

Significance
The photoreductive dissolution of Mn oxides governs the biogeochemical cycle of Mn and the fate of organic and inorganic species associated with Mn oxides in the euphotic zones of marine and freshwater systems. Mn oxide minerals also have garnered interest as water oxidation catalysts inspired by the Mn4CaO4 cluster of photosystem II. However, the mechanism of water oxidation by MnO2 and the rate limiting steps for this reaction are unknown. In this study, we couple flow-through experiments and ultrafast pump–probe optical and X-ray absorption spectroscopy to develop a photoreduction model that includes the mechanism and timescales for the initial electron transfer steps in the oxidation of water by MnO2.
Keywords: manganese oxide, photoreduction, band-gap excitation, pump–probe spectroscopy, water oxidation
Abstract
The photoreductive dissolution of Mn(IV) oxide minerals in sunlit aquatic environments couples the Mn cycle to the oxidation of organic matter and fate of trace elements associated with Mn oxides, but the intrinsic rate and mechanism of mineral dissolution in the absence of organic electron donors is unknown. We investigated the photoreduction of δ-MnO2 nanosheets at pH 6.5 with Na or Ca as the interlayer cation under 400-nm light irradiation and quantified the yield and timescales of Mn(III) production. Our study of transient intermediate states using time-resolved optical and X-ray absorption spectroscopy showed key roles for chemically distinct Mn(III) species. The reaction pathway involves (i) formation of Jahn–Teller distorted Mn(III) sites in the octahedral sheet within 0.6 ps of photoexcitation; (ii) Mn(III) migration into the interlayer within 600 ps; and (iii) increased nanosheet stacking. We propose that irreversible Mn reduction is coupled to hole-scavenging by surface water molecules or hydroxyl groups, with associated radical formation. This work demonstrates the importance of direct MnO2 photoreduction in environmental processes and provides a framework to test new hypotheses regarding the role of organic molecules and metal species in photochemical reactions with Mn oxide phases. The timescales for the production and evolution of Mn(III) species and a catalytic role for interlayer Ca2+ identified here from spectroscopic measurements can also guide the design of efficient Mn-based catalysts for water oxidation.
Manganese is a key element in environmental processes, catalytic materials, and biological systems due to its rich redox chemistry and ability to form species with a high oxidizing potential. Photochemical processes can enhance significantly the cycling of Mn between the +4, +3, and +2 valence states (1–3). Photoreduction of Mn(IV) is the first step in the reductive dissolution of birnessite minerals in the euphotic zone of marine and lacustrine environments (4–6). This process couples the biogeochemical cycle of Mn to the redox cycling of carbon and trace metals associated with Mn oxide phases. In addition, the greater role of Mn(IV) photoreduction relative to microbial Mn(II) oxidation leads to the predominance of dissolved over particulate Mn in the photic zone of natural waters (1). Thermodynamic calculations predict that direct photoexcitation of Mn oxides in water by visible light will lead to net metal reduction over a wide range of environmentally relevant pH values (7). However, experimental evidence of direct photoexcitation of MnO2 and subsequent photoreduction of Mn(IV) in the absence of organic electron donors is currently lacking. Experimental studies on the photochemical cycling of Mn have incorporated natural organic ligands that can enhance metal reduction via multiple pathways (5, 8, 9). These studies have identified aqueous Mn(II) as a reaction end product but have not investigated the fate of Mn(III) in the dissolution process, even though Mn(III) is a necessary intermediate in the reduction of Mn(IV) to Mn(II) (10) and an important component of environmental systems (11).
The photochemistry of Mn also enables solar energy harvesting (12) and water oxidation catalysis in synthetic and biological systems (3, 13, 14). Mn-based cluster compounds (15, 16) and disordered birnessite nanoparticles (2) can exhibit analogous reactivity to the water-oxidizing center of photosystem II. Metal reduction is a key step in water oxidation using Mn oxide catalysts (2, 15, 17, 18) with evidence that Mn(III) plays an important role in O2 generation (19). However, no information on the intrinsic kinetics or efficiency of Mn(IV) reduction has been reported to date. The structural and chemical constraints on the mechanism of Mn photoreduction are not known for any Mn phase (17, 18), although a recent study of MnO2-based water oxidation showed that the substitution of Na with Ca in the interlayer of MnO2 greatly enhances reactivity (15). The mineralogy literature suggests that the interlayer cations, which balance the excess charge in the MnO2 sheet, may influence its photoreactivity because the interlayer cations are known to bind water molecules to the neighboring MnO2 octahedral sheets via hydrogen bonding, with the strength of the interactions dependent on the cation valence (20–22). However, the specific role of Ca in the photoreduction process is unknown (15).
The current work combines laboratory-based experiments and ultrafast pump–probe spectroscopy to investigate the photoreduction of δ-MnO2, a fully oxidized synthetic analog of natural birnessites, which is comprised of randomly stacked MnO2 nanosheets that extend only a few nanometers in the ab plane. The first objective was to measure the photoreduction efficiency of δ-MnO2 in flow-through experiments by 400-nm illumination of aqueous suspensions of δ-MnO2, with Na (Na-MnO2) or Ca (Ca-MnO2) as the interlayer cation. The second objective was to elucidate the mechanism of photoreduction by following the coupled changes in Mn valence and coordination that follow photon absorption over picosecond-to-microsecond timescales using time-resolved optical (23) and X-ray (24) absorption spectroscopy. Pyrophosphate was used in the flow-through experiments to quantitate Mn(III) but was not added during spectroscopic experiments because the timescale for Mn(III) production could be determined directly from the transient X-ray absorption data.
Results
Efficiency of MnO2 Photoreduction.
Flow-through experiments were carried out to evaluate the irreversibility of MnO2 photoreduction, to identify the reduced Mn species, and to quantify the efficiency of the process. The irradiation of 500 μM δ-MnO2 suspensions at pH 6.5 ± 0.2 in 1-cm path length flow-through cuvettes showed irreversible Mn(III) generation upon 400 nm irradiation under a photon flux of 0.77 μE s−1, which is comparable to that of sunlight between 400 and 600 nm (calculations in SI Text). We measured the steady formation of Mn(III) at a rate of 2.20 ± 0.27 μmol Mn(III)⋅d−1 (95% confidence interval) in Ca-MnO2 suspensions and 1.50 ± 0.19 μmol Mn(III)⋅d−1 in Na-MnO2 suspensions containing pyrophosphate (PP) as a Mn(III) trapping agent (Fig. S1). These rates were corrected for any release of Mn(III) in dark controls (0.2–0.9 μmol Mn(III)⋅d−1). The nonzero dark release is attributed to the extraction of residual Mn(III) not detectable by the method used to determine average Mn oxidation number (AMON). Apparent quantum yields of 8.2 ± 0.1 × 10−4 and 3.1 ± 0.3 × 10−4 (95% confidence interval) were calculated for Ca-MnO2 and Na-MnO2, respectively. Similar rates measured in suspensions where PP was added after irradiation (Fig. S1) indicate that PP does not influence photoreduction. Further evidence that PP and Mn(III)-PP do not influence Mn photoreduction comes from their UV-visible (UV-vis) spectra, which show no absorption at 400 nm (25).
Optical Transient Absorption Spectroscopy.
Figs. 1 and 2 present optical transient absorption (TA) spectra and kinetics, respectively, for Ca-MnO2 acquired at subpicosecond-to-microsecond timescales. Equivalent data for Na-MnO2 are presented in Figs. S2 and S3. All samples were resuspended in 1.5% (mass/vol) Nafion to reduce particle aggregation. Following photoexcitation at 400 nm, the TA spectra acquired at a few picoseconds exhibited a broad excited-state absorption (ESA) feature with maximum intensity at 538 nm (Ca-MnO2; Fig. 1) or 570 nm (Na-MnO2; Fig. S2A). The TA spectra also showed a ground-state bleach extending from the laser wavelength to the lowest wavelength measureable (∼335 nm); the bleach reflects the decrease in the population of valence band electrons that remain in the ground state. After correcting for the laser chirp, the comparison of the TA kinetics at 345 and 550 nm showed a rise in ESA intensity that was delayed relative to the prompt bleach signal. The maximum ESA intensity occurred at ∼0.3 ps for Ca-MnO2 (Fig. 2A) and ∼0.6 ps for Na-MnO2 (Fig. S3A). The ESA and bleach signals decayed with similar dynamics up to ∼10 ps (Fig. 2B). The ESA then decayed completely within 1 ns, but the bleach signal remained detectable on the microsecond timescale. The decay kinetics of the bleach differed for Ca- vs. Na-MnO2 (Fig. 1C), although both samples exhibited short (submicrosecond, τ1) and long (10–30 μs, τ2) decay timescales (Table 1).
Fig. 1.

Optical transient absorption (TA) spectra acquired from Ca-MnO2 in an aqueous suspension containing Nafion at 0.75 ps, 3.6 ps, and 2 ns after photoexcitation at 400 nm. The TA spectra (milli-ΔOD units; left axis) are compared with the ground-state UV-vis absorption spectra (OD units; right axis). Vertical lines at 550 nm (the maximum intensity of the ESA) and at 345 nm (ground-state bleach) indicate wavelengths chosen for kinetics analysis. The data from 390–410 nm are affected by scattering of the excitation beam. Equivalent TA data for Na-MnO2 are given in Fig. S2A.
Fig. 2.

Summary of optical transient absorption kinetics acquired from Ca-MnO2 in an aqueous suspension containing Nafion. (A) Transient kinetic traces show a slight delay in the onset of the ESA at 550 nm relative to the bleach at 345 nm. (B) Decay kinetics of the ESA and bleach up to 8 ns. The time-zero was shifted by 0.2 ps to enable plotting on a logarithmic scale. Equivalent data for Na-MnO2 are given in Fig. S3 A and B. (C) Comparison between the decay kinetics of the bleach recorded at 580 nm for Ca-MnO2 and Na-MnO2 in Nafion, fitted with two-component exponential decay functions.
Table 1.
Time constants obtained from fitting the EOS data with exponential decay functions (Fig. S8)
| τ1 (μs) ± SD | τ2 (μs) ± SD | χ2 | |
| Na-MnO2 | |||
| Water | 0.36 ± 0.03 | 15.31 ± 0.96 | 11.03 |
| TPA | 0.36 ± 0.04 | 14.38 ± 1.32 | 18.68 |
| Nafion | 0.24 ± 0.02 | 10.14 ± 0.63 | 12.68 |
| Ca-MnO2 | |||
| Nafion | 0.81 ± 0.06 | 27.35 ± 1.75 | 6.53 |
A time offset of 0.047 μs was used for all data.
Direct comparison of the fitted first-order decay constants for Ca-MnO2 and Na-MnO2 in Nafion-stabilized suspensions reveals that the recombination rates are three- to fourfold slower in the presence of interlayer Ca. In particular, the τ2 values indicate that the bleach returns to baseline within 50 μs for Na-MnO2 (five half-lives elapsed, 0.02% of initial signal remains), whereas for Ca-MnO2 the bleach does not return to the baseline within the resolution of the experiment (two half-lives elapsed, 8% of initial signal remains). Slightly slower decays were observed for samples prepared without Nafion, possibly indicating an effect of aggregation upon recombination rates. Slower decay could occur if recombination involved diffusion of soluble species, but this effect is impossible to explain confidently and is smaller than the influence of the interlayer cation (SI Text). The addition of the anionic hydroxyl radical scavenger, terephthalic acid (TPA), had no detectable effect on the decay kinetics for Na-MnO2.
Light-Initiated Time-Resolved X-Ray Absorption Spectroscopy.
Selected ground-state Mn K-edge spectra collected during a single 4-h light-initiated time-resolved X-ray absorption spectroscopy (LITR-XAS) experiment with Ca-MnO2 are displayed in Fig. 3. The increase in the absorption intensity at 6,550 eV (Fig. 3, Inset) shows a shift of the Mn K-edge to lower energies, which is consistent with a decrease in the average oxidation state of Mn (26). Thus, 400-nm laser excitation causes the accumulation of reduced Mn. A difference X-ray absorption (ΔXA) spectrum, obtained by subtracting the initial ground-state spectrum from the 3-h ground-state spectrum, is included in Fig. 3 to emphasize the irreversible change in Mn valence and bonding environment after 3 h of irradiation.
Fig. 3.

Ground-state Mn K-edge X-ray absorption (XA) spectra show the steady reduction of Mn in a ∼10 mM Ca-MnO2 suspension at pH 6.5 during a ∼4-h laser pump/X-ray probe study. Spectra are shown for 1 min (solid line), 19 min (dashed line), and 175 min (dotted line) after the onset of the experiment (left vertical axis). The difference (ΔXA) spectrum obtained by subtracting the spectrum at 175 min from the spectrum at 1 min (right vertical axis) is included beneath the XA spectra. (Inset) XA intensity at 6,550 eV as a function of time, which indicates an increase in reduced Mn.
Transient ΔXA spectra were obtained by subtracting the ground-state spectrum from the excited-state spectra at delay times of up to 10 ns (Fig. 4A). The first oscillation between 6,540 and 6,570 eV in the transient ΔXA spectrum at a 50-ps delay indicates a shift in the absorption edge position to lower energies that is consistent with the formation of reduced Mn (Fig. 3). The feature is approximately constant from 50 ps (the temporal resolution of the experiment) to 10 ns (the latest time point studied). As shown in Fig. 4B, transient kinetic data at 6,550 eV confirmed the prompt formation and negligible decay of the signal for reduced Mn. Within 600 ps, we observed modulations at energies above 6,570 eV, the fine-structure portion of the transient ΔXA spectrum, which reflect changes in the coordination environment of Mn. Finally, the close agreement between the transient ΔXA data at 10 ns and the ΔXA data associated with permanent photoreduction at 3 h indicates that at least a fraction of the reduced Mn species observed at 10 ns persists indefinitely.
Fig. 4.

(A) ΔXA spectra at various delay times reveal a prompt change in the Mn oxidation state (feature at 6,550 eV, left arrow) and evolution in fine structure within 1 ns (feature at 6,570 eV, right arrow). The ground-state ΔXA spectrum, obtained by subtracting the ground spectrum at 175 min from the ground spectrum at 1 min (Fig. 3), is shown for comparison with the transient ΔXA spectra. (B) Kinetic traces of the X-ray absorption intensity at 6,552 eV with (solid line) and without (dashed line) the laser pump confirm a prompt change in Mn valence and no decay within 2 ns.
The long-term effect of Mn(III) accumulation on the irradiated Mn oxide was assessed by characterization of the mineral structure. Powder X-ray diffraction (XRD) patterns acquired from samples before laser irradiation show broad hk0 peaks at 2.4 and 1.4 Å and no stacking peak at 7.2 Å (in d-spacing). This pattern is characteristic of δ-MnO2 nanosheets with hexagonal sheet symmetry and fewer than three randomly stacked sheets (26). Following laser irradiation, the XRD patterns showed additional 001 and 002 Bragg reflections (Fig. S4) consistent with increased ordered stacking of the MnO2 sheets (26).
Discussion
Efficiency of Mn(IV) Oxide Photoreduction.
Our flow-through experiments show that the irreversible photoreduction of δ-MnO2 to form Mn(III) occurs readily under environmentally relevant conditions in the absence of any electron donor other than water. In addition, our measurements show no production of Mn(II). The apparent quantum yield of the process at pH 6.5 is up to two orders of magnitude larger than the apparent quantum yield measured for γ-Fe2O3(s) at pH 3.0, ∼10−5 (27), and comparable to the value measured for γ-FeOOH(s) at pH 3.0, ∼10−3 (28). Furthermore, Fe photoreduction in water has only been observed at pH values below 5. These results emphasize the importance of photoreduction in the redox cycling of Mn oxides relative to Fe oxides and support the thermodynamic calculations presented by Sherman (7), which indicate that photoreduction may occur at higher pH values for Mn oxides than for Fe oxides.
Environmental Mn cycling is frequently coupled to the oxidation of dissolved organic matter (DOM). Manganese oxide photodissolution in the presence of 10–40 mg/L DOM has been reported to have a 5–10 times greater efficiency (9) than measured here for MnO2 photoreduction without organics. DOM can increase the accumulation of reduced Mn by acting as the chromophore that either initiates electron transfer to the mineral (9) or photolyzes to generate reactive oxygen species (5); by acting as an electron donor to the photoexcited mineral; or by providing ligands that can complex intermediate Mn3+ as Mn(III)(aq) species (11). Due to the optical properties of DOM, the first mechanism is only important under UV light, which is a minor component of the sun’s irradiance spectrum on the Earth’s surface and has a lower penetration depth (up to ∼25 m) in natural waters than visible wavelengths (∼100 m) (29). The complexation of reduced manganese by DOM is likely to occur independently from the photoreduction mechanism. Thus, our results indicate that Mn photoreduction in which water serves as electron donor must be a significant contributor to any overall environmental rate.
Manganese Redox Dynamics.
We used the time-resolved optical and X-ray data to construct a model for the photoexcitation and evolution of Mn(III) states in δ-MnO2 as visualized in Fig. 5.
Fig. 5.
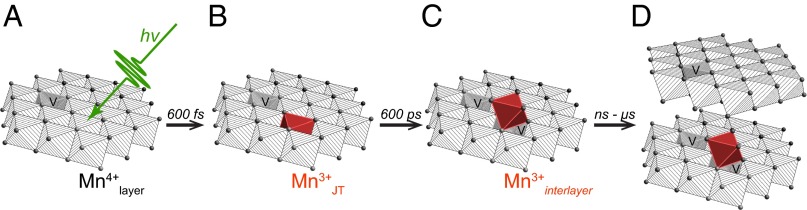
Proposed model for the evolution of metal redox chemistry during δ-MnO2 photoreduction. (A) Absorption of a photon (hv) by a single Mn(IV) oxide nanosheet that includes metal vacancy sites (V). (B) Formation within 600 fs of a Jahn–Teller distorted Mn(III) state within the nanosheet (dark red octahedron). (C) Migration within 1 ns of Mn(III) from the nanosheet to an adsorption site above a new vacancy. (D) Increased nanosheet stacking due to increased compensation of sheet negative charge by interlayer Mn(III). Water molecules are omitted for clarity.
A transient Jahn–Teller-distorted Mn(III) intermediate forms in the nanosheet.
We interpret the transient optical ESA that appears within 0.3–0.6 ps, and that has a lifetime less than 1 ns, as a transient intermediate Mn(III) state that is formed by ligand-to-metal charge-transfer that excites an oxygen-centered valence electron into the metal 3d state (7). This interpretation requires justification because transient electronic excitations observed in TA spectroscopy of metal oxides could be associated with either electrons (e.g., the transitions of electrons promoted to the conduction band) or holes (i.e., new excitations within the valence band). For example, band-gap excitation of hematite (α-Fe2O3) generates a prompt optical ESA centered at 580 nm attributed to hole excitations based on UV-vis spectra of a thin-film hematite electrode under oxidizing vs. reducing conditions (30). In the present case, however, the delayed onset of the ESA is consistent with electronic relaxation into a polaronic state, as observed for photoexcitation of manganites (31). Polaron formation would be expected for the creation of a Mn(III) state, for which Jahn–Teller (JT) distortion of the octahedron can lower the ground-state energy by adopting high-spin d4 electron configuration and splitting of the eg and t2g orbitals (32). In this depiction, the ESA is caused by low-energy excitations of the extra electron localized in the JT state. Additionally, UV-vis spectra from well-characterized MnOx phases show that Mn(III) located in the MnO2 sheets confers absorption features in the visible range between 400 and 600 nm (Fig. S5).
The line shape of the ESA reported here closely matches a feature in the UV-vis spectrum reported for a birnessite anode poised at sufficiently positive potentials to oxidize water (19). Takashima et al. (19) assigned the optical feature between 400 and 600 nm to adsorbed Mn(III) based on pyrophosphate extractions. However, any Mn(III) extracted by pyrophosphate after the electrochemical oxidation of water reflects only the final Mn(III) state. Our kinetic measurements demonstrate that the visible ESA is due to a transient state.
A long-lived interlayer Mn(III) adsorbate between the nanosheets.
We propose that the loss of the optical ESA signal (i.e., the JT state) is caused by two processes occurring on different timescales. Within the first 10 ps, the ESA decay and that of the bleach were closely coupled, indicating electron–hole recombination. Further decay of the ESA signal, however, was not matched by further loss of the bleach. The LITR-XAS also showed that within ∼600 ps there was a change in the Mn bonding environment but no loss of the reduced Mn state. We explain these trends by the displacement of Mn(III) from the nanosheet layer into the hydrated interlayer region, with the formation of a vacancy site beneath (Fig. 5C). Prior studies have established that Mn(III) cations can reside stably within the interlayer of birnessite nanosheets to reduce the sheet steric strain (33, 34). The JT distortion at the Mn(III) site could drive its migration into the interlayer, where it can act as an adsorbed cation that enhances the stacking of the negatively charged sheets (Fig. 5D). In addition, JT-distorted Mn(III) sites have been reported as key species for water oxidation (17). The migration of JT distorted Mn(III) from the MnO2 sheet into the interlayer and change in Mn(III) bonding environment may decrease the overall reactivity of the material for multielectron water oxidation. Thus, our finding has implications regarding the mechanistic constraints on Mn oxide photocatalysis. The formation of new vacancy sites likely also alters the optical absorption properties of the modified nanosheet (35). Our model, however, requires that the putative interlayer Mn(III) ion lack an optical absorption signature in the 335- to 800-nm range; this could not be verified through published studies of the UV-vis absorption spectra of Mn3+ because this species is unstable in aqueous solution (11, 25).
Hole dynamics: Recombination vs. water oxidation chemistry.
Photoexcitation of δ-MnO2 and the generation of Mn(III) must be accompanied by the formation of valence band holes. Hole states in transition metal oxides are typically localized at oxygen sites, but holes that form close to surface sites may participate in reactions with water, including steps in the four-electron oxygen evolution reaction (36) or with aqueous ions such as Cl− (37).
The bleach signal in the TA data shows complete return to the ground state after 50 μs or longer, indicating that most of the hole states are able to recombine with the photoexcited electrons in Mn(III) even after migration of the metal ion into the interlayer. Although we proved that net photoreduction occurs, the apparent quantum yield is too low for the irreversible fraction to be detectable above the statistical noise in the TA data. Recombination may involve exclusively solid-phase species, or the formation of chemical intermediates. Borer et al. (28) used a hydroxyl radical scavenger to demonstrate the generation of hydroxyl radicals (OH•) through hole reaction with surface hydroxyls on γ-FeOOH, but in that case only an anionic scavenger was able to react with the net positively charged surface of iron oxyhydroxides. In the present case, we were unable to identify a cationic hydroxyl scavenger that was stable against dark reaction with Mn(IV), and the anionic TPA may have been unable to interact with the net negatively charged δ-MnO2 surface. Nevertheless, surface-bound hydroxyl groups on birnessite have been identified in a synthetic Mn(III)-bearing microcrystalline birnessite phase from the interpretation of thermogravimetric data (33). Reactions between OH• species could yield H2O2 or other reactive oxygen species that could oxidize Mn(III) back to Mn(IV) and thus reduce the net quantum yield of direct photoreduction by band-gap excitations. Our efforts are currently dedicated to identifying these radical species, which can also enhance microbial Mn oxidation (38) and induce cellular oxidative stress.
Influence of Interlayer Cation on MnO2 Photoreduction.
We observed that the interlayer cations, which balance the negative layer charge, influence the optical properties and photochemistry of layer-type MnO2, as observed previously in water oxidation studies (15). The slower microsecond timescale kinetics for Ca- vs. Na-MnO2 and greater apparent quantum yield, which cannot be explained by aggregation (SI Text), suggest a catalytic role for this ion that reduces the rate at which interlayer Mn(III) species and photoexcited hole states can recombine. Because Ca2+ has a greater hydration enthalpy (22) and slower water exchange rate than Na+ (108 s−1 vs. 109 s−1) (39), its catalytic role may reduce the mobility of water and other species in the interlayer. However, Ca also caused redshifts in both ground-state and transient UV-vis spectra (Figs. S3B and S6A), indicating that this ion has significant chemical interactions with the Mn oxide nanosheets. Further work will be required to fully determine the role of Ca, but the model proposed here provides a basis for future studies.
Environmental Implications
This work provides insight into the mechanism, rates, and chemical controls on the photoreduction of fully oxidized Mn oxide phases that are the initial products of biological manganese oxidation (6) and that may have been precursors to biological water oxidation catalysts (14). The evidence we provide for δ-MnO2 photoreduction in the absence of organic electron donors establishes this pathway as an important component of the Mn cycle. That this reaction occurs at 400 nm indicates that it is important at all depths of the water column to which visible light penetrates, including depths where dissolved organic matter is not photoactive. The broad absorbance of MnO2 suggests that photoreduction can occur across a wide spectrum of solar irradiation, although further experiments will be required to establish the wavelength dependence. Photoreduction creates Mn(III) states that are stabilized by adsorption at interlayer sites and increase nanosheet stacking. Our results also suggest that Mn(IV) photoreduction in sunlit environments may cause initial biogenic Mn(IV) oxides to transform to a phase with varying amounts of Mn(III) (6). We cannot generalize our conclusions to the many other phases of birnessites that can be found in nature, but we anticipate that photochemical transformations of these phases will retain similar mechanistic aspects, although the net rates may be influenced by Mn(III) content.
Materials and Methods
American Chemical Society-grade chemicals were purchased from Sigma-Aldrich or Merck; Nafion was purchased from Fuel Cell Earth LLC. All solutions were prepared with ultrapure water (18 MΩ ⋅ cm). The δ-MnO2 phase used in flow-through photodissolution experiments and pump–probe experiments was synthesized according to Villalobos et al. (26). Synthesis and characterization of the mineral phase are described in SI Text. All experiments were carried out at ambient temperature, on suspensions under aerated conditions or purged with N2(g) or He2(g).
Efficiency of MnO2 Photoreduction.
Flow-through experiments were carried out to verify whether δ-MnO2 photoreduction by band-gap excitation generated reduced Mn ions irreversibly and to measure the rates and efficiency of the process. We prepared oxide suspensions containing 500 μM Mn and 25 mM sodium pyrophosphate (PP) to trap Mn(III) with a final pH of 6.5 (no pH-adjustment was required). The Ca-MnO2 suspensions were prepared by equilibrating the oxide with CaCl2(aq) in a 3:1 molar ratio. All suspensions were equilibrated in the dark for 3 d before irradiation. The suspensions were then divided into two aliquots: one was used as a dark control and the second was recirculated through a flow-through quartz cuvette. Irradiation for 72 h was provided by an array of three 1-W light-emitting diodes at 400 nm (3.1 eV), close to the maximum UV-vis absorbance of a Na-MnO2 suspension in water (Fig. S5A). The photoreactor was screened from ambient light.
Every 24 h, a sample aliquot was collected for inductively coupled plasma optical emission spectrometry (ICP-OES) measurement of [MnTOT] after digestion with 0.05 M H2C2O4 and 3% (vol/vol) HNO3 and another was filtered through a 0.2-μm polyethersulfone syringe filter for ICP-OES measurement of [Mn(aq)] and [Mn(III)-PP] quantification by UV-vis spectrophotometry (ε254 nm = 6,562 L⋅mol−1⋅cm−1) with 1-cm path-length quartz cuvettes using a 25-mM PP solution as a blank. Measurements of [Mn(aq)] and [Mn(III)-PP] were within 10%, with concentrations ranging from 10 to 50 μM. Experiments were conducted in duplicate; suspension pH was 6.5 ± 0.2 before and after irradiation.
Complementary experiments were carried out to confirm that PP did not influence Mn photoreduction. Experiments were conducted as described above but in the absence of PP and with Na-MnO2 supplemented with 10 mM NaCl. After 0, 24, 48, and 72 h of irradiation, 10-mL sample aliquots were collected, mixed with 2 mL of a 100 mM PP stock solution (pH 6.5), and placed on an end-to-end shaker for 24 h in the dark. Measurements of [MnTOT], [Mn(aq)], and [Mn(III)-PP(aq)] were made as described above; suspension pH was 6.5 ± 0.2 before and after irradiation.
The rate of Mn(III) production was determined from linear regression of Mn(III)-PP against time, after correction for any Mn(III) released in dark control experiments. We then calculated the apparent quantum yield for Mn(III) generation, which is defined as the amount of photoproduced Mn(III) per photon absorbed by the MnO2 suspension. To calculate the number of photons absorbed, we measured the photon flux to the photoreactor by chemical actinometry using potassium ferrioxalate and then scaled the photon flux to the ratio of the absorbance of MnO2 to ferrioxalate (SI Text).
Optical Transient Absorption Spectroscopy.
Optical TA spectroscopy can be used to follow excited-state valence electron dynamics in semiconducting metal oxides (23). Optical TA experiments were carried out on a HELIOS femtosecond transient absorption spectrometer and on an EOS subnanosecond transient absorption spectrometer from Ultrafast Systems installed at the Molecular Foundry, Lawrence Berkeley National Laboratory. The laser source for both instruments was a Coherent Libra Amplified Femtosecond Laser System operating at 1 kHz with 45-fs pulse duration. The laser output was split, one arm passing through a Coherent OPerA optical parametric amplifier (OPA) to produce pump pulses at 400 nm, the other arm delivered to the transient absorption system where a white-light probe pulse was generated in a sapphire plate or by a fiber white-light source (Leukos-STM) for the HELIOS and EOS spectrometers, respectively. The intensity of the pump beam was measured to be 800 nJ⋅pulse−1. Time delay was provided by a mechanical delay stage on the HELIOS setup and by instrument electronics on the EOS setup. Spectra were collected between 335 and 900 nm.
Samples of Ca-MnO2 and Na-MnO2 were either kept in their original aqueous suspension or resuspended in an aqueous solution of 1.5% (mass/vol) Nafion in 3% (vol/vol) isopropanol. The Nafion polymer reduced particle aggregation and there is no evidence that it influences the photochemical behavior of birnessite-based photocatalysts (2, 40). Furthermore, the comparison of TA data of Na-MnO2 in water vs. in an aqueous Nafion solution showed no detectable differences on picosecond-to-nanosecond timescales (Fig. S9). The 3% (vol/vol) isopropanol was evaporated in air from the Nafion solution for 12 h before starting the measurements to ensure complete isopropanol evaporation. Additional Na-MnO2 samples were resuspended in 0.1 M TPA, a hydroxyl radical trap (41), and measured on the EOS setup. The samples were measured in 1-mm path-length quartz cuvettes and stirred with a magnetic stir bar or recirculated in a flow-through cuvette. The optimum MnO2 concentration for TA measurements ranged from 3 to 6 mM on a Mn molar basis, corresponding to an OD at 400 nm between 0.8 and 1.6 absorption units on 0.5-mm path-length quartz cuvettes. Spectra were collected from −10 ps to 8 ns (relative to laser pulse) on the HELIOS system and from 1 ns to 50 μs on the EOS system. Custom user routines developed in the IGOR Pro software (WaveMetrics Inc.) were used to process the data, including correction of the time-dependent frequency modulation of the laser (laser chirp) on the signal to within ∼0.15 ps and to extract transient kinetic data at 354 and 550 nm from 2D plots of spectra vs. time. IGOR Pro was also used to fit first-order exponential decay kinetics to the extracted data. Decay kinetics for all samples were adequately reproduced by fitting two exponential time constants (SI Text).
Laser-Initiated Time-Resolved X-Ray Absorption Spectroscopy.
LITR-XAS excites core electrons and yields information on the oxidation state and local bonding environment of the probed atoms following light excitation (24). LITR-XAS experiments were carried out at Beamline 6.0.1 at Advanced Light Source. At this beamline, the X-ray beam pulses are isolated with an X-ray chopper and combined with the output of a femtosecond Ti:Sapphire laser system with a power output of 800 mW measured at the sample position. The X-ray energy was scanned across the Mn K-edge (6,530–6,595 eV). Ground-state and transient X-ray absorption spectra were collected in fluorescence mode with an avalanche photodiode fitted with Soller slits and a Cr filter. The 400-nm laser pulse was obtained by converting the 800-nm output from the 4-kHz Ti:Sapphire laser system with an OPA. The laser pulse was synchronized to a single electron bunch by locking the 62.5-MHz repetition rate of the laser system oscillator to the 499.64-MHz of the synchrotron’s radiofrequency cavity to an accuracy of less than 20 ps. The FWHM of the laser and X-ray pulses were 0.1 and 70 ps, respectively. Finally, the time 0 delay between X-ray and laser pulses was determined using an iron(II) Tris(2,2′-bipyridine) solution, which exhibits distinct changes in absorption at the Fe K-edge upon laser excitation.
The laser and X-ray beams intersected the sample in a closed He2(g) purged chamber. Samples were recirculated from a bottle on a stir plate through a nozzle forming a 600-μm diameter liquid jet. The size of the X-ray beam on the sample was 60 × 60 µm. To begin an experiment, 250-mL suspensions of ∼10 mM Ca- and Na-MnO2 were prepared. Before data acquisition, the chamber and sample were purged with He2(g). Each experimental condition was repeated in duplicate on suspensions recirculated for up to 4 h to obtain transient and kinetic data. For transient X-ray absorption experiments, spectra were collected at a fixed time delay and the monochromator was scanned from 40 eV below to 70 eV above the Mn K-edge (6,539 eV). The transient spectra were three-point smoothed for plotting. Kinetic data were acquired by setting the monochromator at a fixed energy position and varying the time delay between laser and X-ray pulses. In both acquisition modes, data were collected before and after laser irradiation at the chosen time delay to extract ground-state and excited-state spectra. Following each LITR-XAS experiment, the samples were retained for analysis by X-ray diffraction. To assess the possibility of X-ray radiation damage, a conservative Mn:photon ratio was calculated. We estimated that 1014 X-ray photons are delivered to the sample over the 4-h timespan of the experiment, compared with ∼1021 Mn atoms in the sample (SI Text). The Mn:photon ratio was thus estimated to be 106:1, strongly suggesting that X-ray beam damage is negligible.
Supplementary Material
Acknowledgments
We thank Drs. M. Hertlein and T. E. Glover for assistance with the X-ray measurements; Dr. R. W. Schoenlein for equipment and staff assistance; Drs. K. Sand and T. Adatte for the acquisition of high-energy X-ray scattering data and XRD patterns, respectively; and three anonymous reviewers for their valuable comments. F.F.M. and J.P. acknowledge support from the Swiss National Science Foundation (Proposal 200021_143742) and a grant from the Sandoz Family Foundation. B.G. was supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy (BES-DOE) Contract DE-AC02-05CH11231. Research at the Molecular Foundry and the Advanced Light Source was supported by BES-DOE Contract DE-AC02-05CH11231. Pair distribution function data were acquired at the Advanced Photon Source, operated for BES-DOE by Argonne National Laboratory under Contract DE-AC02-06CH11357.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
1Present address: Beam Line Development, SLAC National Accelerator Laboratory, Menlo Park, CA 94025.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1421018112/-/DCSupplemental.
References
- 1.Sunda WG, Huntsman SA. Diel cycles in microbial manganese oxidation and manganese redox speciation in coastal waters of the Bahama Islands. Limnol Oceanogr. 1990;35(2):325–338. [Google Scholar]
- 2.Hocking RK, et al. Water-oxidation catalysis by manganese in a geochemical-like cycle. Nat Chem. 2011;3(6):461–466. doi: 10.1038/nchem.1049. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong FA. Why did Nature choose manganese to make oxygen? Philos Trans R Soc Lond B Biol Sci. 2008;363(1494):1263–1270, discussion 1270. doi: 10.1098/rstb.2007.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan JJ. Manganese in natural waters and earth’s crust: Its availability to organisms. Met Ions Biol Syst. 2000;37:1–34. [PubMed] [Google Scholar]
- 5.Sunda WG, Huntsman SA. Photoreduction of manganese oxides in seawater. Mar Chem. 1994;46(1-2):133–152. [Google Scholar]
- 6.Spiro TG, Bargar JR, Sposito G, Tebo BM. Bacteriogenic manganese oxides. Acc Chem Res. 2010;43(1):2–9. doi: 10.1021/ar800232a. [DOI] [PubMed] [Google Scholar]
- 7.Sherman DM. Electronic structures of iron(III) and manganese(IV) (hydr)oxide minerals: Thermodynamics of photochemical reductive dissolution in aquatic environments. Geochim Cosmochim Acta. 2005;69(13):3249–3255. [Google Scholar]
- 8.Kim K, Yoon HI, Choi W. Enhanced dissolution of manganese oxide in ice compared to aqueous phase under illuminated and dark conditions. Environ Sci Technol. 2012;46(24):13160–13166. doi: 10.1021/es302003z. [DOI] [PubMed] [Google Scholar]
- 9.Waite TD, Wrigley IC, Szymczak R. Photoassisted dissolution of a colloidal manganese oxide in the presence of fulvic acid. Environ Sci Technol. 1988;22(7):778–785. doi: 10.1021/es00172a006. [DOI] [PubMed] [Google Scholar]
- 10.Luther GW. Manganese(II) oxidation and Mn(IV) reduction in the environment—Two one-electron transfer steps versus a single two-electron step. Geomicrobiol J. 2005;22(3-4):195–203. [Google Scholar]
- 11.Madison AS, Tebo BM, Mucci A, Sundby B, Luther GW., 3rd Abundant porewater Mn(III) is a major component of the sedimentary redox system. Science. 2013;341(6148):875–878. doi: 10.1126/science.1241396. [DOI] [PubMed] [Google Scholar]
- 12.Sakai N, Ebina Y, Takada K, Sasaki T. Photocurrent generation from semiconducting manganese oxide nanosheets in response to visible light. J Phys Chem B. 2005;109(19):9651–9655. doi: 10.1021/jp0500485. [DOI] [PubMed] [Google Scholar]
- 13.Kolling DR, Cox N, Ananyev GM, Pace RJ, Dismukes GC. What are the oxidation states of manganese required to catalyze photosynthetic water oxidation? Biophys J. 2012;103(2):313–322. doi: 10.1016/j.bpj.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson JE, et al. Manganese-oxidizing photosynthesis before the rise of cyanobacteria. Proc Natl Acad Sci USA. 2013;110(28):11238–11243. doi: 10.1073/pnas.1305530110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiechen M, Zaharieva I, Dau H, Kurz P. Layered manganese oxides for water-oxidation: Alkaline earth cations influence catalytic activity in a photosystem II-like fashion. Chem Sci. 2012;3(7):2330–2339. [Google Scholar]
- 16.Hsu YK, Chen YC, Lin YG, Chen LC, Chen KH. Birnessite-type manganese oxides nanosheets with hole acceptor assisted photoelectrochemical activity in response to visible light. J Mater Chem. 2012;22(6):2733–2739. [Google Scholar]
- 17.Robinson DM, et al. Photochemical water oxidation by crystalline polymorphs of manganese oxides: Structural requirements for catalysis. J Am Chem Soc. 2013;135(9):3494–3501. doi: 10.1021/ja310286h. [DOI] [PubMed] [Google Scholar]
- 18.Birkner N, et al. Energetic basis of catalytic activity of layered nanophase calcium manganese oxides for water oxidation. Proc Natl Acad Sci USA. 2013;110(22):8801–8806. doi: 10.1073/pnas.1306623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takashima T, Hashimoto K, Nakamura R. Mechanisms of pH-dependent activity for water oxidation to molecular oxygen by MnO2 electrocatalysts. J Am Chem Soc. 2012;134(3):1519–1527. doi: 10.1021/ja206511w. [DOI] [PubMed] [Google Scholar]
- 20.Johnson EA, Post JE. Water in the interlayer region of birnessite: Importance in cation exchange and structural stability. Am Mineral. 2006;91(4):609–618. [Google Scholar]
- 21.Drits VA, Lanson B, Gorshkov AI, Manceau A. Substructure and superstructure of four-layer Ca-exchanged birnessite. Am Mineral. 1998;83(1-2):97–118. [Google Scholar]
- 22.Cygan RT, Post JE, Heaney PJ, Kubicki JD. Molecular models of birnessite and related hydrated layered minerals. Am Mineral. 2012;97(8-9):1505–1514. [Google Scholar]
- 23.Gilbert B, et al. Ultrafast electron and energy transfer in dye-sensitized iron oxide and oxyhydroxide nanoparticles. Phys Chem Chem Phys. 2013;15(40):17303–17313. doi: 10.1039/c3cp53368a. [DOI] [PubMed] [Google Scholar]
- 24.Katz JE, et al. Electron small polarons and their mobility in iron (oxyhydr)oxide nanoparticles. Science. 2012;337(6099):1200–1203. doi: 10.1126/science.1223598. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Stone AT. Phosphonate- and carboxylate-based chelating agents that solubilize (hydr)oxide-bound MnIII. Environ Sci Technol. 2008;42(12):4397–4403. doi: 10.1021/es7032668. [DOI] [PubMed] [Google Scholar]
- 26.Villalobos M, Toner B, Bargar J, Sposito G. Characterization of the manganese oxide produced by Pseudomonas putida strain MnB1. Geochim Cosmochim Acta. 2003;67(14):2649–2662. [Google Scholar]
- 27.Litter MI, Blesa MA. Photodissolution of iron-oxides. 4. A comparative-study on the photodissolution of hematite, magnetite, and maghemite in EDTA media. Can J Chem. 1992;70(9):2502–2510. [Google Scholar]
- 28.Borer P, Sulzberger B, Hug SJ, Kraemer SM, Kretzschmar R. Photoreductive dissolution of iron(III) (hydr)oxides in the absence and presence of organic ligands: Experimental studies and kinetic modeling. Environ Sci Technol. 2009;43(6):1864–1870. doi: 10.1021/es801352k. [DOI] [PubMed] [Google Scholar]
- 29.Smith RC, Baker KS. Optical properties of the clearest natural waters (200-800 nm) Appl Opt. 1981;20(2):177–184. doi: 10.1364/AO.20.000177. [DOI] [PubMed] [Google Scholar]
- 30.Barroso M, Pendlebury SR, Cowan AJ, Durrant JR. Charge carrier trapping, recombination and transfer in hematite (α-Fe2O3) water splitting photoanodes. Chem Sci. 2013;4(7):2724–2734. [Google Scholar]
- 31.Wu KH, et al. Ultrafast optical probes of polaron dynamics in La0.7Ca0.3MnO3 thin films. J Appl Phys. 2009;105(4) [Google Scholar]
- 32.Burns RG. Mineralogical Applications of Crystal Field Theory. 2nd Ed Cambridge Univ Press; New York: 1993. [Google Scholar]
- 33.Gaillot AC, et al. Structure of synthetic K-rich birnessite obtained by high-temperature decomposition of KMnO4. I. Two-layer polytype from 800 °C experiment. Chem Mater. 2003;15(24):4666–4678. [Google Scholar]
- 34.Manceau A, et al. Short-range and long-range order of phyllomanganate nanoparticles determined using high-energy X-ray scattering. J Appl Cryst. 2013;46(1):193–209. [Google Scholar]
- 35.Kwon KD, Refson K, Sposito G. On the role of Mn(IV) vacancies in the photoreductive dissolution of hexagonal birnessite. Geochim Cosmochim Acta. 2009;73(14):4142–4150. [Google Scholar]
- 36.Tang J, Durrant JR, Klug DR. Mechanism of photocatalytic water splitting in TiO2. Reaction of water with photoholes, importance of charge carrier dynamics, and evidence for four-hole chemistry. J Am Chem Soc. 2008;130(42):13885–13891. doi: 10.1021/ja8034637. [DOI] [PubMed] [Google Scholar]
- 37.Grätzel M, Kiwi J, Morrison CL, Davidson RS, Tseung AC. Visible-light-induced photodissolution of α-Fe2O3 powder in the presence of chloride anions. J Chem Soc Faraday Trans 1. 1985;81(8):1883–1890. [Google Scholar]
- 38.Hansel CM, Zeiner CA, Santelli CM, Webb SM. Mn(II) oxidation by an ascomycete fungus is linked to superoxide production during asexual reproduction. Proc Natl Acad Sci USA. 2012;109(31):12621–12625. doi: 10.1073/pnas.1203885109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stumm W, Morgan JJ. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters. Wiley; New York: 1996. [Google Scholar]
- 40.Young KJ, Gao Y, Brudvig GW. Photocatalytic water oxidation using manganese compounds immobilized in Nafion polymer membranes. Aust J Chem. 2011;64(9):1221–1228. doi: 10.1071/CH11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barreto JC, Smith GS, Strobel NH, McQuillin PA, Miller TA. Terephthalic acid: A dosimeter for the detection of hydroxyl radicals in vitro. Life Sci. 1995;56(4):PL89–PL96. doi: 10.1016/0024-3205(94)00925-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.