

Significance
Our study defines the crucial role of CD14-high bladder cancer (BC) cells in orchestrating multiple hallmarks of cancer in the early stages of BC. Inflammatory factors produced by this subpopulation of tumor cells activate angiogenesis to support establishment and maintenance of an immune-suppressive, inflammatory tumor microenvironment. Additionally, this subpopulation is able to drive tumor growth by producing factors that drive autocrine and paracrine proliferative stimulation. Here we show that a tumor-cell subpopulation establishes a tumor microenvironment orchestrating tumor-promoting inflammation and tumor-cell proliferation. Collectively, this study highlights the need to explore the broader role of CD14-expressing neoplastic cells in other solid tumors. It is noteworthy that CD14 expression is critical for IL6 secretion by these cells. Therefore, therapeutic targeting of CD14 might represent a strategy for treating cancer.
Keywords: bladder cancer, CD14, inflammation, microenvironment
Abstract
Nonresolving chronic inflammation at the neoplastic site is consistently associated with promoting tumor progression and poor patient outcomes. However, many aspects behind the mechanisms that establish this tumor-promoting inflammatory microenvironment remain undefined. Using bladder cancer (BC) as a model, we found that CD14-high cancer cells express higher levels of numerous inflammation mediators and form larger tumors compared with CD14-low cells. CD14 antigen is a glycosyl-phosphatidylinositol (GPI)-linked glycoprotein and has been shown to be critically important in the signaling pathways of Toll-like receptor (TLR). CD14 expression in this BC subpopulation of cancer cells is required for increased cytokine production and increased tumor growth. Furthermore, tumors formed by CD14-high cells are more highly vascularized with higher myeloid cell infiltration. Inflammatory factors produced by CD14-high BC cells recruit and polarize monocytes and macrophages to acquire immune-suppressive characteristics. In contrast, CD14-low BC cells have a higher baseline cell division rate than CD14-high cells. Importantly, CD14-high cells produce factors that further increase the proliferation of CD14-low cells. Collectively, we demonstrate that CD14-high BC cells may orchestrate tumor-promoting inflammation and drive tumor cell proliferation to promote tumor growth.
Solid tumors represent a complex mass of tissue composed of multiple distinct cell types (1, 2). Cells within the tumor produce a range of soluble factors to create a complex of signaling networks within the tumor microenvironment (3–7). One of the outcomes of this crosstalk is tumor-promoting inflammation (TPI) (8, 9). TPI can modulate the functions of tumor-infiltrating myeloid lineage cells including macrophages (10–12). Tumor-associated macrophages (TAMs) consistently display an alternatively activated phenotype (M2) commonly found in sites of wound healing (13–18). These macrophages promote tumor growth while suppressing the host immune response locally (19–22). Polarization and subversion of tumor-infiltrating macrophages is accomplished via immune mediators in the tumor microenvironment (23, 24). Adding to the complexity of solid tumors is the heterogeneity of the cancer cells (2). Tumor cells of varying differentiation states and different characteristics coexist within a tumor (25–29). However, the different roles of each tumor cell subset during cancer progression remain undefined.
Bladder cancer (BC) represents a growing number of solid tumors characterized by the infiltration of a significant number of myeloid cells in the neoplastic lesion (30, 31). We have previously determined that keratin 14 (KRT14) expression marks the most primitive differentiation state in BC cells (32). KRT14 expression is significantly associated with poor overall patient survival. However, the mechanisms used by KRT14-expressing cells to promote tumor growth remain unclear. In the current study, we found that KRT14+ basal BC cells also express higher levels of CD14. Here, we investigate the strategies used by KRT14+ CD14-high BC cells to promote tumor growth.
Results
KRT14+ Basal BC Cells Express Higher Levels of CD14.
We have previously identified KRT14 expression as a marker specific for the primitive/basal differentiation state in BC (32). We further determined that CD90+ cells, which express higher levels of KRT14, represent the tumorigenic subpopulation in primary patient basal BC. Interestingly, macrophage-associated markers including CD14 were also enriched in KRT14+ BC. Flow cytometry and histology of patient BC samples indicate that BC cells expressing the epithelial lineage marker EpCAM coexpress CD14 (Fig. S1A). Quantitative real-time PCR also revealed that CD90+ basal BC cells express higher levels of CD14 (Fig. S1B).
CD14-High BC Cells Express Higher Levels of Inflammatory Mediators.
CD14 expression in CD90+ BC cells may potentially play a functional role in tumor cells. To specifically study the role of CD14 expression, we used a human BC cell line, 639V. 639V is a human tumor cell line established from a grade III bladder transitional cell carcinoma (33). 639V cells uniformly expresses CD90 on their surface, indicating that they were originally derived from basal BC cells. Mirroring our observations with primary patient cancer samples, tumor cells express CD14 on their surface (Fig. S1C). CD14 is expressed in a continuous fashion from negative to high, complicating the delineation of distinct cell populations. However, to investigate the role of CD14 expression on tumor cells, we determined the gate for CD14-low cells based on the isotype control, whereas the gate for CD14-high cells was designed to enrich for cells that had the highest expression of CD14. We isolated CD14-high and CD14-low subpopulations from the bulk 639V tumor cell line using serial FACS sorting and passaging (Fig. 1A and Fig. S1 D and E). Interestingly, KRT14 expression is significantly increased in the CD14-high cells (Fig. 1B), indicating that CD14-high 639V cells possibly represent the basal KRT14+ BC subpopulation observed in primary BC samples. Furthermore, CD14-high cells lose CD14 expression gradually during in vitro passaging and in vivo tumor formation, indicating possible differentiation of the CD14-high cells into the CD14-low phenotype.
Fig. 1.

CD14-high BC cells express higher levels of inflammation factors. (A) Serial FACS isolation of CH14-high and CD14-low subpopulations from bulk 639V tumor cells and flow cytometry analysis of CD14 levels on each 639V subpopulation. (B) Quantitative PCR (qPCR) of KRT14 mRNA in CD14-low and CD14-high 639V cells (n = 4) (mean and SEM; P < 0.0001). (C) Cytokine profiling of CD14-high and CD14-low 639V cells using Luminex arrays (n = 4) (mean and SEM; IL6, P < 0.0001; IL8, P = 0.0028; M-CSF, P = 0.0035; VEGF, P = 0.0341; FGF-2, P = 0.0250). (D) Real-time PCR of inflammatory factors in CD90− and CD90+ tumor cells from basal patient BC samples (n = 3). Tumor cells were gated based on live, lineage (CD45, CD31)-negative cells (mean and SEM; IL6, P = 0.0245; IL8, P = 0.0077; M-CSF, P = 0.0266; VEGF, P = 0.0010; FGF-2, P = 0.0207).
We evaluated the cytokine production profile of CD14-high, -low, and bulk-unsorted 639V cells. CD14-high cells produce higher levels of various inflammation mediators, including cytokines, chemokines, growth factors, and angiogenic factors in the absence of lipopolysaccharide (LPS) stimulation (Fig. S1F). Specifically, differences in IL6, IL8, M-CSF, VEGF-A, and FGF-2 production were found to be most dramatic in the CD14-high vs. -low cells (Fig. 1C) (2, 34, 35). Bulk unsorted cells generally have a cytokine production profile that resembles CD14-low cells. The augmented expression of these mediators was also observed in primary human samples (Fig. 1D). These results revealed that CD14 expression in BC marks cells that express higher levels of various inflammation mediators.
CD14-High BC Tumors That Are More Vascularized and Have Higher Frequencies of Myeloid Cell Infiltration.
To evaluate the functional role of CD14 expression in BC cells, CD14-high and -low 639V cells were transplanted s.c. into immune-deficient NOD scid gamma (NSG) mice that allow the engraftment of human cells. CD14-high 639V cells formed smaller tumors than CD14-low cells (Fig. S1G). Interestingly, the cellular composition of the tumors was significantly different. CD14-high tumors were characterized by higher frequencies of total blood lineage cells (CD45+) and endothelial cells (CD31+) (Fig. S1 H and I). Further examination of different myeloid cell compartments demonstrated that the frequencies of macrophage (CD11b+ F4/80+) and dendritic cell (DC) (CD11b+ CD11c+) infiltrations were higher in CD14-high tumors (Fig. S1 J–L).
Inflammatory mediators produced by CD14-high tumor cells possibly exert wide-ranging effects on different immune cell compartments during tumor growth (23). Therefore, lack of B, T, and natural killer (NK) cells in severe combined immunodeficiency mice and potential cross-reactivity issues between human cytokines in mouse immune cells complicates the interpretation of experiments with 639V cells. Accordingly, we developed a syngeneic BC model in wild-type mice. We focused on the MB49 model, which shares several interesting similarities with human BC regarding cell-surface markers, sensitivity to apoptosis, and immunological profile (36–38). Similar to 639V, CD14 is expressed in a continuous fashion from negative to high on MB49 cells (Fig. S2A). Using serial FACS sorting and passaging, we isolated CD14-high and CD14-low subpopulations from the bulk MB49 cell line (Fig. 2A and Fig. S2 B and C). The gate for CD14-low cells was based on the isotype control, whereas the gate for CD14-high cells was designed to enrich for MB49 cells that had the highest expression of CD14. Interestingly, CD14 expression on CD14-high MB49 cells was stable over time through multiple passages.
Fig. 2.

CD14-high BC tumors that are more vascularized and have higher frequencies of myeloid cell infiltration. (A) Serial FACS isolation of CD14-high and CD14-low subpopulations from bulk MB49 tumor cells and flow cytometry analysis of CD14 levels in each MB49 subpopulation. (B) Differences in inflammatory small-molecule, cytokine, and chemokine production between CD14-high and CD14-low mouse BC cells measured by ELISA (mean and SEM; NO, P < 0.0001; PGE2, P = 0.0124; IL6, P = 0.0062; CXCL2, P < 0.0001; CXCL5, P < 0.0001; CXCL1, P < 0.0001; CCL3, P = 0.0001; G-CSF, P < 0.0001; LIF, P < 0.0001). (C) Growth of ectopic tumors formed by s.c. injection of CD14-high (n = 8) and CD14-low (n = 9) MB49 mouse BC subpopulations into syngeneic wild-type C57BL/6 mice after 4 wk (mean and SEM; P = 0.0011). (D and E) Frequencies of total hematopoietic cell (CD45+) and endothelial cell (CD31+) infiltration in tumors formed by CD14-high and CD14-low MB49 subpopulations (n = 5) (mean and SEM; Hematopoietic cells, P = 0.0020; Endothelial cells, P = 0.0014). (F–H) Frequencies of myeloid cell infiltration in tumors formed by CD14-high and CD14-low MB49 subpopulations in syngeneic wild-type C57BL/6 (n = 5). Macrophages/monocytes (CD11b+ F4/80+); DCs (CD11b+ CD11c+); Granulocytes (CD11b− Gr1+) (mean and SEM; Macrophages/monocytes, P = 0.0122; DC, P = 0.0153; Granulocytes, P = 0.0001). (I) IL6 and CCL3 production by CD14 KO cells measured by ELISA (n = 3) (mean and SEM; IL6, P < 0.0001; CCL3 < 0.0001). (J) Growth of ectopic tumors formed by s.c. injection of CD14-low (n = 5), CD14-high (n = 5), and CD14 KO (n = 8) cells into syngeneic wild-type mice after 4 wk (mean and SEM; P < 0.0001; P = 0.4653, not significant).
CD14-high MB49 cells produce higher levels of numerous inflammatory mediators compared with CD14-low and bulk-unsorted MB49 cells (Fig. S2D). CXCL1, CXCL2, CXCL5 (mouse functional homologs of IL8), and IL6 production is higher in CD14-high cells (Fig. 2B and Fig. S2E). Additionally, CD14-high cells secrete higher levels of inflammatory small molecules (nitric oxide, prostaglandin E2), myeloid cell chemokines (CCL3), and cytokines (G-CSF, LIF). Consistent with the cytokine production profile, mRNA gene expression profiling and gene set enrichment analysis (GSEA) revealed that CD14-high MB49 cells express higher levels of several genes associated with inflammation and hypoxia (Fig. S3 A and B).
We compared tumor growth of CD14-high and -low MB49 cells by s.c. implantation into syngeneic wild-type C56BL/6 mice. CD14-high tumors are more vascularized and have a higher frequency of blood lineage cell infiltration (Fig. 2 D and E and Fig. S2F). Frequencies of granulocyte, macrophage, and DC infiltration are also increased in CD14-high tumors (Fig. 2 F–H). However, contrary to the human BC cell line model, mouse CD14-high cells formed larger ectopic tumors (Fig. 2C).
The discrepancy of tumor growth between the xenotransplantation and syngeneic model could possibly be caused by the absence of B, T, and NK cells in the former model. To evaluate this possibility, tumor growth of different MB49 subpopulations was assessed after implantation into syngeneic C57BL/6 RAG−/− γc−/− mice lacking B, T, and NK cells. Tumor growth for both MB49 subpopulations is significantly faster in RAG−/− γc−/− mice compared with wild-type hosts, providing evidence that an antitumor response is typically mounted in immune-competent hosts to suppress tumor growth (Fig. S2G). However, CD14-high and -low subpopulations show no significant difference in tumor growth, indicating that the larger tumors formed by CD14-high cells in an immune-competent host could be due to its enhanced ability to suppress an antitumor response. Additionally, both CD14-high and -low tumor cells express many T and NK cell ligands, including H2-K1, CD276 (B7-H3), and Rae-1 with notable differences in CD40 expression. Collectively, the differences in human and mouse in vivo tumor growth can be reconciled by the differential ability of the tumor cell subpopulations in modulating their respective hosts’ immune cells. Therefore, subsequent studies exploring the functional role CD14-high BC cells focused on the MB49 BC model in syngeneic immune-competent C57BL/6 mice.
CD14-Mediated Signaling Is Required for IL6 Production and Larger Tumor Formation.
To evaluate the role of CD14 expression on cytokine secretion, CD14 expression on CD14-high MB49 cells was knocked out by transcription-activator–like effector nucleases (TALENs) (Fig. S2H). We isolated CD14-high MB49 cells that have lost CD14 expression (CD14 KO) by FACS and found that these cells produce drastically reduced levels of IL6 and CCL3 (Fig. 2I). Furthermore, when implanted into mice, CD14 KO cells form significantly smaller tumors that are comparable to CD14-low cells (Fig. 2J). These results demonstrate that CD14 expression is essential for cytokine production and tumor growth of CD14-high MB49 cells.
CD14 has been shown to be critically important in the signaling pathways of Toll-like receptor (TLR) 2, 3, 4, 7, and 9 on macrophages (39, 40). To identify upstream signaling components facilitating cytokine secretion, CD14-high MB49 cells were treated with a series of compounds that target different components of TLR signaling (Fig. S2I). Inhibition of CD14 by a blocking antibody or OxPAPC results in reduction of IL6 production. Blocking of TLR2, 3, 4, 7, and 9 by monoclonal antibodies or chloroquine similarly results in inhibition of IL6 production. The role of adaptor molecules downstream of TLRs was also evaluated using blocking peptides. Inhibition of MyD88 or TIR domain-containing adaptor protein (TIRAP), but not TIR domain-containing adapter-inducing interferon β (TRIF), inhibited IL6 production. Taken together, these results suggest that CD14-induced IL6 secretion can be mediated by multiple TLR-signaling pathways.
Inflammation Mediators from CD14-High BC Cells Promote Immune Suppression.
To evaluate whether soluble factors produced by CD14-high cells might have a direct role in promoting increased myeloid cell recruitment in CD14-high tumors, conditioned media (CM) from different MB49 subpopulations was collected, filtered, and assessed for their relative abilities to induce chemotaxis of different myeloid cells in vitro. CD14-high CM was more efficient in inducing migration of macrophages, monocytes, and neutrophils across a transwell, indicating that factors secreted by CD14-high cells can act directly on these myeloid cells to promote their recruitment (Fig. 3 A–C). In contrast, CD14-high CM inhibited migration of bone-marrow–derived dendritic cells in vitro, possibly reflecting the ability of CD14-high BC cells in retaining the DCs at a neoplastic lesion in vivo and preventing their migration to draining lymph nodes to activate T cells (Fig. S4A).
Fig. 3.
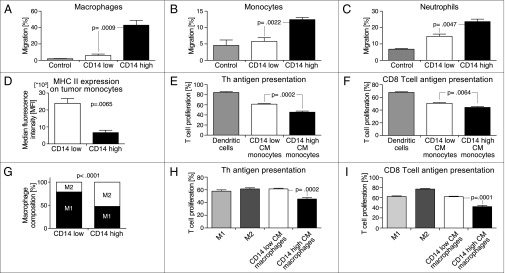
Inflammation mediators from CD14-high BC cells promote immune suppression. (A–C) Chemotaxis of macrophages, monocytes, and neutrophils across a transwell in response to different tumor CMs (n = 4) (mean and SEM; Macrophages, P = 0.0009; Monocytes, P = 0.0022; Neutrophils, P = 0.0047). (D) MHC II expression on tumor-infiltrating monocytic cells (Gr-1+ CD11b+) from each BC subpopulation (n = 4) (mean and SEM; P = 0.0065). (E) Presentation of OVA to TCR transgenic CD4+ Th cells by tumor CM-differentiated monocytes at 1:2.5 APC:T-cell ratio. Frequency of proliferating cells was determined by gating for CFSELO T cells (n = 5) (mean and SEM; P = 0.0002). (F) Presentation of OVA to TCR transgenic CD8+ CTL cells by tumor CM-differentiated monocytes at 1:5 APC:T-cell ratio. Frequency of proliferating cells was determined by gating for CFSELO T cells (n = 5) (mean and SEM; P = 0.0064). (G) Percentage of M2 macrophages over total TAMs in MB49 tumors (n = 5) (mean; P < 0.0001. (H) Presentation of OVA to TCR transgenic CD4+ Th cells by tumor CM-polarized macrophages at 1:10 APC:T-cell ratio (n = 5) (mean and SEM; P = 0.0002). (I) Presentation of OVA to TCR transgenic CD8+ CTL cells by tumor CM-polarized macrophages at 1:10 APC:T-cell ratio (n = 5) (mean and SEM; P = 0.0001).
Previous studies have demonstrated that tumor-infiltrating myeloid progenitors such as monocytes are immune-suppressive (41, 42). Flow cytometry analysis revealed that tumor-infiltrating monocytic cells (Gr-1+ CD11b+) from MB49 CD14-high tumors have significantly reduced expression of MHC II, indicating possible impairment in antigen presentation to CD4 T cells (Fig. 3D and Fig. S4B). Monocytes differentiated in CD14-high CM also have lower MHC II expression compared with those in the CD14-low CM group (Fig. S4C). To assess T-cell stimulation, monocytes pulsed with the chicken ovalbumin OVA OT-I or OT-II peptide were also assayed for their relative abilities to present antigen-to-transgenic OVA-specific CD8 T lymphocytes (CD8) and CD4 helper T (Th) cells (Fig. 3 E and F). Monocytes differentiated in CD14-high CM were impaired in their ability to stimulate T-cell proliferation.
Recent studies have revealed that TAMs are polarized into protumorigenic M2 macrophages (17, 19). We analyzed BC TAMs (Gr-1- CD11b+ F4/80+) for the expression of CD206 and CD301, two markers that are expressed on M2 macrophages. CD14-high tumors were characterized by a higher relative frequency of CD206+ CD301+ M2-like macrophages (Fig. 3G and Fig. S4D). CD14-high CM-polarized bone-marrow–derived macrophages showed reduced production of nitric oxide and increased arginase activity compared with CD14-low CM-polarized macrophages, indicating that factors secreted by CD14-high BC cells more efficiently polarize macrophages to an M2-like phenotype (Fig. S4 E and F). Furthermore, CD14-high CM-polarized macrophages are less efficient at presenting antigens to stimulate T-cell proliferation (Fig. 3 H and I). Accordingly, the frequency of CD8+ T-cell infiltration in CD14-low BC tumors is higher than in CD14-high tumors, even though MHC I expression in both subpopulations of tumor cells is similar (Figs. S4G and S5).
Inflammation Mediators from CD14-High BC Cells Promote Tumor Proliferation.
The gene expression profile of CD14-low cells corresponds to genes associated with cell cycle and proliferation (Fig. S3 C and D). Accordingly, the frequency of tumor cells in CD14-low MB49 tumors was higher compared with CD14-high tumors (Fig. S6). Measurement of BrdU incorporation by different BC subpopulations in vitro revealed that CD14-low BC cells proliferate faster than CD14-high cells (Fig. 4A). Bulk, unsorted MB49 cells also proliferate at a similar rate to the CD14-low subpopulation. To compare the proliferation rates of different BC subpopulations in vivo, equal numbers of each subpopulation were implanted into mice and analyzed after 1 mo. As predicted, CD14-low BC cells have a higher proliferation rate and composed ∼90% of the MB49 cells within the tumor (Fig. 4B).
Fig. 4.

Inflammation mediators from CD14-high BC cells promote tumor proliferation. (A) Proliferation rate of BC subpopulations measured by BrdU incorporation (n = 6) (mean and SEM; P < 0.0001). (B) Equal numbers of CD14-high and CD14-low subpopulations were implanted into mice. Relative frequency of each subpopulation in the ectopic tumors after 1 mo is shown (n = 4) (mean; P < 0.0001). (C) Proliferation rate of CD14-high and -low BC subpopulations in exogenously added tumor CMs (n = 6) (mean and SEM; P < 0.0001). (D) Growth of ectopic tumors formed by s.c. injection of CD14-high (n = 5), CD14-low (n = 10), and bulk-unsorted MB49 subpopulations into syngeneic wild-type mice (n = 7) after 4 wk (mean and SEM; P = 0.0344).
In vitro, both BC subpopulations show increased proliferation upon culture in exogenously added CD14-high CM, with CD14-low BC cells still proliferating at a higher rate compared with CD14-high cells (Fig. 4C). CM from CD14-low cells also induces an increase in proliferation of CD14-high BC cells. Collectively, these results suggest that inflammatory factors secreted by both BC subpopulations can act on each other in an autocrine and paracrine fashion to promote tumor proliferation. To evaluate the possible cooperative effects of paracrine signaling between different BC subpopulations on tumor growth, both MB49 subpopulations and bulk, unsorted MB49 cells were respectively implanted into mice and evaluated after 1 mo. Tumor mass of bulk, unsorted MB49 tumors was significantly higher than tumors formed by either subpopulation alone, further suggesting that tumor growth is most efficient when cellular heterogeneity of the tumor is preserved (Fig. 4D).
Discussion
TPI is a consistent hallmark of the tumor microenvironment (43, 44). Here, we reveal CD14-high tumor cells produce many signaling mediators that act on a range of host cells to establish the inflammatory tumor microenvironment. These factors include small molecules, cytokines, chemokines, growth factors, and angiogenic factors that can promote tumor growth through multiple different mechanisms. IL6 and IL8/CXCL1 have protumorigenic effects on tumor, stromal, and immune cells (45–47). Overexpression of IL6 or IL8 in many tumors has been associated with poor prognosis (48, 49).
We used the MB49 mouse BC cell line to examine the BC tumor microenvironment in a syngeneic, immune-competent host that mimics physiological conditions during tumor growth. In our study, we found that CD14-high BC cells consistently form larger tumors that have higher frequencies of vessel-forming endothelial cell infiltration. Tumors formed by CD14-high cells are also characterized by higher frequencies of myeloid cell infiltration, including monocytes, macrophages, neutrophils, and DCs. This is an important feature frequently observed during chronic inflammation in BC, hepatocellular carcinoma, and pancreatic adenocarcinoma (12, 20, 30). Our study further determined that immune mediators produced by CD14-high tumor cells effectively recruit myeloid cell infiltration and retention.
Using CD14 knockouts, we found that CD14 plays a central role in cytokine production by CD14-high BC cells and the consequent protumorigenic effects of the cytokines. Nevertheless, even in the absence of LPS stimulation, we observed that both CD14-high human and mouse BC cell lines produce higher levels of IL6 and other factors. Furthermore, LPS stimulation did not significantly increase cytokine secretion by both BC subpopulations, suggesting that tumor cells might be constitutively producing endogenous ligands for CD14 at significant levels. Accordingly, MB49 BC subpopulations have high expression levels of numerous reported endogenous ligands for CD14 and TLRs, including HSP60, peroxiredoxin 1 (Prdx1), and high mobility group box 1 (HMGB1) proteins (Fig. S5). However, further studies beyond the scope of this report are needed to identify the ligand for CD14 in bladder cancers.
TLRs have long been implicated in promoting tumor growth through divergent mechanisms. Recent studies in gastric cancer (GC) found that TLR2 expression in GC cells promotes tumor cell survival and proliferation, but not inflammation (50). In contrast, another study proposed that TLR2 expression on melanoma cells mediated the production of immune-suppressive cytokines such as IL6 (51). Moreover, knockdown of TLR4 expression in colon cancer cells resulted in the loss of IL6 and blockade of tumor growth (52). In our model, TLR expression is similar across different MB49 populations (Fig. S5). However, CD14 is known to be critically important in the signaling pathways of TLR 2, 3, 4, 7, and 9, possibly providing a crucial link between studies evaluating the role of different TLRs (39, 40). We found that CD14 signaling in CD14-high BC cells is indeed mediated through multiple TLRs except TLR3. This redundancy ensures that the crucial function of cytokine secretion is preserved even when individual TLRs are blocked. Detailed dissection of the crosstalk between different TLR-signaling pathways will have to be explored in future studies.
Tumor-associated macrophages are a major component of TPI (23). TAMs promote tumor growth by producing soluble factors that activate neo-angiogeneis and stimulate tumor proliferation. In our study, we observed that CD14-high tumor-factor–polarized monocytes and macrophages are more immune-suppressive and are impaired in their ability to stimulate T-cell proliferation. Inflammation factors secreted by CD14-high BC cells are consistently more efficient at downregulating MHC II on monocytes and polarizing macrophages toward an M2-like phenotype.
Heterogeneity of cancer cells within a neoplastic lesion is a well-documented phenomenon (26, 29). In both the 639V and MB49 models, heterogeneity of CD14 expression is preserved, allowing for CD14-high and -low cell isolation by FACS. A key finding of our study was that CD14-low BC cells proliferate faster than CD14-high BC cells, thus representing the BC subpopulation constituting the vast majority of tumor mass in established tumors. However, CD14-high tumor cells produce higher levels of factors that act on itself and the CD14-low subpopulation in an autocrine and paracrine fashion to increase tumor cell proliferation. The observation that unsorted MB49 cells, which preserve the heterogeneity of the cancer, form larger tumors than CD14-high or -low cells alone lends support to this. Interestingly, the cytokine production profile of bulk unsorted cells (consisting mainly of CD14-low cells) closely resembles that of CD14-low cells, with notable exceptions in IL8 (639V) and IL12p40 (MB49).
The experimental models used in our studies do not exhibit invasive or metastatic characteristics. Given this important limitation in our experimental models, we were unable to study whether TPI and increased tumor cell proliferation induced by CD14-high cells ultimately leads to tumor progression and metastases. Further studies using metastatic tumor cell models are needed to investigate the role of CD14 expression in these processes.
In summary, our study defines the crucial role of CD14-high tumor cells in orchestrating TPI to promote tumor growth and proliferation in BC. Inflammatory factors produced by this subpopulation of tumor cells activate angiogenesis to support the establishment and maintenance of an immune-suppressive, inflammatory tumor microenvironment. Additionally, this subpopulation is able to drive tumor growth by producing factors that drive autocrine and paracrine proliferative stimulation. Collectively, this study highlights the need to explore the broader role of CD14-expressing neoplastic cells in other solid tumors. Given that CD14 expression is critical for IL6 production by CD14-high cells, therapeutic targeting of CD14 might possibly represent a novel and efficient strategy to mitigate TPI and treat cancer.
In a broader context, it has recently been shown that inflammation in the mouse bladder can result in damage to the umbrella cell layer, which stimulates basal cells to secrete sonic hedgehog (SHH) (53). This signals SHH-responsive subbasal stromal cells with SHH receptors to activate Gli-1 and transcribe Gli-1–responsive genes, including WNT 2 and 4, which in turn stimulate basal bladder epithelial cells to proliferate and begin bladder repair (53). The complex cellular composition of bladder cancer cell subsets, including KRT14+CD14+CD90+ tumor-initiating bladder cancer cells and stromal cells of as-yet-undefined lineages, emphasizes the need to dissect the pathways that support bladder cancer growth and progression to develop rational combination therapies for this disease.
Materials and Methods
The Stanford University Institutional Review Board approved the enrollment of human subjects under protocol 1512. The Stanford Administrative Panel on Laboratory Animal Care approved the mouse studies under protocol 26270.
Bladder Tumor Tissue Dissociation, Flow Cytometry Analysis and Cell Sorting, and Tumor Engraftment.
Dissociation, FACS analysis, sorting, and engraftment were preformed as previously described (28). See SI Materials and Methods for further details.
Quantification of Soluble Factors.
See SI Materials and Methods for further details.
Microarray Analysis of BC Cell Lines.
See SI Materials and Methods for further details.
Immune Cell Isolation, Culture, and Assays.
See SI Materials and Methods for further details.
Tumor Cell Proliferation.
See SI Materials and Methods for further details.
TALEN Design, Construction, and Transfection.
See SI Materials and Methods for further details.
Blocking of CD14, TLRs, and Adaptor Molecules.
See SI Materials and Methods for further details.
Supplementary Material
Acknowledgments
The research reported in this article was supported by the National Cancer Institute of the National Institutes of Health under Grants P01CA139490 and R01CA86017 (to I.L.W.), the Siebel Foundation, and the Virginia and D. K. Ludwig Fund for Cancer Research. M.T.C. was supported by a Smith Stanford Graduate Fellowship. F.A.S. was supported by a fellowship from the Dutch Cancer Society and by a seed grant of the organization My Blue Dots.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424795112/-/DCSupplemental.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 4.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev Cell. 2010;18(6):884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dirat B, Bochet L, Escourrou G, Valet P, Muller C. Unraveling the obesity and breast cancer links: A role for cancer-associated adipocytes? Endocr Dev. 2010;19:45–52. doi: 10.1159/000316896. [DOI] [PubMed] [Google Scholar]
- 6.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 7.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010;29(2):309–316. doi: 10.1007/s10555-010-9223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6(1):24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 12.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 13.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 14.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur J Cancer. 2006;42(6):717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 16.Johansson M, Denardo DG, Coussens LM. Polarized immune responses differentially regulate cancer development. Immunol Rev. 2008;222:145–154. doi: 10.1111/j.1600-065X.2008.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat Immunol. 2010;11(10):889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 18.Fridlender ZG, Albelda SM. Tumor-associated neutrophils: Friend or foe? Carcinogenesis. 2012;33(5):949–955. doi: 10.1093/carcin/bgs123. [DOI] [PubMed] [Google Scholar]
- 19.Allavena P, Mantovani A. Immunology in the clinic review series; Focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012;167(2):195–205. doi: 10.1111/j.1365-2249.2011.04515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zumsteg A, Christofori G. Corrupt policemen: Inflammatory cells promote tumor angiogenesis. Curr Opin Oncol. 2009;21(1):60–70. doi: 10.1097/CCO.0b013e32831bed7e. [DOI] [PubMed] [Google Scholar]
- 21.De Palma M, Murdoch C, Venneri MA, Naldini L, Lewis CE. Tie2-expressing monocytes: Regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 2007;28(12):519–524. doi: 10.1016/j.it.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Gordon S, Martinez FO. Alternative activation of macrophages: Mechanism and functions. Immunity. 2010;32(5):593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 23.Balkwill FR, Mantovani A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22(1):33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Melnikova VO, Bar-Eli M. Bioimmunotherapy for melanoma using fully human antibodies targeting MCAM/MUC18 and IL-8. Pigment Cell Res. 2006;19(5):395–405. doi: 10.1111/j.1600-0749.2006.00331.x. [DOI] [PubMed] [Google Scholar]
- 25.Cho RW, Clarke MF. Recent advances in cancer stem cells. Curr Opin Genet Dev. 2008;18(1):48–53. doi: 10.1016/j.gde.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy KM, Dewhirst MW. Tumor metabolism of lactate: The influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 2010;6(1):127–148. doi: 10.2217/fon.09.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishimoto T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 28.Chan KS, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci USA. 2009;106(33):14016–14021. doi: 10.1073/pnas.0906549106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–699. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 30.Eruslanov E, et al. Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer. Int J Cancer. 2012;130(5):1109–1119. doi: 10.1002/ijc.26123. [DOI] [PubMed] [Google Scholar]
- 31.Michaud DS. Chronic inflammation and bladder cancer. Urol Oncol. 2007;25(3):260–268. doi: 10.1016/j.urolonc.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Volkmer JP, et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc Natl Acad Sci USA. 2012;109(6):2078–2083. doi: 10.1073/pnas.1120605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elliott AY, Bronson DL, Stein N, Fraley EE. In vitro cultivation of epithelial cells derived from tumors of the human urinary tract. Cancer Res. 1976;36(2 Pt 1):365–369. [PubMed] [Google Scholar]
- 34.Lippitz BE. Cytokine patterns in patients with cancer: A systematic review. Lancet Oncol. 2013;14(6):e218–e228. doi: 10.1016/S1470-2045(12)70582-X. [DOI] [PubMed] [Google Scholar]
- 35.Bünger S, Laubert T, Roblick UJ, Habermann JK. Serum biomarkers for improved diagnostic of pancreatic cancer: A current overview. J Cancer Res Clin Oncol. 2011;137(3):375–389. doi: 10.1007/s00432-010-0965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen F, Zhang G, Cao Y, Hessner MJ, See WA. MB49 murine urothelial carcinoma: Molecular and phenotypic comparison to human cell lines as a model of the direct tumor response to bacillus Calmette-Guerin. J Urol. 2009;182(6):2932–2937. doi: 10.1016/j.juro.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 37.Loskog A, et al. Adenovirus CD40 ligand gene therapy counteracts immune escape mechanisms in the tumor microenvironment. J Immunol. 2004;172(11):7200–7205. doi: 10.4049/jimmunol.172.11.7200. [DOI] [PubMed] [Google Scholar]
- 38.O’Donnell MA, et al. Interleukin-12 immunotherapy of murine transitional cell carcinoma of the bladder: Dose dependent tumor eradication and generation of protective immunity. J Urol. 2004;171(3):1330–1335. doi: 10.1097/01.ju.0000109742.88380.a2. [DOI] [PubMed] [Google Scholar]
- 39.Akashi-Takamura S, Miyake K. TLR accessory molecules. Curr Opin Immunol. 2008;20(4):420–425. doi: 10.1016/j.coi.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Baumann CL, et al. CD14 is a coreceptor of Toll-like receptors 7 and 9. J Exp Med. 2010;207(12):2689–2701. doi: 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umansky V, Sevko A. Tumor microenvironment and myeloid-derived suppressor cells. Cancer Microenviron. 2013;6(2):169–177. doi: 10.1007/s12307-012-0126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138(2):105–115. doi: 10.1111/imm.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dvorak HF. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 44.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naugler WE, Karin M. The wolf in sheep’s clothing: The role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14(3):109–119. doi: 10.1016/j.molmed.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Bollrath J, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15(2):91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 47.Bencsáth M, Blaskovits A, Borvendég J. Biomolecular cytokine therapy. Pathol Oncol Res. 2003;9(1):24–29. doi: 10.1007/BF03033710. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, et al. Interleukin-6 signaling regulates anchorage-independent growth, proliferation, adhesion and invasion in human ovarian cancer cells. Cytokine. 2012;59(2):228–236. doi: 10.1016/j.cyto.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, et al. Interleukin-8 secretion by ovarian cancer cells increases anchorage-independent growth, proliferation, angiogenic potential, adhesion and invasion. Cytokine. 2012;59(1):145–155. doi: 10.1016/j.cyto.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 50.Tye H, et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell. 2012;22(4):466–478. doi: 10.1016/j.ccr.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 51.Yang HZ, et al. Blocking TLR2 activity attenuates pulmonary metastases of tumor. PLoS ONE. 2009;4(8):e6520. doi: 10.1371/journal.pone.0006520. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Huang B, et al. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005;65(12):5009–5014. doi: 10.1158/0008-5472.CAN-05-0784. [DOI] [PubMed] [Google Scholar]
- 53.Shin K, et al. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature. 2011;472(7341):110–114. doi: 10.1038/nature09851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.