

Abstract
To develop new anticancer drug candidates from 2-arylnaphthyridin-4-one (AN), we have designed and synthesized a series of 3′-hydroxy and 6-hydroxy derivatives of AN. The results of cytotoxicity screening indicated that the replacement of the 3′-methoxy moiety on the C-ring phenyl group of AN (6a–e) with 3′-hydroxy (7a–e) made no significant effect on the inhibitory activity against HL-60, Hep3B and NCI-H460 cancer cell lines. On the other hand, replacing the 6-methoxy group on the A-ring of AN (6g–i) with a 6-hydroxy group (7g–i) resulted in reduced inhibitory activity against the above three cancer cell lines. Among the above-mentioned target compounds, 2-(3-hydroxyphenyl)-5-methyl-1,8-naphthyridin-4(1H)-one (7a) demonstrated the greatest potency and the best selectivity toward tumorigenic cancer cell lines. In a 7a preliminary mechanism of action study in Hep3B hepatoma cells, 7a showed the effects on microtubules followed by cell cycle arrest and sequentially led to apoptosis.
In addition, a phosphate prodrug (11) of 7a exhibited significant antitumor activity when tested in a Hep3B xenograft nude mice model. Since compound 11 has demonstrated good development potential, it is recommended for further preclinical studies.
Keywords: 2-Arylnaphthyridin-4-ones, Antitumor agents, Phosphate prodrug
1. Introduction
During our continuing effort to obtain new anticancer drug candidates, 2-phenylquinolin-4-one (PQ-1)[1–3]3 was used as a lead compound to develop several series of related analogs, including 2-arylquinolin-4-one (AQ) [2–4], 2-arylnaphthyridin-4-one (AN) [5–7], and 2-arylquinazolin-4-one (AQZ) [8] (Chart 1). In structure-activity relationship (SAR) studies of the AQ series compounds, AQ-A (Chart 2) exhibited potent antitumor activity when functional groups with lone pair electrons (e.g., OR, F, Cl, NRR′) were present on the A-ring 6-position [1–3]. At the same time, replacement of the C-ring phenyl with a 3′-aryl group afforded AQ-B (Chart 2) with potent anticancer activity [3, 4]. Furthermore the two most potent antitumor AQ derivatives were converted into their corresponding monophosphate prodrugs [9–11] (AQ-P1, AQ-P3, Chart 3), which demonstrated excellent antitumor activity in vivo and currently are in preclinical studies. The SAR of AN resembles, but does not exactly equal, that of AQ. Similarly to AQ-A, potent antitumor activity was observed if functional groups with lone pair electrons, such as OCH3, F, and Cl, were placed on the C-ring 3′-position of AN-A. However, unlike the SAR observed in AQ, placement of CH3, Br, and Cl substituents on the A-ring of AN-A did not noticeably enhance the antitumor activity [5–7, 12, 13]. Meanwhile, replacement of the C-ring 3′-phenyl with a 3′-aryl group in AN-B also resulted in potent antitumor activity [6, 7].
Chart 1.
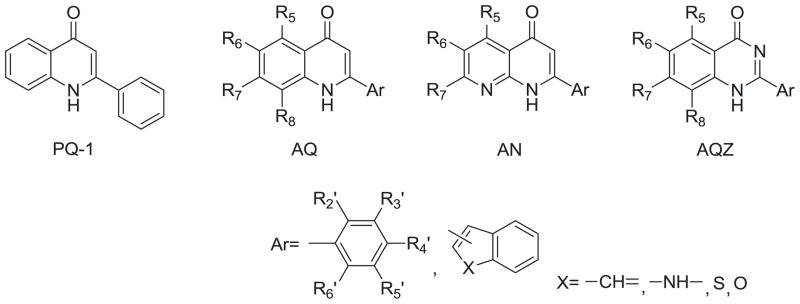
Three series of structural skeletons of 2-phenylquinolin-4-one analogs (PQ-1): 2-arylquinolin-4-ones (AQ), 2-arylnaphthyridin-4-ones (AN) and 2-arylquinazolin-4-ones (AQZ).
Chart 2.
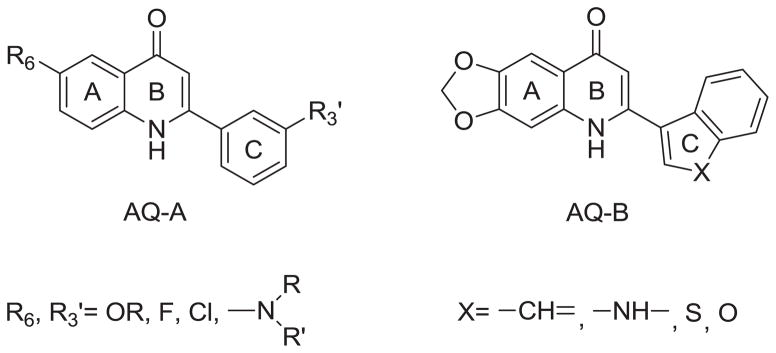
Structures of AQ-A and AQ-B.
Chart 3.
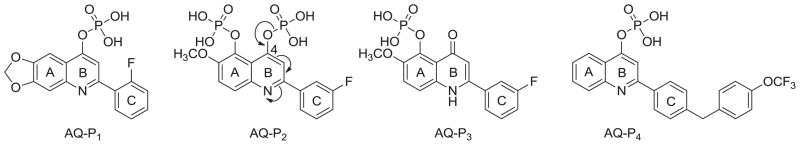
Structures of AQ-P1, AQ-P2, AQ-P3 and AQ-P4.
Overall, we have identified many ANs with potent anticancer activity. Recently, Shedden and coworkers analyzed 34,909 antimitotic agents and identified four ANs that selectivity and efficiently inhibited the growth of tumorigenic cells [14]. Among these four compounds, 2-(3-methoxyphenyl)-5-methyl-1,8-naphthyridin-4(1H)-one (AN-1, Chart 5) exhibited the most promise. From a therapeutic point of view, the use of antitumor agents targeting those cells with the highest tumorigenic potential may be the most effective cancer therapy. With an aim of developing anticancer drug candidates that inhibit the growth of cancer cells with the highest tumorigenic potential, we selected 2-(3-methoxyphenyl)-5-methyl-1,8-naphthyridin-4(1H)-one (AN-1, Chart 5) from the AN series, and attempted to prepare its corresponding phosphate prodrug (AN-P1) (Chart 3). Unfortunately, the same procedure used to produce prodrugs AQ-P1 and AQ-P4 (Chart 3) failed to produce AN-P1. We reported previously that a phosphate group on the para position of the AQ pyridine ring can be unstable, as demonstrated by the fact that AQ-P2 was readily converted to a monophosphate (AQ-P3) by loss of one of two phosphate groups [10]. Therefore, in the present study, a hydroxy group was introduced at the 3′-position of the C-ring or the 6-position of the A-ring of AN. Among the new AN derivatives screened for in vitro anticancer activity, 2-(3-hydroxyphenyl)-5-methyl-1,8-naphthyridin-4(1H)-one (7a) demonstrated the best anticancer activity, and was converted into its corresponding phosphate prodrug (11) for evaluation in an antitumor animal model. This paper reports our detailed results in this investigation.
Chart 5.
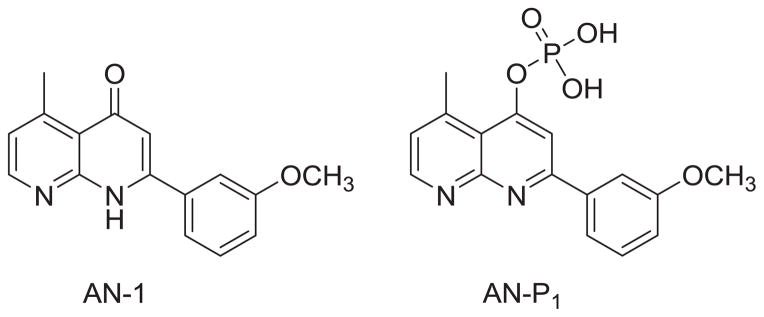
Structures of AN-1 and AN-P1.
2. Results and discussion
2.1. Chemistry
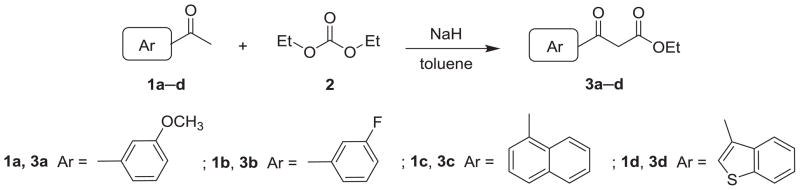
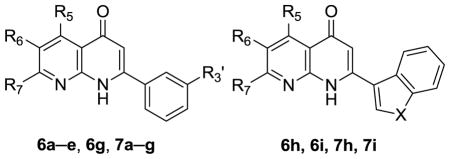
The syntheses of 2-(3-hydroxyphenyl) AN derivatives (7a–e) are illustrated in Schemes 1 and 2. As shown, starting substituted ethanones (1a–d) were reacted separately with diethyl carbonate (2), in the presence of NaH, to yield the desired ethyl 3-substituent-3-oxopropanoates (3a–d). Next, compound 3a was condensed with various 2-aminopyridines (4a–e) in polyphosphoric acid (PPA) at 115 ± 5 °C to afford 2-substituted-4H-pyrido[1,2-a]pyrimidin-4-ones (5a–e). Compounds 5a–e then were converted to their corresponding 2-substituted-1,8-naphthyridin-4(1H)-ones (6a–e) after thermal rearrangement in mineral oil at 350 ± 5 °C. Compounds 6a–e were subsequently heated in acetic acid, in a sealed bottle and in the presence of HBr, at 140 ± 5 °C to obtain the target 2-(3-hydroxyphenyl) AN derivatives (7a–e).
Scheme 1.
Scheme 2.

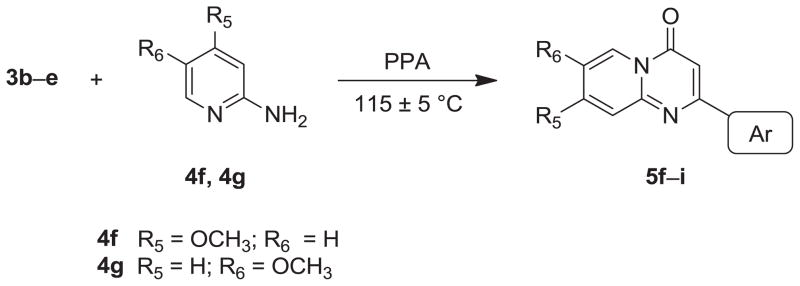
As shown in Schemes 3 and 4, intermediates 5f–i were prepared by the same procedure for 5a–e, except for the use of different starting materials (3b–d). Compounds 3b–d underwent condensation with 2-aminopyridines (4f–g) to yield intermediates 5f–i. Following the same procedure for preparation of 7a–e, 5-hydroxy (7f) and 6-hydroxy AN derivatives (7g–i) were prepared from intermediates 5f and 5g–i, respectively.
Scheme 3.
Scheme 4.
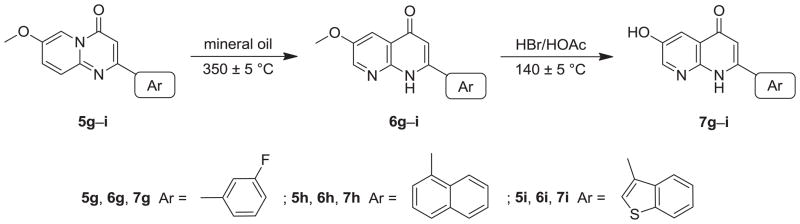
In the thermal rearrangement of 5f in mineral oil at 350 ± 5 °C, we failed to obtain our expected product 5-methoxy-2-(3-fluorophenyl)-1,8-naphthyridin-4(1H)-one (6f), but rather obtained its demethylated product, 5-hydroxy-2-(3-fluorophenyl)-1,8-naphthyridin-4(1H)-one (7f) (Scheme 5). The chemical structure of target compound 7f was confirmed by NMR and HRMS analysis. Compound 7f has the molecular formula C14H10FO2N2 [M+H]+ as determined by HRMS. In the proton spectrum of 7f, no methoxy proton signal was seen, but a hydroxy proton signal (δ 15.47) at very low-field indicated the presence of an intramolecular hydrogen bond.
Scheme 5.
In vitro screening of all target compounds 7a–i revealed that compound 7a showed the best developmental potential. Hence, it was converted into its corresponding phosphate prodrug 11 according to Scheme 6. First, compound 7a was reacted with tetrabenzyl pyrophosphate in THF, in the presence of NaH, to yield a bis(dibenzylphosphate) intermediate 9. Without purification, intermediate 9 was then treated with MeOH to afford monodibenzylphosphate 10, whose chemical structure was confirmed by the presence of the H-3 signal (δ 6.22) in its 1H NMR spectrum. Subsequently, compound 10 was catalytically hydrogenated in MeOH to yield stable monophosphoric acid derivative 11.
Scheme 6.

2.2. Growth inhibitory activity of 6a–e, 6g–i and 7a–i against human cancer cell lines
Compounds 6a–e, 6g–i, and 7a–i were evaluated for inhibitory activity against HL-60 leukemia, Hep3B hepatoma, NCI-H460 non-small cell lung cancer, and Detroit 551 human skin fibroblast cells. The results in Table 1 indicated that compounds 6a–e and 6g–i, with a methoxy group on the C-ring 3′-position or A-ring 6-position, exhibited potent inhibitory activity against HL-60, Hep3B and NCI-H460 cells, with IC50 values from 0.02 to 0.86 μM. Meanwhile, they showed low inhibition against the Detroit 551 cell line.
Table 1.
In vitro cytotoxic activities of hydroxy derivatives of 2-arylnaphthyridin-4-ones (6a–e, 6g–i, 7a–i).
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
IC50a,b (μM)
|
|||||||
| Cmpd | R5 | R6 | R7 | R3′ | X | HL-60c | Hep3Bc | NCI-H460c | Detroit551c |
|
|
|
|
|||||||
| 6a | CH3 | H | H | OCH3 | - | 0.03 | 0.13 | 0.18 | >5.0 |
| 7a | CH3 | H | H | OH | - | 0.03 | 0.17 | 0.20 | >50 |
| 6b | H | CH3 | H | OCH3 | - | 0.06 | 0.12 | 0.14 | >2.5 |
| 7b | H | CH3 | H | OH | - | 0.08 | 0.44 | 0.45 | >2.5 |
| 6c | CH3 | H | CH3 | OCH3 | - | 0.33 | 0.73 | 0.75 | >50 |
| 7c | CH3 | H | CH3 | OH | - | 0.29 | 0.68 | 1.30 | >100 |
| 6d | H | Cl | H | OCH3 | - | 0.03 | 0.10 | 0.10 | >50 |
| 7d | H | Cl | H | OH | - | 0.11 | 0.34 | 0.20 | >100 |
| 6e | H | F | H | OCH3 | - | 0.07 | 0.20 | 0.24 | >5.0 |
| 7e | H | F | H | OH | - | 0.14 | 0.38 | 0.70 | >100 |
| 7f | OH | H | H | F | - | 0.82 | 1.82 | 2.48 | >100 |
| 6g | H | OCH3 | H | F | - | 0.02 | 0.02 | 0.05 | >10 |
| 7g | H | OH | H | F | - | 0.08 | 0.16 | 0.86 | >100 |
| 6h | H | OCH3 | H | - | –CH=CH- | 0.04 | 0.05 | 0.85 | >25 |
| 7h | H | OH | H | - | –CH=CH- | 0.48 | 0.46 | 1.34 | >100 |
| 6i | H | OCH3 | H | - | S | 0.04 | 0.09 | 0.16 | >10 |
| 7i | H | OH | H | - | S | 0.57 | 1.07 | 0.39 | 10 |
| Taxol | - | - | - | - | - | 0.002 | 0.50 | 0.005 | NTd |
Data was presented as IC50 (μM, the concentration of 50% proliferation-inhibitory effect).
Human tumor cells were treated with different concentrations of samples for 48 h.
HL-60, human leukemia cell line; Hep3B, human hepatoma cell line; NCI-H460, non-small cell lung cancer cell line; Detroit 551, human skin fibroblast cell line.
NT = not tested.
In comparison, when the methoxy groups of compounds 6a–e and 6g–i were demethylated, the resulting hydroxy substituted compounds 7a–i were generally less potent against the above three cancer cell lines tested, with the exception of 7a, which retained inhibitory activity equal to its 3′-methoxy compound (6a).
From the above data, we noticed that replacement of the 3′-methoxy of compounds 6a–e with 3′-hydroxy (7a–e) made no significant effect on the inhibitory activity against HL-60, Hep3B, NCI-H460 cell lines. However, substitution of the 6-methoxy group (6g–i) with 6-hydroxy (7g–i) resulted in markedly reduced inhibitory activity against the above three cancer lines.
Five compounds (7a–c, 7g, 7h) were selected from nine target compound 7a–i and submitted to NCI for evaluation against the NCI-60 human cancer cell line panel. The detailed anticancer screening results are provided as supporting information. Herein, only the anticancer effects of the representative compound 7a are discussed.
Compound 7a exhibited significant inhibitory activity against many cancer cell lines, with an average logGI50 (MID) of −6.30. In particular, its logGI50 values were less than −7.00 against several cell lines, including K562 and SR leukemia, NCI-522 non-small cell lung cancer and SW-620 colon cancer, as well as M14 and MDA-MB-435 melanoma. Judging from the total growth inhibition (TGI) values, compound 7a showed the great inhibitory activity against NCI-H522 (TGI= −6.63), a lung cancer cell line with high tumorigenic potential, and thus, compound 7a merited further investigation.
2.3. Selective targeting of tumorigenic cancer cell lines by compounds 7a–c, 7g and 7h
The cytotoxic activity (−logGI50) of target compounds 7a–c, 7g and 7h against NCI-60 cancer cell lines was analyzed using the statistical approach reported by Shedden11 and was scatter plotted in relation to the four categories of tumorigenic potential (Figure 1), in which compound 7a showed a Pearson correlation coefficient of 0.355. These results suggested that, among the five compounds, 7a exhibited better growth inhibitory activity toward the most tumorigenic cell lines. Furthermore, the selectivity window of the five compounds was also plotted against −logGI50 between tumorigenic and non-tumorigenic cancer cells (Figure 2). Overall, compound 7a appeared to strongly and selectively inhibit the most tumorigenic cancer cell lines and, thus, deserved further investigation.
Figure 1.

Data of 7a c, 7g and 7h against NCI-60 cancer cell lines analyzed and scatter plotted in relation to the four categories of tumorigenic potential.
Figure 2.

Selectivity window of 7a c, 7g and 7h plotted against -logGI50 between tumorigenic and non-tumorigenic cancer cells.
2.4. In vivo antitumor activity of phosphate prodrug of 7a (11)
Since as described above, compound 7a demonstrated strong and selective growth inhibition of cancer cells with high tumorigenicity, the tumorigenic Hep3B hepatoma cell line was chosen as thein vivo tumor target. Compound 11, the water soluble monophosphate of 7a, was evaluated in a Hep3B xenograft nude mice model by I.V. administration at doses of 10, 20 and 40 mg/kg/day. As shown in Figure 3A–C, compound 11 induced dose- and time-dependent inhibition of Hep3B tumor growth. At 20 mg/kg, the Hep3B inhibitory activity of 11 exceeded that of 5 mg/kg doxorubicin, and at 40 mg/kg of 11, the Hep3B tumor weight was reduced to 78.6% of the control (Figure 3A–B). During the course of antitumor evaluation, no significant body weight changes were detected in either the test or control mice (Figure 3C).
Figure 3.



Figure 3A. Mean tumor volume-time profile in Hep3B xenograft nude mice (n = 8) following i.v. dosing of doxorubicin at 2 mg/kg and 11 at 10, 20, 40 mg/kg five days per week for three consecutive weeks.
Figure 3B. Mean tumor weight-time profile in Hep3B xenograft nude mice (n = 8) following i.v. dosing of doxorubicin at 2 mg/kg and 11 at 10, 20, 40 mg/kg five days per week for three consecutive weeks.
Figure 3C. Mean body weight-time profile in Hep3B xenograft nude mice (n = 8) following i.v. dosing of doxorubicin at 2 mg/kg and 11 at 10, 20, 40 mg/kg five days per week for three consecutive weeks.
2.5. Antitumor mechanistic study of 7a
The antitumor mechanism of 7a was further investigated in Hep3B cells. Based on the structural comparison of 7a with our previous developed 2-phenylquinolin-4-one series (PQ-1) (Wang et al., 2008), we postulated that 7a may act as an antimitotic agent. Cell cycle arrest at G2/M phase is the main characteristic response to antimitotic agents (Bhattacharyya et al., 2008; Jordan and Wilson, 2004). To determine whether the inhibition in response to 7a was due to cell cycle alteration, we examined the effect of 7a on DNA profile of Hep3B cells using flow cytometry. As shown in Figure 4A, 7a caused a significant cell population accumulation at G2/M phase in a concentration-dependent manner. Moreover, 7a displayed an equivalent effect on cell cycle arrest compared with other known antimitotic agents, such as vinblastine, colchicine, and paclitaxel (Figure 4A). A predominant increase in sub-G1 phase was observed in 7a-treated Hep3B cells (Figure 4B), suggesting that 7a could elicit apoptotic cell death. We next determined whether 7a could induce cell apoptosis. The cleavage of caspases, a hallmark in apoptosis activation, was used as an indicator for the next study. Western blot analysis was performed to detect the cleaved caspases in 7a-treated Hep3B cells. As shown in Figure 4C, after 48 h of treatment, 7a induced a concentration-dependent increase in the cleavage of caspase-8, -9, -3, and PARP. These data indicated that 7a could effectively induce cell apoptosis. Furthermore, 7a could trigger both extracellular and intrinsic apoptotic pathways to cause cell death. These results indicated that 7a induced cell cycle arrest at G2/M phase and followed by cell apoptosis.
Figure 4.

Effects of 7a on cell cycle progression in Hep3B hepatoma cells. (A,B) Hep3B cells were incubated with vehicle (DMSO, as CTL), 0.1, 0.3, 1, 3 μM 7a or 1 μM of vinblastine, colchicine, or paclitaxel for 24 h and collected for cell cycle analysis. Data are expressed as the mean ± SEM of three independent. *P < 0.05; **P < 0.01, compared with CTL. (C) After 48 h-treatment of 7a, cells were harvested and subjected to western blot analysis to detect the cleavage of caspase-8, caspase-9, caspase-3, and PARP. β-Actin was loading control.
Microtubule dynamics are important in spindle formation and chromosomal separation during mitosis (Meunier and Vernos, 2012). Disruption of microtubule dynamics leads to cell cycle arrest at G2/M phase and consequent apoptotic cell death (Pasquier and Kavallaris, 2008). Most antimitotic agents achieve G2/M phase arrest by interfering with tubulin polymerization. Accordingly, we reasoned that 7a may exert antimitotic activity by interfering with microtubule dynamics. Consequently, in vivo tubulin assembly in response to 7a was determined. Hep3B cells were treated with 7a and three known antimitotic agents, vinblastine, colchicine, and paclitaxel, for 24 h followed by isolation of polymerized tubulins. Results from western blot analysis showed that 7a decreased both α-tubulin and β-tubulin contents in the cytoskeleton fraction (Figure 5A). The levels of tubulin polymerization inhibition by 7a were comparable to those by vinblastine, vincristine, and colchicine. In contrast, paclitaxel induced tubulin polymerization (Figure 5A). We next determined whether 7a could interfere directly with tubulin polymerization. 7a was incubated with pure tubulin at 37 °C and the kinetics of tubulin polymerization were evaluated. 7a exhibited a concentration-dependent inhibition on tubulin polymerization with an IC50 value of 1.0 μM (Figure 5B). 7a displayed a similar interference pattern like vinblastine, vincristine, and colchicine, but unlike paclitaxel (Figure 5B). Thus, both in vivo and in vitro tubulin assembly assays suggested that 7a is a tubulin-disrupting antimitotic agent and can interact directly with tubulin.
Figure 5.

Effects of 7a on the microtubule assembly in vivo and in vitro. (A) Hep3B cells were treated with vehicle (DMSO, as CTL), 1 μM 7a, 1 μM vinblastine, 1 μM vincristine, 1 μM colchicine, or 1 μM paclitaxel for 24 h. Cytosolic (S, soluble) and cytoskeletal (P, polymerized tubulin) fractions were separated and followed by western blot analysis to detect α-tubulin, β-tubulin and β-actin protein expression. (B) GPEM buffer containing pure tubulin protein was incubated with vehicle (DMSO, as control) or 0.3, 1, 3, 10 μM 7a, 10 μM vinblastine, 10 μM vincristine, 10 μM colchicine, or 10 μM paclitaxel at 37 °C. Signals from compound-effected tubulin polymerization were recorded kinetically by microplate reader at the absorbance of 340 nm.
3. Conclusion
Several new hydroxyl derivatives of AN were designed and synthesized in this study. Most of the target compounds exhibited significant in vitro cytotoxicity. After establishing initial SAR, five compounds were selected and submitted to NCI for evaluation against the NCI-60 cancer cell line panel to explore their inhibitory activity and selectivity against tumorigenic cancer cell lines. Based on the results, compound 7a was identified as the most promising antitumor agent for further study. Subsequently, compound 7a was also converted into its corresponding phosphate (11). In an animal study, compound 11 exhibited significant inhibitory activity in vivo against the growth of the highly tumorigenic Hep3B cancer cell line. We believe that compound 11 is an antihepatoma drug candidate with excellent developmental potential. In addition, compound 7a demonstrated potent inhibitory activity against NCI-H522 non-small cell lung cancer cells and merits further investigation.
4. Experimental
4.1. Chemistry
All reagents and solvents were obtained commercially and used without further purification. Products were purified by flash column chromatography on silica gel (Merck, Silica Gel 60, 40–63 μm) using a mixture of CH2Cl2, CHCl3 and EtOAc as eluent. The progress of reactions was monitored by TLC on 2 × 6 cm pre-coated silica gel 60 F254 plate of thickness 0.25 mm (Merck). The chromatograms were visualized under UV light at 254 and 366 nm. Melting points were determined on a Yanaco MP-500D melting point apparatus and were uncorrected. Proton NMR spectra were recorded on a Bruker Avance 400 MHz or Avance III 500 MHz spectrometer at room temperature, and chemical shifts are reported in ppm (δ). The following abbreviations are used: s, singlet; d, doublet; t, triplet; dd, double doublet; and m, multiplet. Carbon NMR spectra were obtained on a Bruker DPX-200 FT-NMR, Avance 400 MHz or Avance III 500 MHz spectrometer at room temperature in CDCl3, DMSO-d6, or D2O solutions. Tested compounds 6a–e, 6g–i, 7a–i and 11 were analyzed on a HPLC (Shimadzu LC-10AT) apparatus equipped with Shimadzu SPD-M10AVP diode-array detector and Shimadzu and Shimadzu SIL-10A autoinjector. Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) column was used for analytical purposes. Purity of tested compounds was determined by HPLC and, in all cases, reached at least 95%. EIMS spectra were measured with HP 5995 GC-MS. Low-resolution mass (ESIMS) spectra were obtained using LC-ESI-MS performed on a Thermo Accela UPLC apparatus equipped with a Thermo TSQ Quantum Access MAX mass spectrometer. High-resolution mass (HRMS) spectra were measured with Finnigan/Thermo QustMAT95XL at National Chung Hsing University, Taichung, Taiwan.
4.1.1. General procedure for the synthesis of 3b–d
To a stirred suspension of NaH and CO(OEt)2 (2) in toluene was added dropwise a solution of substituted acetophenone (1b–d) in toluene under reflux. The mixture was allowed to reflux and was stirred for 20 min after the addition was complete. When cooled to room temperature, the mixture was acidified with glacial HOAc. After ice-cold water was added, the mixture was extracted with toluene. The organic extract was then dried over anhydrous MgSO4. After the toluene was evaporated under vacuum, the corresponding substituted ethyl 3-phenyl-3-oxopropanoate was obtained.
4.1.1.1. Ethyl 3-(3-fluorophenyl)-3-oxopropanoate (3b)
Obtained from 1b and 2, 58% yield, oil; 1H NMR (500 MHz, CDCl3): δ 1.29 (t, J = 7.0 Hz, 3H, -OCH2CH3), 4.00 (s, 2H, -CH2), 4.25 (q, J = 7.0 Hz, 2H, -OCH2CH3), 7.31–7.35 (m, 1H, Ar-H), 7.48–7.52 (m, 1H, Ar-H), 7.67 (d, J = 9.5 Hz, 1H, H-2), 7.75 (d, J = 8.0 Hz, 1H, H-6). 13C NMR (50 MHz, CDCl3): δ 14.0, 46.0, 61.6, 115.2 (d, J = 22.5 Hz), 120.8 (d, J = 21.0 Hz), 121.6, 124.3, 130.5 (d, J = 7.5 Hz), 162.9 (d, J = 247.0 Hz), 191.3; HRMS m/z calculated for C11H12FO3, [M+H]+, 211.0765, found 211.0762.
4.1.1.2. Ethyl 3-(naphthalen-1-yl)-3-oxopropanoate (3c)
Obtained from 1c and 2, 80% yield, oil; 1H NMR (400 MHz, CDCl3): δ 1.23 (t, J = 6.8 Hz, 3H, -OCH2CH3), 4.13 (s, 2H, -CH2), 4.22 (q, J = 6.8 Hz, 2H, -OCH2CH3), 7.47–7.61 (m, 3H, H-3, H-6, H-7), 7.90 (d, J = 8.0 Hz, 1H, H-5), 7.94 (d, J = 6.8 Hz, 1H, H-2), 8.05 (d, J = 8.0 Hz, 1H, H-4), 8.78 (d, J = 8.0 Hz, 1H, H-8). 13C NMR (50 MHz, CDCl3): δ 14.0, 48.8, 61.5, 124.3, 125.9, 126.7, 128.4, 128.8, 130.4, 133.7, 134.0, 134.2, 167.6, 195.7; HRMS m/z calculated for C15H15O3, [M+H]+, 243.1016, found 243.1014.
4.1.1.3. Ethyl 3-(benzo[b]thiophen-3-yl)-3-oxopropanoate (3d)
Obtained from 1d and 2, 43% yield, oil; 1H NMR (400 MHz, CDCl3): δ 1.29 (t, J = 7.2 Hz, 3H, -OCH2CH3), 4.03 (s, 2H, -CH2), 4.25 (q, J = 7.2 Hz, 2H, -OCH2CH3), 7.44–7.55 (m, 2H, H-5, H-6), 7.88 (d, J = 8.0 Hz, 1H, H-7), 8.38 (s, 1H, H-4), 8.79 (d, J = 8.0 Hz, 1H, H-2). 13C NMR (50 MHz, CDCl3): δ 14.1, 47.7, 61.6, 122.3, 125.7, 126.1, 134.3, 136.4, 138.6, 139.7, 167.4, 187.0; HRMS m/z calculated for C13H13O3S, [M+H]+, 249.0580, found 249.0583.
4.1.2. General procedure for the synthesis of 5a–i
A mixture of substituted 2-aminopyridine (4a–g), substituted ethyl 3-phenyl-3-oxopropanoate (3a–e), and polyphosphoric acid was heated at 115 ± 5 °C with stirring. The reaction was monitored by TLC. After the reaction was complete, the mixture was quenched by ice-cold water. After extraction with CH2Cl2, the organic layer was evaporated under vacuum. The residue was passed through a silica gel column to give the 2-arylpyrido[1,2-a]pyrimidin-4-one.
4.1.2.1. 2-(3-Methoxyphenyl)-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (5a)
Obtained from 3a and 4b, 45% yield, light-yellow solid; mp 150–152 °C; 1H NMR (500 MHz, CDCl3): δ 2.50 (s, 3H, -CH3), 3.90 (s, 3H, -OCH3), 6.81 (s, 1H, H-3), 6.95 (dd, J = 7.2, 1.7 Hz, 1H, H-7), 7.02–7.04 (m, 2H, Ar-H), 7.50 (s, 1H, H-9), 8.08–8.09 (m, 2H, Ar-H), 8.94 (d, J = 7.2 Hz, 1H, H-6). 13C NMR (50 MHz, CDCl3): δ 21.4, 55.3, 99.4, 112.5, 116.5, 117.9, 119.7, 124.8, 126.5, 129.7, 138.9, 148.2, 150.9, 158.6, 159.9, 162.1; EIMS (70eV) m/z 266 [M]+.
4.1.2.2. 2-(3-Methoxyphenyl)-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (5b)
Obtained from 3a and 4a, 47% yield, white solid; mp 163–164 °C; 1H NMR (400 MHz, CDCl3): δ 2.39 (s, 3H, -CH3), 3.86 (s, 3H, -OCH3), 6.83 (s, 1H, H-3), 6.99 (dd, J = 8.2, 2.3 Hz, 1H, H-8), 7.36 (t, J = 7.8 Hz, 1H, H-5′), 7.57–7.68 (m, 4H, H-9, H-2′, H-4′, H-6′), 8.83 (s, 1H, H-6). 13C NMR (100 MHz, CDCl3): δ 18.3, 55.4, 99.9, 112.5, 116.5, 119.7, 124.7, 125.6, 126.0, 129.7, 138.6, 139.2, 149.7, 158.3, 159.9, 161.2; EIMS (70eV) m/z 266 [M]+.
4.1.2.3. 2-(3-Methoxyphenyl)-6,8-dimethyl-4H-pyrido[1,2-a]pyrimidin-4-one (5c)
Obtained from 3a and 4c, 9% yield, light-yellow solid; mp 125–126 °C; 1H NMR (500 MHz, CDCl3): δ 2.36 (s, 3H, -CH3), 3.07 (s, 3H, -CH3), 3.91 (s, 3H, -OCH3), 6.50 (s, 1H, H-3), 6.66 (s, 1H, H-7), 7.02–7.04 (m, 1H, Ar-H), 7.29–7.30 (m, 1H, Ar-H), 7.40 (t, J = 8.0 Hz, 1H, H-5′), 7.61–7.64 (m, 2H, Ar-H). 13C NMR (50 MHz, CDCl3): δ 20.9, 24.5, 55.3, 101.5, 112.2, 116.3, 119.5, 120.8, 123.8, 129.6, 138.5, 142.9, 146.8, 153.4, 159.9, 160.2, 162.8; HRMS m/z calculated for C17H16N2O2, [M]+, 280.1212, found 280.1202.
4.1.2.4. 7-Chloro-2-(3-methoxyphenyl)-4H-pyrido[1,2-a]pyrimidin-4-one (5d)
Obtained from 3a and 4d, 16% yield, light-yellow solid; mp 176–177 °C; 1H NMR (500 MHz, CDCl3): δ 3.93 (s, 3H, -OCH3), 6.95 (s, 1H, H-3), 7.07 (dd, J = 8.0, 2.0 Hz, 1H, H-8), 7.44 (t, J = 8.0 Hz, 1H, H-5′), 7.65–7.70 (m, 4H, Ar-H), 9.10 (s, 1H, H-6). 13C NMR (50 MHz, DMSO-d6): δ 55.7, 99.9, 112.8, 117.0, 120.0, 123.6, 125.1, 128.3, 130.3, 138.2, 138.4, 149.6, 157.3, 160.1, 160.4; HRMS m/z calculated for C15H11ClN2O2, [M]+, 286.0509, found 286.0519.
4.1.2.5. 7-Fluoro-2-(3-methoxyphenyl)-4H-pyrido[1,2-a]pyrimidin-4-one (5e)
Obtained from 3a and 4e, 21% yield, light-yellow solid; mp 140–141 °C; 1H NMR (200 MHz, CDCl3): δ 3.85 (s, 3H, -OCH3), 6.86 (s, 1H, H-3), 7.01 (ddd, J = 8.3, 2.2, 1.5 Hz, H-4′), 7.36 (t, J = 8.0 Hz, 1H, H-5′), 7.56–7.70 (m, 4H, Ar-H), 8.91 (dd, J = 4.7, 2.5 Hz, 1H, H-6). 13C NMR (50 MHz, CDCl3): δ 55.4, 100.0, 112.6, 113.4 (d, J = 41.0 Hz), 116.6, 119.7, 128.5 (d, J = 3.0 Hz), 128.9 (d, J = 14.0 Hz), 129.8, 138.3, 148.9, 154.0 (d, J = 244.0 Hz), 158.6, 160.0, 161.4; HRMS m/z calculated for C15H11FN2O2, [M]+, 270.0805, found 270.0795.
4.1.2.6. 2-(3-Fluorophenyl)-8-methoxy-4H-pyrido[1,2-a]pyrimidin-4-one (5f)
Obtained from 3b and 4f, 11% yield, white solid; mp 151–152 °C; 1H NMR (400 MHz, DMSO-d6): δ 4.02 (s, 3H, -OCH3), 6.89 (s, 1H, H-3), 7.07 (d, J = 8.0 Hz, 1H, H-7), 7.15 (s, 1H, H-9), 7.37–7.38 (m, 1H, Ar-H), 7.56–7.58 (m, 1H, Ar-H), 8.01–8.07 (m, 2H, Ar-H), 8.87 (d, J = 7.2 Hz, 1H, H-6). 13C NMR (50 MHz, CDCl3): δ 56.4, 98.3, 102.4, 110.5, 114.4 (d, J = 24.0 Hz), 117.3 (d, J = 21.0 Hz), 122.9, 128.4, 130.2 (d, J = 7.5 Hz), 139.9 (d, J = 7.5 Hz), 153.2, 158.6, 160.4, 161.3, 165.5; HRMS m/z calculated for C15H12FN2O2, [M+H]+, 271.0877, found 271.0874.
4.1.2.7. 2-(3-Fluorophenyl)-7-methoxy-4H-pyrido[1,2-a]pyrimidin-4-one (5g)
Obtained from 3b and 4g, 48% yield, white solid; mp 181–182 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.95 (s, 3H, -OCH3), 7.09 (s, 1H, H-3), 7.37 (dd, J = 8.0, 2.0 Hz, 1H, H-8), 7.57–7.58 (m, 1H, Ar-H), 7.78–7.84 (m, 2H, Ar-H), 8.01–8.08 (m, 2H, Ar-H), 8.48 (d, J = 2.4 Hz, 1H, H-6). 13C NMR (50 MHz, CDCl3): δ 56.5, 99.6, 106.8, 114.3 (d, J = 23.0 Hz), 117.2 (d, J = 21.0 Hz), 122.7, 127.4, 130.2 (d, J = 8.0 Hz), 131.7, 139.7 (d, J = 7.5 Hz), 148.1, 150.9, 158.4, 159.3, 163.1 (d, J = 244.0 Hz); HRMS m/z calculated for C15H11FN2O2, [M]+, 270.0805, found 270.0799.
4.1.2.8. 7-Methoxy-2-(naphthalen-1-yl)-4H-pyrido[1,2-a]pyrimidin-4-one (5h)
Obtained from 3c and 4g, 18% yield, white solid; mp 207–208 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.98 (s 3H, -OCH3), 6.63 (s, 1H, H-3), 7.56–7.64 (m, 3H, Ar-H), 7.70 (d, J = 6.8 Hz, 1H, Ar-H), 7.78 (d, J = 9.2 Hz, 1H, Ar-H), 7.85 (dd, J = 9.2, 2.4 Hz, 1H, H-8), 8.03 (d, J = 7.6 Hz, 1H, Ar-H), 8.06 (d, J = 8.4 Hz, 1H, Ar-H), 8.22 (d, J = 7.6 Hz, 1H, Ar-H), 8.56 (d, J = 2.4 Hz, 1H, H-6). 13C NMR (50 MHz, DMSO-d6): δ 57.0, 103.5, 107.2, 125.8, 126.0, 126.6, 127.0, 127.6, 127.8, 128.7, 129.9, 130.6, 132.6, 133.8, 137.1, 148.1, 151.2, 157.4, 163.0; HRMS m/z calculated for C19H14N2O2, [M]+, 302.1047, found 302.1055.
4.1.2.9. 2-(Benzo[b]thiophen-3-yl)-7-methoxy-4H-pyrido[1,2-a]pyrimidin-4-one (5i)
Obtained from 3d and 4g, 32% yield, white solid; mp 162–164 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.97 (s, 3H, -OCH3), 6.94 (s, 1H, H-3), 7.46–7.51 (m, 2H, Ar-H), 7.87 (s, 2H, Ar-H), 8.09–8.11 (m, 1H, Ar-H), 8.54 (s, 1H, Ar-H), 8.59 (s, 1H, Ar-H), 8.87 (s, 1H, H-2′). 13C NMR (50 MHz, CDCl3): δ 56.5, 101.2, 106.9, 122.8, 124.2, 124.8, 124.9, 127.3, 129.2, 131.7, 134.7, 136.7, 141.0, 148.2, 150.8, 158.0, 158.2; HRMS m/z calculated for C17H13N2O2S, [M+H]+, 309.0692, found 309.0695.
4.1.3. General procedure for the synthesis of 6a–e and 6g–i
The 2-arylpyrido[1,2-a]pyrimidin-4-one (5a–e and 5g–i) was added to liquid paraffin at 350 ± 5 °C with stirring. The oil was maintained at 350 ± 5 °C for 2 h. The reaction was monitored by TLC. After the reaction was complete, the mixture cooled to room temperature, and liquid paraffin was removed by filtration. The residue was passed through a silica gel column chromatography to give the desired 2-aryl-1,8-naphthyridin-4(1H)-one.
4.1.3.1. 2-(3-Methoxyphenyl)-5-methyl-1,8-naphthyridin-4(1H)-one (6a)
Obtained from 5a, 40% yield, white solid; mp 196–198 °C; 1H NMR (200 MHz, CDCl3): δ 2.99 (s, 3H,-CH3), 3.88 (s, 3H, -OCH3), 6.54 (s, 1H, H-3), 6.96 (d, J = 5.0 Hz,1H, H-6), 7.12 (dd, J = 8.2, 2.6 Hz, 1H, H-4′), 7.23–7.34 (m, 2H, H-2′ and H-6′), 7.42–7.50 (m, 1H, H-5′), 7.89 (d, J = 5.0 Hz,1H, H-7), 10.73 (s, 1H, NH). 13C NMR (50 MHz, CDCl3): δ 22.6, 55.2, 100.1, 110.9, 112.2, 116.3, 119.0, 122.3, 130.2, 135.2, 148.9, 150.9, 152.0, 152.7, 160.0, 181.1; EIMS (70eV) m/z 266.2 [M]+, ESIMS m/z 289.02 [M+Na]+, HRMS m/z calculated for C16H15N2O2, [M+H]+, 267.1128, found 267.1129.
4.1.3.2. 2-(3-Methoxyphenyl)-6-methyl-1,8-naphthyridin-4(1H)-one (6b)
Obtained from 5b, 40% yield, white solid; mp 196–198 °C; 1H NMR (400 MHz, CDCl3): δ 2.38 (s, 3H, -CH3), 3.84 (s, 3H, -OCH3), 6.64 (s, 1H, H-3), 7.12 (dd, J = 8.2, 2.0 Hz, 1H, H-4′), 7.24 (s, 1H, H-2′), 7.30 (d, J = 7.8 Hz, 1H, H-6′), 7.44 (t, J = 8.2 Hz, 1H, H-5′), 7.95 (d, J = 1.5 Hz, 1H, H-5), 8.50 (d, J = 1.5 Hz, 1H, H-7). 13C NMR (100 MHz, CDCl3): δ 18.0, 55.4, 109.4, 112.8, 116.5, 119.5, 119.6, 129.8, 130.4, 135.4, 135.7, 149.1, 151.2, 153.5, 160.1, 178.5; EIMS (70eV) m/z 266 [M]+, ESIMS m/z 289.02 [M+Na]+, HRMS m/z calculated for C16H15N2O2, [M+H]+, 267.1128, found 267.1127.
4.1.3.3. 2-(3-Methoxyphenyl)-5,7-dimethyl-1,8-naphthyridin-4(1H)-one (6c)
Obtained from 5c, 88% yield, white solid; mp 178–179 °C; 1H NMR (500 MHz, DMSO-d6): δ 2.51 (s, 3H, -CH3), 2.80 (s, 3H, -CH3), 3.87 (s, 3H, -OCH3), 6.32 (s,1H, H-3), 7.02 (s, 1H, H-6), 7.09–7.10 (m, 1H, H-4′), 7.37–7.45 (m, 3H, H-2′, H-5′ and H-6′), 12.02 (s,1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 22.5, 24.3, 55.7, 110.3, 112.7, 116.4, 117.1, 120.1, 122.6, 130.3, 135.1, 149.3, 150.7, 152.7, 159.8, 161.3, 180.1; ESIMS m/z303.03 [M+Na]+, HRMS m/z calculated for C17H17N2O2, [M+H]+, 281.1285, found 281.1287.
4.1.3.4. 6-Chloro-2-(3-methoxyphenyl)-1,8-naphthyridin-4(1H)-one (6d)
Obtained from 5d, 28% yield, white solid; mp 253–255 °C; 1H NMR (500 MHz, DMSO-d6): δ 3.88 (s, 3H, -OCH3), 6.50 (s, 1H, H-3), 7.14 (dd, J = 8.0, 1.5 Hz, 1H, H-′4), 7.41–7.42 (m, 2H, H-2′ and H-6′), 7.48 (t, J = 4.0 Hz, 1H, H-5′), 8.45 (d, J = 3.0 Hz, 1H, H-5), 8.86 (d, J = 2.5 Hz, 1H, H-7), 12.59 (s, 1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 55.8, 109.1, 113.1, 117.4, 120.3, 120.4, 126.6, 130.5, 133.5, 135.2, 150.1, 152.1, 159.8, 176.5; ESIMS m/z 286.60 [M]+, 309.06 [M+Na]+, HRMS m/z calculated for C15H12N2O2Cl, [M+H]+, 287.0582, found 287.0576.
4.1.3.5. 6-Fluoro-2-(3-methoxyphenyl)-1,8-naphthyridin-4(1H)-one (6e)
Obtained from 5e, 59% yield, white solid; mp 254–255 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.87 (s, 3H, -OCH3), 6.46 (s, 1H, H-3), 7.13 (d, J = 8.0 Hz, 1H, H-4′), 7.42–7.47 (m, 3H, H-2′, H-5′ and H-6′), 8.24 (d, J = 5.2 Hz, 1H, H-5), 8.89 (s, 1H, H-7), 12.56 (s, 1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 55.8, 108.2, 113.1, 117.3, 119.4 (d, J = 18.5 Hz), 120.4, 130.5, 135.3, 143.0 (d, J = 11.0 Hz), 148.4, 152.0, 156.4 (d, J = 249.0 Hz), 159.8, 177.2; HRMS m/z calculated for C15H12FN2O2, [M+H]+, 271.0877, found 271.0877.
4.1.3.6. 2-(3-Fluorophenyl)-6-methoxy-1,8-naphthyridin-4(1H)-one (6g)
Obtained from 5g, 40% yield, white cotton fiber solid; mp 281–283 °C; 1H NMR (200 MHz, DMSO-d6 + HCl): δ 3.99 (s, 3H, -OCH3), 7.33 (s, 1H, H-3), 7.47–7.81 (m, 4H, H-2′, H-4′, H-5′ and H-6′), 8.01 (d, J = 3.2 Hz, 1H, H-5), 8.88 (d, J = 3.2 Hz, 1H, H-7). 13C NMR (50 MHz, DMSO-d6 + HCl): δ 56.9, 106.6, 111.8, 116.0 (d, J = 23.5 Hz), 117.4, 118.9 (d, J = 21.0 Hz), 125.3, 131.8 (d, J = 8.5 Hz), 135.1, 145.4, 148.5, 152.8, 154.6, 163.0 (d, J = 246.5 Hz), 171.7; HRMS m/z calculated for C15H11FN2O2, [M]+, 270.0805, found 270.0814. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 6g was 2.40 min; the purity of 6g was 95.3%.
4.1.3.7. 6-Methoxy-2-(naphthalen-1-yl)-1,8-naphthyridin-4(1H)-one (6h)
Obtained from 5h, 35% yield, beige solid; mp > 300 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.96 (s, 3H, -OCH3), 6.16 (s,1H, H-3), 7.60–7.70 (m, 3H, H-2′, H-3′, and H-6′), 7.86 (d, J = 7.6 Hz, 1H, H-8′), 7.96 (d, J = 2.4 Hz, 1H, H-5), 8.07 (d, J = 7.2 Hz, 1H, H-4′), 8.12 (d, J = 7.6 Hz, 1H, H-5′), 8.58 (d, J = 2.4 Hz, 1H, H-7), 12.58 (s, 1H, NH). 13C NMR (50 MHz, DMSO-d6 + HCl): δ 56.4, 110.8, 113.8, 120.2, 125.3, 125.8, 126.9, 127.6, 127.7, 128.9, 130.3, 130.9, 133.0, 133.6, 144.8, 146.0, 150.6, 153.4, 177.1; HRMS m/z calculated for C19H14N2O2, [M]+, 302.1055, found 302.1060. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 6h was 3.30 min; the purity of 6h was 96.8%.
4.1.3.8. 2-(Benzo[b]thiophen-3-yl)-6-methoxy-1,8-naphthyridin-4(1H)-one (6i)
Obtained from 5i, 42% yield, white solid; mp 287–289 °C; 1H NMR (500 MHz, DMSO-d6): δ 3.96 (s, 3H, -OCH3), 6.32 (s, 1H, H-3), 7.50–7.53 (m, 2H, H-5 and H-6′), 7.94–7.97 (m, 2H, H-7′, and H-5′), 8.15 (d, J = 7.5 Hz, 1H, H-4′), 8.31 (s, 1H, H-2′), 8.60 (s, 1H, H-7), 12.50 (s, 1H, NH). 13C NMR (125 MHz, DMSO-d6): δ 56.5, 109.2, 113.9, 120.3, 123.1, 123.8, 125.6, 130.4, 137.0, 140.2, 144.7, 145.8, 146.2, 153.4, 177.4; HRMS m/z calculated for C17H13N2O2S, [M+H]+, 309.0692, found 309.0693. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 6i was 2.67 min; the purity of 6i was 95.8%.
4.1.4. General procedure for the synthesis of 7a–i
A mixture of methoxy substituted 2-aryl-1,8-naphthyridin-4(1H)-one (5a–e and 5g–i), glacial HOAc, and HBr in HOAc (33%) was heated at 140 ± 5 °C with stirring in a sealed bottle. The reaction was monitored by TLC. After the reaction was complete, the mixture was cooled to room temperature and neutralized with saturated aqueous Na2CO3 solution. The precipitate was collected by filtration, and passed through a silica gel column to give the corresponding hydroxyl substituted 2-aryl-1,8-naphthyridin-4(1H)-one.
4.1.4.1. 2-(3-Hydroxyphenyl)-5-methyl-1,8-naphthyridin-4(1H)-one (7a)
Obtained from 6a, 67% yield, white solid; mp 287–289 °C; 1H NMR (500 MHz, DMSO-d6): δ 2.85 (s, 3H, -CH3), 6.22 (s, 1H, H-3), 6.96 (dd, J = 8.5, 2.0 Hz, 1H, H-4′), 7.14 (d, J = 4.5 Hz, 1H, H-6), 7.16 (t, J = 2.0 Hz, 1H, H-2′), 7.23 (d, J = 8.0 Hz, 1H, H-6′), 7.34 (t, J = 8.0 Hz, 1H, H-5′), 8.52 (d, J = 4.5 Hz, 1H, H-7), 9.77 (s, 1H, -OH), 11.99 (s, 1H, NH). 13C NMR (125 MHz, DMSO-d6): δ 22.6, 110.2, 114.8, 117.9, 118.5, 118.7, 122.7, 130.4, 135.2, 150.3, 150.9, 152.0, 152.9, 158.1, 180.3; ESIMS m/z 252.67 [M]+, HRMS m/z calculated for C15H13N2O2, [M+H]+, 253.0972, found 253.0978. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 7a was 3.04 min; the purity of 7a was 96.0%.
4.1.4.2. 2-(3-Hydroxyphenyl)-6-methyl-1,8-naphthyridin-4(1H)-one (7b)
Obtained from 6b, 87% yield, white solid; mp > 300 °C; 1H NMR (400 MHz, DMSO-d6): δ 2.44 (s, 3H, -CH3), 6.30 (s, 1H, H-3), 6.95 (d, J = 7.6 Hz, 1H, H-4′), 7.20–7.33 (m, 3H, H-2′, H-5′, and H-6′), 8.27 (s, 1H, H-5), 8.64 (s, 1H, H-7), 10.22 (br, s, 1H, -OH), 12.06 (br, s, 1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 18.0, 108.5, 114.8, 117.9, 118.7, 119.3, 129.7, 130.4, 134.0, 135.5, 149.9, 151.7, 154.3, 158.0, 177.6; ESIMS m/z 253.03 [M+H]+, HRMS m/z calculated for C15H13N2O2, [M+H]+, 253.0972, found 253.0979. The purity analysis was detected by reversed- phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The Na2HPO4 retention time of 7b was 2.66 min; the purity of 7b was 96.1%.
4.1.4.3. 2-(3-Hydroxyphenyl)-5,7-dimethyl-1,8-naphthyridin-4(1H)-one (7c)
Obtained from 6c, 50% yield, light yellow solid; mp 287–288 °C; 1H NMR (400 MHz, DMSO-d6): δ 2.50 (s, 3H, -CH3), 2.79 (s, 3H, -CH3), 6.17 (s, 1H, H-3), 6.92–7.31 (m, 4H, H-2′, H-4′, H-5′ and H-6′), 7.01 (s, 1H, H-6), 9.77 (s, 1H, -OH), 11.95 (s, 1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 22.6, 24.4, 110.1, 114.6, 116.4, 117.8, 118.6, 122.6, 130.4, 135.2, 149.8, 150.7, 152.6, 158.0, 161.4, 180.0; HRMS m/z calculated for C16H14N2O2, [M]+, 266.1055, found 266.1049. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 7c was 3.04 min; the purity of 7c was 98.4%.
4.1.4.4. 6-Chloro-2-(3-hydroxyphenyl)-1,8-naphthyridin-4(1H)-one (7d)
Obtained from 6d, 32% yield, light yellow solid; mp > 300 °C; 1H NMR (400 MHz, DMSO-d6): δ 6.36 (s, 1H, H-3), 6.97 (s, 1H, H-4′), 7.17 (s, 1H, H-2′), 7.23 (s, 1H, H-6′), 7.34–7.36 (m, 1H, H-5′), 8.44 (s, 1H, H-5), 8.85 (s, 1H, H-7), 9.87 (s, 1H, OH), 12.53 (s, 1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 108.9, 114.9, 118.1, 118.9, 120.3, 126.6, 130.5, 133.4, 135.3, 150.1, 152.1, 152.6, 158.1, 176.5; HRMS m/z calculated for C14H9ClN2O2, [M]+, 272.0353, found 272.0349. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 7d was 2.26 min; the purity of 7d was 98.7%.
4.1.4.5. 6-Fluoro-2-(3-hydroxyphenyl)-1,8-naphthyridin-4(1H)-one (7e)
Obtained from 6e, 54% yield, light yellow solid; mp 285–287 °C; 1H NMR (400 MHz, DMSO-d6): δ 6.32 (s, 1H, H-3), 6.97 (d, J = 7.2 Hz, 1H, H-4′), 7.16 (s, 1H, H-2′), 7.24 (d, J = 6.8 Hz, 1H, H-6′), 7.34–7.37 (m, 1H, H-5′), 8.21 (s, 1H, H-5), 8.87 (s, 1H, H-7), 9.85 (s, 1H, OH), 12.50 (s, 1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 108.0, 114.9, 118.1, 118.9, 119.3 (d, J = 18.5 Hz), 120.3, 130.5, 135.4, 143.0 (d, J = 27.8 Hz), 148.4, 152.4, 156.3 (d, J = 248.5 Hz), 158.1, 177.1; HRMS m/z calculated for C14H10FN2O2, [M+H]+, 257.0721, found 257.0723. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 7e was 2.27 min; the purity of 7e was 95.8%.
4.1.4.6. 2-(3-Fluorophenyl)-5-hydroxy-1,8-naphthyridin-4(1H)-one (7f)
Obtained from 5f, 8% yield, light brown solid; mp 255–257 °C; 1H NMR (400 MHz, DMSO-d6): δ 6.24 (s, 1H, H-3), 7.36–7.38 (m, 2H, H-6, H-2′), 7.55–7.59 (m, 1H, H-4′), 7.97–8.11 (m, 3H, H-5′, H-6′ and H-7), 12.76 (s, 1H, NH), 15.47 (s, 1H, OH). 13C NMR (50 MHz, DMSO-d6): δ 103.1, 108.4, 114.3 (d, J = 23.0 Hz), 117.5 (d, J = 21.0 Hz), 123.8, 131.2 (d, J = 8.5 Hz), 140.6 (d, J = 7.3 Hz), 143.0, 151.4, 159.6, 163.0 (d, J = 242.5 Hz), 170.6, 182.7; HRMS m/z calculated for C14H10FN2O2, [M+H]+, 257.0721, found 257.0722. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 7f was 2.90 min; the purity of 7f was 97.4%.
4.1.4.7. 2-(3-Fluorophenyl)-6-hydroxy-1,8-naphthyridin-4(1H)-one (7g)
Obtained from 6g, 38% yield, light yellow solid; mp > 300 °C; 1H NMR (500 MHz, DMSO-d6): δ 6.37 (s, 1H, H-3), 7.41 (t, J = 8.3 Hz, 1H, H-5′), 7.57–7.60 (m, 1H, H-4′), 7.69–7.74 (m, 2H, H-2′, H-6′), 7.76 (d, J = 2.5 Hz, 1H, H-5), 8.45 (s, 1H, H-7), 10.31 (s, 1H, OH), 12.28 (s, 1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 106.0, 115.0, 115.8 (d, J = 23.0 Hz), 118.0, 118.7 (d, J = 21.0 Hz), 125.1, 131.7 (d, J = 8.0 Hz), 135.4 (d, J = 8.0 Hz), 144.4, 147.4, 152.6, 153.1, 162.5 (d, J = 238.0 Hz), 171.3; HRMS m/z calculated for C14H9FN2O2, [M]+, 256.0648, found 256.0645. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 7g was 2.20 min; the purity of 7g was 99.0%.
4.1.4.8. 6-Hydroxy-2-(naphthalen-1-yl)-1,8-naphthyridin-4(1H)-one (7h)
Obtained from 6h, 43% yield, white solid; mp > 300 °C; 1H NMR (500 MHz, DMSO-d6): δ 6.09 (s, 1H, H-3), 6.50 (s, 1H, H-5), 7.60–7.68 (m, 3H, H-3′, H-6′ and H-7′), 7.80–7.83 (m, 1H, H-2′), 7.88 (d, J = 8.5 Hz, 1H, H-8′), 8.06 (d, J = 8.0 Hz, 1H, H-4′), 8.11 (d, J = 7.5 Hz, 1H, H-5′), 8.43 (s, 1H, H-7), 10.41 (s, 1H, OH), 12.41 (s, 1H, NH). 13C NMR (50 MHz, DMSO-d6): δ 110.3, 116.7, 120.7, 125.2, 125.8, 126.9, 127.6, 127.7, 128.9, 130.3, 130.9, 133.0, 133.5, 144.1, 145.1, 150.4, 151.4, 177.3; HRMS m/z calculated for C18H12N2O2, [M]+, 288.0899, found 288.0895.
4.1.4.9. 2-(Benzo[b]thiophen-3-yl)-6-hydroxy-1,8-naphthyridin-4(1H)-one (7i)
Obtained from 6i, 32% yield, white solid; mp > 300 °C; 1H NMR (500 MHz, DMSO-d6): δ 6.27 (s, 1H, H-3), 7.50 (s, 2H, H-5, and H-6′), 7.79 (s, 1H, H-5′), 8.03–8.05 (m, 1H, H-7′), 8.13 (d, J = 7.5 Hz, 1H, H-4′), 8.27 (s, 1H, H-2′), 8.44 (s, 1H, H-7), 10.35 (s, 1H, OH), 12.30 (s, 1H, NH). 13C NMR (125 MHz, DMSO-d6): δ 106.9, 115.0, 118.0, 119.4, 123.1, 124.0, 125.9, 129.1, 133.2, 136.6, 140.4, 144.6, 147.5, 149.0, 153.1, 171.2; HRMS m/z calculated for C16H11N2O2S, [M+H]+, 295.0535, found 295.0535. The purity analysis was detected by reversed-phase HPLC on a Nucleodur® C18 HTec (5 μm, 250 × 4.6 mm i.d) using a MeOH/0.1 mM Na2HPO4 (99:1) mixture as eluent. The flow rate was 0.8 mL/min and UV detector was set at 254 nm. The retention time of 7h was 2.25 min; the purity of 7h was 97.0%.
4.1.5. Procedure for the synthesis of phosphate prodrug of 7a (11)
4.1.5.1. Dibenzyl (3-(5-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-2-yl)phenyl) phosphate (10)
To a stirred solution of 7a in dry THF was added NaH at room temperature. After the mixture was stirred for 1h, tetrabenzyl pyrophosphate (8) was added and stirring was continued for 25 min. The reaction mixture was filtered and washed with CH2Cl2. The filtrate was concentrated under vacuum at a temperature below 30 °C. A suspension of the residue in anhydrous MeOH was stirred at 25 °C for 48 h. The mixture was evaporated and purified by column chromatography (SiO2, CH2Cl2/EtOAc = 3:1) to afford 10 with yield 62%; yellow solid; mp 152–153 °C; 1H NMR (400 MHz, CDCl3): δ 2.99 (s, 3H, -CH3), 5.17 (d, J = 9.0 Hz, 2H, -CH2), 5.18 (d, J = 9.4 Hz, 2H, -CH2), 6.45 (s, 1H, H -3), 7.03 (d, J = 4.7 Hz, 1H, H-6), 7.30 (d, J = 7.4 Hz, 1H, H-3′), 7.34–7.40 (m, 10H, Ar-H), 7.44 (t, J = 7.8 Hz, 1H, H-5′), 7.50 (d, J = 7.4 Hz, 1H, H-6′), 8.30 (d, J = 4.7 Hz, 1H, H-7), 9.70 (s, 1H, -NH). 13C NMR (100 MHz, CDCl3): δ 22.7, 70.3 (d, J = 6.1 Hz), 111.0, 118.4 (d, J = 4.6 Hz), 118.9, 122.4 (d, J = 4.6 Hz), 122.8, 123.3, 128.1, 128.6, 128.8, 130.7, 135.1 (d, J = 6.1 Hz), 135.5, 147.5, 151.0 (d, J = 7.7 Hz), 151.4, 151.9, 152.6, 181.2; HRMS m/z calculated for C29H26N2O5P, [M+H]+, 513.1579, found 513.1572.
4.1.5.2. 3-(5-Methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-2-yl)phenyl dihydrogen phosphate (11)
A suspension of 10 in anhydrous MeOH was hydrogenated in the presence of 10% Pd/C at 25 °C for 1 h. The catalyst and precipitate were collected and dissolved in 10% NaHCO3 solution and then filtered. The filtrate was acidified with dilute HCl and the precipitate was then collected and washed with acetone to give 11 with yield 73%; white solid; mp 232–235 °C; 1H NMR (500 MHz, D2O + NaOH): δ 2.83 (s, 3H, -CH3), 6.87 (s, 1H, H-3), 7.00 (d, J = 4.8 Hz, 1H, H-6), 7.13 (d, J = 7.2 Hz, 1H, H-4′), 7.24–7.27 (m, 2H, H-2′, H-6′), 7.39 (t, J =8.0 Hz, 1H, H-5′), 8.44 (d, J = 4.8 Hz, 1H, H-7). 13C NMR (100 MHz, D2O + NaOH): δ 22.8, 110.3, 118.4 (d, J = 5.7 Hz), 119.7, 120.8, 121.3 (d, J = 4.5 Hz), 121.4, 121.5, 129.4, 140.7, 151.0, 151.5, 153.9, 154.1 (d, J = 5.4 Hz), 158.8, 178.4; HRMS m/z calculated for C15H12N2O5P, [M-H]-, 331.0484, found 331.0492. The purity analysis was detected by reversed-phase HPLC on a Thermo® ODS Hypersil (5 μm, 150 × 4.6 mm i.d) using a MeOH/0.01M NaHCO3 (70:30) mixture as eluent. The flow rate was 0.3 mL/min and UV detector was set at 254 nm. The retention time of 11 was 4.08 min; the purity of 11 was 99.49%.
4.2. Biological evaluation
4.2.1. Reagents and antibodies
DMEM/Ham’s F12 medium, RPMI 1640 medium, fetal bovine serum (FBS), penicillin and streptomycin were purchased from Hyclone Laboratories (Logan, UT, USA). Other reagents were purchase from Sigma-Aldrich Chemicals (St. Louis, MO, USA). Antibodies against caspase-8, caspase-3, caspase-9, and PARP were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against α-tubulin and β-tubulin were obtained from Santa Cruz (Biotechnology (Santa Cruz, CA, USA). Antibody against β-actin was obtained from EMD Millipore Corporation (Billerica, MA, USA). Other reagents were purchased from Sigma-Aldrich Chemicals (St. Louis, MO, USA).
4.2.2. Cell culture
Human leukemia HL-60 and non-small-cell lung cancer H460 cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (GIBCO/BRL), penicillin (100 U/mL)/streptomycin (100 μg/mL) (GIBCO/BRL) and 1% L-glutamine (GIBCO/BRL) at 37 °C in a humidified atmosphere containing 5% CO2. Human hepatocellular carcinoma cell line Hep3B was obtained from America Type Culture Collection (Manassas, VA, USA). Hep3B cells were cultured in DMEM/F12 medium supplemented with 10% FBS and penicillin (100 U/mL)/streptomycin (100 μg/mL) and maintained in a humidified incubator containing 5% CO2. Normal skin Detroit 551 cells were maintained in DMEM medium supplemented with 10% fetal bovine serum (GIBCO/BRL), penicillin (100 U/mL)/streptomycin (100 μg/mL) (GIBCO/BRL) and 1% L-glutamine (GIBCO/BRL) at 37 °C in a humidified atmosphere containing 5% CO2. Logarithmically growing cancer cells were used for all experiments.
4.2.3. Cell viability assay
Cell viability was detected by [3′-(4,5-dimethythiazol-2-yl)-2,5-diphenyl] tetrazolium bromide (MTT) assay. Cells were cultured in 96-well plates at 37 °C and incubated with complete medium containing the vehicle (DMSO) or compound for indicated times and concentrations. After treatment, cells were incubated with MTT solution (1 mg/mL in 1× PBS) at 37 °C for 2 h. Absorbance of the samples was read at wavelength of 570 nm and corrected for inference at 630 nm.
4.2.4. In vivo antitumor activity assay
4.2.4.1. Cell line and culture media
The Hep3B tumor cell line was purchased from American Type Culture Collection (ATCC HB-8064, human hepatocellular carcinoma cells). The culture medium contained 90% DMEM and 10% fetal bovine serum, supplemented with 1% penicillin-streptomycin. Tumor cells were incubated in an atmosphere containing 5% CO2 at 37 °C.
4.2.4.2. Animals
Balb/c nude mice used in this study were male, 4–6 weeks in age, weighing 18–20g and provided by National Animal Center. All animals were housed in Individually Ventilated Cages Racks (IVC Racks, 36 Mini Isolator system) under Specific Pathogen-Free (SPF) conditions throughout the experiment. Each cage (in cm, 26.7 length × 20.7 width × 14.0 height) was sterilized with autoclave and contained eight mice. The animals were maintained in a hygienic environment under controlled temperature (20–24 °C) and humidity (40%–70%) with 12 hour light/dark cycle. The animals were given free access to sterilized lab chow and sterilized distilled water ad libitum. All aspects of this work, i.e., housing, experimentation and disposal of animals were performed in general accordance with the Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, D. C., 1996).
In the xenograft tumor model of human hepatocellular carcinoma cell lines (Hep3B Cell ATCC HB-8064) in male Balb/c Nude mice, compound 11 was administered i.v. at doses of 10, 20 and 40 mg/kg five times/week until Day 21. The tumor size and body weight were monitored and recorded for 21 days. Human hepatocellular carcinoma cells (Hep3B Cell ATCC HB-8064) with 2 × 106 cells in 0.1 mL were injected subcutaneously into the right flank of the mice. When the tumor growth reached > 100 mm3 in volume (assumed as day 0), the tumor-bearing animals were assigned into several groups (8 animals in each group) for study. The body weight and tumor size were measured and recorded every seven days during the experiment period of 21 days. Tumor volume (mm3) was estimated according to the formula of length × (width)2 × 0.5 in mm3. Tumor growth inhibition was calculated as T/C (treatment/control) by the following formula: (T0: Tumor volume of treated group in Day 0.; Tn: Tumor volume of treated group in Day n.; C0: Tumor volume of control group in Day 0.; Cn: Tumor volume of control group in Day n.)
4.2.5. Antitumor mechanistic study
4.2.5. 1. Cell cycle distribution
Cells were stained with propidium iodide and analyzed by FACS Calibur flow cytometer (BD Biosciences, Mountain View, CA, USA).
4.2.5. 2. Western blot analysis
Cells were suspended in PBS containing proteinase inhibitors and sonicated. Lysate protein was separated by electrophoresis of SDS-PAGE and transferred to Immobilon P membrane (EMD Millipore Corp.). The immunoblots were incubated with the appropriate primary antibodies and horseradish peroxidase-conjugated secondary antibodies (EMD Millipore Corp). Signaling was visualized using a chemiluminescent substrate kit (EMD Millipore Corp.) and detected with a luminescent image analyzer (LAS4000, Fuji Photo Film Co, Tokyo, Japan). The band intensities were quantified by Multi Gauge software (Fuji Photo Film).
4.2.5.3. In vivo tubulin assembly assay
After treatments, cells were washed with ice-cold PBS and lysed in hypotonic buffer (1 mM MgCl2, 2 mM EGTA, 0.5% IGEPAL CA-630, 2 mM PMSF, 200 Units/mL aprotinin, 5 mM amino caproic acid, 1 mM benzamidine, and 20 mM Tris-HCl, pH 6.8) for 5 min at 37 °C. The cell lysates were centrifugated at 15,000 ×g for 10 min at 25 °C. The supernatant contained cytosolic tubulin. The pellets were polymerized tubulin, which was resuspended in hypotonic buffer and sonicated. Both fractions were subjected to western blot analysis to detect the tubulin contents by using anti-α-tubulin, β-tubulin, or β-actin antibodies.
4.2.5.4. In vitro tubulin polymerization
The Tubulin Polymerization Assay Kit (Cat #BK006P; Cytoskeleton Inc, Denver, CO, USA) was used for in vitro assay of microtubule polymerization according to the manufacturer’s instruction. Briefly, pure tubulin protein (300 μg) was suspended in 100 μL of GPEM buffer (80 mM PIPES, pH 6.9, 2 mM MgCl2, 0.5 mM EGTA, 1 mM GTP, and 5% glycerol) followed by incubation with test compounds at 37 °C. The alternation of tubulin polymerization was measured at the absorbance of 340 nm every 30 sec for 30 min (Synergy 2; BioTek, Winooski, VT, USA).
Supplementary Material
Chart 4.
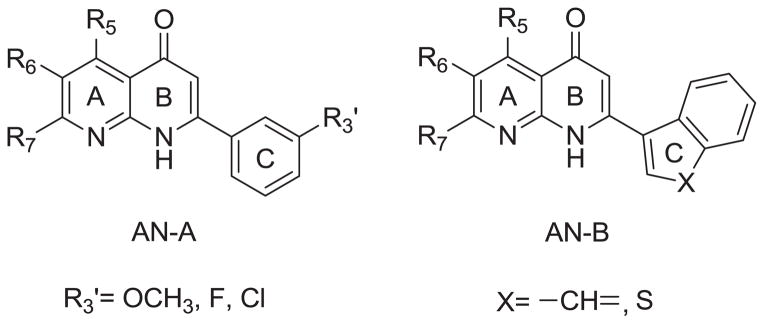
Structures of AN-A, AN-B.
RESARCH HIGHLIGHTS.
2-Arylnaphthyridin-4-ones were synthesized as potent cytotoxic agents.
2-(3-Hydroxyphenyl)-5-methyl-1,8-naphthyridin-4(1H)-one (7a) was most potent.
A phosphate prodrug (11) of 7a exhibited significant antitumor activity in vivo.
Compound 11 has good development potential as an antitumor agent.
Acknowledgments
This investigation was supported by research grants from the National Science Council of the Republic of China awarded to S.-C. Kuo (NSC101-2320-B-039-008). Thanks are also due to support by grant CA177584 from the National Cancer Institute, NIH awarded to K.-H. Lee.
Footnotes
Supplementary material related to this article can be found at
These data include National Cancer Institute, Developmental Therapeutics Program mean graphs of 7a–c, 7g, 7h
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuo SC, Lee HZ, Juang JP, Lin YT, Wu TS, Chang JJ, Lednicer D, Paull KD, Lin CM, Hamel E, Lee KH. J Med Chem. 1993;36:1146–1156. doi: 10.1021/jm00061a005. [DOI] [PubMed] [Google Scholar]
- 2.Li L, Wang HK, Kuo SC, Wu TS, Lednicer D, Lin CM, Hamel E, Lee KH. J Med Chem. 1994;37:1126–1135. doi: 10.1021/jm00034a010. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Wang HK, Kuo SC, Wu TS, Mauger A, Lin CM, Hamel E, Lee KH. J Med Chem. 1994;37:3400–3407. doi: 10.1021/jm00046a025. [DOI] [PubMed] [Google Scholar]
- 4.Chang YH, Hsu MH, Wang SH, Huang LJ, Qian K, Morris-Natschke SL, Hamel E, Kuo SC, Lee KH. J Med Chem. 2009;52:4883–4891. doi: 10.1021/jm900456w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen K, Kuo SC, Hsieh MC, Mauger A, Lin CM, Hamel E, Lee KH. J Med Chem. 1997;40:2266–2275. doi: 10.1021/jm960858s. [DOI] [PubMed] [Google Scholar]
- 6.Chen K, Kuo SC, Hsieh MC, Mauger A, Lin CM, Hamel E, Lee KH. J Med Chem. 1997;40:3049–3056. doi: 10.1021/jm970146h. [DOI] [PubMed] [Google Scholar]
- 7.Zhang SX, Bastow KF, Tachibana Y, Kuo SC, Hamel E, Mauger A, Narayanan VL, Lee KH. J Med Chem. 1999;42:4081–4087. doi: 10.1021/jm990208z. [DOI] [PubMed] [Google Scholar]
- 8.Hour MJ, Huang LJ, Kuo SC, Xia Y, Bastow K, Nakanishi Y, Hamel E, Lee KH. J Med Chem. 2000;43:4479–4487. doi: 10.1021/jm000151c. [DOI] [PubMed] [Google Scholar]
- 9.Chou LC, Chen CT, Lee JC, Huang SM, Huang LJ, Kuo SC, Qian K, Morris-Natschke SL, Lee KH, Way TD, Huang CH, Teng CM, Yamori T, Wu TS, Sun CM, Chien DS. J Med Chem. 2010;53:1616–1626. doi: 10.1021/jm901292j. [DOI] [PubMed] [Google Scholar]
- 10.Chou LC, Tsai MT, Hsu MH, Lin HY, Huang LJ, Kuo SC, Wang SH, Way TD, Huang CH, Qian K, Dong Y, Lee KH. J Med Chem. 2010;53:8047–8058. doi: 10.1021/jm100780c. [DOI] [PubMed] [Google Scholar]
- 11.Kuo SC, Teng CM, Lee KH, Huang LJ, Chou LC, Chang CS, Sun CM, Wu TS, Pan SL, Way TD, Lee JC, Chung JG, Yang JS, Chen CT, Huang CC, Huang SM. WO 2008/070176 A1 (PCT) Novel Hydrophilic Derivatives of 2-Aryl-4-quinolones as Antitumor Agents. 2008 [Patent Applied in Taiwan (96146890), U.S. (12/448088), Australia (2007328034), Canada (2670292), China (200780044796.9), EU (07853279.3), India (3052/EHENP/2009), Japan (2009-540310), Korea (10-2009-7014196), New Zealand (577130), Russian Federation (2009124622), South Africa (2009/03694).]
- 12.Lee KH, Chen K, Kuo SC. WO9839332 A1. 2-Aryl-naphthyridin-4-ones as Antitumor Agents. 1998
- 13.Lee KH, Chen K, Kuo SC. US5994367. Method for treating tumors using 2-aryl-naphthyridin-4-ones. 1999; US6071930. 2000; US6229016. 2001
- 14.Abdullah NM, Rosania GR, Shedden K. Plos One. 2009;4:1–8. doi: 10.1371/journal.pone.0004470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang SW, Pan SL, Huang YC, Guh JH, Chiang PC, Huang DY, Kuo SC, Lee KH, Teng CM. Mol Cancer Ther. 2008;7:350–360. doi: 10.1158/1535-7163.MCT-07-2000. [DOI] [PubMed] [Google Scholar]
- 16.Bhattacharyya B, Panda D, Gupta S, Banerjee M. Med Res Rev. 2008;28:155–183. doi: 10.1002/med.20097. [DOI] [PubMed] [Google Scholar]
- 17.Jordan MA, Wilson L. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 18.Meunier S, Vernos I. J Cell Sci. 2012;125:2805–2814. doi: 10.1242/jcs.092429. [DOI] [PubMed] [Google Scholar]
- 19.Pasquier E, Kavallaris M. IUBMB Life. 2008;60:165–170. doi: 10.1002/iub.25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.