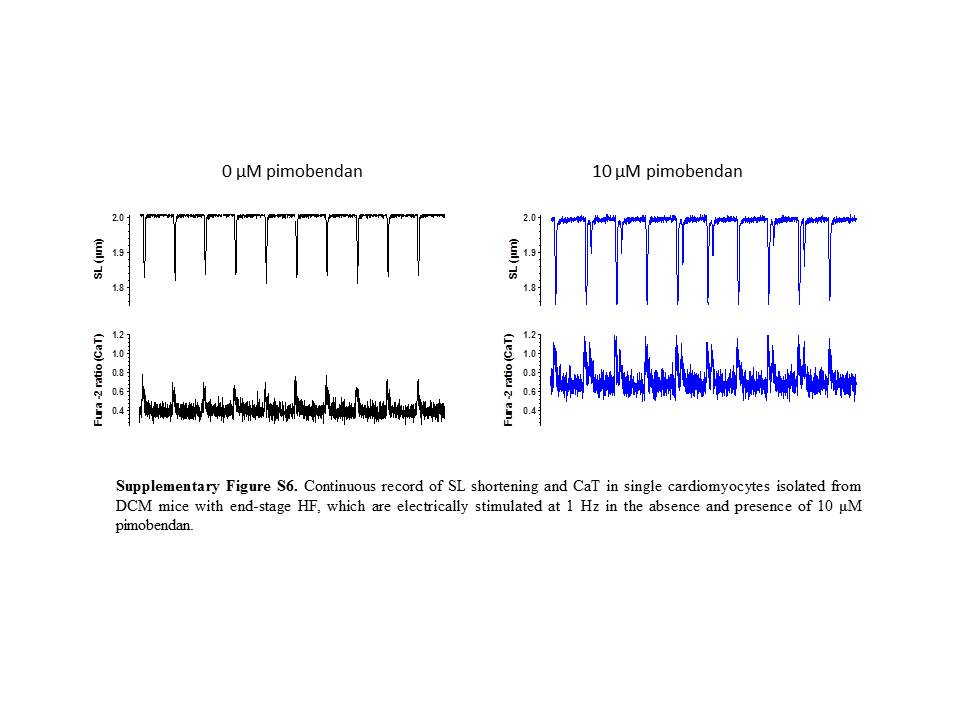{kind=link}
Figure S7 Time course of changes in locomotor activity in DCM mice on the genetic background of C57B1/6 and BALB/c. *P < 0.05 vs. C57B1/6 at the same age (unpaired t-test).
{kind=link}
Table S1 Echocardiography data of DCM mice with compensated or end-stage HF treated with pimobendan 10 and 100 mg·kg−1·day−1 p.o.
References
- Ahmed A, Gondi S, Cox C, Wang S, Stupin IV, Shankar KJ, et al. Circadian body temperature variability is an indicator of poor prognosis in cardiomyopathic hamsters. J Card Fail. 2010;16:268–274. doi: 10.1016/j.cardfail.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013a;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013c;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsallem E, Kasparian C, Haddour G, Boissel JP, Nony P. Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst Rev. 2005;(1) doi: 10.1002/14651858.CD002230.pub2. CD002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M, Matsui H, Periasamy M. Sarcoplasmic reticulum gene expression in cardiac hypertrophy and heart failure. Circ Res. 1994;74:555–564. doi: 10.1161/01.res.74.4.555. [DOI] [PubMed] [Google Scholar]
- Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- Bohm M, Morano I, Pieske B, Ruegg JC, Wankerl M, Zimmermann R, et al. Contribution of cAMP-phosphodiesterase inhibition and sensitization of the contractile proteins for calcium to the inotropic effect of pimobendan in the failing human myocardium. Circ Res. 1991;68:689–701. doi: 10.1161/01.res.68.3.689. [DOI] [PubMed] [Google Scholar]
- Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–981. doi: 10.1016/j.jacc.2004.11.066. [DOI] [PubMed] [Google Scholar]
- Chugun A, Taniguchi K, Murayama T, Uchide T, Hara Y, Temma K, et al. Subcellular distribution of ryanodine receptors in the cardiac muscle of carp (Cyprinus carpio. Am J Physiol Regul Integr Comp Physiol. 2003;285:R601–R609. doi: 10.1152/ajpregu.00419.2002. [DOI] [PubMed] [Google Scholar]
- CIBIS-II. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9–13. [PubMed] [Google Scholar]
- Dash R, Frank KF, Carr AN, Moravec CS, Kranias EG. Gender influences on sarcoplasmic reticulum Ca2+-handling in failing human myocardium. J Mol Cell Cardiol. 2001;33:1345–1353. doi: 10.1006/jmcc.2001.1394. [DOI] [PubMed] [Google Scholar]
- DiPaola NR, Sweet WE, Stull LB, Francis GS, Schomisch Moravec C. Beta-adrenergic receptors and calcium cycling proteins in non-failing, hypertrophied and failing human hearts: transition from hypertrophy to failure. J Mol Cell Cardiol. 2001;33:1283–1295. doi: 10.1006/jmcc.2001.1390. [DOI] [PubMed] [Google Scholar]
- Du CK, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, et al. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res. 2007;101:185–194. doi: 10.1161/CIRCRESAHA.106.146670. [DOI] [PubMed] [Google Scholar]
- Endoh M. Amrinone, forerunner of novel cardiotonic agents, caused paradigm shift of heart failure pharmacotherapy. Circ Res. 2013;113:358–361. doi: 10.1161/CIRCRESAHA.113.301689. [DOI] [PubMed] [Google Scholar]
- Effects of Pimobendan on Chronic Heart Failure Study (EPOCH Study) Effects of pimobendan on adverse cardiac events and physical activities in patients with mild to moderate chronic heart failure: the effects of pimobendan on chronic heart failure study (EPOCH study) Circ J. 2002;66:149–157. doi: 10.1253/circj.66.149. [DOI] [PubMed] [Google Scholar]
- Fischer TH, Herting J, Tirilomis T, Renner A, Neef S, Toischer K, et al. Ca2+/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation. 2013;128:970–981. doi: 10.1161/CIRCULATIONAHA.113.001746. [DOI] [PubMed] [Google Scholar]
- Flesch M, Schwinger RH, Schiffer F, Frank K, Sudkamp M, Kuhn-Regnier F, et al. Evidence for functional relevance of an enhanced expression of the Na+-Ca2+ exchanger in failing human myocardium. Circulation. 1996;94:992–1002. doi: 10.1161/01.cir.94.5.992. [DOI] [PubMed] [Google Scholar]
- Fujino K, Sperelakis N, Solaro RJ. Sensitization of dog and guinea pig heart myofilaments to Ca2+ activation and the inotropic effect of pimobendan: comparison with milrinone. Circ Res. 1988;63:911–922. doi: 10.1161/01.res.63.5.911. [DOI] [PubMed] [Google Scholar]
- Houser SR, Piacentino V, 3rd, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubalova Z, Gyorke I, Terentyeva R, Viatchenko-Karpinski S, Terentyev D, Williams SC, et al. Modulation of cytosolic and intra-sarcoplasmic reticulum calcium waves by calsequestrin in rat cardiac myocytes. J Physiol. 2004;561:515–524. doi: 10.1113/jphysiol.2004.073940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Morimoto S, Take S, Zhan DY, Du CK, Wang YY, et al. Role of brain serotonin dysfunction in the pathophysiology of congestive heart failure. J Mol Cell Cardiol. 2012;53:760–767. doi: 10.1016/j.yjmcc.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Lubsen J, Just H, Hjalmarsson AC, La Framboise D, Remme WJ, Heinrich-Nols J, et al. Effect of pimobendan on exercise capacity in patients with heart failure: main results from the Pimobendan in Congestive Heart Failure (PICO) trial. Heart. 1996;76:223–231. doi: 10.1136/hrt.76.3.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- McAlister FA, Wiebe N, Ezekowitz JA, Leung AA, Armstrong PW. Meta-analysis: β-blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784–794. doi: 10.7326/0003-4819-150-11-200906020-00006. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERIT-HF. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF) Lancet. 1999;353:2001–2007. [PubMed] [Google Scholar]
- Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–390. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- Morimoto S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res. 2008;77:659–666. doi: 10.1093/cvr/cvm084. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, Ohta M, et al. Ca2+-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc Natl Acad Sci U S A. 2002;99:913–918. doi: 10.1073/pnas.022628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai K, Seino Y, Kimata N, Inami T, Murakami D, Abe J, et al. Efficacy and limitations of oral inotropic agents for the treatment of chronic heart failure. Int Heart J. 2013;54:75–81. doi: 10.1536/ihj.54.75. [DOI] [PubMed] [Google Scholar]
- Nakaura H, Morimoto S, Yanaga F, Nakata M, Nishi H, Imaizumi T, et al. Functional changes in troponin T by a splice donor site mutation that causes hypertrophic cardiomyopathy. Am J Physiol. 1999;277:C225–C232. doi: 10.1152/ajpcell.1999.277.2.C225. [DOI] [PubMed] [Google Scholar]
- Odagiri F, Inoue H, Sugihara M, Suzuki T, Murayama T, Shioya T, et al. Effects of candesartan on electrical remodeling in the hearts of inherited dilated cardiomyopathy model mice. PLoS ONE. 2014;9:e101838. doi: 10.1371/journal.pone.0101838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovaska M, Taskinen J. A model for human cardiac troponin C and for modulation of its Ca2+ affinity by drugs. Proteins. 1991;11:79–94. doi: 10.1002/prot.340110202. [DOI] [PubMed] [Google Scholar]
- Packer M, Coats AJ, Fowler MB, Katus HA, Krum H, Mohacsi P, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651–1658. doi: 10.1056/NEJM200105313442201. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacentino V, 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, et al. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- Reinecke H, Studer R, Vetter R, Holtz J, Drexler H. Cardiac Na+/Ca2+ exchange activity in patients with end-stage heart failure. Cardiovasc Res. 1996;31:48–54. [PubMed] [Google Scholar]
- Scheld HH, Fritsche R, Schlepper M, van Meel JC. Pimobendan increases calcium sensitivity of skinned human papillary muscle fibers. J Clin Pharmacol. 1989;29:360–366. doi: 10.1002/j.1552-4604.1989.tb03342.x. [DOI] [PubMed] [Google Scholar]
- Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E. Reduced Ca2+-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999;31:479–491. doi: 10.1006/jmcc.1998.0897. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Bito V, Antoons G, Volders PG, Vos MA. Na/Ca exchange and cardiac ventricular arrhythmias. Ann N Y Acad Sci. 2007;1099:339–348. doi: 10.1196/annals.1387.066. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Fujino K, Sperelakis N. The positive inotropic effect of pimobendan involves stereospecific increases in the calcium sensitivity of cardiac myofilaments. J Cardiovasc Pharmacol. 1989;14(Suppl. 2):S7–S12. [PubMed] [Google Scholar]
- Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, et al. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- Summerfield NJ, Boswood A, O'Grady MR, Gordon SG, Dukes-McEwan J, Oyama MA, et al. Efficacy of pimobendan in the prevention of congestive heart failure or sudden death in Doberman Pinschers with preclinical dilated cardiomyopathy (the PROTECT Study) J Vet Intern Med. 2012;26:1337–1349. doi: 10.1111/j.1939-1676.2012.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc Natl Acad Sci U S A. 2003;100:11759–11764. doi: 10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Morimoto S, Du CK, Lu QW, Zhan DY, Tsutsumi T, et al. Up-regulation of type 2 iodothyronine deiodinase in dilated cardiomyopathy. Cardiovasc Res. 2010;87:636–646. doi: 10.1093/cvr/cvq133. [DOI] [PubMed] [Google Scholar]
- Zhan DY, Morimoto S, Du CK, Wang YY, Lu QW, Tanaka A, et al. Therapeutic effect of β-adrenoceptor blockers using a mouse model of dilated cardiomyopathy with a troponin mutation. Cardiovasc Res. 2009;84:64–71. doi: 10.1093/cvr/cvp168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Time course of changes in locomotor activity and body temperature in WT and DCM mice on the genetic background of BALB/c. (A) Locomotor activity in WT (n = 3) and DCM mice (n = 5). (B) Body temperature in DCM mice (n = 6). **P < 0.01 versus 4 weeks old (one-way anova and post hoc Dunnett's comparison test).
Figure S2 Blood plasma concentrations of pimobendan and UD-CG 212 CL in 8-week-old WT male mice after low-dose (10 mg·kg−1) and high-dose (100 mg·kg−1) p.o. administration of pimobendan.
Figure S3 Effects of pimobendan treatment on locomotor activity in DCM mice in different stages of HF.
Figure S4 Effects of pimobendan on SL shortening and CaT in single cardiomyocytes isolated from WT mice.
Figure S5 Effects of pimobendan on relaxation in single cardiomvocytes isolated from compensated and end-stage HF mice.
Figure S6 Continuous record of SL shortening and CaT in single cardiomyocytes isolated from DCM mice with end-stage HF, which are electrically stimulated at 1 Hz in the absence and presence of 10 μM pimobendan.
Figure S7 Time course of changes in locomotor activity in DCM mice on the genetic background of C57B1/6 and BALB/c. *P < 0.05 vs. C57B1/6 at the same age (unpaired t-test).
Table S1 Echocardiography data of DCM mice with compensated or end-stage HF treated with pimobendan 10 and 100 mg·kg−1·day−1 p.o.