

Abstract
Background and Purpose
The activation of the metabotropic glutamate receptor 2 (mGlu2) reduces glutamatergic transmission in brain regions where excess excitatory signalling is implicated in disorders such as anxiety and schizophrenia. Positive allosteric modulators (PAMs) can provide a fine-tuned potentiation of these receptors' function and are being investigated as a novel therapeutic approach. An extensive set of mutant human mGlu2 receptors were used to investigate the molecular determinants that are important for positive allosteric modulation at this receptor.
Experimental Approach
Site-directed mutagenesis, binding and functional assays were employed to identify amino acids important for the activity of nine PAMs. The data from the radioligand binding and mutagenesis studies were used with computational docking to predict a binding mode at an mGlu2 receptor model based on the recent structure of the mGlu1 receptor.
Key Results
New amino acids in TM3 (R635, L639, F643), TM5 (L732) and TM6 (W773, F776) were identified for the first time as playing an important role in the activity of mGlu2 PAMs.
Conclusions and Implications
This extensive study furthers our understanding of positive allosteric modulation of the mGlu2 receptor and can contribute to improved future design of mGlu2 PAMs.
Tables of Links
| TARGETS | |
|---|---|
| β2-adrenoceptor | mGlu5 receptor |
| mGlu1 receptor | mGlu6 receptor |
| mGlu2 receptor | mGlu7 receptor |
| mGlu3 receptor | mGlu8 receptor |
| mGlu4 receptor |
| LIGANDS | |
|---|---|
| BINA | MNI-135 |
| Glutamate | MPEP |
| JNJ-40068782 | [35S]-GTPγS |
| LY2607540 (THIIC) | VU-71 |
| LY487379 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Glutamate is the major excitatory neurotransmitter in the mammalian CNS and is crucial for physiological functions including learning and memory, development of synaptic plasticity and motor control. Because of this ubiquitous activity in the CNS, abnormal glutamate neurotransmission has been linked to a number of neurological and psychiatric diseases (Niswender and Conn, 2010; Chiechio and Nicoletti, 2012; Vinson and Conn, 2012).
Glutamate binds to and activates metabotropic glutamate receptors (mGlu) through the extracellular amino terminal domain, the orthosteric binding site. This induces a conformational change in the receptor, resulting in activation of a G-protein and intracellular signalling pathways (Huang et al., 2011). To date, eight mGlu receptors have been identified and are divided into three groups according to their amino acid sequence homology, pharmacology and their preferred signal transduction mechanism. Group I mGlu receptors (mGlu1/5) preferentially activate PLCβ and groups II (mGlu2/3) and III (mGlu4/6/7/8) inhibit AC activity (Conn and Pin, 1997).
High levels of mGlu2 receptor are seen in brain areas such as the prefrontal cortex, hippocampus and amygdala where glutamate hyperfunction may be implicated in diseases such as anxiety and schizophrenia (Gu et al., 2008; Ghose et al., 2009). Receptor activation has been shown to dampen excitatory neurotransmission, and hence, the mGlu2 receptor is being investigated as a potential target for the treatment of these diseases (Niswender and Conn, 2010). A number of group II mGlu receptor agonists have been identified from analogy with glutamate (Monn et al., 1997) and have shown anxiolytic-like and antipsychotic-like effects in animal models (Shigeyuki and Hirohiko, 2011; Vinson and Conn, 2012). Efficacy has been observed in clinical studies in patients with schizophrenia or generalized anxiety disorder (Patil et al., 2007; Dunayevich et al., 2008).
Selectivity between mGlu2 and mGlu3 receptors at the conserved orthosteric site is a challenge. This has motivated the search for positive allosteric modulators (PAMs) that specifically potentiate glutamate-induced mGlu2 receptor activity by binding to an alternative site from the orthosteric agonist. Early examples of mGlu2 receptor PAMs include BINA and LY487379 (Schaffhauser et al., 2003; Galici et al., 2006). These compounds are selective for mGlu2 receptors and have efficacy in animal models of antipsychotic activity similar to that seen with mGlu2/3 receptor agonists (Galici et al., 2005). The number of reports on mGlu2 receptor PAM chemical series has increased over recent years (Trabanco et al., 2011a, Trabanco and Cid, 2013) including multiple lead series from our laboratories (Cid-Nuñez et al., 2008b; 2010b,; Tresadern et al., 2010; Trabanco et al., 2011c; 2012,; Cid et al., 2012a,b,).
The interaction of allosteric ligands with different mGlu receptors has been shown to be disturbed by mutation of seven-transmembrane (7-TM) amino acids (Litschig et al., 1999; Pagano et al., 2000; Knoflach et al., 2001; Malherbe et al., 2003; Hemstapat et al., 2006; 2007,; Lundström et al., 2011). This suggests that allosteric modulators bind within the 7-TM region and a binding site involving TM4 and TM5 has been hypothesized (Schaffhauser et al., 2003). The same mutations often affect the activity of all ligands, although there are exceptions. For example, V757 was important for the mGlu1 receptor PAMs Ro 67-7476 and VU-71, but the very close analogue VU-48 was not affected by the same mutation (Knoflach et al., 2001; Hemstapat et al., 2006). In the case of the mGlu2 receptor, the two PAMs BINA and LY487379 were both affected by mutation N735D whereas the negative allosteric modulator (NAM) MNI-135 was not (Hemstapat et al., 2007). Similarly, S688, G689 and N735 mutants all disrupted LY487379 PAM activity; however, no alteration of the MNI-135 NAM activity was seen (Schaffhauser et al., 2003; Hemstapat et al., 2007). For the mGlu5 receptor, a recent study elegantly demonstrated the molecular interactions that contribute to PAM versus NAM pharmacology. Using a combination of site-directed mutagenesis and computational modelling six residues (P654, Y658, T780, W784, S808 and A809) were identified as key affinity determinants across several allosteric modulator scaffolds of the mGlu5 receptor (Gregory et al., 2013; 2014,). The 7-TM binding site for allosteric modulators was recently confirmed by X-ray crystallographic structures of the mGlu1 and mGlu5 receptor 7-TM domains bound to NAMs (Dore et al., 2014; Wu et al., 2014).
In the present study, we set out to clarify where our internal mGlu2 PAM lead series bind on the receptor and which amino acids are critical for their activity. Six molecules representing different chemical series were selected for study (Figure 1). Three mGlu2 PAM reference compounds were also included: LY487379, BINA, LY2607540 (THIIC) (Johnson et al., 2003; Bonnefous et al., 2005; Fell et al., 2011, respectively; Figure 1). A total of 39 mutant mGlu2 receptors were prepared by site-directed mutagenesis, and the effect of the receptor mutations on the functional activity of the nine PAM molecules was evaluated. Binding displacement experiments using a tritiated radioligand were also performed to evaluate whether the molecules act at a common site. We identified several new amino acids crucial for the activity of mGlu2 PAMs. We discuss these new findings in the context of a potential allosteric binding site at the mGlu2 receptor.
Figure 1.
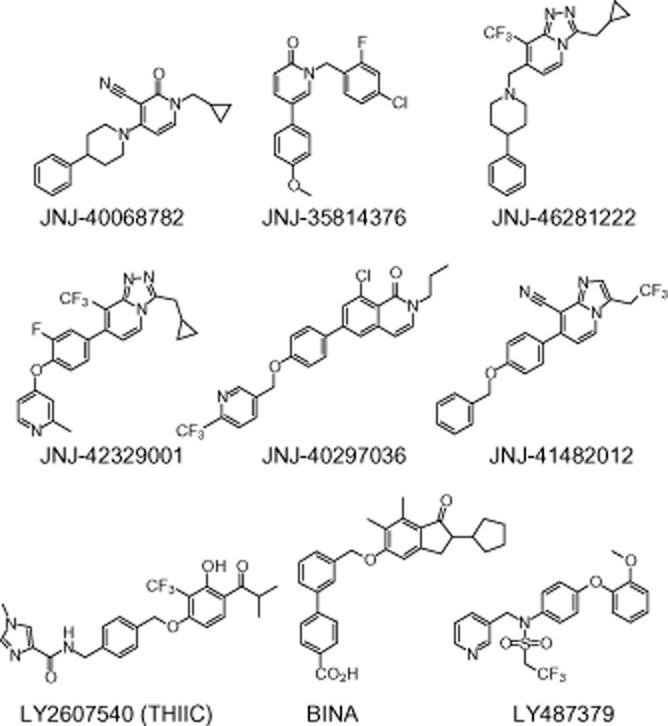
Structures of mGlu2 PAMs used in this study.
Methods
Plasmids, cell transfection and cell culture
cDNA encoding human non-mutated and mutated mGlu2 receptors was synthesized by GeneArt® (Life Technologies, Carlsbad, CA, USA) and subcloned to the mammalian expression vector pcDNA3.1(+) (Life Technologies). Plasmid DNA was amplified through Escherichia coli transformation and used for transient transfection in CHO-K1 cells. All transfections were performed using the cationic lipid transfection reagent Lipofectamine™ (Life Technologies). Cells expressing mutated and non-mutated mGlu2 receptors were maintained in DMEM supplemented with 10% heat-inactivated FBS, penicillin G, streptomycin sulphate, pyruvic acid and L-glutamine.
Membrane preparation
Confluent cells were washed in ice-cold 50 mM Tris-HCl buffer, pH 7.4, and stored at −20°C until membrane preparation. After thawing, cells were resuspended in 50 mM Tris-HCl buffer, pH 7.4, and the cell suspension was centrifuged for 10 min at 23 500× g at 4°C. The resulting cell pellet was resuspended and homogenized in ice-cold 5 mM hypotonic Tris-HCl, pH 7.4, using an Ultra Turrax homogenizer (IKA-Werke GmbH & Co. KG, Staufen, Germany) and the homogenate was centrifuged again for 20 min at 30 000× g at 4°C. The final pellet was resuspended and homogenized in 50 mM Tris-HCl. Protein concentration was measured by the Bio-Rad Protein Assay (Bio-Rad Laboratories, Munich, Germany) using BSA as standard and membranes were stored at −20°C.
[3H]-LY341495 binding
Membranes were thawed on ice and diluted in ice-cold binding buffer containing 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2 and 2 mM CaCl2. Assay mixtures contained 10 μg of membrane protein and 3 nM of [3H]-LY341495 in a total volume of 0.5 mL. In order to measure the non-specific binding, 1 mM glutamate was used. Assay mixtures were incubated for 60 min at room temperature (RT). The incubation was stopped by filtration using Unifilter-96 GF/B filter plates in a 96-well Perkin Elmer filtermate harvester, and the plates were dried overnight at RT.
[3H]-JNJ-40068782 binding
After thawing, membranes were homogenized using an Ultra Turrax homogenizer and suspended in ice-cold binding buffer containing 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, and 2 mM CaCl2; 75 μg membrane protein, test compound and 10 nM [3H]-JNJ-40068782 were incubated for 60 min at RT, in a total volume of 0.5 mL. Non-specific binding (about 30% of total binding) was determined in the presence of 10 μM JNJ-40264796 (a mGlu2 PAM structurally related to JNJ-40068782, molecule 34 from Cid et al., 2012a). Filtration was performed using Unifilter-96 GF/C filters pre-soaked in 0.1% PEI and a 40-well manifold or 96-well Brandell harvester.
For both [3H]-LY341495 and [3H]-JNJ-40068782, assay conditions including radioligand concentrations were chosen such that <10% of the free radioligand concentration was receptor-bound. After the addition of scintillation liquid, radioactivity on the filters was counted in a Microplate Scintillation and Luminescence Counter or Liquid Scintillation Analyzer from Perkin Elmer.
[35S]-GTPγS binding
Membranes were thawed on ice and diluted in assay buffer (10 mM HEPES acid and 10 mM HEPES salt, pH 7.4, containing 100 mM NaCl, 3 mM MgCl2, 10 μM guanosine 5′-diphosphate and 14.3 μg·mL−1 saponin). Assay mixtures containing 10 μg of membrane protein and test compound were pre-incubated with buffer (to detect agonist effects) or an EC20-equivalent concentration of glutamate (to detect PAM effects) for 30 min at 30°C. Finally, 0.1 nM [35S]-GTPγS was added and the assay mixture was incubated for another 30 min at 30°C. The reaction was stopped through filtration using Unifilter-96 GF/B filter plates (Perkin Elmer Life Sciences) in a 96-well Perkin Elmer filtermate harvester. The filters were washed three times with ice-cold 10 mM NaH2PO4/10 mM Na2HPO4 buffer, pH 7.4, and dried overnight at RT. The remaining radioactivity was counted in a Microplate scintillation and luminescence counter from Packard.
Data analysis
Data analysis was performed using GraphPad Prism version 4.02 for Windows (GraphPad Software, San Diego, CA, USA). Concentration–response curves were fitted using non-linear regression analysis fitting the equation: Y = Bottom + (Top − Bottom)/{1 + 10∧[(LogEC50 − X) × Hill Slope]}. pEC50 values obtained at the non-mutated receptor were compared with those at the mutant receptors by one-way anova analysis.
Amino acid selection for site-directed mutagenesis
Amino acids previously shown to be important for only mGlu2 receptor allosteric modulators were prioritized for mutation in our experiments. Therefore, the following amino acids were chosen: R635, R636, F643, S688, G689, H723, M728, S731, L732, N735, V736, W773 and F780.
The compounds being studied have been reported as selective for mGlu2 versus mGlu3 receptors. The mGlu2 and mGlu3 sequences were compared and different amino acids were identified. These are potentially important for the selective binding of mGlu2 receptor PAMs. Amino acids from the extracellular side of TM2, TM4, TM5 and ECL2 were selected for mutation. This resulted in many potential mutations so preference was given to amino acids with a more significant change in properties between mGlu2 and mGlu3: C616, I622, T641, A642, A681, I693, V695, A696, G706, E708, A710, P711, V716, T718, A726, G730, A733 and A740. In these cases, all amino acids were mutated to their mGlu3 equivalent.
At the time of initiating this work, the newly solved structures of mGlu1 and mGlu5 receptors were not available. Hence, an initial mGlu2 receptor model was built based on precedent for the alignment of class C GPCRs with class A. The β2-adrenoceptor X-ray structure 2RH1 was used as a template and this model used for docking to suggest amino acids for mutation. Molecules such as JNJ-40068782 were docked and plausible interacting amino acids were identified: R636, F643, L732, W773, F780, L639, D725, F776, S644, V700 and H723. Some of these had not been selected by the approaches discussed earlier and were therefore added to the list for experimental mutation. Subsequently, a second mGlu2 receptor model was built from the mGlu1 X-ray structure template. This model was used for interpretation of the experimental results and further docking to propose binding modes. The method for building the mGlu2 receptor homology model and performing protein ligand docking are provided in Supporting Information Appendices S1 and S2 respectively. We highlight that caveats exist for GPCR homology modelling. The difference in sequence identity, loop lengths, loop conformations and correct amino acid side chain orientation present a challenge for structure prediction. Use of the inactive states of the β2-adrenoceptor and mGlu1 receptors to model the mGlu2 PAM bound receptor is also problematic. The activated state of the β2-adrenoceptor (Rasmussen et al., 2011a,b,) showed large structural shifts in the intracellular G-protein binding region but only subtle changes in the 7-TM binding site, suggesting the effect may be small. The recent mGlu1 X-ray suggests interactions at the intracellular side of helices 3 and 6 play a role in stabilizing the inactive state, but a full understanding of the activation mechanism and plausible active state is some way off. Overall, modelling an mGlu2 receptor structure is improved by the availability of the mGlu1 receptor crystal structure but the limitation of relying upon an inactive receptor conformation remains.
In summary, 36 amino acids were identified as potentially important for the PAM-mGlu2 receptor interaction and chosen for site-directed mutagenesis. We prepared 36 single-point mutations along with one double and two triple mutations (Table 1).
Table 1.
mGlu2 receptor mutagenesis constructs
| WT aa | Position | Mutation | WT codon | Mutant codon | Name | Region |
|---|---|---|---|---|---|---|
| Cys | 616 | Ser | TGC | TCC | C616S | TM2 |
| Ile | 622 | Phe | ATC | TTC | I622F | |
| Arg | 635 | Ala | AGA | GCG | R635A | TM3 |
| Arg | 636 | Ala | CGT | GCT | R636A | |
| Leu | 639 | Ala | TTG | GCG | L639A | |
| Thr | 641 | Ser | ACC | TCC | T641S | |
| Ala | 642 | Ser | GCC | TCC | A642S | |
| Phe | 643 | Ala | TTC | GCC | F643A | |
| Ser | 644 | Ala | TCT | GCT | S644A | |
| Ala | 681 | Phe | GCC | TTC | A681F | TM4 |
| Ser | 688 | Leu | TCG | TTG | S688L | |
| Gly | 689 | Val | GGC | GTC | G689V | |
| Ile | 693 | Met | ATT | ATG | I693M | |
| Val | 695 | Ser | GTG | TCG | V695S | |
| Ala | 696 | Val | GCC | GTC | A696V | |
| Val | 700 | Leu | GTC | CTC | V700L | |
| Gly | 706 | Arg | GGA | CGA | G706R | ECL2 |
| Glu | 708 | Tyr | GAG | TAC | E708Y | |
| Ala | 710 | Leu | GCC | CTC | A710L | |
| Pro | 711 | Ala | CCC | GCC | P711A | |
| Val | 716 | Thr | GTG | ACG | V716T | |
| Thr | 718 | Ile | ACC | ATC | T718I | |
| His | 723 | Val | CAC | GTC | H723V | |
| Asp | 725 | Ala | GAT | GCT | D725A | TM5 |
| Ala | 726 | Ser | GCC | TCC | A726S | |
| Met | 728 | Ala | ATG | GCG | M728A | |
| Gly | 730 | Ile | GGC | ATC | G730I | |
| Ser | 731 | Ala | AGC | GCC | S731A | |
| Leu | 732 | Ala | CTG | GCG | L732A | |
| Ala | 733 | Thr | GCC | ACC | A733T | |
| Asn | 735 | Asp | AAT | GAT | N735D | |
| Val | 736 | Ala | GTG | GCG | V736A | |
| Ala | 740 | Ile | GCC | ATC | A740I | |
| Trp | 773 | Ala | TGG | GCG | W773A | TM6 |
| Phe | 776 | Ala | TTC | GCC | F776A | |
| Phe | 780 | Ala | TTC | GCC | F780A | |
| Ser/Gly | 688/689 | Leu/Val | TCG/GGC | TTG/GTC | S688L/G689V | TM4 |
| Ser/Val/His | 644/700/723 | Ala/Leu/Val | TCT/GTC/CAC | GCT/CTC/GTC | S644A/V700L/H723V | TM3/4/ECL2 |
| Ser/Gly/Asn | 688/689/735 | Leu/Val/Asp | TCG/GGC/AAT | TTG/GTC/GAT | S688L/G689V/N735D | TM4/5 |
Materials
JNJ-35814376 (Imogai et al., 2006; Cid et al., 2010), JNJ-40068782 (Imogai et al., 2007; Cid-Nuñez et al., 2008a; Lavreysen et al., 2013), JNJ-40297036 (Trabanco et al., 2011b), JNJ-41482012 (Trabanco-Suárez et al., 2009; Tresadern et al., 2010), JNJ-42329001 (Cid-Nuñez et al., 2010a) and JNJ-46281222 (Cid-Nuñez et al., 2010a) are original products of Janssen Research and Development, a division of Janssen Pharmaceutica. LY487379, BINA and LY2607540 (Johnson et al., 2003; Bonnefous et al., 2005; Fell et al., 2011, respectively) were synthesized for in-house use as reference compounds. All PAMs were dissolved in 100% DMSO, and serially diluted in assay buffer with final concentrations of 1% DMSO. L-glutamate was purchased from Aldrich® Chemistry. The radioligand [3H]-LY341495 was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA) and [35S]-GTPγS was obtained from Perkin Elmer® (Boston, MA, USA). The radioligand [3H]-JNJ-40068782 is an original product of Janssen Research and Development, a division of Janssen Pharmaceutica (Lavreysen et al., 2013). All cell culture and transfection reagents were obtained from Invitrogen (Carlsbad, CA, USA), except for FBS, which came from HyClone® (Thermo Scientific, Cramlington, UK). CHO cells (CHO-K1, ATCC: CCL-61) were obtained from ATCC®. Drug and receptor nomenclature follows Alexander et al. (2013).
Results
Expression of mGlu2-wt and mutant mGlu2 receptors in CHO-K1 cells
Radioligand-binding studies using the orthosteric mGlu2/3 receptor antagonist [3H]-LY341495 were performed to assess the expression of non-mutated (defined as ‘mGlu2-wt’ throughout the text) and mutated mGlu2 receptors in membranes of transiently transfected CHO-K1 cells. All mutated receptors showed similar specific [3H]-LY341495 binding (in the range of 90%, data not shown), thereby confirming that the orthosteric site had remained intact. Furthermore, glutamate was able to displace [3H]-LY341495 from mGlu2-wt (stably and transiently transfected) and mutant receptors with similar pIC50 values (pIC50 ∼5, see Supporting Information Appendix S3 and Figure 2A). Proper receptor expression was also confirmed via [3H]-LY341495 Bmax determinations as well as immunoblotting (data not shown).
Figure 2.
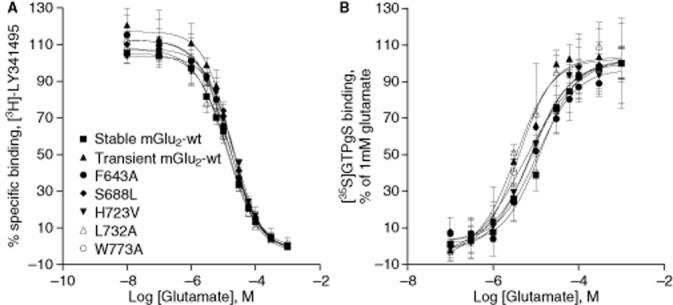
(A) Displacement of [3H]LY341495 binding from mGlu2-wt (stably and transiently transfected) and mGlu2 mutants by glutamate. Results are presented as percentage of specific binding. The affinity of glutamate is not altered by any of the mutations; total binding levels varied for the different membrane pools. (B) Glutamate-induced [35S]-GTPγS binding. Glutamate concentration–response curves were determined on membranes from CHO-K1 cells expressing mGlu2-wt (stably and transiently transfected) and five representative mutant receptors (transiently transfected). Results are expressed as % ± SD of the response to 1 mM glutamate. Both data sets are from one experiment performed in triplicate. Similar data were found for the additional set of mutants.
Glutamate potency for mGlu2-wt and mutated mGlu2 receptors
The potency of glutamate was assessed using [35S]-GTPγS assays permitting further verification of the effect of the mutations on glutamate-mediated receptor signalling. Mutated receptors elicited concentration–response curves for glutamate resulting in potencies that were similar to those of the stably and the transiently transfected mGlu2-wt receptor (see Supporting Information Appendix S3 and Figure 2B). The amplitude of the response to glutamate, that is the ratio between the response obtained under basal conditions (only using assay buffer) and the response obtained with 1 mM glutamate, was however considerably lower in cells transiently expressing the mGlu2 receptor compared with the stable cell line (Supporting Information Appendix S3).
Effect of mGlu2 mutations on the activity of mGlu2 PAMs
Initial screening
Given the large number of compounds and mutants being studied, an initial screening approach was applied to confirm if a mutation was likely to affect PAM activity. In this preliminary step, two concentrations of PAM were tested: a concentration equivalent to the compound's EC50 and a concentration producing a maximal PAM response (typically 3 or 10 μM). Both were previously determined with the use of stably transfected cells. The resulting PAM effect at each concentration was calculated as a percentage of the response to 1 mM glutamate and was then compared between mGlu2 mutants and the transiently transfected mGlu2-wt receptor. The results from these experiments, including details on the reliability assessment of this initial screening are added as Supporting Information Appendix S4. Supporting Information Fig. S1 provides a useful graphical comparison of the pre-screen results at the two concentrations. Mutants showing effects at either of the two concentrations were carried forward for further study.
Through this screening approach, 26 mutations were considered not to have an effect on the activity of any of the compounds. The others were evaluated in more detail by performing concentration–response analyses. Corresponding pEC50 values are shown in Table 2 along with concentration–response curves for a subset in Figure 3.
Table 2.
pEC50 of the enhancement of glutamate-induced [35S]-GTPγS binding by nine PAMs on mGlu2-wt and the mutant receptors that were selected for further evaluation, after transient transfection into CHO-K1 cells
| JNJ-40068782 | JNJ-35814376 | JNJ-46281222 | JNJ-42329001 | JNJ-40297036 | JNJ-41482012 | LY2607540 (THIIC) | BINA | LY487379 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| WT | Stable | 6.9 ± 0.1 | 6.19 ± 0.12 | 8.09 ± 0.23 | 7.41 ± 0.18 | 6.79 ± 0.17 | 6.74 ± 0.23 | 6.97 ± 0.08 | 7.03 ± 0.14 | 6.78 ± 0.13 |
| Transient | 7.1 ± 0.1 | 6.36 ± 0.25 | 8.22 ± 0.29 | 7.55 ± 0.2 | 6.75 ± 0.15 | 6.83 ± 0.15 | 6.97 ± 0.14 | 7.11 ± 0.30 | 6.85 ± 0.20 | |
| TM3 | R635A | 6.42 ± 0.03*** | 6.07 ± 0.04a*** | n.c. | n.c. | n.c. | n.c. | n.c. | 6.21a*** | n.c. |
| L639A | 6.27 ± 0.16*** | 5.83 ± 0.03** | n.c. | n.c. | 6.28 ± 0.35a*** | n.c. | 6.78 ± 0.06a** | 5.86 ± 0.01a*** | 6.16a*** | |
| F643A | 5.64 ± 0.18*** | n.c. | 6.32 ± 0.25a*** | 5.24 ± 0.08a*** | <5*** | <5*** | 6.08a*** | 5.85 ± 0.08a*** | <5*** | |
| TM4 | G689V | 6.27 ± 0.20*** | 5.74 ± 0.12*** | 7.21 ± 0.30* | 6.84 ± 0.18** | 6.30 ± 0.29* | <5*** | 6.22 ± 0.22** | 6.20 ± 0.37* | 6.58 ± 0.34 |
| ECL2 | H723V | 6.27 ± 0.06*** | 5.79 ± 0.11** | n.c. | n.c. | 6.46 ± 0.12 | 6.72 ± 0.10 | 6.56 ± 0.10 | n.c. | n.c. |
| TM5 | S731A | n.c. | 5.92 ± 0.03* | n.c. | n.c. | n.c. | n.c. | 6.56 ± 0.12 | n.c. | 6.08 ± 0.03*** |
| L732A | 7.03 ± 0.06 | 6.22 ± 0.16 | 7.31 ± 0.33 | 6.25 ± 0.09*** | 6.51 ± 0.18 | <5*** | 6.49 ± 0.24* | 7.80 ± 0.03a | <5*** | |
| N735D | 5.52a*** | <5*** | 6.67 ± 0.23a*** | 5.97 ± 0.06a*** | <5*** | <5*** | 5.53 ± 0.04*** | 5.13a*** | <5*** | |
| TM6 | W773A | <5*** | <5*** | 7.11a, b*** | 5.9a*** | <5*** | <5*** | <5*** | <5*** | <5*** |
| F776A | 6.38 ± 0.23*** | n.c. | n.c. | 7.16 ± 0.07b | n.c. | n.c. | 6.93 ± 0.07 | 6.71 ± 0.06 | <5*** | |
| S688L/G689V | 6.29 ± 0.39*** | 5.82a*** | 7.15 ± 0.41* | 6.54 ± 0.46*** | 6.58 ± 0.06b | 6.70 ± 0.30b | 6.22 ± 0.18** | 6.30 ± 0.23* | 6.48 ± 0.14* | |
| S688L/G689V/N735D | <5*** | <5*** | 6.38 ± 0.27a*** | 5.95 ± 0.51a*** | <5*** | <5*** | <5*** | <5*** | <5*** | |
| S644A/V700L/H723V | 5.91 ± 0.05a*** | 5.67 ± 0.11a*** | 7.10 ± 0.24* | 6.96 ± 0.12* | 6.40 ± 0.05 | 6.64 ± 0.08 | 6.21 ± 0.05** | 7.11 ± 0.15 | 6.18 ± 0.06*** | |
pEC50 ± SD values were determined based on three independent experiments, each performed in triplicate. Activity at transient mGlu2-wt receptors was always tested in parallel as a control.
n.c., no change (in the pre-screening, the mutation did not have an effect on the activity of the compound).
For one or two experiments, pEC50 was <5.
n = 2.
*P < 0.05, **P < 0.01, ***P < 0.001 significantly different from transient mGlu2-wt value, one-way anova. It is of note that by applying the Dunnett's post-test, some results lost statistical significance (e.g. mutant G689V and compound JNJ-46281222).
Figure 3.

Effect of the PAMs used in this study on glutamate-induced [35S]-GTPγS binding in mGlu2-wt and a subset of mGlu2 mutants. Concentration-dependent enhancement of 4 μM glutamate-induced [35S]-GTPγS binding by nine PAMs used in this study. Results are expressed as % ± SD of the response to 1 mM glutamate, and refer to one experiment performed in triplicate.
Overall results for all mutations (for screening data see Supporting Information Appendix S4)
Mutations located in TM2
The mutations located in this receptor region (C616S and I622F), which were selected based on sequence alignment and comparison of mGlu2 and mGlu3 receptor sequence, did not affect the response to either concentration of compound tested in the screening phase.
Mutations located in TM3
The mutations located in TM3 caused distinct effects on the activity of the compounds tested. Mutation F643A showed the most prominent results (Table 2, Figure 3). For the different compounds tested, the effects of this mutation ranged from no change (for JNJ35814376) to a complete loss of compound's activity for JNJ-40297036, JNJ-41482012 and LY487379 (i.e. pEC50 < 5; Table 2). Mutation L639A affected the activity of JNJ-40068782, JNJ-35814376, JNJ-40297036, LY2607540, BINA and LY487379 albeit to a lesser extent compared with mutation F643A (2- to 18-fold potency decrease, Table 2); no change was observed for compounds JNJ-46281222, JNJ-42329001 and JNJ-41482012. Mutation R635A had no effect on the majority of the compounds tested; it did cause a slight decrease in potency of JNJ-40068782 (fivefold), JNJ-35814376 (twofold) and BINA (eightfold) (Table 2). Finally, mutations S644A, R636A, T641S and A642S had no effect on the responses of any of the molecules tested.
Mutations located in TM4
Mutations A681F, I693M, V695S, A696V and V700L showed no effect on the activity of the tested compounds. In addition, mutation S688L yielded similar results as the mGlu2-wt receptor for every compound. With the exception of LY487379, all the other compounds seemed to be affected by G689V (Table 2).
Mutations located in ECL2
In ECL2, mutations G706R, E708Y, A710L, P711A, V716T and T718I did not change the response to the two concentrations of compound tested initially; only H723V caused an increase in EC50 of JNJ-40068782 (sevenfold) and JNJ-35814376 (fourfold) (Table 2).
Mutations located in TM5
Mutants D725A, A726S, M728A, G730I, A733T, A740I and V736A yielded similar responses as the mGlu2-wt receptor. Mutation S731A also had a minor impact on the activity of the compounds tested, with the exception of JNJ-35814376 (threefold decrease in potency) and LY487379 (sixfold decrease in potency). Mutation L732A affected the potency of JNJ-46281222, JNJ-42329001 and LY2607540 and caused a complete loss of activity of JNJ-41482012 and LY487379. It is of note that changing this amino acid lowered the efficacy of these and other compounds (see e.g. LY2607540 in Figure 3). Finally, mutation N735D dramatically impacted the activity and potency of all the molecules (Table 2, Figure 3).
Mutations located in TM6
For the mutations localized in TM6, changing the tryptophan 773 residue (W773A) seemed to affect the potency of all compounds (Table 2, Figure 3). Although to a lesser extent, mutation F776A also significantly affected the activity of JNJ-40068782, and led to the complete loss of the receptor's ability to respond to increasing concentrations of LY487379. Mutation F780A showed similar results as the mGlu2-wt receptor.
Double and triple mutations
Mutation G689V/S688L caused significant shifts in the activity of all the tested molecules (3- to 11-fold potency decrease) with the exception of JNJ-40297036, JNJ-41482012 and LY487379. Mutation G689V/S688L/N735D had a major impact on the concentration–response curves for all the compounds. Finally, the triple mutation S644A/V700L/H723V decreased the activity of the majority of the tested compounds, with the exception of JNJ-40297036, JNJ-41482012 and BINA (Table 2).
Binding displacement of mGlu2 PAMs on mGlu2-wt receptor
We showed previously that JNJ-40068782, BINA, LY487379 and LY2607540 can fully displace [3H]-JNJ-40068782 (Lavreysen et al., 2013) and likely bind within the same pocket. We tested the additional mGlu2 PAMs included in this study for competition with [3H]-JNJ-40068782. Membranes prepared from stably mGlu2-wt receptor transfected CHO-K1 cells were used. JNJ-42329001 and JNJ-46281222 fully displaced [3H]-JNJ-40068782 with relatively high affinity (IC50 values of 23 and 30 nM, n = 2 for JNJ-42329001 and 19.5 ± 0.4 nM, n = 3 for JNJ-46281222), in line with these compounds being most potent in the [35S]-GTPγS assay (pEC50 values of 7.41 ± 0.18 for JNJ-42329001 and 8.09 ± 0.23 for JNJ-46281222; Table 2). Although the least potent JNJ-35814376 also inhibited binding with lower affinity (IC50 values of 417 and 676 nM, n = 2), the moderately potent JNJ-41482012 and JNJ-40297036 displaced the radioligand 36 ± 13% (n = 3) and 58 ± 9% (n = 2) at 10 μM respectively (Figure 4). So while some compounds may reside in an overlapping binding site, this suggests potential differences in binding for other allosteric modulators.
Figure 4.
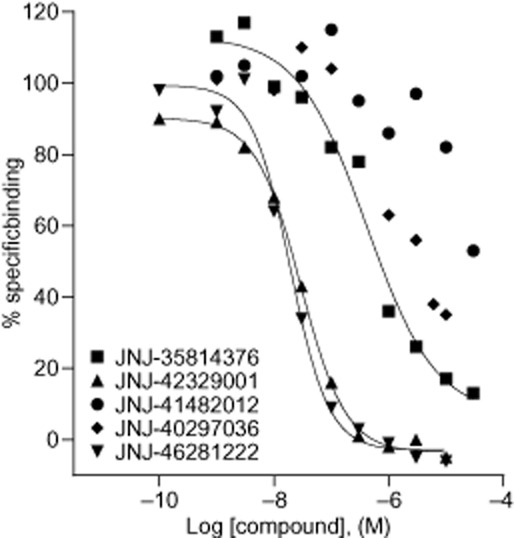
Displacement curves of [3H]-JNJ-40068782 binding to membranes prepared from CHO-K1 cells stably expressing the hmGlu2 receptor. Results are expressed as a % of specific binding and are from one representative experiment. The same results were found in at least one independent experiment.
Docking mGlu2 PAMs
The alignment of the transmembrane domains of human mGlu receptors 1–8 as well as rat and mouse mGlu2 receptor sequences are provided as Supporting Information Figure S2.
Molecules were docked into the homology model of the mGlu2 receptor built using mGlu1 as template (Figure 5). Important amino acids identified in the mutagenesis experiments are seen to cluster around the 7-TM binding site (Figure 5A). A consistent binding mode was identified among the top-ranked docking poses and four examples JNJ-40068782, JNJ-40297036, JNJ-46281222 and LY2607540 are shown in Figures 5B to E.
Figure 5.

(A) Amino acids, R635, L639, F643, S644, S688, G689, V700, H723, L732, N735, W773, F776, highlighted in the mGlu2 receptor model built using the mGlu1 receptor X-ray structure as template, the docked binding mode of JNJ-40068782 shown in sphere representation, coloured turquoise. View between TM5 and TM6 (cut-away) of the docked binding modes of (B) JNJ-40068782, (C) JNJ-40297036, (D) JNJ-46281222, (E) LY2607540 (THIIC). Selected important amino acids from experimental mutagenesis are shown in green. Amino acids and TM annotation is provided in (B) and consistent for (C), (D) and (E).
JNJ-40068782 (Figure 5B) binds in the 7-TM site interacting with the hydrophobic cluster formed between amino acids L639, F643, L732, W773 and F776 from TM3, TM5 and TM6. The cyclopropyl methyl substituent enters furthest into the receptor towards TM3 and TM5. The hydrophobic residues cluster tightly around the pyridone scaffold and the cyclopropyl methyl substituent. Space exists beyond the cyclopropyl methyl to go deeper towards W773 located on TM5, allowing larger groups in other mGlu2 PAMs such as the halogenated benzyl in JNJ-35814376. The carbonyl of the pyridone scaffold forms a hydrogen bond with N735. S644 forms an H-bond to the sidechain carbonyl oxygen of N735 suggesting that mutation of this residue may alter the position or orientation of N735 and disturb its interaction with the ligand. The 4-phenylpiperidine substituent in JNJ-40068782 is directed towards the extracellular side of the binding site and the distal phenyl sits between R635 and H723. As the binding site becomes larger in this region it permits bigger groups seen in this position for many PAMs. This greater space and flexibility is consistent with not all ligands being affected by the same mutations in this region. Also, the presence and importance of positively charged amino acids such as R635 in this vicinity is in agreement with structure–activity relationships (SAR), which allow distal carboxylic acids such as in BINA.
LY2607540 (Figure 5E) occupies the same 7-TM binding site interacting above the hydrophobic cluster of amino acids L639, F643, L732, W773 and F776. The lipophilic scaffold substituent, in this case isopropyl, again goes deepest into the receptor towards TM3 and TM5. It extends on top of F643 and forms a hydrophobic interaction with the edge of the phenylalanine. The phenolic scaffold sits in a similar space to the pyridone of JNJ-40068782 and also forms an H-bond with N735. The large ether substituent with the distal imidazole is directed perpendicular to the 7-TM helices towards the extracellular side. The active site has a bent shape and sufficient space, which is again consistent with the greater structural diversity in this region of the molecules.
Discussion
The present work represents the most extensive study to date of mGlu2 receptor amino acids important for allosteric activity. The activity of nine mGlu2 receptor PAMs was assessed on 39 human mGlu2 receptors carrying 7-TM mutations. The mutations were introduced outside of the extracellular domain and no change was observed in the affinity or potency of glutamate compared with mGlu2-wt receptor, confirming the integrity of the orthosteric site and the receptor. Amino acids N735 and W773 were identified as crucial for the activity of all the PAMs tested. In addition, F643, R635, L639, H723, L732 and F776 were shown to be important for multiple mGlu2 PAMs.
Effect on functional activity
Many factors can influence the potency of PAMs, including the binding affinity of the PAM, the affinity cooperativity between the PAM and glutamate, the efficacy cooperativity between PAM and glutamate and the level of receptor expression. Our data show that all mutant receptors get expressed at the cell membrane, but do not allow quantitative comparison of expression. Changes in [35S]-GTPγS effect because of either compound binding affinity and/or cooperativity can also not be excluded. Efforts are ongoing to assess potential effects on binding affinity. Unfortunately, [3H]-JNJ-40068782 binding to transiently transfected cells was too weak and novel radioligands are being evaluated. In addition, validating an operational model (according to Leach et al., 2007; Gregory et al., 2012) to analyze shifts of glutamate concentration–response curves by mGlu2 modulators may help delineate determinants for cooperativity versus affinity.
With regards to differences in functional or binding effects induced by the mutants it is of interest to highlight S688 and G689. These amino acids have been reported to influence binding of allosteric modulators at mGlu2 receptors, for instance LY487379 (Schaffhauser et al., 2003) and MRSLD-650 (a BINA analogue) (Rowe et al., 2008). However, it is clear from the homology model and the docking results (Figure 5) that S688 and G689 are not directly in the allosteric binding site. This is consistent with a study of mGlu2 receptor allosteric antagonists (Lundström et al., 2011) and suggests these amino acids cause indirect effects related to signalling. Alternatively, they may play a role in dimerization as the adjacent amino acids, I687 and Q690, may be at the dimer interface (Bruno et al., 2009).
Previously identified important amino acids were also crucial for the mGlu2 receptor PAMs tested in this study (G689V, N735D and S688L/G689V/N735D, Schaffhauser et al., 2003). Interestingly, mutations R635A (TM3), F643A (TM3), H723V (ECL2) and W773A (TM6), which were shown to affect NAM activity (Lundström et al., 2011), also affected the activity of the PAMs tested. In addition, residues in TM3 (L639), TM5 (L732) and TM6 (F776) are reported for the first time as affecting the activity of mGlu2 PAMs.
Common and unique amino acids important for allosteric mGlu modulation
Allosteric binding studies have been performed for other mGlu receptors. Mutation of two residues in TM3 of mGlu1 receptors (S668, C671) led to a loss of effect of the mGlu1 receptor PAMs diphenylacetyl-carbamic acid ethyl ester (Ro 01-6128) and (2S)-2-(4-fluorophenyl)-1-[(4-methylphenyl)sulfonyl]-pyrrolidine (Ro 67-7476). These amino acids were also relevant from the corresponding mGlu5 study by Pagano et al. (2000). A residue in TM5 (V757 in rat mGlu1 equivalent to L757 in human mGlu1) was also shown to be crucial for the binding of these compounds (Knoflach et al., 2001). Residues in TM3 (P655, S658) and TM7 (A810) were shown to be important for the binding of MPEP at mGlu5 (Pagano et al., 2000). Malherbe et al. (2003) identified eight additional residues in TM3, TM5, TM6 and TM7 crucial for MPEP binding to mGlu5. Y658, T780, S808, P654 and A809 in TM3, TM6 and TM7 were found to be important for mGlu5 PAM affinity (Gregory et al., 2013; 2014,), and a common allosteric binding site was proposed. The mGlu receptor sequence alignment in Supporting Information Fig. S2 facilitates comparison of these amino acids with mGlu2 receptor. Our data with [3H]-JNJ-40068782 also support an overlapping binding site for some mGlu2 allosteric modulators. In our study, mutations of residues F643 and L732, equivalent to residues P655 in mGlu5 and L757 in human mGlu1, respectively, were also seen to have a great impact on the compounds tested.
The exchange of mGlu2 receptor amino acid residues H723, G689 and N735 with the correspondent residues of mGlu3 receptor caused a significant impact on the activity of multiple PAMs tested in this study, which suggests, as seen previously for G689 and N735 (Schaffhauser et al., 2003) the importance of these residues for subtype specificity between mGlu2 and mGlu3.
Structural interpretation and binding mode of mGlu2 PAMs
The homology model of the mGlu2 receptor built from the recently solved mGlu1 X-ray structure was used to dock the mGlu2 receptor PAMs and suggest a possible binding mode. The mGlu1 structure offers the chance to build a class C GPCR atomic model based on a similar class C structural template. The most significant structural difference arising from the mGlu1 template is a tighter transmembrane binding site formed by the inwards movement of TM5 and TM7 compared with class A GPCR templates (Wu et al., 2014). Previous reports of mutagenesis studies on the mGlu2 receptor used rat sequence in the case of Lundström et al., 2011 and human in the cases of Schaffhauser et al., 2003 and Hemstapat et al., 2007. The sequence alignment (Supporting Information Appendix S5) demonstrates only three amino acids differ between species therefore permitting direct comparison of results.
We can be quite certain in the location of amino acids in the mGlu2 receptor model given the facile sequence alignment to the mGlu1 X-ray structure. Highlighting the location of the important amino acids in the mGlu2 receptor structure shows clearly how the majority: L639, F643, S644, L732, N735, W773 and F776 are closely clustered on the deep interior of the 7TM binding site (Figure 5A). However, R635 and H723 are located at the top of the binding site on the extracellular side. As commented earlier, S688, G689 and V700 do not make direct interactions with the ligand. Previous reports (Schaffhauser et al., 2003; Hemstapat et al., 2007; Rowe et al., 2008) have suggested a role for S688 and G689 in the binding of allosteric modulators at the mGlu2 receptor. All three studies assume direct interaction between these amino acids and the allosteric ligands. Lundström et al. (2011) subsequently constructed an mGlu2 receptor homology model based on the β2-adrenoceptor template and revealed that S688 and G689 (Ser4.44 and Gly4.45 in their work) were located outside the characterized 7-TM binding cavity. Our results based on a more accurate structural template using the mGlu1 receptor structure confirm this. Interestingly, functional activity of all the PAMs was affected by amino acids S688, G689 and N735, which are not conserved across the mGlu family. Therefore, they may still contribute to the subtype selectivity albeit in an indirect manner for S688 and G689.
The predicted binding mode was seen to be in good agreement with known SAR. Important pharmacophoric features and consistent interactions were found. A key feature of mGlu2 PAMs is the hydrogen bond acceptor on the scaffold. For each of the four compounds shown in Figure 5 this is satisfied by the interaction with the sidechain of N735. This is in agreement with mutation of N735 impacting the activity of all the molecules in this study. Three additional SAR features are consistent for all four molecules and present in the docked binding poses. Firstly, the requirement for a lipophilic substituent such as the isopropylmethyl from JNJ-40068782 is consistent with the binding of this group in the hydrophobic pocket formed by amino acids F643, W773 and F776. Secondly, the small scaffold substituents adjacent to the scaffold acceptor (e.g. CF3, -Cl, -CN, -Me) all occupy a similar space. Thirdly, the scope for broad substitution in the region of the distal phenyl in JNJ-40068782 is in accordance with this part of the binding site being larger and more open than the tighter binding pocket in the deeper 7TM region. JNJ-40068782, LY2607540 and JNJ-46281222 all showed potent displacement of [3H]JNJ-40068782 in agreement with the overlapping binding modes presented. However, some caution may be needed because the computational approach predicts a similar binding mode for JNJ-40297036 (Figure 5C), which only showed weak displacement of [3H]-JNJ-40068782, 58 ± 9% at 10 μM. Overall, the docking poses were consistent with the overlap of shared pharmacophoric features and SAR from our previously reported ligand overlay hypothesis and QSAR work (Tresadern et al., 2010; 2014).
In conclusion, the present study provides further additional insight on the interactions that drive the activity of PAMs towards the mGlu2 receptor and enabled the identification of amino acids that play a crucial role in PAM pharmacology. Multiple amino acids in TM3 (R635, L639, F643), TM5 (L732) and TM6 (W773, F776) were for the first time shown to have an effect on mGlu2 PAM activity, with many consistent between molecules suggesting a common binding site. The knowledge obtained from the mapping of allosteric modulators can contribute to guide research towards the achievement of more selective and potent drugs.
Acknowledgments
We thank Luc Gabriels for support with cell culture and transfection and Jean-Marc Neefs for assistance with sequence alignment. We would also like to acknowledge Addex Therapeutics, our partner involved in the discovery of multiple of the mGlu2 PAM compounds.
Glossary
- 7-TM
seven-transmembrane
- BINA
4-[3-[(2-cyclopentyl-6,7-dimethyl-1-oxo-2,3-dihydroinden-5-yl)oxymethyl]phenyl]benzoic acid
- JNJ-35814376
1-(4-chloro-2-fluorobenzyl)-5-(4-methoxyphenyl)pyridin-2(1H)-one
- JNJ-40068782
3-cyano-1-cyclopropylmethyl-4-(4-phenyl-piperidin-1-yl)pyridine-2(1H)-one
- JNJ-40264796
1-butyl-4-[4-(2-methylpyridin-4-yloxy)phenyl]-2-oxo-1,2-dihydropyridine-3-carbonitrile
- JNJ-40297036
2-propyl-8-chloro-6-[4-[(6-trifluoromethyl-3-pyridinyl)methoxy]phenyl]-1(2H)-isoquinolinone
- JNJ-41482012
7-(4-benzyloxy)-phenyl-3-(2,2,2-trifluoro-ethyl)-imidazo[1,2-a]pyridine-8-carbonitrile
- JNJ-42329001
7-[3-fluoro-4-(2′-methyl-pyridin-4-yloxy)-phenyl]-8-trifluoromethyl-3-cyclopropylmethyl-1,2,4-triazolo[4,3-a]pyridine
- JNJ-46281222
3-(cyclopropylmethyl)-7-[(4-phenyl-1-piperidinyl)methyl]-8-(trifluoromethyl)-1,2,4-triazolo[4,3-a]pyridine
- LY2607540 (THIIC)
N-({4-[3-hydroxy-4-(2-methylpropanoyl)-2-(trifluoromethyl)phenoxymethyl]phenyl}methyl)-1-methyl-1H-imidazole-4-carboxamide
- LY341495
(1S,2S)-2-[(2S)-2-amino-3-(2,6-dioxo-3H-purin-9-yl)-1-hydroxy-1-oxopropan-2-yl]cyclopropane-1-carboxylic acid
- LY487379
2,2,2-trifluoro-N-[4-(2-methoxyphenoxy)phenyl]-N-(pyridin-3-ylmethyl)ethanesulfonamide
- mGlu receptor
metabotropic glutamate receptor
- MNI-135
3-(8-iodo-5-oxo-3,4,5,6-tetrahydro-1,6-benzodiazocin-2-yl)benzonitrile
- MPEP
2-methyl-6-(2-phenylethynyl)pyridine
- PAM
positive allosteric modulator
- VU-48
4-nitro-N-(1-(2-bromophenyl)-3-phenyl-1H-pyrazol-5-yl) benzamide
Author contributions
A. F., L. P., B. R., H. L. and G. T. participated in research design. A. F., L. P., B. R. and G. T. conducted experiments. A. A. T., J. C. and S. M. contributed new reagents or analytic tools. A. F., L. P. and B. R. performed data analysis. A. F., H. L., G. T., L. P., B. R., S. M., A. A. T and J. C. wrote or contributed to the writing of the paper.
Conflict of interest
None.
Supporting Information
Appendix S1 Building the mGlu2 receptor homology model.
Appendix S2 Protein ligand docking.
Appendix S3 Data confirming the integrity of the orthosteric receptor binding site.
Appendix S4 Details on the initial screening of each PAM on all 39 mutant mGlu2 receptors.
Appendix S5 Sequence alignment.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefous C, Vernier J-M, Hutchinson JH, Gardner MF, Cramer M, James JK, et al. Biphenyl-indanones: allosteric potentiators of the metabotropic glutamate subtype 2 receptor. Bioorg Med Chem Lett. 2005;15:4354–4358. doi: 10.1016/j.bmcl.2005.06.062. [DOI] [PubMed] [Google Scholar]
- Bruno A, Guadix AE, Costantino G. Molecular dynamics simulation of the heterodimeric mGluR2/5HT2A complex. An atomistic resolution study of a potential new target in psychiatric conditions. J Chem Inf Model. 2009;49:1602–1616. doi: 10.1021/ci900067g. [DOI] [PubMed] [Google Scholar]
- Chiechio S, Nicoletti F. Metabotropic glutamate receptors and the control of chronic pain. Curr Opin Pharmacol. 2012;12:28–34. doi: 10.1016/j.coph.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Cid JM, Duvey G, Cluzeau P, Nhem V, Macary K, Raux A, et al. Discovery of 1,5-disubstituted pyridones: a new class of positive allosteric modulators of the metabotropic glutamate 2 receptor. ACS Chem Neurosci. 2010;1:788–795. doi: 10.1021/cn1000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid JM, Duvey G, Tresadern G, Nhem V, Funary R, Cluzeau P, et al. Discovery of 1,4-disubstituted 3-cyano-2-pyridones: a new class of positive allosteric modulators of the metabotropic glutamate 2 receptor. J Med Chem. 2012a;55:2388–2405. doi: 10.1021/jm2016864. [DOI] [PubMed] [Google Scholar]
- Cid JM, Tresadern G, Vega JA, de Lucas AI, Matesanz E, Iturrino L, et al. Discovery of 3-cyclopropylmethyl-7-(4-phenylpiperidin-1-yl)-8-trifluoromethyl[1,2,4]triazolo[4,3-a]pyridine (JNJ-42153605): a positive allosteric modulator of the metabotropic glutamate 2 receptor. J Med Chem. 2012b;55:8770–8789. doi: 10.1021/jm3010724. [DOI] [PubMed] [Google Scholar]
- Cid-Nuñez JM, Trabanco-Suárez AA, MacDonald GJ, Duvey GAJ, Luetjens RJ. 2008a. Preparation of 3-cyano-4-(4-phenylpiperidin-1-yl)-pyridin-2-one derivatives for treating and preventing neurol. and psychiatric diseases associated with glutamate dysfunction. PCT Int. Appln. WO 2008/107479 A1.
- Cid-Nuñez JM, Trabanco-Suárez AA, MacDonald GJ, Duvey GAJ, Lutjens RJ. 2008b. 1,4-Disubstituted 3-cyano-pyridone derivatives and their use as positive mgluR2-receptor modulators. PCT Int. Appln. WO 2008/107480 A1.
- Cid-Nuñez JM, Oehlrich D, Trabanco-Suárez AA, Tresadern GJ, Vega Ramiro JA, Macdonald GJ. 2010a. Preparation of 1,2,3-triazolo[4,3-a]pyridine for the treatment or prevention of neurological and psychiatric disorders. PCT Int. Appln. WO 2010/130424 A1.
- Cid-Nuñez JM, Trabanco-Suárez AA, Macdonald GJ. 2010b. Preparation of indole and benzoxazine derivatives as modulators of metabotropic glutamate receptors for treating neurological and psychiatric disorders. PCT Int. Appln. WO 2010/060589 A1.
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Dore AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–562. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD. Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology. 2008;33:1603–1610. doi: 10.1038/sj.npp.1301531. [DOI] [PubMed] [Google Scholar]
- Fell MJ, Witkin JM, Falcone JF, Katner JS, Perry KW, Hart J, et al. N-(4-( (2-(trifluoromethyl)-3-hydroxy-4-(isobutyryl)phenoxy)methyl)benzyl)-1-methyl-1H-imidazole-4-carboxamide (THIIC), a novel metabotropic glutamate 2 potentiator with potential anxiolytic/antidepressant properties: in vivo profiling suggests a link between behavioral and central nervous system neurochemical changes. J Pharmacol Exp Ther. 2011;336:165–177. doi: 10.1124/jpet.110.172957. [DOI] [PubMed] [Google Scholar]
- Galici R, Echemendia NG, Rodriguez AL, Conn PJ. A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J Pharmacol Exp Ther. 2005;315:1181–1187. doi: 10.1124/jpet.105.091074. [DOI] [PubMed] [Google Scholar]
- Galici R, Jones CK, Hemstapat K, Nong Y, Echemendia NG, Williams LC, et al. Biphenyl-indanone A, a positive allosteric modulator of the metabotropic glutamate receptor subtype 2, has antipsychotic- and anxiolytic-like effects in mice. J Pharmacol Exp Ther. 2006;318:173–185. doi: 10.1124/jpet.106.102046. [DOI] [PubMed] [Google Scholar]
- Ghose S, Gleason KA, Potts BW, Lewis-Amezcua K, Tamminga CA. Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: a mechanism for antipsychotic drug action? Am J Psychiatry. 2009;166:812–820. doi: 10.1176/appi.ajp.2009.08091445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, et al. Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure–activity relationships. Mol Pharmacol. 2012;82:860–875. doi: 10.1124/mol.112.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, et al. Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a ‘molecular switch’ in PAM pharmacology. Mol Pharmacol. 2013;83:991–1006. doi: 10.1124/mol.112.083949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Malosh C, Mendenhall JL, Zic JZ, Bates BS, et al. Identification of specific ligand–receptor interactions that govern binding and cooperativity of diverse modulators to a common metabotropic glutamate receptor 5 allosteric site. ACS Chem Neurosci. 2014;5:282–295. doi: 10.1021/cn400225x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Lorrain DS, Wei H, Cole RL, Zhang X, Daggett LP, et al. Distribution of metabotropic glutamate 2 and 3 receptors in the rat forebrain: implication in emotional responses and central disinhibition. Brain Res. 2008;1197:47–62. doi: 10.1016/j.brainres.2007.12.057. [DOI] [PubMed] [Google Scholar]
- Hemstapat K, de Paulis T, Chen Y, Brady AE, Grover VK, Alagille D, et al. A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol Pharmacol. 2006;70:616–626. doi: 10.1124/mol.105.021857. [DOI] [PubMed] [Google Scholar]
- Hemstapat K, Da Costa H, Nong Y, Brady AE, Luo Q, Niswender CM, et al. A novel family of potent negative allosteric modulators of group II metabotropic glutamate receptors. J Pharmacol Exp Ther. 2007;322:254–264. doi: 10.1124/jpet.106.117093. [DOI] [PubMed] [Google Scholar]
- Huang S, Cao J, Jiang M, Labesse G, Liu J, Pin J-P, et al. Interdomain movements in metabotropic glutamate receptor activation. Proc Natl Acad Sci U S A. 2011;108:15480–15485. doi: 10.1073/pnas.1107775108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imogai HJ, Cid-Nuñez JM, Duvey GAJ, Bolea CM, Nhem V, Finn TP, et al. 2006. Novel pyridinone derivatives and their use as positive allosteric modulators of mgluR2-receptors. PCT Int. Appln. WO 2006/030032 A1.
- Imogai HJ, Cid-Nuñez JM, Andrés-Gil JI, Trabanco-Suárez AA, Oyarzábal-Santamarina J, Dautzenberg FM, et al. 2007. 1,4-Disubstituted 3-cyano-pyridone derivatives and their use as positive mgluR2-receptor modulators. PCT Int. Appln. WO 2007/104783 A2.
- Johnson MP, Baez M, Jagdmann GE, Jr, Britton TC, Large TH, Callagaro DO, et al. Discovery of allosteric potentiators for the metabotropic glutamate 2 receptor: synthesis and subtype selectivity of N-(4-(2-Methoxyphenoxy)phenyl)-N-(2,2,2-trifluoroethylsulfonyl)pyrid-3-ylmethylamine. J Med Chem. 2003;46:3189–3192. doi: 10.1021/jm034015u. [DOI] [PubMed] [Google Scholar]
- Knoflach F, Mutel V, Jolidon S, Kew JNC, Malherbe P, Vieira E, et al. Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci U S A. 2001;98:13402–13407. doi: 10.1073/pnas.231358298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavreysen H, Langlois X, Ahnaou A, Drinkenburg W, te Riele P, Biesmans I, et al. Pharmacological characterization of JNJ-40068782, a new potent, selective and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H]JNJ-40068782. J Pharmacol Exp Ther. 2013;346:514–527. doi: 10.1124/jpet.113.204990. [DOI] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, et al. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol. 1999;55:453–461. [PubMed] [Google Scholar]
- Lundström L, Bissantz C, Beck J, Wettstein JG, Woltering TJ, Wichmann J, et al. Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: mechanistic studies with new potent negative allosteric modulators. Br J Pharmacol. 2011;164:521–537. doi: 10.1111/j.1476-5381.2011.01409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, et al. Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-Methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol. 2003;64:823–832. doi: 10.1124/mol.64.4.823. [DOI] [PubMed] [Google Scholar]
- Monn JA, Valli MJ, Massey SM, Wright RA, Salhoff CR, Johnson BG, et al. Design, synthesis, and pharmacological characterization of (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic Acid (LY354740): a potent, selective, and orally active group 2 metabotropic glutamate receptor agonist possessing anticonvulsant and anxiolytic roperties. J Med Chem. 1997;40:528–537. doi: 10.1021/jm9606756. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn JP. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, et al. The non-competitive antagonists 2-methyl-6-(phenethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group 1 metabotropic glutamate receptors. J Biol Chem. 2000;275:33750–33758. doi: 10.1074/jbc.M006230200. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011a;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011b;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe BA, Schaffhauser H, Morales S, Lubbers LS, Bonnefous C, Kamenecka TM, et al. Transposition of three amino acids transforms the human metabotropic glutamate receptor (mGluR)-3 positive allosteric modulation site to mGluR2, and additional characterization of the mGluR2 positive allosteric modulation site. J Pharmacol Exp Ther. 2008;326:240–251. doi: 10.1124/jpet.108.138271. [DOI] [PubMed] [Google Scholar]
- Schaffhauser H, Rowe BA, Morales S, Chavez-Noriega LE, Yin R, Jachec C, et al. Pharmacological characterization and identification of amino acids involved in the positive modulation of metabotropic glutamate receptor subtype 2. Mol Pharmacol. 2003;64:798–810. doi: 10.1124/mol.64.4.798. [DOI] [PubMed] [Google Scholar]
- Shigeyuki C, Hirohiko H. Targeting of metabotropic glutamate receptors for the treatment of schizophrenia. Curr Pharm Des. 2011;17:94–102. doi: 10.2174/138161211795049570. [DOI] [PubMed] [Google Scholar]
- Trabanco AA, Cid JM, Lavreysen H, Macdonald GJ, Tresadern G. Progress in the development of positive allosteric modulators of the metabotropic glutamate receptor 2. Curr Med Chem. 2011a;18:47–68. doi: 10.2174/092986711793979706. [DOI] [PubMed] [Google Scholar]
- Trabanco AA, Duvey G, Cid JM, Macdonald GJ, Cluzeau P, Nhem V, et al. New positive allosteric modulators of the metabotropic glutamate receptor 2 (mGluR2): identification and synthesis of N-propyl-8-chloro-6-substituted isoquinolones. Bioorg Med Chem Lett. 2011b;21:971–976. doi: 10.1016/j.bmcl.2010.12.048. [DOI] [PubMed] [Google Scholar]
- Trabanco AA, Duvey G, Cid JM, Macdonald GJ, Cluzeau P, Nhem V, et al. New positive allosteric modulators of the metabotropic glutamate receptor 2 (mGluR2): identification and synthesis of N-propyl-5-substituted isoquinolones. MedChemComm. 2011c;2:132–139. doi: 10.1016/j.bmcl.2010.12.048. [DOI] [PubMed] [Google Scholar]
- Trabanco AA, Tresadern G, Macdonald GJ, Vega JA, de Lucas AI, Matesanz E, et al. Imidazo[1,2-a]pyridines: orally active positive allosteric modulators of the metabotropic glutamate 2 receptor. J Med Chem. 2012;55:2688–2701. doi: 10.1021/jm201561r. [DOI] [PubMed] [Google Scholar]
- Trabanco AA, Cid JM. mGluR2 positive allosteric modulators: a patent review (2009–present) Expert Opin Ther Pat. 2013;23:629–647. doi: 10.1517/13543776.2013.777043. [DOI] [PubMed] [Google Scholar]
- Trabanco-Suárez AA, Tresadern GJ, Vega Ramiro JÁ, Cid-Nuñez JM. 2009. Preparation of imidazopyridine derivatives for use as mGluR2 receptor modulators. PCT Int. Appln. WO 2009/062676 A2.
- Tresadern G, Cid JM, Macdonald GJ, Vega JA, de Lucas AI, García A, et al. Scaffold hopping from pyridones to imidazo[1,2-a]pyridines. New positive allosteric modulators of metabotropic glutamate 2 receptor. Bioorg Med Chem Lett. 2010;20:175–179. doi: 10.1016/j.bmcl.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Tresadern G, Cid JM, Trabanco AA. QSAR design of triazolopyridine mGlu2 receptor positive allosteric modulators. J Mol Graph Model. 2014;53:82–91. doi: 10.1016/j.jmgm.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2012;62:1461–1472. doi: 10.1016/j.neuropharm.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344:58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Building the mGlu2 receptor homology model.
Appendix S2 Protein ligand docking.
Appendix S3 Data confirming the integrity of the orthosteric receptor binding site.
Appendix S4 Details on the initial screening of each PAM on all 39 mutant mGlu2 receptors.
Appendix S5 Sequence alignment.