

Abstract
Electroporation has proven to be a highly effective technique for the in vivo delivery of genes to a number of solid tissues. In most of the reported methods, DNA is injected into the target tissue and electrodes are placed directly on or in the tissue for application of the electric field. While this works well for solid tissues, there are many tissues and organs that are not amenable to such an approach. In this review I will focus on the development of electroporation protocols for two such tissues: the vasculature and the lung. Several methods for in vivo electroporation of the vasculature have been developed in recent years that deliver DNA to vessel segments from either the inside or outside of the vessel. The advantages and disadvantages of each are discussed, as are the applications for which they have been used. In more recent work, our laboratory has developed a novel method to deliver genes to the rodent lung that results in high level, uniform, gene expression throughout all cell types of the lung. Most importantly, this technique is safe, and causes no inflammatory response or alterations in normal physiology of the organs. Taken together, these studies demonstrate the utility of electroporation for gene transfer to noninjectible tissues.
INTRODUCTION
Although Electroporation has been routinely used to transfer DNA to bacteria, yeast, and mammalian cells in culture for the past 20 years (Ausubel et al., 1999), it has only relatively recently been applied to intact tissues in living animals. Electroporation uses electrical fields to create transient pores in the cell membrane that allow the entry of normally impermeable macromolecules into the cytoplasm (Somiari et al., 2000). Surprisingly, at the appropriate field strengths, the application of these fields to tissues results in little, if any, damage or trauma. More importantly, by simply applying the electric field to tissues that have received DNA, the levels of gene transfer and expression increase on the order of 100- to 1000- fold compared to DNA in the absence of electroporation (Mir et al., 1999). Although DNA electroporation protocols have been developed for, and work well in, solid tissues such as skeletal muscle or tumors in which the DNA can be injected, such an approach is impossible in other tissues, including blood vessels and the lung. However, electroporation has been used in these tissues very efficiently. The remainder of this report will review the work of several groups that have targeted the lung and vasculature using electroporation.
DNA delivery and field application
The successful use of electroporation to transfer genes to tissues requires two components: DNA, and an electric field. The major target organs for electroporation-mediated gene delivery are those into which DNA can be delivered by injection. These include skeletal muscle, the heart, liver, cornea, kidney, and tumors (R. Heller et al., 1996; Aihara and Miyazaki, 1998; Harrison et al., 1998; Oshima et al., 1998; Suzuki et al., 1998; Mathiesen, 1999; Mir et al., 1999; L. Heller et al., 2000; Tsujie et al., 2001; Blair-Parks et al., 2002). In all cases, purified plasmids are injected into the tissue using some sort of syringe and needle. For effective gene transfer to all cells within the target tissue, the injected DNA must be evenly distributed throughout the tissue. Further, because electroporation-mediated gene delivery and expression are dependent on DNA dose, as are other gene delivery methods, the concentration of DNA delivered to the tissue must be above an experimentally derived threshold level. If the concentration of DNA is too low, very little gene expression will be obtained. Similarly, if the DNA is not distributed throughout the tissue, some areas will receive DNA and express while others will not. Plasmids can be suspended at relatively high concentrations, and large volumes can be injected into one or multiple places within the target tissues to aid distribution. To this end, injection works very well.
The second component that is crucial to electroporation is the electric field itself. Both exponential decay and square waves can be used to mediate gene transfer, although it is widely accepted that square waves are less damaging to tissues. It has been shown in skeletal muscle and tumors that several different combinations of pulse lengths and field strengths can be used for electroporation (Lucas et al., 2002; Satkauskas et al., 2002). In the seminal papers using electroporation, high field strengths (1000 to 2000 V/cm) and multiple short pulses (<100 μsec) were used, but more recently, the trend has shifted to lower field strengths (200 V/cm) with multiple longer pulses (10 to 20 msec). A recent report from Mir has suggested that a combination of the two is even more effective for gene transfer (Satkauskas et al., 2002). In all of the studies using solid tissues, the electric fields are delivered using either plate electrodes placed directly on the tissue that received the DNA or needle electrodes that are inserted into the DNA-containing tissue (Somiari et al., 2000). Again, as for delivery of DNA by injection, these electrode configurations will only work for solid tissues or tissues that are accessible to direct placement of the electrodes. Thus, for many internal organs, this may be problematic, and may require surgery to make the technique possible.
While injection of plasmids has proven to be an effective and reliable method to deliver DNA to solid tissues, other tissues cannot be injected, and plate and needle electrodes may be inappropriate. The two tissues that our laboratory has focused on are the vasculature and the lung. Both of these tissues are not amenable to either DNA delivery by injection or standard electrode design and use.
VASCULAR ELECTROPORATION
The vasculature presents a challenge to electroporation-mediated gene delivery using standard approaches. With the exception of a few large vessels, such as the ascending aorta, the walls of arteries and veins are too thin to be injected with DNA. Moreover, even if DNA could be delivered to the walls by injection, the architecture of the vessel wall would prevent the even distribution of the DNA throughout it. Indeed, with multiple elastic lamina in large vessels and the tube-like structure of the vessel, injected DNA cannot freely diffuse throughout the wall. As such, other approaches are necessary.
There are two ways to deliver genes to the vessel wall: from the inside, and from the outside. Numerous viral and nonviral approaches to transfer genes to the vasculature have used transfer from within the lumen (Gunnett and Heistad, 2002; Young and Dean, 2002). The advantage to this method is that with the appropriate device (e.g., double balloon catheter), vectors can be delivered to defined regions of the vasculature with relatively simple methods that are clinically routine. The disadvantages are that for vectors to be delivered to the vessel lumen using double balloon catheters, blood flow must be restricted, which may cause ischemia of the downstream vessels and tissues. Alternatively, vectors can be administered from the adventitial surface of the vessel. The advantage of this approach to target defined regions of the vessel is that blood flow is not restricted, allowing no potential for ischemic injury. However, the major disadvantage is that the region of the vessel to be targeted for gene delivery must be exposed, requiring surgery. While this may be appropriate for ex vivo gene transfer for vein grafts where surgery is inevitable, it may not be appropriate for routine clinical application. However, it is a highly effective method for gene transfer in experimental animal models.
Electroporation following luminal delivery of DNA
Several different laboratories have reported the use of electroporation to transfer either DNA or heparin to the vessel wall following delivery of the molecule via the lumen (Dev et al., 1998, 2000; Matsumoto et al., 2001). Although the transfer of heparin to the vessel wall is not exactly gene delivery, many of the physical properties of heparin resemble those of DNA, so one may assume that heparin is a good model for DNA. Thus, the findings for electroporation-mediated transfer of heparin may be extended to DNA as well. In two separate studies, Dev and colleagues used a novel electrode that is contained within a porous catheter (Fig. 1A) (Dev et al., 1998, 2000). The catheter is inserted into the artery over a previously inserted guide wire and placed at the desired location within the vessel. The porous balloon is inflated so that blood flow is transiently obstructed, and a solution of heparin is infused into the balloon. Because of the porous nature of the balloon, the heparin can be delivered to the vessel wall. The electrode system uses the guide wire as one electrode and an internal wrapped wire contained entirely within the balloon as the second electrode. When a voltage is applied, an electric field develops between the two electrodes, causing electroporation of the vessel wall and delivery of heparin to cells within the wall. Immediately following electroporation, the balloon is deflated and the catheter is removed. As such, this procedure is very similar to those used daily by interventional cardiologists for percutaneous coronary transluminal angioplasty (PCTA), but without the injury associated with movement of the balloon.
FIG. 1.

Approaches for vascular electroporation. (A) Porous catheter-based electrode (Genetronics) (Dev et al., 1998, 2000). E1 and E2 indicate the electrodes. (B) Double balloon catheter and external electrodes (Matsumoto et al., 2001). (C) Bath electrode (Martin et al., 2000).
Using this catheter-based electrode, the authors infused a solution of heparin and applied a series of four pulses of 10- to 20-msec duration each at a field strength of approximately 250 V/cm over several seconds. When the distribution of a fluorescein-labeled heparin was studied, very little labeled heparin was detected in the vessel walls of animals that were not electroporated. By contrast, significant levels of the fluorescein-labeled heparin could be detected throughout the endothelium and the tunica media and tunica adventitia following electroporation (Dev et al., 1998). Thus, the electric field mediated the movement of the heparin into the vessel wall and through the elastic lamina into several layers of cells. However, the distribution of the heparin was heterogeneous, and was not detected completely circumferentially in the vessel wall. In a subsequent study, the authors also looked at the therapeutic benefits of heparin delivery by electroporation to vessels that had been injured by angioplasty (Dev et al., 2000). Rat carotid arteries were injured by angioplasty, and immediately following injury the catheter electrode was inserted and used to transfer heparin to the injured segment. At 4 weeks postinjury and treatment, animals that were electroporated with heparin showed a marked reduction in intimal wall thickness compared to animals that received heparin by the same delivery method but without electroporation. Thus, this approach effectively delivers heparin, and presumably DNA, to multiple cell layers within the vasculature.
Another group has used a slightly different approach to deliver DNA by electroporation to the vessel wall (Matsumoto et al., 2001). Instead of using a combined catheter-electrode device for simultaneous DNA delivery and electroporation, this group delivered DNA using a double balloon catheter and applied the electric field from the adventitial surface of the vessel. Using rabbits, the common carotid was exposed, and a 18-gauge double-balloon catheter was inserted via the external carotid and positioned just downstream. By inflating the distal balloon, the lumen was rinsed with saline prior to addition of a luciferase-expressing plasmid solution. Once the lumen was filled with DNA, two “T”-shaped stainless steel electrodes (2.5 cm long × 0.5 cm wide) were placed on either side of the vessel segment containing the DNA, and square-wave electric pulses were delivered (Fig. 1B). Similar to the experiments using heparin, the optimal field strength used was 200 V/cm delivered by 10 10- to 20-msec pulses. Immediately following electroporation, the electrodes and catheter were removed, the external carotid was ligated, and the animal was allowed to recover. When luciferase gene expression was assayed 2 days after electroporation, it was found to be dependent on field strength and pulse length. Similar to findings from multiple groups in different tissues, gene transfer and expression increased as the field strength was raised from 0 to 100 to 200 V/cm and then decreased by 300 and 400 V/cm (Martin et al., 2000; Blair-Parks et al., 2002; Dean et al., 2003). However, in contrast to most other studies, gene expression was not entirely dose dependent. Expression increased as the dose of DNA was raised from 50 to 200 μg/ml, but then decreased when the DNA concentration was raised further to 300 and 400 μg/ml. This is unusual, based on the fact that we and others have found that gene expression continues to increase as the dose of administered DNA is raised, even up to levels above 2 mg/ml, suggesting that the vessel’s ability to take up and express DNA is not saturated (Mir et al., 1999; Blair-Parks et al., 2002).
Electroporation of downstream target organs following luminal delivery of DNA
The use of the vasculature to deliver DNA to downstream target organs has been used for many viral and nonviral approaches to gene delivery. For example, if a large bolus of DNA is administered rapidly via the tail vein, high level gene transfer and expression will be obtained in the liver (a first pass organ) without any applied electric field to DNA carrier (Liu et al., 1999). Similarly, tail vein injection of DNA–liposome complexes results in high level expression in the hepatic and pulmonary endothelium (Uyechi et al., 2001). The use of the vasculature to target DNA to an organ has also been combined with electroporation to target several tissues, including the kidney and liver (Tsujie et al., 2001; Kobayashi et al., 2003). In all cases, DNA is delivered by injection into the vessels feeding the target organ: the renal artery in the case of the kidney and the portal vein for the liver. Following injection of DNA, electrodes are placed on either side of the exposed and exteriorized organ and a series of pulses are delivered. Maximal gene expression was obtained using similar pulse and field parameters as for other studies (six pulses at 100 to 200 V/cm and 50-msec duration). In both cases, the highest levels of gene transfer and expression were achieved in perivascular cells, including glomerular mesangial cells and hepatocytes. Although this method is highly effective at gene delivery, it is not without problems. One disadvantage to this method is that the half-life of noncomplexed DNA within the blood is very short due to the presence of numerous nucleases and serum components (Lew et al., 1995; Barron et al., 1999). Thus, unless electroporation occurs very shortly after DNA administration, the effective concentration of DNA within the blood and the target organ will decrease to the point where gene transfer and expression will not be detected. Regardless, it is a straightforward approach that may be applicable to other target organs as well.
Electroporation following adventitial delivery of DNA
Our laboratory has taken a different approach to vascular electroporation by developing an electrode system to deliver DNA to the vessel wall from the adventitial surface (Fig. 1C). The electrode resembles a spoon, and contains two parallel wires as electrodes. Different sized electrodes can be easily made using rubber O-rings, epoxy, and nichrome wire, to accommodate any sized vessel segment. Most of our studies have been focused on the rat mesenteric vasculature due to its ease of isolation, size, and architecture (Fig. 2A). Because of this, the electrode that we have used in most studies is approximately 1 cm in diameter with a 3-mm electrode gap, and holds 50 μl of plasmid solution. However, we have also used this approach successfully in the rat carotid and the mouse femoral arteries (J. L. Young and D. A. Dean, unpublished). To carry out gene transfer by electroporation using this model system, the small intestine and the mesenteric vascular tree are exteriorized through a midline incision in isoflurane anesthetized male Sprague-Dawley rats (150–400 g) and the vessels to be treated are identified based on their position relative to the illeo–cecal junction. The thin mesentery between vessels is cut away on either side of the segment to be treated and the entire neurovascular bundle (containing artery, vein, nerve, adventitial tissue, fat, and other tissue) is laid into the electrode, draping over each end. The DNA solution (in 10 mM Tris, pH 8, 1 mM EDTA, and 140 mM NaCl) is pipetted into the electrode (Fig. 2B and C), and a series of square wave electric pulses is applied. We routinely use Qiagen Gigaprep kits to purify plasmids, and have found that both endotoxin-free and standard kits work equally as well for gene transfer without inflammation. Immediately following electroporation, the vessel is removed from the electrode and another vessel is treated. One advantage to this method is that the DNA solution can be “reused” for subsequent vessels. Only small amounts of the solution are lost due to wicking by the vessels (<20%), so that less than 100 μl of plasmid are needed to electroporate eight vessel segments using a 50-μl electrode. Finally, after all desired vessels are treated, the intestines are replaced, the incision sutured, and the animal is allowed to recover. The only disadvantage to this approach is that it requires exposure of the vessels being treated. Thus, it may not be appropriate for many clinical applications, but it is definitely useful for animal studies, ex vivo applications (e.g., vein grafts), and certain in vivo clinical procedures.
FIG. 2.
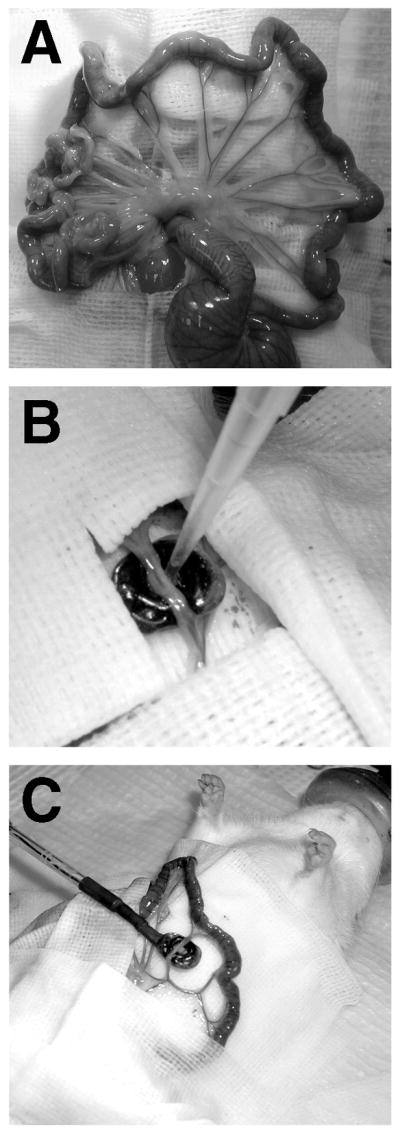
Gene transfer and electroporation of the mesenteric vasculature. (A) The mesenteric vascular tree. (B) DNA solution being pipetted into the electrode. The vessel is draped into the bath electrode, which is surrounded by surgical drapes to maintain a sterile field. At the end of the electroporation of this vessel, the DNA solution can be removed and reused for the next vessel. (C) Placement of electrode during surgery.
Using this method, we have shown that gene transfer is dependent on the electric field and the DNA dose. To assess these parameters, plasmids expressing GFP (pEGFP-C1, Clontech, Palo Alto, CA) and/or luciferase (pCMV-Lux-DTS; Vacik et al., 1999) were used. As has been found in other in vivo electroporation studies in other tissues, essentially no gene transfer and expression is detected in the absence of an electric field. Our lab routinely uses a BTX ECM 830 square wave electroporator (BTX, San Diego, CA), but we have also had success using a Grass stimulator (Grass Instruments, West Warwick, RI) and a Biorad GenePulser (Biorad, Richmond, CA). When the field was increased to 100 V/cm, high levels of gene expression were detected in some of the vessels, but not in others. However, when the field was raised to 200 V/cm, higher and much more uniform gene expression was detected in vessels from multiple animals. Using eight square-wave pulses at 10 msec each, an average of 400 pg of luciferase per cm of vessel (with a maximum of 2 ng per cm of vessel) are obtained 2 days postelectroporation using this field strength and 2 mg/ml DNA. When the field is raised further to 400 V/cm, expression levels drop off dramatically. This could be due in part to tissue damage that is seen at this high field strength (see below). We also found that gene transfer was dependent on the concentration of DNA used. At concentrations of plasmid less than 0.2 mg/ml, essentially no gene expression was detected in vessels. However, between 50 and 100 pg of luciferase per cm of vessel was expressed when the concentration was raised to 0.5 mg/ml, and 400 to 500 pg of product per cm of vessel were routinely obtained when 2 mg/ml DNA was used. Although higher concentrations were not tested, it is likely that expression will continue to increase as the DNA concentration is raised, based on results in other tissues, including skeletal muscle, the lung, and the cornea (Mir et al., 1999; Blair-Parks et al., 2002; Dean et al., 2003). Further, multiple plasmids can be transferred at the same time, each of which will express gene product. We routinely transfer experimental plasmids along with a GFP-expressing reporter plasmid to identify segments of vessels that were electroporated. One potential problem to delivering multiple expression constructs is promoter competition, which may decrease transgene expression from one or both administered plasmids (Manthorpe et al., 1993; Hartikka et al., 1996). While this may be a problem with certain promoter combinations, we have not found this to be a major issue when using two or more plasmids using the CMV immediate early promoter/enhancer (CMViep), or the CMViep and the SV40 early promoter (Martin et al., 2000; Shirasawa et al., 2003; Young et al., 2003).
This approach has been used to deliver multiple genes, including several reporter and functional genes. For example, a dominant-negative PKCε mutant gene was electroporated into the vasculature to study vasoconstriction in a rat model (Shirasawa et al., 2003). Studies have suggested that PKCε may play a role in regulating agonist-induced vascular smooth muscle contraction (Masuo et al., 1994; Buus et al., 1998). To examine the role of PKCε in adrenoreceptor-mediated contraction of mesenteric arteries, a dominant-negative PKCε (PKCε-KN) (Genot et al., 1995) was transferred to vessels by electroporation as described above. Two days posttransfer, vessels were excised and mounted on a myograph for functional studies of phenylephrine-induced vasoconstriction. A separate group of vessels was treated with the isoform-nonspecific PKC inhibitor, chelerythrine (2.5 μM) for comparison. PKCε-KN significantly attenuated phenylephrine responses (EC50 = 4.99 ± 1.07 μM) compared to control, nonelectroporated vessels (2.81 ± 0.17 μM). Vasoconstrictor responses to KCl did not differ between the groups. Inhibition of all PKC isoforms by chelerythrine attenuated vasoconstrictor function in normal vessels from 2.80 ± 0.31 to 4.76 ± 0.75 μM.
Phenylephrine responses between chelerythrine and PKCε-KN/chelerythrine vessels were similar. These results clearly show that delivery of plasmids using electroporation is a viable method for achieving high level, functional gene expression.
Finally, our laboratory has also used this method to transfer oligonucleotides to the vasculature in vivo (Nunamaker et al., 2003). Using the same electrode design, we have shown that catalytic DNA oligonucleotides (termed “DNAzymes”) can be transferred to the mesenteric vessels as effectively as plasmids. These oligos are designed to act as catalytic molecules that selectively bind to an RNA substrate by Watson-Crick base pairing and cleave phosphodiester bonds, resulting in decreased target mRNA levels and subsequent protein production (Khachigian, 2000; Young and Dean, 2002). As for plasmid electroporation, electroporation-mediated transfer of 31-mer single-stranded deoxyoligonucleotides was field strength and oligonucleotide dose dependent. Maximal oligonucleotide transfer was achieved at 100 μM oligo, as determined by catalytic activity of the transferred oligonucleotide on target mRNA and protein levels. By comparison, a solution of plasmids at 2 mg/ml corresponds to a concentration of approximately 1 μM. At 1 μM oligonucleotide, no activity could be detected, suggesting that insufficient transfer had occurred. However, at 100 μM, a 60% reduction in target protein level was detected in the vessels at 24 h following delivery. Thus, electroporation can be used to deliver both plasmids and oligonucleotides efficiently to the vasculature.
Localization of delivered DNA and gene expression
To determine where the plasmids localized following electroporation using the bath electrode, several methods were used, including observation of transgene expression (both by direct GFP fluorescence and by immunohistochemistry for gene product), and in situ hybridization of the electroporated DNA itself. When neurovascular bundles that had been electroporated with a GFP-expressing plasmid were excised from the animals, strong GFP expression was detected within an approximately 1-cm segment of vessel but dropped off rapidly outside this area. Presumably, this corresponds to the area contained within the electrode which sees the vast majority of the electric field. When looked at in whole bundles, the GFP fluorescing vessels can be seen buried within layers of fat, most of which is non-expressing, although a few bright GFP+ cells can be seen that are not part of the vessels themselves. This is rather surprising, since most of the cells within the neurovascular bundle, including most of those outside the artery and vein, have detectable levels of plasmid in their nuclei following electroporation, as detected by in situ hybridization (Young et al., 2003). Why these cells do not express gene product is unclear.
When cross-sections of the vessels are analyzed, high levels of GFP expression are detected throughout all layers of the vessels (both artery and vein), including the adventitial cells, smooth muscle cells, and endothelial cells. The highest levels of gene expression appear to be in endothelial cells lining the lumen, despite the fact that the DNA is administered from the outside of the vessel. Whether this expression is due to the fact that these cells take up more DNA or are more active for gene expression is unclear. We are currently performing experiments to determine this.
Because the average neurovascular bundle is about 1 to 1.5 mm in diameter and the vessels, which are 100 to 200 μm in diameter, are embedded in the middle of the bundle, the DNA in the bath must move between 500 and 750 μm to reach the cells. It has been proposed that DNA delivery to tissues using electroporation has an electrophoretic and a permeabilization component (Pucihar et al., 2002; Puc et al., 2003). The first would allow the DNA to be driven into a tissue, and the second would allow for entry of the DNA into the cell through the plasma membrane. It is thought that short pulses of high field strength can cause permeabilization, while longer pulses of lower voltage mediate the movement of the DNA. Indeed, several recent papers suggest that by combining a short high-energy pulse with subsequent longer lower energy pulses, even greater degrees of gene transfer can be achieved (Pucihar et al., 2002; Puc et al., 2003). We have used pulses of uniform duration for our studies which are sufficient for movement and cell entry in this system, but whether we can increase gene transfer using this dual parameter approach remains to be seen.
Duration of expression
Following electroporation of DNA into the vasculature, we can detect gene expression as early as 6 h postelectroporation. The levels of expression maximize by 24 h and remain relatively constant for 3 days, after which time they decrease to baseline by 7 days (Martin et al., 2000; Young et al., 2003). This gradual rise in gene expression between 6 and 24 h is paralleled by the nuclear localization of the electroporated DNA, as detected by in situ hybridization (Young et al., 2003). At 8 h postelectroporation, large amounts of plasmid can be seen throughout the neurovascular bundle, largely in extracellular and cytoplasmic locations, although some nuclear accumulation can be detected. This small amount of nuclear DNA probably accounts for the low level of gene expression at this time. By 24 h postelectroporation, much of the extracellular and cytoplasmic DNA has disappeared, while the amount of plasmid localized to the nucleus has increased. No increases in nuclear DNA can be detected after this time, resulting in maximal gene expression by 24 h. The disappearance of extracellular and cytoplasmic plasmid is due to a combination of degradation by DNases and active nuclear transport of plasmids carrying DNA nuclear targeting sequences. However, why gene expression ceases after 3 to 5 days is unclear. Using PCR of extracted tissue, we have found that significant amounts of plasmid remain at 7 days postelectroporation, yet no gene expression is detected. At present, it is unclear why transgene expression ceases, but it may be due in part to the promoter used (CMViep). Similar time courses of gene expression using this promoter have been detected in most tissues in vivo, regardless of delivery method (liposome, polyethyleneimine, electroporation, hydrodynamic delivery), with the exception of skeletal muscle, which expresses for the lifetime of the host (Wolff et al., 1992). It has been suggested that promoter inactivation, plasmid methylation, subnuclear partitioning, or other mechanisms may be responsible for the silencing of gene expression. Indeed, it has been reported that other promoters may express longer. One such promoter is the UbC promoter, which has been reported to express for periods up to several months in the lung compared to the CMViep, which expresses for less than 10 days (Gill et al., 2001). Thus, with the appropriate promoter, long-term gene expression may be a possibility in the vessel wall.
Physiological changes induced by electroporation and gene transfer
The health of electroporated tissues have been investigated on multiple levels. It has been demonstrated in numerous tissues that at field strengths less than 200 V/cm (10 pulses of 20 msec duration), very little histological damage can be detected (Aihara and Miyazaki, 1998; Suzuki et al., 1998; Martin et al., 2000; Matsumoto et al., 2001; Blair-Parks et al., 2002; Gehl et al., 2002). Although electroporated vessels appear healthy at the time of harvest by visual inspection and subsequent histological analysis of sections, direct evidence was desired to demonstrate that they maintained unaltered vascular function after electroporation. Therefore, the responses of electroporated and control arteries to vasoconstricting stimuli were measured using intravital microscopy (Martin et al., 2000). Measurements were made on vessels harvested within the window of gene expression (day 2), or well after transgene expression had ceased (day 40). In both cases, control arteries constricted in a dose-dependent manner to increasing concentrations of phenylephrine. Similarly, the responses of the electroporated vessels were indistinguishable from those of the control vessels. Further, addition of either 0.1 mM adenosine or 50 μM isoproterenol to maximally constricted electroporated or control vessels resulted in nearly identical degrees of vessel dilation. These results demonstrate that vascular function, in terms of constriction and dilation, is unaltered by electroporation or the expression of the reporter genes in the vessels, for at least 40 days postprocedure.
To investigate the effects of electroporation on the global pattern of gene expression, DNA microarray analyses were performed (Young et al., 2003). Multiple vessels from two animals were electroporated with a solution of TE-saline (no DNA). One day postelectroporation, these vessels, and an equal number of untreated vessels from the same animals, were removed and RNA was isolated, labeled, and hybridized to Affymetrix Rat Genome DNA Microarrays. Out of the 8700 genes and ESTs represented on the arrays, the expression patterns of less than 100 changed. All changes were between 1.4- and 3.6-fold, and no single class of genes were affected more than others (e.g., transcriptional machinery, cell cycle, etc), rather the genes appeared to be largely obscure and not indicative of any negative side effects. In fact, changes were not detected in any genes involved in cell cycle, proliferation, transcription, apoptosis, or inflammation. These results further support the use of electroporation in the vasculature as a safe method that does not cause unwanted harmful effects.
PULMONARY ELECTROPORATION
The lung is an attractive target for gene therapy. Multiple genetic, acquired, and infectious diseases are manifested in the lung, and it is quite amenable to different delivery strategies (West and Rodman, 2001). Because of this, multiple techniques for gene delivery to the lung have been developed, including the use of adenoviruses, adeno-associated viruses, lipoplex, and polyethyleneimine (West and Rodman, 2001; Weiss, 2002). Further, vector administration has been achieved either by tracheal delivery to target the pulmonary epithelium or by vascular delivery to target the pulmonary endothelium. However, all of these approaches have limitations, including inefficiency of gene transfer, immunological responses, inflammation, non-specificity of cell targeting, and low levels of gene expression. Based on the successful application of electroporation to multiple tissues in vivo, we developed electroporation methods to transfer genes to the lung, both ex vivo and in vivo (Dean et al., 2003).
Ex vivo gene delivery to the lung
To determine whether electroporation could also be used in the rodent lung, we first tested whether genes could be delivered ex vivo to the tissue, by placing flat electrodes on either side of isolated lung lobes from mice and rats. Animals were euthanized and a solution of plasmid DNA in 10 mM Tris, pH 8, 1 mM EDTA, and 140 mM NaCl was administered into the bronchi of the lungs immediately following removal en bloc. For mice (female Balb/c, 15–20 g), 200 μl were delivered and for rats (male Sprague-Dawley, 150–400 g), 500 μl were used. Within 30 sec of DNA delivery, 0.7-cm diameter Tweezertrodes (Genetronics, San Diego, CA) were placed on either side of each lobe and an electric field of 200 V/cm was applied in a series of eight pulses of 10-msec duration each using an ECM830 electroporator (BTX), based on our previous studies in other tissues (Martin et al., 2000; Blair-Parks et al., 2002; Shirasawa et al., 2003; Young et al., 2003). After electroporation of one lobe, the electrodes were moved and successive lobes were treated similarly. Upon completion of electroporation, the lobes were separated, placed in growth medium, and incubated overnight at 37°C. Gene expression at 24 h post-electroporation was dose dependent in both mouse and rat lungs, but was much more efficient in the mouse lung. Indeed, just 20 μg of plasmid gave over 5 ng of gene product per g wet weight of lung in the mouse, whereas 0.5 mg of DNA were required to produce 1 ng of gene product per g wet weight in the rat (Fig. 3A). One possible explanation for this difference is that the volume of plasmid delivered to the mouse lung was much higher relative to the lung itself than in the rat, and thus greater plasmid distribution could be achieved and result in transfer to a greater number of cells. Ongoing experiments are addressing this possibility.
FIG. 3.

Comparison of gene expression in vivo and ex vivo following gene transfer to the lungs using electroporation. (A) Dose–response curve for ex vivo gene transfer in mice and rats. pCMV-lux-DTS plasmid at the indicated doses was administered to excised mouse (closed circles: female Balb/c, 15–20 g) or rat (open circles: male Sprague–Dawley, 150–400 g) lungs via the bronchi and the individual lobes were electroported by direct placement of electrodes on the lobes using eight square wave pulses of 10 msec duration each at 200 V/cm. Mouse lungs received 200 μl of plasmid and rat lungs received 500 μl. Lungs were placed into medium and luciferase gene expression was measured 24 h later (mean ± SEM, n = 3). (B) Comparison of in vivo and ex vivo gene transfer and expression in the mouse lung. Ex vivo transfer was performed as described in (A). For in vivo electroporation, 20 μg of pCMV-Lux-DTS in 100 μl were injected intratracheally and varying field strengths were applied to the chest of mice (eight pulses of 10-msec duration each). The levels of luciferase gene expression were measured at 2 days posttreatment as previously described (Dean et al., 2003) (mean ± SEM; n = 4 animals/point). Values from (A) are shown for ex vivo expression at the same DNA dose (Dean et al., 2003).
In vivo gene delivery to the lung
Although gene transfer to lungs by electroporation using direct placement of the electrodes on the lungs was very efficient, the procedure was much too invasive to be used easily for in vivo studies. To circumvent this, DNA was delivered directly to the lungs of sodium pentobarbital-anesthetized mice by either injection of 100 μl into the mouse trachea using a needle or administration of 100 μl of plasmid through an endotracheal tube. Qiagen Gigaprep-purified DNA was delivered in 10 mM Tris, pH 8, 1 mM EDTA, and 140 mM NaCl, and the animals were allowed to regain normal breathing patterns, typically within 30 sec. Immediately following this, a series of eight square-wave electric pulses of 10-msec duration each were administered to the animals using 7-mm Tweezertrodes. The electrodes were placed on either side of the chest, which had been wetted with 70% ethanol. The animals recovered and survived with no apparent trauma until the experiments were terminated at the desired times, between 1 and 7 days posttreatment. We have had no mortality due to electroporation alone (n = 30 animals) and less than 12% mortality due to drugs, surgery, endotracheal tube placement, or fluid delivery (n = 187 animals). However, we have detected the greatest mortality when the interval between fluid delivery and electroporation is decreased to less than 15 sec. Consequently, electroporation is performed 30 sec following DNA delivery.
With no applied electric field, essentially no gene transfer and expression was detected in vivo (Dean et al., 2003). As for the vasculature, the optimal field strength for gene transfer is 200 V/cm, using eight, 10-msec square-wave pulses. However, unlike the vasculature, where significant, although variable, gene expression was detected at a field strength of 100 V/cm, essentially no gene transfer and expression were detected below 200 V/cm. Although expression was detected at 400 V/cm, the level was lower than at 200 V/cm. Further, at 400 V/cm, tissue damage is observed in lungs and other tissues (Martin et al., 2000; Blair-Parks et al., 2002; Dean et al., 2003). Electroporation-mediated gene transfer to the lung was also DNA dose dependent. At doses of DNA equal to or less than 10 μg per mouse (0.1 μg/ml) less than 10 pg of luciferase were produced per g wet weight of tissue. As the dose was increased, levels of gene expression increased dramatically to approximately 200 pg product per g wet weight using 20 μg of plasmid and over 10 ng of product using 100 μg of DNA. When corrected for the weight of the lungs, this corresponds to several hundred pg per animal or 10 to 100 pg product per μg of administered DNA per gram of tissue at high doses of DNA (100 μg per animal). These levels are very high compared to those achieved by other nonviral methods, including polyethyleimine or DNA–liposome complexes (Li and Huang, 1997; Gautam et al., 2000).
It is rather interesting that the levels of gene transfer achieved in vivo are much less than those achieved in our ex vivo studies. For example, a 20-μg dose of DNA gave approximately 200 pg per g wet weight in vivo, but over 5 ng per g wet weight following ex vivo delivery (Fig. 3B). One possible reason for this is that the distribution and relative strength of the electric field are very different in these two settings. In ex vivo delivery, the electrodes are placed directly on the lungs so that the entire field is delivered to the target tissue. By contrast, during in vivo electroporation, the electrodes are placed on the chest. Thus, the field must travel through multiple layers of different tissues, including skin, fat, muscle, bone, cartilage, and interstitial fluid, all of which have different resistive properties, prior to reaching the lungs. As such, it is not clear what field strength the lungs themselves actually see, but it is very possible that it is less than 200 V/cm. We are currently modeling the electric fields through the chest to develop testable hypotheses to address this.
Distribution of gene transfer and expression in the lung
To investigate the distribution of gene transfer in the lung and determine which cell types were being electroporated, we transferred a plasmid expressing the lacZ gene that encodes β-galactosidase (pCMVβ-DTS (Dean et al., 1999)). Two days following electroporation, lungs were fixed and reacted with the chromogenic β-galactosidase substrate X-gal. Despite the high levels of luciferase expression detected following electroporation in the lung, there were relatively few X-gal reactive cells in the lung, suggesting that gene transfer was limited to relatively few cells. Further, when whole lungs were observed, these X-gal reactive cells clustered in dots (clusters of alveoli) that were concentrated in a 0.5-cm diameter area in the upper lobes. Microscopic examination of thin sections revealed that most of the cells that were β-galactosidase positive were alveolar type I and type II epithelial cells within individual alveoli. These cells were also clustered so that when one cell within an alveolus was β-galactosidase positive by X-gal, all the cells in that alveolus expressed the gene. One reason for this is that these alveoli may have received more of the DNA solution, and thus the DNA was concentrated in these regions when the electric field was applied. Another possibility that takes into account the distribution of the expressing cells throughout the lung is that the electric field is concentrated in this area. Thus, cells within this region, which roughly corresponds in size to that of the Tweezertrode electrode, may receive the full 200 V/cm, while other regions of the lung may receive a lower field strength.
To determine whether this distribution was due to nonuniform expression or to a technical problem with the X-gal staining procedure limiting penetration of X-gal to all areas of the lungs, we performed immunohistochemistry on thin sections with antibodies directed against β-galactosidase. In animals that received no lacZ plasmid, there was no immunoreactivity with an antibody against β-galactosidase above that seen in the absence of primary antibody. By contrast, animals that were electroporated with the lacZ plasmid showed considerable immunoreactivity against β-galactosidase in multiple cell types throughout all sections of the lungs, including airway and alveolar epithelial cells, airway smooth muscle cells, and endothelial cells. This expression pattern mimicked that seen when antibodies against luciferase were used to stain paraffin-embedded thin sections of lungs that had been electroporated with plasmid encoding luciferase. One reason for this apparent discrepancy between X-gal reactivity and immunohistochemistry is that X-gal reactivity underestimates transfection efficiency during transient transfections of lacZ-expressing plasmids (Bebok et al., 1996). While it is thought that several thousand β-galactosidase molecules are needed to cause a cell to be X-gal positive, fewer than several hundred β-galactosidase molecules are needed for visualization by immunohistochemistry using an amplification system such as the Vector ABC system (Vector Laboratories, Burlingame, CA). This makes immunohistochemistry a more reliable indicator of real transfection efficiency, as long as the appropriate controls are performed (Zhang et al., 1996). Thus, it is possible that more DNA is delivered to the alveolar epithelium or that these cells may express more protein than other pulmonary cells, but all cells in the lung do receive and express the transgene. Taken together, these results suggest that electroporation can be used for high-level gene transfer and expression throughout the lung.
Safety of pulmonary electroporation
One of the main drawbacks to many methods of gene delivery to the lung is the induction of inflammatory and immune responses following vector administration. This is very true in the case of viral vectors, such as adenovirus, in which many times the degree of inflammation is so great that the results of gene transfer cannot be assessed for 7 to 10 days following viral administration due to severe inflammatory responses and the presence of many infiltrating cells. Although nonviral vectors have been hailed as eliciting no adverse inflammatory responses, it is now known that this is not entirely true; even liposome–DNA complexes can cause inflammation (Tan and Huang, 2002). By contrast, pulmonary electroporation at 200 V/cm (10-msec pulses) across the chest causes no trauma or damage, either macro- or microscopically (Dean et al., 2003). Histological analysis of the lungs showed no inflammatory infiltrates or other pathological changes induced by electroporation. Electroporated lungs were indistinguishable from untreated lungs, and showed no increases in vascular congestion, hyaline membranes, polymorphonuclear cell infiltrates, interstitial infiltrates, peribronchial inflammation, perivascular edema and inflammation, alveolar hemorrhage, or nuclear debris.
One concern that has been raised is the perceived problem associated with delivering electric fields to the chest. Having watched numerous episodes of “ER” on the television, everyone instantly recognizes the effects of cardioversion following delivery of electricity to the chest (“Clear!”). However, because of the relatively low energies used to deliver DNA to cells of the lung by electroporation, this is not an issue. The optimal field strength for pulmonary gene transfer (200 V/cm) corresponds to much less than 1 J of energy (W · s), and is well below that used to defibrillate the heart (200 to 360 J). We have examined the animals for signs of cardiac dysfunction, and have detected none following electroporation. Thus, the procedure is well tolerated by mice and rats, and is safe both to the lung and the rest of the animal.
CONCLUSIONS AND FUTURE DIRECTIONS
These studies demonstrate that electroporation can be used effectively to deliver DNA to tissues that cannot be readily injected with DNA solutions. The techniques are rapid, easy to perform, and highly reproducible. They mediate the highly efficient delivery of DNA (and oligonucleotides) to tissues, and result in levels of gene expression that approach and even surpass those achieved with the best viral and nonviral means. In contrast to all other techniques developed to date, electroporation promotes gene transfer to multiple cell layers within both large (e.g., carotid) and small (e.g., mesenteric) blood vessels without the need to damage the vessel itself, and to multiple cell types within the lung. Thus, based on the ease, efficiency, and nontraumatic nature of these electroporation methods for pulmonary and vascular gene transfer, their use may be of great experimental and clinical potential.
Acknowledgments
This work was supported in part by grants from the Crane Asthma Fund, the Sandler Program for Asthma Research, and NIH Grants HL59956 and HL71643.
References
- AIHARA H, MIYAZAKI J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998;16:867–870. doi: 10.1038/nbt0998-867. [DOI] [PubMed] [Google Scholar]
- AUSUBEL FM, BRENT R, KINGSTON RE, MOORE DD, SEIDMAN JG, SMITH JA, STRUHL K, editors. Short protocols in molecular biology. New York: John Wiley & Sons; 1999. [Google Scholar]
- BARRON L, UYECHI L, SZOKA FC. Cationic lipids are essential for gene delivery mediated by intravenous administration of lipoplexes. Gene Ther. 1999;6:1179–1183. doi: 10.1038/sj.gt.3300929. [DOI] [PubMed] [Google Scholar]
- BEBOK Z, ABAI AM, DONG JY, KING SA, KIRK KL, BERTA G, HUGHES BW, KRAFT AS, BURGESS SW, SHAW W, et al. Efficiency of plasmid delivery and expression after lipid-mediated gene transfer to human cells in vitro. J Pharmacol Exp Ther. 1996;279:1462–1469. [PubMed] [Google Scholar]
- BLAIR-PARKS K, WESTON BC, DEAN DA. Gene delivery to the cornea by plasmid injection and electroporation. J Gene Med. 2002;4:92–100. doi: 10.1002/jgm.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUUS CL, AALKJAER C, NILSSON H, JUUL B, MOLLER JV, MULVANY MJ. Mechanisms of Ca2+ sensitization of force production by noradrenaline in rat mesenteric small arteries. J Physiol. 1998;510:577–590. doi: 10.1111/j.1469-7793.1998.577bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEAN BS, BYRD JN, Jr, DEAN DA. Nuclear targeting of plasmid DNA in human corneal cells. Cur Eye Res. 1999;19:66–75. doi: 10.1076/ceyr.19.1.66.5344. [DOI] [PubMed] [Google Scholar]
- DEAN DA, MACHADO-ARANDA D, BLAIR-PARKS K, YELDANDI AV, YOUNG JL. Electroporation as a method for high-level non-viral gene transfer to the lung. Gene Ther. 2003;10:1608–1615. doi: 10.1038/sj.gt.3302053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEV NB, HOFMANN GA, DEV SB, RABUSSAY DP. Intravascular electroporation markedly attenuates neointima formation after balloon injury of the carotid artery in the rat. J Intervent Cardiol. 2000;13:331–338. [Google Scholar]
- DEV NB, PREMINGER TJ, HOFMANN GA, DEV SB. Sustained local delivery of heparin to the rabbit arterial wall with an electroporation catheter. Cathet Cardiovasc Diagn. 1998;45:337–345. doi: 10.1002/(sici)1097-0304(199811)45:3<337::aid-ccd28>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- GAUTAM A, DENSMORE CL, XU B, WALDREP JC. Enhanced gene expression in mouse lung after PEI–DNA aerosol delivery. Mol Ther. 2000;2:63–70. doi: 10.1006/mthe.2000.0087. [DOI] [PubMed] [Google Scholar]
- GEHL J, SKOVSGAARD T, MIR LM. Vascular reactions to in vivo electroporation: Characterization and consequences for drug and gene delivery. Biochim Biophys Acta. 2002;1569:51–58. doi: 10.1016/s0304-4165(01)00233-1. [DOI] [PubMed] [Google Scholar]
- GENOT EM, PARKER PJ, CANTRELL DA. Analysis of the role of protein kinase C-alpha, -epsilon, and -zeta in T cell activation. J Biol Chem. 1995;270:9833–9839. doi: 10.1074/jbc.270.17.9833. [DOI] [PubMed] [Google Scholar]
- GILL DR, SMYTH SE, GODDARD CA, PRINGLE IA, HIGGINS CF, COLLEDGE WH, HYDE SC. Increased persistence of lung gene expression using plasmids containing the ubiquitin C or elongation factor 1alpha promoter. Gene Ther. 2001;8:1539–1546. doi: 10.1038/sj.gt.3301561. [DOI] [PubMed] [Google Scholar]
- GUNNETT CA, HEISTAD DD. Virally mediated gene transfer to the vasculature. Microcirculation. 2002;9:23–33. doi: 10.1038/sj.mn.7800119. [DOI] [PubMed] [Google Scholar]
- HARRISON RL, BYRNE BJ, TUNG L. Electroporation-mediated gene transfer in cardiac tissue. FEBS Lett. 1998;435:1–5. doi: 10.1016/s0014-5793(98)00987-9. [DOI] [PubMed] [Google Scholar]
- HARTIKKA J, SAWDEY M, CORNEFERT-JENSEN F, MARGALITH M, BARNHART K, NOLASCO M, VAHLSING HL, MEEK J, MARQUET M, HOBART P, et al. An improved plasmid DNA expression vector for direct injection into skeletal muscle. Hum Gene Ther. 1996;7:1205–1217. doi: 10.1089/hum.1996.7.10-1205. [DOI] [PubMed] [Google Scholar]
- HELLER L, JAROSZESKI MJ, COPPOLA D, POTTINGER C, GILBERT R, HELLER R. Electrically mediated plasmid DNA delivery to hepatocellular carcinomas in vivo. Gene Ther. 2000;7:826–829. doi: 10.1038/sj.gt.3301173. [DOI] [PubMed] [Google Scholar]
- HELLER R, JAROSZESKI M, ATKIN A, MORADPOUR D, GILBERT R, WANDS J, NICOLAU C. In vivo gene elctroinjection and expression in rat liver. FEBS Lett. 1996;389:225–228. doi: 10.1016/0014-5793(96)00590-x. [DOI] [PubMed] [Google Scholar]
- KHACHIGIAN LM. Catalytic DNAs as potential therapeutic agents and sequence-specific molecular tools to dissect biological function. J Clin Invest. 2000;106:1189–1195. doi: 10.1172/JCI11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOBAYASHI S, DONO K, TAKAHARA S, ISAKA Y, IMAI E, ZHENHUI L, NAGANO H, TOMOAKI K, UMESHITA K, NAKAMORI S, et al. Electroporation-mediated ex vivo gene transfer into graft not requiring injection pressure in orthotopic liver transplantation. J Gene Med. 2003;5:510–517. doi: 10.1002/jgm.370. [DOI] [PubMed] [Google Scholar]
- LEW D, PARKER SE, LATIMER T, ABAI AM, KUWAHARA-RUNDELL A, DOH SG, YANG ZY, LAFACE D, GROMKOWSKI SH, NABEL GJ, et al. Cancer gene therapy using plasmid DNA: pharmacokinetic study of DNA following injection in mice. Hum Gene Ther. 1995;6:553–564. doi: 10.1089/hum.1995.6.5-553. [DOI] [PubMed] [Google Scholar]
- LI S, HUANG L. In vivo gene transfer via intravenous administration of cationic lipid–protamine–DNA (LPD) complexes. Gene Ther. 1997;4:891–900. doi: 10.1038/sj.gt.3300482. [DOI] [PubMed] [Google Scholar]
- LIU F, SONG Y, LIU D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- LUCAS ML, HELLER L, COPPOLA D, HELLER R. IL-12 plasmid delivery by in vivo electroporation for the successful treatment of established subcutaneous B16.F10 melanoma. Mol Ther. 2002;5:668–675. doi: 10.1006/mthe.2002.0601. [DOI] [PubMed] [Google Scholar]
- MANTHORPE M, CORNEFERT-JENSEN F, HARTIKKA J, FELGNER J, RUNDELL A, MARGALITH M, DWARKI V. Gene therapy by intramuscular injection of plasmid DNA: studies on firefly luciferase gene expression in mice. Hum Gene Ther. 1993;4:419–431. doi: 10.1089/hum.1993.4.4-419. [DOI] [PubMed] [Google Scholar]
- MARTIN JB, YOUNG JL, BENOIT JN, DEAN DA. Gene transfer to intact mesenteric arteries by electroporation. J Vasc Res. 2000;37:372–380. doi: 10.1159/000025753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASUO M, REARDON S, IKEBE M, KITAZAWA T. A novel mechanism for the Ca(2+)-sensitizing effect of protein kinase C on vascular smooth muscle: Inhibition of myosin light chain phosphatase. J Gen Physiol. 1994;104:265–286. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHIESEN I. Electropermeabilization of skeletal muscle enhances gene transfer in vivo. Gene Ther. 1999;6:508–514. doi: 10.1038/sj.gt.3300847. [DOI] [PubMed] [Google Scholar]
- MATSUMOTO T, KOMORI K, SHOJI T, KUMA S, KUME M, YAMAOKA T, MORI E, FURUYAMA T, YONEMITSU Y, SUGIMACHI K. Successful and optimized in vivo gene transfer to rabbit carotid artery mediated by electronic pulse. Gene Ther. 2001;8:1174–1179. doi: 10.1038/sj.gt.3301502. [DOI] [PubMed] [Google Scholar]
- MIR LM, BUREAU MF, GEHL J, RANGARA R, ROUY D, CAILLAUD JM, DELAERE P, BRANELLEC D, SCHWARTZ B, SCHERMAN D. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc Natl Acad Sci USA. 1999;96:4262–4267. doi: 10.1073/pnas.96.8.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUNAMAKER EA, ZHANG HY, SHIRASAWA Y, BENOIT JN, DEAN DA. Electroporation mediated delivery of catalytic oligodeoxynucleotides for manipulation of vascular gene expression. Am J Physiol. 2003;285:H2240–H2247. doi: 10.1152/ajpheart.00350.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSHIMA Y, SAKAMOTO T, YAMANAKA I, NISHI T, ISHIBASHI T, INOMATA H. Targeted gene transfer to corneal endothelium in vivo by electric pulse. Gene Ther. 1998;5:1347–1354. doi: 10.1038/sj.gt.3300725. [DOI] [PubMed] [Google Scholar]
- PUC M, KOTNIK T, MIR LM, MIKLAVCIC D. Quantitative model of small molecules uptake after in vitro cell electropermeabilization. Bioelectrochemistry. 2003;60:1–10. doi: 10.1016/s1567-5394(03)00021-5. [DOI] [PubMed] [Google Scholar]
- PUCIHAR G, MIR LM, MIKLAVCIC D. The effect of pulse repetition frequency on the uptake into electropermeabilized cells in vitro with possible applications in electrochemotherapy. Bioelectrochemistry. 2002;57:167–172. doi: 10.1016/s1567-5394(02)00116-0. [DOI] [PubMed] [Google Scholar]
- SATKAUSKAS S, BUREAU MF, PUC M, MAHFOUDI A, SCHERMAN D, MIKLAVCIC D, MIR LM. Mechanisms of in vivo DNA electrotransfer: Respective contributions of cell electropermeabilization and DNA electrophoresis. Mol Ther. 2002;5:133–140. doi: 10.1006/mthe.2002.0526. [DOI] [PubMed] [Google Scholar]
- SHIRASAWA Y, RUTLAND TJ, YOUNG JL, DEAN DA, JOSEPH BN. Modulation of protein kinase C (PKC)-mediated contraction and the possible role of PKcepsilon in rat mesenteric arteries. Front Biosci. 2003;8:133–138. doi: 10.2741/1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOMIARI S, GLASSPOOL-MALONE J, DRABICK JJ, GILBERT RA, HELLER R, JAROSZESKI MJ, MALONE RW. Theory and in vivo application of electroporative gene delivery. Mol Ther. 2000;2:178–187. doi: 10.1006/mthe.2000.0124. [DOI] [PubMed] [Google Scholar]
- SUZUKI T, SHIN BC, FUJIKURA K, MATSUZAKI T, TAKATA K. Direct gene transfer into rat liver cells by in vivo electroporation. FEBS Lett. 1998;425:436–440. doi: 10.1016/s0014-5793(98)00284-1. [DOI] [PubMed] [Google Scholar]
- TAN Y, HUANG L. Overcoming the inflammatory toxicity of cationic gene vectors. J Drug Target. 2002;10:153–160. doi: 10.1080/10611860290016757. [DOI] [PubMed] [Google Scholar]
- TSUJIE M, ISAKA Y, NAKAMURA H, IMAI E, HORI M. Electroporation-mediated gene transfer that targets glomeruli. J Am Soc Nephrol. 2001;12:949–954. doi: 10.1681/ASN.V125949. [DOI] [PubMed] [Google Scholar]
- UYECHI LS, GAGNE L, THURSTON G, SZOKA FC., Jr Mechanism of lipoplex gene delivery in mouse lung: Binding and internalization of fluorescent lipid and DNA components. Gene Ther. 2001;8:828–836. doi: 10.1038/sj.gt.3301461. [DOI] [PubMed] [Google Scholar]
- VACIK J, DEAN BS, ZIMMER WE, DEAN DA. Cell-specific nuclear import of plasmid DNA. Gene Ther. 1999;6:1006–1014. doi: 10.1038/sj.gt.3300924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEISS D. Delivery of gene transfer vectors to lung: Obstacles and the role of adjunct techniques for airway administration. Mol Ther. 2002;6:148–152. doi: 10.1006/mthe.2002.0662. [DOI] [PubMed] [Google Scholar]
- WEST J, RODMAN DM. Gene therapy for pulmonary diseases. Chest. 2001;119:613–617. doi: 10.1378/chest.119.2.613. [DOI] [PubMed] [Google Scholar]
- WOLFF JA, LUDTKE JJ, ACSADI G, WILLIAMS P, JANI A. Long-term persistence of plasmid DNA and foreign gene expression in mouse muscle. Hum Mol Genet. 1992;1:363–369. doi: 10.1093/hmg/1.6.363. [DOI] [PubMed] [Google Scholar]
- YOUNG JL, DEAN DA. Non-viral gene transfer strategies for the vasculature. Microcirculat Res. 2002;9:35–50. doi: 10.1038/sj/mn/7800120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOUNG JL, BENOIT JN, DEAN DA. Effect of a DNA nuclear targeting sequence on gene transfer and expression of plasmids in the intact vasculature. Gene Ther. 2003;10:1465–1470. doi: 10.1038/sj.gt.3302021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG G, GURTU V, KAIN SR. An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem Biophys Res Commun. 1996;227:707–711. doi: 10.1006/bbrc.1996.1573. [DOI] [PubMed] [Google Scholar]