

Significance
The primary cilium is an organelle protruding from most postmitotic vertebrate cells. A growing body of data supports the crucial role of primary cilia in developmental signaling pathways. Recent studies describe the main stages in ciliogenesis at the morphological level and components of some of the mechanisms involved, including the selective acetylation of tubulin. How this acetylation is modulated in cilia nonetheless remains poorly understood. Here we show that the death inducer-obliterator (dido) gene product, which regulates histone deacetylase 6 deacetylase activity, is necessary for orofacial development in the mouse embryo and influences brain patterning and neuromuscular activity. Mice deficient in dido function present neonatal mortality and various ciliopathies including cleft palate and hydrocephalus, as well as hippocampal and commissural dysplasia.
Keywords: ciliopathies, brain patterning, perinatal lethality
Abstract
Nearly all vertebrate cells have a single cilium protruding from their surface. This threadlike organelle, once considered vestigial, is now seen as a pivotal element for detection of extracellular signals that trigger crucial morphogenetic pathways. We recently proposed a role for Dido3, the main product of the death inducer-obliterator (dido) gene, in histone deacetylase 6 delivery to the primary cilium [Sánchez de Diego A, et al. (2014) Nat Commun 5:3500]. Here we used mice that express truncated forms of Dido proteins to determine the link with cilium-associated disorders. We describe dido mutant mice with high incidence of perinatal lethality and distinct neurodevelopmental, morphogenetic, and metabolic alterations. The anatomical abnormalities were related to brain and orofacial development, consistent with the known roles of primary cilia in brain patterning, hydrocephalus incidence, and cleft palate. Mutant mice that reached adulthood showed reduced life expectancy, brain malformations including hippocampus hypoplasia and agenesis of corpus callosum, as well as neuromuscular and behavioral alterations. These mice can be considered a model for the study of ciliopathies and provide information for assessing diagnosis and therapy of genetic disorders linked to the deregulation of primary cilia.
The primary cilium is a unique, mainly nonmotile, microtubule-based organelle found in nearly all noncycling cells in vertebrates. Its main function is to transduce extracellular signals that regulate a wide range of functions from fluid flow to cell proliferation, differentiation, and migration; hence, it alters cell polarity and tissue development. Defects in cilia growth, resorption, or stability lead to deregulation of these pathways, which results in a number of functional defects (ciliopathies) that affect diverse organs, giving rise to complex pleiotropic phenotypes such as those of Joubert, Meckel-Gruber, or Bardet-Bield syndromes (1). In many cases, these pathologies are associated with embryonic or perinatal lethality (2). Mechanically or chemically stimulated signaling pathways are associated with the cilium; hedgehog (Hh), canonical and noncanonical Wnt, Notch, fibroblast growth factor, and platelet-derived growth factor are some of the pathways involved in development and are altered in organisms with dysfunctional cilia (3, 4).
The cognitive impairment found in human ciliopathies is manifested as behavioral changes in the mouse. Anatomical changes in the brain are usual in both species, reflecting alterations in central nervous system (CNS) development (2) that include defects in neural tube patterning and closure (5, 6) and in hippocampal neurogenesis, leading to neuropsychiatric phenotypes (7, 8). All these processes are closely linked to Hh signaling, and most CNS-related ciliopathies appear to be associated with altered Hh signaling (9).
Morphogenetic activity of sonic hedgehog (Shh) in the neural tube requires coordinated antagonistic expression of bone morphogenetic protein (BMP) (10, 11) and involves a physical interaction between Smad and Gli proteins (12). Death inducer-obliterator (dido) is a BMP4-specific Smad-regulated target gene (13), and we recently demonstrated a role for Dido3, the main dido product, in actin-dependent histone deacetylase 6 (HDAC6) delivery to the primary cilium (14). HDAC6 counteracts the activity of α-tubulin acetyl transferase (Atat1) (15), which acetylates tubulin and thus stabilizes primary cilium structure. Recent studies also established a relationship between HDAC and tumors as well as neurological and immunological diseases. HDAC6 in particular appears to be involved in mood control, in tau-driven neurological disorders, in the progression of Alzheimer's and Huntington's diseases, in immune synapse formation, and in regulatory T-cell homeostasis (16).
In humans and mice, the dido locus encodes three isoproteins, Dido1, Dido2, and Dido3, by alternative splicing (17). We reported generation of a mouse mutant that expresses an N-terminal–truncated Dido protein that lacks the initial 422 amino acids (Fig. S1). Studies in homozygous dido∆Nt/∆Nt mice on a mixed genetic background showed an essential role for Dido3 in the regulation of the spindle assembly checkpoint (SAC), control of centrosome number, chromosome stability, and cytokinesis. Pups were nonetheless born at Mendelian frequencies and reached weaning with no obvious anatomical or behavioral abnormalities, although they developed a myelodysplasia/myeloproliferative-like syndrome (MDS/MPD) (17).
We also generated a dido mouse mutant that lacks exon 16, which encodes the Dido3 isoform-specific 1080 C-terminal amino acids. This dido3∆Ct allele is lethal in homozygosis at Theiler embryonic stage ts10–11 and is associated with DNA damage, apoptosis, and growth arrest during embryonic stem (ES) cell differentiation in vitro (18). The dido∆Nt allele rescued embryonic lethality of the dido3∆Ct mutation as well as the ability of dido3∆Ct mutant mouse ES cells to differentiate (18). During these studies, we identified a synthetic phenotype in the dido∆Nt/∆Ct double heterozygotes that drives severe perinatal lethality. Here we describe this phenotype, which identifies an important role for the dido gene in craniofacial development. The few dido∆Nt/∆Ct mice that reached adulthood had metabolic and neuromuscular alterations that highlight dido involvement in many physiological functions, including behavioral abnormalities.
Results
Dido Function Is Necessary in Vivo for Primary Cilium Control.
We previously reported the ability of the dido∆Nt allele to rescue the embryonic lethality of the dido3∆Ct mutation (18). In addition, we described dissociation between Dido function in stem cell differentiation and its known role in the SAC, centrosome amplification, and cytokinesis (17, 19).
We also previously identified the role of Dido3 in the control of cilium size. To analyze the phenotype of the dido∆Nt allele-rescued embryonic lethality of the dido3∆Ct mutation in vivo, we crossed dido∆Nt/+ × dido∆Ct/+ mice. To test the relevance of normal Dido activity in cilia development and maintenance, we quantified primary cilia in the hippocampal granular cell layer of adult mouse brain. Adenylyl cyclase III (ACIII) is a prominent ciliary marker (20); to compare brain sections from 3-mo-old wild type (WT) and dido∆Nt/∆Ct mice, we used anti-ACIII staining and confocal microscopy (Fig. 1 A and B). The percentage of ciliated cells was reduced in dido∆Nt/∆Ct mice (Fig. 1C), although mean cilium length did not differ significantly (Fig. 1D). Distribution of cilium length is altered in dido∆Nt/∆Ct brains, with increased frequency of longer and shorter cilia (Fig. 1E and Fig. S2).
Fig. 1.

Quantification of primary cilia in the adult hippocampal granular cell layer. (A and B) Immunofluorescent visualization of ACIII-positive primary cilia (green) of WT (A) and didoΔNT/ΔCT (B) mice (z-projection of confocal stack; DAPI-stained nuclei in blue) (Scale bar, 30 μm.) (C) didoΔNT/ΔCT genotype is associated with a lower percentage of ciliated cells. Cilia were counted manually in 30 microscopy fields of 5-μm-thick brain sections from two WT and two didoΔNT/ΔCT mice. Individual values are shown; means are calculated for each genotype. Unpaired Student’s t test with Welch's correction, ***P < 0.001. (D and E) Length of individual cilia for WT (n = 62) and didoΔNT/ΔCT mice (n = 54) were determined (Materials and Methods and Fig. S2). (D) The median value and the 50th (boxes) and 10–90th percentile range (whiskers) are shown. Student's t test indicates no significant differences. (E) Distribution of cilia length in didoΔNT/ΔCT brain is different from WT. Snedecor's F distribution, P = 0.01.
Perinatal Lethality in dido∆Nt/∆Ct Mice.
To analyze the phenotype of the dido∆Nt allele-rescued embryonic lethality of the dido3∆Ct mutation in vivo, we crossed dido∆Nt/+ × dido∆Ct/+ mice. After a 1-y follow-up of the colony, dido∆Nt/∆Ct, dido∆Nt/+, dido∆Ct/+, and WT genotypes appeared at frequencies of 2.0, 37.2, 30.7, and 30.2% at weaning, respectively (Fig. 2A), indicating selective loss of most double-heterozygous mice (P < 10−5). Genotype analysis at various days postcoitum (dpc) of embryos in a dido∆Nt/+ × dido∆Ct/+ cross showed expected Mendelian frequencies up to birth (Fig. 2B), but most dido∆Nt/∆Ct mice died shortly thereafter.
Fig. 2.
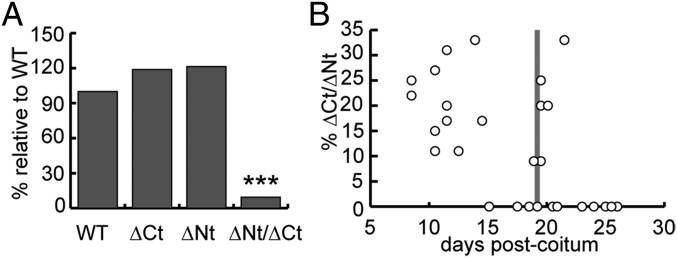
Viability of didoΔNT/ΔCT mutant mice. (A) The N- and C-terminal Dido3 deletions studied have no effect on viability when each allele is in heterozygosis, and only a few didoΔNT/ΔCT mutant mice reached weaning. Relative frequency of each genotype is shown (n = 409); the χ2 test was used to compare frequency for each group frequency with that of WT. ***P < 0.001. (B) Double heterozygotes did not show imbalanced proportions during the embryonic or perinatal periods, but dropped abruptly by 24–48 h postpartum (pp). Genotype results are shown for 15 litters between 9 and 19 dpc, 3 litters at ∼24 h pp, and 7 litters between 2 and 7 d (d)pp. Mean litter size ± SD was 10.47 ± 2.0.
Neonatal death after 24 h postpartum is usually associated with inability to suckle rather than with respiratory failure or homeostatic deficiencies (21). We found no gross morphological alterations or hemorrhage in dido∆Nt/∆Ct pups, nor were there obvious breathing defects in most dido∆Nt/+ × dido∆Ct/+ litters during the first 24 h after birth, although some dido∆Nt/∆Ct mice showed cyanosis by days 1 and 2 after birth.
Another cause of perinatal lethality is linked to macroautophagy defects (22). As a cilium-associated protein, Dido3 is potentially linked to the autophagosome (23); knockout mice for another cilium-associated protein, autophagy-related 5 (Atg5), show shortened primary cilia (24). We tested the Dido/autophagosome relationship by monitoring microtubule-associated protein 1 light chain 3B (LC3B). Cytosolic LC3B is processed for autophagosome formation; Apg7p and Apg3p activate cytosolic LC3B-I, which is phospholipid conjugated and forms membrane-bound, autophagosome-associated LC3B-II (25). We studied WT and dido∆Nt/∆Ct embryos obtained by cesarean section at 19 dpc (full term). At time 0 and after 6 h in starvation conditions, heart protein was isolated and analyzed in Western blot for LC3BI/II expression. We found no differences between WT and mutant embryos at any time (Fig. 3A). Adult primary fibroblasts and EBV-immortalized murine embryonic fibroblasts (MEFs) cultured in starvation medium showed no LC3B processing defect, either in primary lung (Fig. 3B) or in immortalized fibroblasts (Fig. 3C).
Fig. 3.

Autophagy test in primary cells, cell lines, and fetuses. (A) Fetuses at 19.5 dpc were starved for 4 h and killed. Heart protein extracts were analyzed by Western blot and by LC3BI and LC3BII detection. The LC3BII/LC3BI ratio was quantified and showed no differences. (B) Adult lung primary fibroblasts and (C) immortalized embryonic fibroblasts were cultured in rich (r) or starvation (s) medium and analyzed as in A; no LC3B processing deficiency was found. All experiments were repeated twice.
dido∆Nt/∆Ct pups showed no size differences compared with WT or heterozygous littermates at birth (Fig. 4A), whereas those that survived >24 h showed reduced body size, suggesting a nutrition defect (Fig. 4B). Mice were inspected visually through the semitransparent skin; from ∼12 h postpartum onwards all dido∆Nt/∆Ct mice showed little or no milk in the stomach (Fig. 4 A and B), which was dilated and air filled (Fig. 4C). A possible glucose mobilization deficiency was ruled out by glycemia measurement and by estimation of glycogenolysis activity by RT-PCR of hepatic glucose-6-phosphatase (Fig. S3).
Fig. 4.

Phenotypic alterations associated with didoΔNT/ΔCT. (A) At 0.5 dpp, mutant pups were similar in size to WT littermates, but their stomachs contained little or no milk. (B) At 1.5–2 dpp, the size of surviving mutant pups was approximately two-thirds that of WT littermates, with very little milk in the stomach. Grid lines = 2 mm. (C) didoΔNT/ΔCT mutant pups showed accumulated air in the stomach. A peritoneal incision to expose the stomach shows inflation due to excess air pressure. (Inset; Scale bar, 5 mm.) (D) Reduced body size was maintained throughout life. At 13–17 mo of age, weight of didoΔNT/ΔCT mice was about one-half that of littermates, whereas didoΔCt/+ and didoΔNt/+ showed no size differences compared with WT mice. (E) Life expectancy was greatly reduced for didoΔNT/ΔCT mice, again with no effect for didoΔCt/+ and didoΔNt/+ genotypes. (F) Turnbull's blue stain shows depletion of ferrous iron (II) storage in spleen of didoΔNT/ΔCT compared with WT mice. (Scale bar, 100 μm.) Original magnification, 6×. (G) Abdominal fat tissue (arrows) is virtually absent in 9-mo-old female didoΔNT/ΔCT.
The few dido∆Nt/∆Ct mice that reached adulthood showed no external morphological abnormalities, except that size and weight were approximately one-half to two-thirds that of WT or heterozygous littermates (Fig. 4D), and life expectancy at birth was <1 y (Fig. 4E). Adults were infertile and showed limited cachexia. Hemograms, blood biochemistry profiles, and proteinograms of dido∆Nt/∆Ct mice showed normal values, with only slight hypoproteinemia compared with those of littermates (Fig. S4). Histochemical studies showed anemia in some individuals as assessed by the Turnbull blue reaction (Fig. 4F), and visual examination showed differences in body fat accumulation (Fig. 4G), implying metabolic problems. Various nonrecurrent tumors were occasionally found in necropsies, which suggests nonspecific activity that facilitates tumor growth.
Delayed Closure of the Palate Primordium During dido∆Nt/∆Ct Embryo Development.
The snout of some dido∆Nt/∆Ct fetuses was shorter than in WT mice (Fig. 5A). We dissected the oral cavity of dido∆Nt × dido∆Ct F1 pups at 19.5 dpc (full-term pregnancy) to expose tongue, throat, and palate. The secondary palate was 15% shorter in dido∆Nt/∆Ct than in WT mice (Fig. 5 B and C), in agreement with the consistent lack of ruga 7b (the last-formed ruga in normal development), indicative of abnormal palate growth. The few double-heterozygous dido∆Nt/∆Ct mice that survived to adulthood showed normal palatal rugae number and palate size (Fig. S5), again suggesting defective palate development as a cause of perinatal death.
Fig. 5.

Craniofacial defects in didoΔNT/ΔCT neonates. (A) didoΔNT/ΔCT mice showed altered craniofacial development at birth, with a shortened snout compared with WT littermates. Representative images of both genotypes are shown, with a false-color merged image for comparison. (B and C) Palate was fully closed at birth, but the distance between rugae 2 and 8 was 15% shorter in didoΔNT/ΔCT than in WT pups (WT, n = 4; didoΔNT/ΔCT, n = 2; Student's t test P = 0.0016). (D) Delayed palate closure in didoΔNT/ΔCT embryos. At 15.5 dpc, embryos were extracted and the lower jaw and tongue removed to expose the developing palate. The secondary palate was fully closed in WT embryos (Left), whereas didoΔNT/ΔCT embryos showed varying degrees of delay in palate closure, with partial (Center) or almost no (Right) horizontal growth of palatal halves (arrows) toward the midline at the time analyzed (n = 8). (White scale bar, 5 mm.)
To determine the origin of the craniofacial and palate defects in newborn dido∆Nt/∆Ct mice, we analyzed palate features in dido∆Nt × dido∆Ct F1 embryos. At 15.5 dpc, the palatal shelves had already fused in WT embryos, whereas the mouth roof in dido∆Nt/∆Ct embryos remained open, with some degree of variability at this age (Fig. 5D). No palatal clefts were observed in dido∆Nt/∆Ct neonates, however, suggesting that the mutation causes delayed growth that gives rise to a shorter, defectively ossified palate.
Brain and Neurobehavioral Abnormalities in Surviving Adult dido∆Nt/∆Ct Mice.
Previous reports of dido expression in the CNS, in embryo brain (www.emouseatlas.org/emage/, EMAGE:2631) and adult cerebellum (Allen Mouse Brain Atlas, mouse.brain-map.org/, experiment 69262318), and in gray matter of the spinal cord (Allen Mouse Spinal Cord Atlas, mouse.brain-map.org/, experiment 100014937), prompted us to analyze the effect of dido mutations on CNS development and neuron function. Histological examination of the brain showed enlarged ventricles with proportional reduction of caudoputamen and lateral striatum, agenesis of the corpus callosum, and hippocampus dysplasia (Fig. 6A).
Fig. 6.

Neural anomalies in dido mutant mice. (A) Sagittal sections of brain from a representative didoΔNT/ΔCT mouse, showing ventricle enlargement and dysplasia of corpus callosum (cc) and hippocampus (hp). cp, caudoputamen; ls, lateral striatum; lv, lateral ventriculum; Pb, Probst bundles. (B) Horizontal and (C) vertical spontaneous locomotor activity in surviving adult didoΔNT/ΔCT mice. Each mouse was tested three times over a 4-mo period. One-tailed Student's t test, P = 0.04 for vertical differences. (D) Plantar sensitivity to thermal stimulus in surviving adult didoΔNT/ΔCT mice. (B–D) As one WT and two didoΔNT/ΔCT mice died during this period, two values for n are given for each group; n = 10 (9) for WT, n = 7 (5) for didoΔNT/ΔCT mice. (E) Toluidine-stained myelin was photographed in cross-sections of left and right sciatic nerves from adult didoΔNT/ΔCT mice and WT littermates, and myelin sheet thickness was measured with Adobe Photoshop (n = 6 for WT, n = 4 for didoΔNT/ΔCT); 20 axons per nerve were evaluated in a blind manner. (F) Peripheral nerve α-tubulin deacetylation in didoΔNT/ΔCT and WT mice (n = 4 + 4). Optical density of acetylated tubulin bands in Western blots of sciatic nerve extracts, normalized to total α- + β-tubulin levels. One-tailed paired Student's t test; P = 0.02.
Whereas WT mice displayed a normal grasping reflex, dido∆Nt/∆Ct mice normally stretched their forelimbs toward the wires and gripped them effectively, but tucked in their hind limbs and maintained them clasped.
Spontaneous locomotor activity of the mice was measured in a chamber equipped with infrared sensors. There was no notable difference in horizontal activity, defined as exploratory movements around and across the base of the chamber (Fig. 6B); in contrast, dido∆Nt/∆Ct mice showed reduced vertical activity (Fig. 6C), which assesses exploratory movements while standing upright, usually on the walls of the chamber. Pawprint tests along a covered straight path showed no marked gait or stride differences between dido∆Nt/∆Ct and WT mice. To further test sensorimotor performance, we used a thermal plantar test to assess adult dido∆Nt/∆Ct and WT mice. Latency to paw withdrawal following plantar heating with an infrared beam was similar in the two mouse groups (Fig. 6D); results were similar at different heat potencies, as well as after hand or foot stimulus. The results on the whole suggested mild distal motor neuropathy.
Differences in α-Tubulin Deacetylation of the Sciatic Nerve in dido∆Nt/∆Ct Mutant Mice.
Sciatic nerves were dissected from thighs of dido∆Nt/∆Ct and WT mice of ages between 7 and 15 mo. A sample of each nerve was processed for histological study and another used to prepare whole protein extracts. Axon number and average myelin thickness were determined (Fig. S6); no differences were found in axon density or myelin integrity (Fig. 6E). Relative amounts of total and acetylated α-tubulin were evaluated by Western blot (Fig. S7). In age-matched mice, we found a significant difference in the amount of acetylated α-tubulin normalized to total amounts; each dido∆Nt/∆Ct mouse had lower levels of acetylated α-tubulin than an age-matched WT littermate (Fig. 6F). No significant differences were observed in total α- + β-tubulin levels between mutant and WT mice.
Discussion
Dido3 is a key determinant of cilium size (14), and Dido3 mutations cause chromosome segregation defects, meiosis prophase alterations, and stem cell differentiation blockade (17, 19, 26, 27). Misregulation of the dido gene is linked to increased incidence of genomic instability, myelodysplasia, and myeloproliferation, melanoma, and infertility (13, 17, 26, 28). Elucidating the mechanisms that govern the complex activities could help clarify the role of Dido in promoting stem cell differentiation, cilia size, tumorigenesis and, as shown here, life expectancy, brain development, and neural behavior alterations.
Homozygous dido∆Nt/∆Nt, heterozygous dido∆Nt/+, and dido∆Ct/+ mice are viable, although mice of the first two genotypes occasionally develop MDS/MPD. We anticipated dido∆Nt complementation of the severe early embryonic lethality associated with the dido∆Ct allele in homozygosis (18), but low dido∆Nt/∆Ct frequency in weaned offspring suggested complementation deficiencies. This lack of complementation affects ciliogenesis in vivo, as noted by abnormal cilium size and numbers in the adult brain.
Normal mouse pups gain 50–70% body weight from birth to day 2 (29), which highlights the importance of efficient nutrition in the first 48 h. We thus infer that the primary cause of death of dido∆Nt/∆Ct pups is an inability to feed adequately, although the metabolic anomalies detected in adult mutants could contribute to lethality. Inadequate feeding could be due to the mechanics of suckling itself, but also to deficient recognition or processing of pheromonal and/or complex odor signals (30). Although primary cilia in olfactory ganglion cells are reported to be necessary for pheromone detection (31), we suspect that the inability of dido∆Nt/∆Ct mice to feed is a consequence of their craniofacial abnormalities or/and neurological behavior defects.
Palatal shelves grow horizontally and fuse to each another by 15.5 dpc (32). At this age, dido∆Nt/∆Ct fetuses showed a marked delay in this process, albeit with considerable individual variation. Because of its frequency in man, the genetic basis of cleft palate has been studied extensively. Although the list of genes with a role in this condition is long (33), to our knowledge it does not include dido. Major alterations in Dido proteins might be too deleterious, as for the homozygous dido∆Ct condition in mice, and only specific allelic combinations on a given genetic background would result in this ciliopathy. It is also possible that dido does not primarily drive human cleft palate-like syndromes, but mediates the effects of other genes. Alterations in BMP are among those most often found in orofacial malformations (34); Braig and Bosserhoff (13) reported that dido is a notable mediator of BMP downstream effects. The dido gene is a target of the BMP-dependent Smad family of transcriptional activators, which are implicated directly in experimental cleft palate induced by all-trans retinoic acid.
Dido also regulates expression of integrin αV (13), which probably has a role in correct precursor cell migration from the neural crest of the developing embryo toward the zone that becomes the jaw primordium. Mouse dido expression correlates negatively with body weight and jaw length in several strains (35, 36). Further work is needed to determine how Dido mutations act to produce the defects in mouse palate development, as well as the mechanisms by which dido mediates or prompts action in palate development by molecules such as BMP or integrin αV. Specific focus on human Dido gene status in studies of cleft palate etiology will help to elucidate its clinical relevance.
The few surviving adult dido∆Nt/∆Ct mice showed hindlimb dyskinesia. As clasping behavior is common in mouse models of various human central and peripheral neuromuscular pathologies, we analyzed the possibility of a neuromuscular system disorder in dido∆Nt/∆Ct mice. Equilibrium and neuromuscular tests (tightrope walking, rotarod running, or grip strength) did not indicate consistent differences, arguably due to the disparity in body weight between mice of each genotype. It is unclear whether the reduced vertical activity observed in dido∆Nt/∆Ct mice is the result of peripheral neuropathy, a muscular defect, or both, but it coincides with clasping behavior to imply spinal and/or hindlimb neuromuscular disease. If so, it would be essentially a motor pathology, because study of plantar thermal sensitivity by the Hargreaves method showed no defect in sensorial competence.
The normal axon count and myelin coating of sciatic nerves from dido∆Nt/∆Ct mice are consistent with the mild nature of the hindlimb motor defect. Peripheral neuromotor defects in mice can be associated with decreased axon tubulin content or an abnormal acetylated-to-total α-tubulin ratio (15). Tubulin is the main component of microtubules, which are crucial for efficient transport of organelles and effector molecules along axons. Microtubule stability is especially important for the function of long axons such as those of the sciatic nerve and depends on the acetylation of α-tubulin Lys40. As we found increased HDAC6 activity in embryo fibroblasts from dido∆Nt/∆Ct mice (14), we reasoned that increased deacetylation could destabilize axon microtubules along the dido∆Nt/∆Ct mouse sciatic nerve, leading to hindlimb motor defects of the type observed. Indeed, all dido∆Nt/∆Ct mice had less acetylated α-tubulin in the sciatic nerve. Deacetylation-driven microtubule destabilization would impair nervous transmission along the sciatic and other peripheral nerves, contributing to neuromotor defects that affect the hindlimbs.
Increased tubulin deacetylation has a major role in murine distal neuropathies related to human diseases. The symptoms of Charcot-Marie-Tooth disease (CMT), the most common inherited peripheral nervous system disorder in man, were reversed by HDAC6 inhibition in a mouse model of CMT (15). Correct control of cytoskeleton stability is crucial for balanced proliferation, neurogenesis, migration, differentiation, and connectivity in the brain, and mutations in genes such as TUBA1a, which encodes α-tubulin, are responsible for neurodevelopmental disorders, such as cortical digenesis (37), usually associated with mental retardation, epilepsy, or autism. A recent genome-wide association study (38) identified polymorphisms of genes such as TUBA1a, TUBA4a, and KIF1B (kinesin family member 1B, associated with CMT). An exome-wide rare variant analysis (39) also associated TUBA4a mutations with familial amyotrophic lateral sclerosis, which supports the destabilization of cytoskeleton as a cause of neurodegenerative diseases. Whether dido is involved in these or other distal neuropathies in humans requires further study, which will also identify factors other than increased tubulin deacetylation that affect the distal neuropathy in dido∆Nt/∆Ct mice. Whereas the limited survival of these mice to adulthood poses difficulties in their use, we are currently generating conditional dido mutants, which will be valuable for identifying the many roles of this gene.
It remains to be determined whether the phenotype associated with dido mutations is related not only to primary, nonmotile cilia regulation but is also modified by effects on motile cilia, which could account for phenotypic characteristics such as infertility or adult hydrocephalus.
The previously demonstrated role of Dido in the control of cilium length, based on its interaction with HDAC6, might be the basis for understanding the brain alterations observed in dido∆Nt/∆Ct mutant mice. Lack of Atat1, whose action is opposite that of HDAC6, leads to hydrocephalus and hippocampal dysplasia (40), brain alterations similar to those associated with the dido∆Nt/∆Ct genotype. Although Atat1 and HDAC6 are respectively the major α-tubulin acetyltransferase (41) and deacetylase (40) in mice, atat1−/− and hdac6−/− mice are viable and fertile (42). This is not the case for dido (dido∆Ct/∆Ct mice show early embryonic lethality), which suggests that Dido3 function is not restricted to the control of α-tubulin acetylation/deacetylation or that of other HDAC6 targets.
In a number of cases, dido mutation shows a distinct manifestation of cilia defects during brain development, namely agenesis of corpus callosum. It is tempting to speculate on a relationship of this gene with autism, as this aplasia is a distinctive characteristic of both the human condition (43) and its murine model (44). Of particular interest in human patients are subtelomeric microdeletions involving band 20q13.33, where the Dido gene is located. In a study of six subjects with this deletion (45), only one showed a thin corpus callosum and autistic disorders (46); this subject differed from others with normal MRI in the deletion of just 12 loci. These loci did not include Dido, but the breakpoint mapped only ∼60 kb downstream of the Dido transcription unit. This is compatible with neotelomere formation that could affect control of Dido expression, supporting the hypothesis of Dido involvement in human autistic behavior.
In summary, we describe various neurodevelopmental and morphogenetic phenotypes associated with murine dido mutations. Our model will help to understand the pathogenic mechanisms of some genetic or epigenetic disorders and could improve diagnosis and choice of treatment.
Materials and Methods
Mice.
Heterozygous dido∆Nt and dido∆Ct mice were generated as described (17, 18) and maintained on a mixed genetic background (62.5% Sv129, 37.5% CD1). Double heterozygous dido∆Nt/∆Ct offspring were obtained from heterozygous parents, and sex-matched WT littermates were used as controls. Mice were handled according to national and European Union guidelines, and experiments were approved by the Comité Ético de Experimentación Animal, Centro Nacional de Biotecnología, Consejo Superior de Investigaciones Científicas.
Histology.
Mouse brains were fixed in PBS/4% (wt/vol) paraformaldehyde (PFA; 24 h) and cryoprotected in PBS/30% (wt/vol) sucrose (>48 h). Trimmed brains (cut along the longitudinal axis) were embedded in OCT (Sakura). Floating sections (30 μm) were stained in 0.1% cresyl violet in 1% acetic acid (5 min).
For cilia analysis, brains were fixed in PBS/4% PFA, 24 h, and paraffin embedded. Sections (5 μm thick) were stained with rabbit anti-rat ACIII antibody (1:500; Santa Cruz) and goat anti-rabbit Alexa-488 (BD Systems); cell nuclei were DAPI stained. Confocal microscopy was performed using an IX81 microscope (Olympus). For 3D reconstruction and cilium measurement, we used Imaris 7 software.
For Turnbull staining, 4-μm-thick paraffin sections were hydrated, placed in 0.06 N potassium ferricyanide staining solution (1 h), washed in 1% acetic acid, and counterstained with nuclear-fast red (5 min). Sciatic nerve portions (7–8 mm long) were collected. Proximal (next to spinal segments L5–6) and distal segments (∼1 mm) were used for histochemistry; central segments were processed for protein analysis. Cryosections (10 μm) were stained with 0.5% toluidine blue.
Autophagy Assay.
Progesterone (2.5 mg Depo-Provera; Upjohn) was administered to pregnant females at 17.5 and 18.5 dpc. Fetuses were extracted by cesarean section at 19.5 dpc and placed in a humidified chamber (30 °C). Heart samples were extracted at time 0 and 4 h.
Immortalized (19) or primary fibroblasts (obtained from 3-mo-old mice by mechanical disaggregation and trypsin digestion) were cultured in rich medium (DMEM + 10% FBS). At second passage, cells were plated at 75% confluence, cultured overnight, washed twice with HBSS, and incubated in starvation (HBSS) or rich medium (4 h).
Cell and tissue samples were lysed in cold PBS, 1% SDS with protease inhibitors. Equalized samples were fractionated in NuPAGE 12% Bis-Tris gels (Life Technologies) and transferred to Hybond-ECL nitrocellulose membranes (General Electric). LC3B I/II was detected with rabbit polyclonal antibody ab51520 (Abcam), anti-rabbit Ig-HRP (Dako), and Amersham-ECL (General Electric). Images were captured with X-ray film or a Proxima 2700 device (Isogen). Autograph density was analyzed with Adobe Photoshop CS5.
Behavioral Tests.
Spontaneous locomotor activity was assessed in a flat 40 × 40 cm Perspex cage with two sets of IR emitter/sensor arrays for automatic monitoring of horizontal and vertical activity (Ugo Basile). Each mouse was tested for 5 min on 3 different days, and scores were averaged.
Latency to paw withdrawal after limb heating was assessed by the Hargreaves method using a plantar test instrument (Ugo Basile). All limbs were tested daily on each mouse for 4 d, and scores for fore- and hindlimbs were averaged separately.
Acetylated Tubulin Measurement.
Left and right sciatic nerves from five WT and four mutant mice were dissected (see Histology). Protein extracts (8 μg) were loaded on 12% polyacrylamide gels. Acetylated α-tubulin was assessed by Western blot using mouse anti–α-acetylated tubulin mAb (clone 6-11B-1, Sigma) and normalized to total tubulin using DM1A + DM1B anti–α- and β-tubulin mAb (ab44928 Abcam) and GAPDH (anti-GAPDH (ab8245 Abcam). Luminescent signals were developed with HRP-conjugated rabbit anti-mouse IgG (Dako) and ECL reagents (Amersham/General Electric). Luminescence was detected and quantified with a Proxima 2700 device (Isogen).
Supplementary Material
Acknowledgments
We thank Dr. J. R. Naranjo for advice and help with neurobehavioral tests, A. Alonso-Guerrero for technical assistance, R. Gutiérrez for animal handling, and C. Mark for editorial assistance. This work is financed by Grants SAF2010-21205, PIB2010BZ-00564, and SAF2013-42289-R from the Spanish Ministerio de Economía y Competitividad.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1419300112/-/DCSupplemental.
References
- 1.Zaki MS, Sattar S, Massoudi RA, Gleeson JG. Co-occurrence of distinct ciliopathy diseases in single families suggests genetic modifiers. Am J Med Genet A. 2011;155A(12):3042–3049. doi: 10.1002/ajmg.a.34173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Norris DP, Grimes DT. Mouse models of ciliopathies: The state of the art. Dis Model Mech. 2012;5(3):299–312. doi: 10.1242/dmm.009340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh EC, Katsanis N. Cilia in vertebrate development and disease. Development. 2012;139(3):443–448. doi: 10.1242/dev.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basten SG, Giles RH. Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia. 2013;2(1):6. doi: 10.1186/2046-2530-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horner VL, Caspary T. Disrupted dorsal neural tube BMP signaling in the cilia mutant Arl13b hnn stems from abnormal Shh signaling. Dev Biol. 2011;355(1):43–54. doi: 10.1016/j.ydbio.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murdoch JN, Copp AJ. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res A Clin Mol Teratol. 2010;88(8):633–652. doi: 10.1002/bdra.20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breunig JJ, et al. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci USA. 2008;105(35):13127–13132. doi: 10.1073/pnas.0804558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennouna-Greene V, et al. Hippocampal dysgenesis and variable neuropsychiatric phenotypes in patients with Bardet-Biedl syndrome underline complex CNS impact of primary cilia. Clin Genet. 2011;80(6):523–531. doi: 10.1111/j.1399-0004.2011.01688.x. [DOI] [PubMed] [Google Scholar]
- 9.Goetz SC, Anderson KV. The primary cilium: A signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eom DS, Amarnath S, Fogel JL, Agarwala S. Bone morphogenetic proteins regulate neural tube closure by interacting with the apicobasal polarity pathway. Development. 2011;138(15):3179–3188. doi: 10.1242/dev.058602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz i Altaba A, Nguyên V, Palma V. The emergent design of the neural tube: Prepattern, SHH morphogen and GLI code. Curr Opin Genet Dev. 2003;13(5):513–521. doi: 10.1016/j.gde.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Liem KF, Jr, Jessell TM, Briscoe J. Regulation of the neural patterning activity of sonic hedgehog by secreted BMP inhibitors expressed by notochord and somites. Development. 2000;127(22):4855–4866. doi: 10.1242/dev.127.22.4855. [DOI] [PubMed] [Google Scholar]
- 13.Braig S, Bosserhoff AK. Death inducer-obliterator 1 (Dido1) is a BMP target gene and promotes BMP-induced melanoma progression. Oncogene. 2013;32(7):837–848. doi: 10.1038/onc.2012.115. [DOI] [PubMed] [Google Scholar]
- 14.Sánchez de Diego A, Alonso Guerrero A, Martínez-A C, van Wely KH. Dido3-dependent HDAC6 targeting controls cilium size. Nat Commun. 2014;5:3500. doi: 10.1038/ncomms4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.d’Ydewalle C, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- 16.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 17.Fütterer A, et al. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J Clin Invest. 2005;115(9):2351–2362. doi: 10.1172/JCI24177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fütterer A, et al. Ablation of Dido3 compromises lineage commitment of stem cells in vitro and during early embryonic development. Cell Death Differ. 2012;19(1):132–143. doi: 10.1038/cdd.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trachana V, van Wely KHM, Guerrero AA, Fütterer A, Martínez-A C. Dido disruption leads to centrosome amplification and mitotic checkpoint defects compromising chromosome stability. Proc Natl Acad Sci USA. 2007;104(8):2691–2696. doi: 10.1073/pnas.0611132104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bishop GA, Berbari NF, Lewis J, Mykytyn K. Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J Comp Neurol. 2007;505(5):562–571. doi: 10.1002/cne.21510. [DOI] [PubMed] [Google Scholar]
- 21.Turgeon B, Meloche S. Interpreting neonatal lethal phenotypes in mouse mutants: Insights into gene function and human diseases. Physiol Rev. 2009;89(1):1–26. doi: 10.1152/physrev.00040.2007. [DOI] [PubMed] [Google Scholar]
- 22.Kuma A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 23.Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33(1):109–122. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- 24.Tang JY, et al. Immunopositivity of Beclin-1 and ATG5 as indicators of survival and disease recurrence in oral squamous cell carcinoma. Anticancer Res. 2013;33(12):5611–5616. [PubMed] [Google Scholar]
- 25.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prieto I, et al. Synaptonemal complex assembly and H3K4Me3 demethylation determine DIDO3 localization in meiosis. Chromosoma. 2009;118(5):617–632. doi: 10.1007/s00412-009-0223-7. [DOI] [PubMed] [Google Scholar]
- 27.Ozlü N, et al. Binding partner switching on microtubules and aurora-B in the mitosis to cytokinesis transition. Mol Cell Proteomics. 2010;9(2):336–350. doi: 10.1074/mcp.M900308-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Douet-Guilbert N, et al. Chromosome 20 deletions in myelodysplastic syndromes and Philadelphia-chromosome-negative myeloproliferative disorders: Characterization by molecular cytogenetics of commonly deleted and retained regions. Ann Hematol. 2008;87(7):537–544. doi: 10.1007/s00277-008-0462-3. [DOI] [PubMed] [Google Scholar]
- 29.Eisen EJ. Results of growth curve analyses in mice and rats. J Anim Sci. 1976;42(4):1008–1023. doi: 10.2527/jas1976.4241008x. [DOI] [PubMed] [Google Scholar]
- 30.Bussell JJ, Vosshall LB. Behavioral neuroscience: Learning to suckle with signature odor. Curr Biol. 2012;22(21):R907–R909. doi: 10.1016/j.cub.2012.09.034. [DOI] [PubMed] [Google Scholar]
- 31.Brechbühl J, Klaey M, Broillet MC. Grueneberg ganglion cells mediate alarm pheromone detection in mice. Science. 2008;321(5892):1092–1095. doi: 10.1126/science.1160770. [DOI] [PubMed] [Google Scholar]
- 32.Lin C, et al. The inductive role of Wnt-β-Catenin signaling in the formation of oral apparatus. Dev Biol. 2011;356(1):40–50. doi: 10.1016/j.ydbio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gritli-Linde A. Molecular control of secondary palate development. Dev Biol. 2007;301(2):309–326. doi: 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 34.Liu W, et al. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005;132(6):1453–1461. doi: 10.1242/dev.01676. [DOI] [PubMed] [Google Scholar]
- 35.Tabakoff BBS, Bhave S, Saba L. 2012. Whole brain gene expression in males of 20 inbred strains of mice. Available at phenome.jax.org. Accessed December 15, 2013.
- 36. Schadt EE, Su SW (2010) Whole liver gene expression in 22 inbred strains of mice. Available at phenome.jax.org. Accessed December 15, 2013.
- 37.Jaglin XH, Chelly J. Tubulin-related cortical dysgeneses: Microtubule dysfunction underlying neuronal migration defects. Trends Genet. 2009;25(12):555–566. doi: 10.1016/j.tig.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Mahurkar S, Moldovan M, Suppiah V, O’Doherty C. Identification of shared genes and pathways: A comparative study of multiple sclerosis susceptibility, severity and response to interferon beta treatment. PLoS ONE. 2013;8(2):e57655. doi: 10.1371/journal.pone.0057655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith BN, et al. SLAGEN Consortium Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84(2):324–331. doi: 10.1016/j.neuron.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim GW, Li L, Gorbani M, You L, Yang XJ. Mice lacking α-tubulin acetyltransferase 1 are viable but display α-tubulin acetylation deficiency and dentate gyrus distortion. J Biol Chem. 2013;288(28):20334–20350. doi: 10.1074/jbc.M113.464792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalebic N, et al. αTAT1 is the major α-tubulin acetyltransferase in mice. Nat Commun. 2013;4:1962. doi: 10.1038/ncomms2962. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28(5):1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul LK, et al. Agenesis of the corpus callosum: Genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8(4):287–299. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- 44.Stephenson DT, et al. Histopathologic characterization of the BTBR mouse model of autistic-like behavior reveals selective changes in neurodevelopmental proteins and adult hippocampal neurogenesis. Mol Autism. 2011;2(1):7. doi: 10.1186/2040-2392-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Traylor RN, et al. A genotype-first approach for the molecular and clinical characterization of uncommon de novo microdeletion of 20q13.33. PLoS ONE. 2010;5(8):e12462. doi: 10.1371/journal.pone.0012462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Béri-Deixheimer M, et al. Genotype-phenotype correlations to aid in the prognosis of individuals with uncommon 20q13.33 subtelomere deletions: A collaborative study on behalf of the ‘association des Cytogénéticiens de langue Française’. Eur J Hum Genet. 2007;15(4):446–452. doi: 10.1038/sj.ejhg.5201784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.