

Some researchers believe that a tumor's heterogeneity provides crucial clues about how cancers respond to treatment.
Looking down from an airplane window, it’s hard to appreciate the diversity of plants that thrive in a lush forest thousands of feet below. The shade of green might hint at a dominant species, but what else is mixed in? And what’s growing below those highest treetops? Cataloguing the trees at the edge of the forest is also little help. Is the other side of the forest the same? It’s nearly impossible to tell what’s really sustaining this complex ecosystem.
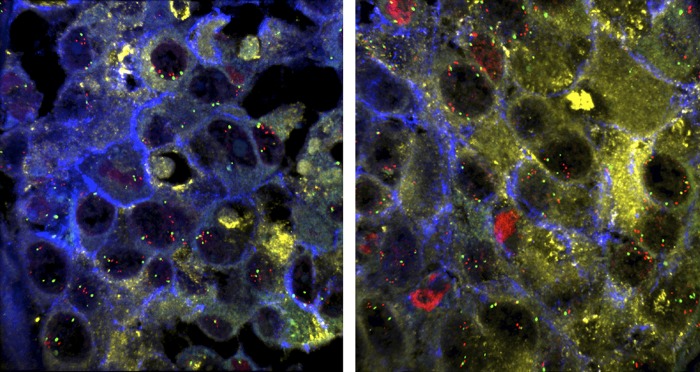
The images reveal tumor heterogeneity by comparing distant metastases in the same patient, in the spleen (Left) and lung (Right). Images courtesy of Kornelia Polyak (Harvard Medical School, Boston) (10).
For oncologists and cancer researchers trying to determine what kinds of cells are thriving and multiplying to keep tumors growing, every patient’s cancer is a dense forest of sorts. Over the past decade, it’s become increasingly clear not only that no two patients’ cancers are exactly the same, but even within one person’s tumor there is a wild diversity of cells with tangled, branching pedigrees. One section of a tumor might be dense with cells containing a particular cancer-causing mutation, whereas another section—or stray cells scattered throughout the whole tumor—might have vastly different mutations driving their growth or enabling their spread. Like surveying the edge of a forest, studying a single biopsied sliver of a tumor can fail to illuminate what lies within.
And yet, in the spirit of “personalized medicine,” physicians are increasingly turning toward such biopsies to guide their treatment decisions for a cancer. On first blush, this seems a reasonable approach: a specific genetic mutation in a tumor’s cells may make it vulnerable to a particular targeted drug. However, if the chosen drug works only against some subset of cells, or against some fraction of the tumor metastases that have cropped up throughout the body, the unaffected cells can continue to thrive, helping the cancer grow and adapt to resist the drug. If clinicians could predict this response—by gauging the tumor’s heterogeneity before treatment—they might be able to plan a treatment strategy that blocks resistance from developing in the first place. Before doctors can begin using tumors’ heterogeneity as a weapon against them, however, the first hurdle is just finding ways to measure that diversity.
“Every cell in a tumor could theoretically be different,” says oncologist Charles Swanton of the London Research Institute. “If you take a metastatic tumor with millions of cells, and assume that every single cell is different, you’re starting to get a sense of the enormity of this challenge.”
To tackle the problem, Swanton and others are pursuing a variety of ways to quantify and describe a tumor’s heterogeneity. They’re relying on gene sequencing that’s cheaper and faster than ever before, high-throughput methods to analyze the properties of single cells, powerful computational approaches, and novel techniques that can reveal previously undetected differences in the way cells act and interact.
The Extent of Diversity
A decade ago, Swanton was becoming increasingly frustrated by how often his cancer patients developed resistance to drugs that seemed at first to be working. The most logical explanation, he reasoned, was a kind of natural selection: as tumors grow and pick up new genetic mutations, some of the new cell lines they generate, inevitably, are resistant to the drugs. To get a sense of the origins and extent of this resistance, Swanton started taking multiple biopsies from individual cancer patients.
In a 2012 New England Journal of Medicine paper, Swanton’s group reported their first analyses of duplicate biopsies collected from advanced kidney cancers (1). When they compared any two tumor biopsies from the same patient, the scientists found the samples shared only about a third of their total mutations. In other words, two-thirds of the mutations seen in any given biopsy were unique to that particular spot in the tumor. It was some of the first quantifiable genetic evidence of just how diverse tumors are.
More recently, Swanton analyzed lung cancers removed from seven different patients. He and his collaborators took a whopping 25 biopsies from each tumor and its metastases and sequenced the full genomes from each biopsy. The results, published in Science in October 2014, were similar to what they’d seen in kidney cancers: 30% of the mutations from any one biopsy were not shared between samples (2). However, Swanton says, even with 25 biopsies it’s unlikely they’re capturing the real degree of diversity. If a mutation is present in only one percent of cells—or less—it can be easily missed.
“At the moment, we don’t have a sense of how close we are to gauging the full extent of the heterogeneity,” he says. “We don’t know how deep this rabbit hole is.”
Doing the MATH
Even without cataloging every mutation in every cell of every tumor, finding other ways to quantify the extent of a tumor’s heterogeneity can hint at how hard it will be to treat. “If you have a tumor with very low heterogeneity, you’re likely to get a great response, or even a complete cure, when you target it with drugs,” explains Jim Rocco, a surgeon at Massachusetts Eye and Ear who specializes in head and neck cancers. A tumor that’s homogeneous, he says, is statistically less likely to have rare drug-resistant variations.
Rocco has collaborated with computational biologists to come up with a method to derive a single number that quantifies the level of heterogeneity in a tumor. Rocco calls it mutant-allele tumor heterogeneity, or MATH. Rather than single-out individual genes—like what percentage of cells have a mutation in the tumor suppressor gene p53—MATH looks at the full exome sequences of cancer cells and quantifies how much variation, on average, there is between those sequences. Thus, even within a single biopsy, Rocco’s group can detect the level of diversity among cells.
“With MATH, you don’t care what the genes are,” Rocco says. “You don’t have preexisting ideas like, ‘let’s look at the estrogen receptor because we think that might be important.’ You’re blinding yourself to all that.”
In their first application of MATH to human tumors, Rocco and his collaborators determined the MATH scores of 74 head and neck squamous cell carcinomas, with results reported in 2013 (3). The MATH values for the tumors ranged from 19—the least heterogeneous—to 55. The levels, they found, correlated with common prognostic factors; people who had smoked heavily, for example, and are known to have highly drug-resistant cancers and poorer outcomes, also had the most heterogeneous tumors. When they followed up with the patients over time, Rocco’s group found something else: the patients with the highest MATH values fared the worst after surgery and, on average, had the shortest survival. The results were presented at the 2014 Multidisciplinary Head and Neck Cancer Symposium in February.
Rocco is now collaborating with other clinicians to test MATH’s prospective power in trials. If the technique’s utility holds true, he says, patients with the highest MATH scores at the time of diagnosis could be shuttled into more aggressive or experimental treatment strategies. “If someone comes in with a basic tongue cancer but it turns out to have a very high MATH score, we might want to consider surgery instead of just starting out with radiation.” Likewise, he says, the scores could help avoid overtreating people with the least-aggressive tumors.
In a technique that’s similarly blind to which genes may be altered, a team at Harvard Medical School recently turned to so-called polyguanine repeats, strings of many guanine nucleotides in a row, to determine how diverse tumor cells were. When cells copy themselves, mutations frequently creep into these repetitive areas of the genome, so
“Until we study tumors as a whole—not as single lines of gene mutations in a dish—we won't be able to fully understand cancer in people.”
Kornelia Polyak
the guanine stretches can vary even among cells that have recently diverged. Without full sequencing, researchers can use a simple PCR-based assay to tell how different these regions are by measuring their length.
The Harvard team focused on colon cancer for their first test run of the technique. With samples of primary tumors and metastases from 22 patients, Kamila Naxerova and her colleagues built lineage trees showing when each metastasis—some in the liver, others in lymph nodes or ovaries—diverged from the primary colon cancer. The more different the polyguanine regions were between two samples, the longer ago these cell populations had separated. The technique was described in PNAS in April (4).
There could be important diagnostic implications. As a cancer spreads, it becomes more heterogeneous; isolated populations of cells pick up new mutations that aren’t shared between metastases. However, if a primary tumor is heterogeneous to begin with, it might also be more prone to spread, or at least more likely to contain scattered cells with the right properties to seed new metastatic tumors. Naxerova’s group is now using polyguanine repeats to build tumor family trees and study basic questions about how tumor heterogeneity may contribute to a cancer’s ability to spread, or perhaps signal its likelihood.
Just the Beginning
When ecologists trek through the woods, counting trees and seedlings and undergrowth, they usually have a larger goal in mind: understanding the underlying processes sustaining the ecosystem. Similarly, researchers homing in on tumor diversity want to know how this heterogeneity helps a cancer grow, how it may change the microenvironment surrounding the tumor or the activity of proteins that mediate key pathways within the cells.
“Genetics is kind of the tip of the iceberg when it comes to tumor heterogeneity,” says breast cancer researcher Kornelia Polyak at Harvard Medical School. She has shown that the interactions among diverse cells could also be part of what makes heterogeneous tumors more aggressive. In an October 2014 Nature paper, Polyak’s laboratory described how they divided a breast cancer tumor into 18 smaller tumors based on the heterogeneous sections of the original (5). Although all of the more homogenous smaller tumors grew on their own, they grew much faster when mixed back together into a more complex assemblage. “Until we study tumors as a whole—not as single lines of gene mutations in a dish—we won’t be able to fully understand cancer in people,” Polyak says.
“The more resolution we get, the more heterogeneity we see,” says Mandana Veiseh, a senior scientist at the Palo Alto Research Center who conducted the work while she was a staff scientist at E. O. Lawrence Berkeley National Laboratories. “We reveal many details that we’re missing with less-sensitive methods.” Veiseh recently used a fluorescent probe technique to detect and quantify the build-up of the sugar hyaluronan in a small percentage of the cells in slow-growing but invasive breast tumor cell lines that were thought to be homogenous; the results were reported last April (6). Eva Turley of the London Health Science Centre had previously linked the accumulation of hyaluronan to the likelihood that a tumor would spread (7). The unexpected heterogeneity they found might not have been picked up with another technique, Veiseh notes.
Improving the methods, she adds, may not only benefit cancer researchers. “We’re focused on cancer, but I wouldn’t be surprised at all if people looking at other diseases start realizing they’re also dealing with heterogeneity,” Veiseh says. The more techniques researchers have to capture heterogeneity in a more analytical and quantitative way, the greater the chances of moving them into the clinic, says Veiseh.
The View From Above
Armed with all of these new approaches, researchers are still faced with a familiar question: How do you see the forest for the trees? Once heterogeneity is measured, what does it mean for cancer patients in the clinic? Most say it’s too soon to know which methods might prove most useful, and what clinical applications of these tests would look like. Just working the idea of intrapatient heterogeneity into clinical trials has been challenging, says Lillian Siu, an oncologist at the University of Toronto (8).
“I think the technology is actually ahead of the clinical trial design science,” says Siu. “We can easily sequence the whole genomes of individual tumor cells. But understanding what that means and then how to integrate that into clinical trials is a different matter.” Trials typically rely on having a group of relatively homogenous patients to give statistical power to any finding. If all of the complexity of heterogeneity is introduced, it’s nearly impossible to find a large group of patients whose cancers match exactly. “If you look at heterogeneity over both space and time, you end up with each patient essentially being their own unique trial,” Siu points out.
What will help integrate measures of heterogeneity into trials, she says, is more basic research into which factors are important to a tumor’s growth. If a forest is dominated by oaks, it might not make a difference to the overall ecosystem whether there are a few daisies on the edges or a few orchids. Similarly, not all heterogeneity will affect the cancer’s biology.
Bert Vogelstein, an oncologist at The Johns Hopkins University, whose group completed 88 of the first 100 full exome sequences of cancers, agrees. “I think many people are being misled by finding lots of heterogeneity among primary tumors,” says Vogelstein. “Oftentimes, there’s little heterogeneity among driver mutations, which is what matters” (9).
Mutated genes that are already known to drive a cancer’s ability to grow, spread, and resist treatment are still the most critical to identify, says Vogelstein. Surgeons can usually cut out a primary tumor, but clinicians frequently turn to drugs to treat metastatic tumors that cannot be reached any other way. Characterizing the molecular heterogeneity of these metastases is where new technology has a chance to make a difference, he says.
In October, more than 200 researchers from across fields gathered at Stanford Medicine for a two-day symposium on tumor heterogeneity, the first of its kind. According to Beverly Mitchell, director of the Stanford Cancer Institute, one of the takeaways was the possibility that quantifying tumor heterogeneity is undermining the current emphasis on personalized cancer treatments that target a particular tumor gene or protein. “We talk more and more about precision medicine, but I think this brought all the complexity to the floor,” she says. “One of the conclusions was that trying to attack one mutation at a time is futile.”
Even putting together personalized mixtures of targeted drugs seems daunting in some cancer types, Mitchell says. Instead, conference attendees were more optimistic about broader treatment strategies, such as therapies that train a patient’s immune cells to recognize and attack the whole tumor. Vogelstein, too, points to the promise of drugs that would undermine an entire pathway containing many of the genes that might be mutated in a heterogeneous tumor. As measures of heterogeneity mature, they could help direct certain patients toward these broader treatment approaches.
“We are at the very, very early stages of starting to see trials that look at whether the heterogeneity of certain markers is relevant or not,” Siu says. “It still seems so complex now, but I think ultimately, as we keep working on this and figuring out new ways of measuring heterogeneity, we’ll be able to put our finger on how to treat cancers with those measures in mind.”
References
- 1.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Bruin EC, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346(6206):251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mroz EA, Rocco JW. MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral Oncol. 2013;49(3):211–215. doi: 10.1016/j.oraloncology.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naxerova K, et al. Hypermutable DNA chronicles the evolution of human colon cancer. Proc Natl Acad Sci USA. 2014;111(18):E1889–E1898. doi: 10.1073/pnas.1400179111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marusyk A, et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514(7520):54–58. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veiseh M, et al. Cellular heterogeneity profiling by hyaluronan probes reveals an invasive but slow-growing breast tumor subset. Proc Natl Acad Sci USA. 2014;111(17):E1731–E1739. doi: 10.1073/pnas.1402383111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamilton SR, et al. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J Biol Chem. 2007;282(22):16667–16680. doi: 10.1074/jbc.M702078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501(7467):355–364. doi: 10.1038/nature12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almendro V, et al. Genetic and phenotypic diversity in breast tumor metastases. Cancer Res. 2014;74(5):1338–1348. doi: 10.1158/0008-5472.CAN-13-2357-T. [DOI] [PMC free article] [PubMed] [Google Scholar]