

Abstract
We report an optimized method to purify and reconstitute histone octamer, which utilizes high expression of histones in inclusion bodies but eliminates the time consuming steps of individual histone purification. In the newly modified protocol, Xenopus laevis H2A, H2B, H3, and H4 are expressed individually into inclusion bodies of bacteria, which are subsequently mixed together and denatured in 8 M guanidine hydrochloride. Histones are refolded and reconstituted into soluble octamer by dialysis against 2 M NaCl, and metal-affinity purified through an N-terminal polyhistidine-tag added on the H2A. After cleavage of the polyhistidine-tag, histone octamer is further purified by size exclusion chromatography. We show that the nucleosomes reconstituted from the purified histone octamer above are fully functional. They serve as effective substrates for the histone methyltransferases DOT1L and MLL1. Small angle X-ray scattering further confirm that reconstituted nucleosomes have correct structural integration of histone octamer and DNA as observed in the X-ray crystal structure. Our new protocol enables rapid reconstitution of histone octamer with an optimal yield. We expect this simplified approach to facilitate research using recombinant nucleosomes in vitro.
Keywords: Histone octamer, Nucleosome, Refolding, Histone methyltransferase, Small angle X-ray scattering
Introduction
Regulation of gene expression is essential for the highly coordinated spatial and temporal patterns of gene expression, critical for cell fate determination and development. There are several distinct mechanisms that govern gene expression at transcriptional, post-transcriptional and post-translational steps. Among them, epigenetic regulation is extensively studied and includes modifications on DNA and histones. Abnormal regulation of epigenetic events leads to numerous human diseases including neurodegeneration, metabolic disorders and cancers [1]. Identification of small molecule inhibitors targeting enzymes responsible for epigenetic changes is a highly sought after therapeutics for the treatment of these diseases.
While synthesized histone tail peptides can be used as a substrate for in vitro assays of some histone modifying enzymes, recombinant nucleosomes are often desirable because certain histone modifying enzymes are enzymatically inactive against peptide substrates and nucleosomes provide a more physiologically relevant substrate [2]. Reconstitution of nucleosomes require separate purifications of histone octamer and DNA template, and final reconstitution of purified histones and DNA [3]. This standard protocol, however, has required multi-step column purifications under denatured conditions. An improved purification method was published recently [4]. However, the labor intensive and time-consuming processes of purifying each histone individually and subsequent reconstitution of octamer is still required in this revised protocol. Alternatively, soluble histone octamers or H2A/H2B dimer and H3/H4 tetramer have been purified from bacteria by coexpression [5,6]. However, overall levels of histone co-expression were low and large portions of histones were still found in inclusion bodies. In this report, we present a novel approach for nucleosome reconstitution that takes advantage of high overexpression levels of histones in inclusion bodies and bypasses all purification steps under denaturation conditions by refolding histone core proteins into octamer in one-pot followed by a simplified purification using metal-affinity chromatography and size exclusion chromatography.
Materials and methods
Expression plasmids
Xenopus laevis H2A was subcloned into the Ligation Independent Cloning vector pMCSG7 [7], which encodes a hexahistidine tag (His6)1 and a TEV recognition sequence followed by SspI restriction enzyme site. PCR amplified H2A was annealed into the vector at the SspI site. After cleavage at the TEV recognition site, additional amino acids Ser-Asn-Ala remain at the N-terminus of the H2A sequence. Intact Xenopus laevis H2B and H3 and H4 were subcloned into the pET3 vector.
Expression and purification of histones
Colonies from a freshly transformed BL21(DE3) E. coli strain were inoculated into starter cultures of LB media with supplement of 0.4% glucose and cultured overnight at 37 °C Cells were 100-fold diluted into fresh LB media and cultured at 37 °C. When OD600 reached 0.6, IPTG was added at a final concentration of 0.4 mM and cells were grown for an additional 3 h for His6-H2A, H2B and H3 and 2 h for H4. Cells were harvested and resuspended in 30 mL/L of culture of 50 mM Tris (pH 7.5) and 100 mM NaCl then stored at −80 °C until purification.
As the expression levels were variable for each histone (Fig. 1), we used different amount of cells for each histone to achieve similar stoichiometry for one-pot refolding. For example, in this report we processed cells from 1 L His6-H2A, 4 L H2B, 2 L H3 and 2 L H4 cultures. After thawing from storage, cells were combined into a mixture and were lysed by sonication for 10min per 30 mL of mixed resuspended cells (output power 9, duty cycle 20%, Branson Sonifier Model 250) and supernatant was removed by centrifugation at 32,000g (F18-12×50 rotor, Fiberlite) at 4 °C. Inclusion bodies were dissolved in 10 mL of 8 M guanidine hydrochloride, 20 mM acetate (pH 5.2) and 10 mM DTT per cells from each liter and incubated for 1 h at room temperature. Then undissolved material was separated by centrifugation at 32,000g for 20 min at 25 °C. Supernatant was transferred into the dialysis membrane of MWCO 8000 (BioDesignDialysis) and dialyzed against the 4 L buffer containing 20 mM Tris (pH 8.0), 2 M NaCl and 2 mM β-mercaptoethanol in 4 L at 4 °C three times for at least 8 h. Precipitates formed heavily during the octamer refolding and were removed by centrifugation at 15,000g for 10 min at 4 °C. Supernatant containing refolded histone octamer was loaded on a 5 mL Ni-NTA resin (Qiagen). After extensive washing with buffer containing 20 mM Tris (pH 8.0), 2 M NaCl, 10 mM imidazole and 2 mM β-mercaptoethanol, histone octamer was eluted stepwise at 30, 60, 90, 120, 150, 210 and 300 mM imidazole (5 mL each). TEV protease (Jennifer Meagher, Center for Structural Biology, University of Michigan) was added to the pooled fractions containing histone octamer and cleavage reaction was carried out at 4 °C for overnight. The TEV protease-cleaved sample was concentrated and loaded on a HiLoad 16/600 Superdex 200 PG column (GE Healthcare). The buffer for the size exclusion chromatography contained 20 mM Tris (pH 7.5), 2 M NaCl and 1 mM DTT. Fractions were analyzed by SDS-PAGE and those containing only histone octamer were pooled and concentrated to >2 mg/mL using a MWCO 30 kDa centrifugal concentrator (EMD Millipore). The sample was dialyzed against 500 mL of 20 mM Tris (pH 7.5), 2 M NaCl, 1 mM DTT and 1 mM EDTA in 50% glycerol (v/v) for overnight at 4 °C and stored at −20 °C.
Fig. 1.

Expression levels of histones in the host bacteria BL21(DE3) are variable. Histones were expressed using the starter cultures grown overnight at 37 °C with a supplement of 0.4% glucose (lanes 1) or its omission (lanes 2). Lanes 3; cells were not saturated before protein induction. M; size markers (kDa).
Purification of 147 bp Widom 601 DNA
Plasmid DNA containing 12 copies of strong positioning Widom 601 DNA sequence with EcoRV sites between repeats [8] was transformed into XL1-Blue bacterial cell. Single colonies were inoculated into 0.5 L of LB media in a 2.8 L flask. A total of 2.5 liters were cultured for plasmid preparation using Qiagen Plasmid Giga Kit. The yield from the kit was about 10 mg of the DNA plasmid. The Widom 601 fragment was excised by digestion with 200 units of EcoRV per 1 mg of DNA plasmid at 37 °C for 16 h. The 601 fragment was purified by PEG fractionation as described in Dyer et al. [3].
Reconstitution of mononucleosome
Histone octamer stock in 50% glycerol was dialyzed against 20 mM Tris (pH 7.5), 2 M NaCl, 1 mM EDTA and 1 mM DTT for overnight at 4 °C. Octamer concentration was measured based on UV absorption at 280 nm with extinction coefficient of 39,020 cm−1 M−1 or 2.78 mg/mL for OD280 = 1. Next, DNA and histone octamer were combined in a 1:1 molar ratio for reconstitution with the concentration of 0.70 mg/mL of the 601 DNA and 0.85 mg/mL of histone octamer. Nucleosome reconstitution was carried out by salt dialysis either by stepwise gradient for small scale or linear gradient for a large scale as described by Dyer et al. [3]. Briefly, for salt dialysis by stepwise gradient, samples were dialyzed against 500 mL of 20 mM Tris (pH 7.5), 1 mM DTT, 1 mM EDTA with 2, 0.85, 0.65, 0.2 and 0.05 M NaCl for at least 1.5 h each at 4 °C. For long-term storage, reconstituted nucleosomes were further dialyzed against to 20 mM cacodylate (pH 6) and 1 mM EDTA and stored at 4 °C as recommended [3].
Histone methyltransferase (HMTase) assay
GST-DOT1L catalytic domain (amino acids 1-420), with N-terminal His6 tag, was recombinantly expressed in E. coli and purified by nickel affinity chromatography and subsequent size exclusion chromatography using a Superdex-75 column with 20 mM Tris (pH 8.0), 200 mM NaCl, 1 mM DTT, 1 mM EDTA buffer. Human MLL1 SET domain, WDR5, RbBP5, and ASH2L were cloned on a pET28a-based vector encoding polyhistidine-SUMO tag, and purified as previously described [9]. HMT assays for human MLL1 were carried out as described previously [10]. The HMTase reaction for DOT1L was carried out by incubation of 125 nM GST-DOT1L with 0.7 μg of either recombinant nucleosomes or HeLa extracted nucleosomes (52015; BPS Bioscience) and 125 nM (0.28 μCi) 3H-S-adenosyl methionine (NET155250UC; Perkin Elmer). S-adenosyl homocysteine (SAH) was added to the reaction at varying concentrations before the addition of 3H-S-adenosyl methionine to inhibit DOT1 HMTase activity. The reaction was carried out in 20 mM Tris (pH 7.9), 4 mM EDTA, 1 mM DTT, 0.01% Triton X-100 at a final volume of 26.5 μL. After 1 h of reaction, 5 μL of reaction mixture was transferred to P81 filter paper. Filter papers were dried and washed three times with 50 mM NaHCO3 (pH 9.0). Filter papers were dried again and radioactivity was measured in vials with 10 mL liquid scintillation fluid on a Tri-Carb 2800 TR liquid scintillation counter (Perkin Elmer).
Small angle X-ray scattering
SAXS data for recombinant nucleosome was collected at the 18-ID BioCAT Beamline (Biophysics Collaborative Access Team, Advanced Photon Source, Argonne National Laboratory). A volume of 200 μL recombinant nucleosome (1.6 mg/mL) was loaded on the inline Superdex 200 increase column (10 × 300 mm, GE Healthcare), which was pre-equilibrated with 20 mM Tris (pH 7.5), 200 mM NaCl and 1 mM DTT. Flow rate was set to 0.7 mL/min during the data collection. The scattering data was collected every 2 s with 1 s exposure between 5 - 24 mL of elution from the column. After data reduction, the strongest scattering data around the sample peak were selected. Several data points with low and flat scattering along the elution near the sample peak were chosen for obtaining buffer scattering. PRIMUS [11] was used for averaging scattering data, background subtraction and calculation of the radius of gyration, Rg. The pair distribution function was calculated by GNOM [12] in the GUI version of PRIMUS. Ab initio models were calculated by DAMMIF [13], and clustered and averaged by DAMCLUST and DAMAVER [14]. SUPCOMB [15] was used to superimpose the averaged model onto the X-ray crystal structure of nucleosome (PDB code 1KX5) [16]. A theoretical SAXS curve for the X-ray crystal structure was calculated using the FOXS server [17] and compared with the experimental SAXS data.
Results
In order to optimize a procedure for one-pot refolding of histone octamer, we first determined expression levels of each histone in the host bacterial strain of BL21(DE3). Three different conditions were tested to find optimal expression levels. Glucose was either added or omitted in the starter cultures at a final concentration of 0.4%. Presence of glucose in growth media represses leaky expressions of target proteins that may be toxic to bacteria [18]. We also tested whether saturation of cells in starter cultures causes adverse effects on protein expression. For all histones, expression levels were maximal either when glucose was supplemented in the growth media or the cells were not saturated before protein expressions were induced (Fig. 1). Expression levels were more than 50% lower with starter cultures grown overnight without glucose. For this reason, we chose to use starter cultures with a supplement of 0.4% glucose.
Each histone provided variable levels of expression (Fig. 1). Histone His6-H2A overexpression level was the highest and the H2B was the lowest. Histone H3 and H4 expression was intermediate. In order to achieve roughly the same stoichiometry of histones, we used 1:4:2:2 (H2A:H2B:H3:H4) ratio of culture volume as described in the methods. Cells from each histone cultures were combined all together for cell disruption. All histones were found in inclusion bodies (Fig. 2A) after cell disruption as expected. However, refolding after denaturation in 8 M guanidine hydrochloride and dialysis against 2 M NaCl resulted in mostly soluble histones (Fig. 2A), suggesting the refolding was highly efficient. Refolded histones were then purified on a Ni-NTA column (Fig. 2B). Coelution of all histones in the same fractions indicates intact H2B, H3 and H4 are bound to the metal-affinity column through integration into a properly folded soluble octamer with His6-H2A. The polyhistidine tag was removed from H2A by TEV protease. The amount of TEV protease was optimized before large-scale cleavage reaction (Fig. 3). Complete cleavage of polyhistidine-tag from H2A was confirmed (Fig. 2C) before further purification by size exclusion chromatography. Two major elution peaks corresponding to histone octamer and H2A/H2B dimer were found during the size exclusion chromatography (Fig. 2D). Fractions containing histone octamer were pooled and dialyzed against the buffer containing 50% glycerol (v/v) for long-term storage. Overall purity was high (Fig. 2D) and the yield of histone octamer was 40 mgs from cultures of 1 L His6-H2A, 4 L H2B, 2 L H3 and 2 L H4.
Fig. 2.
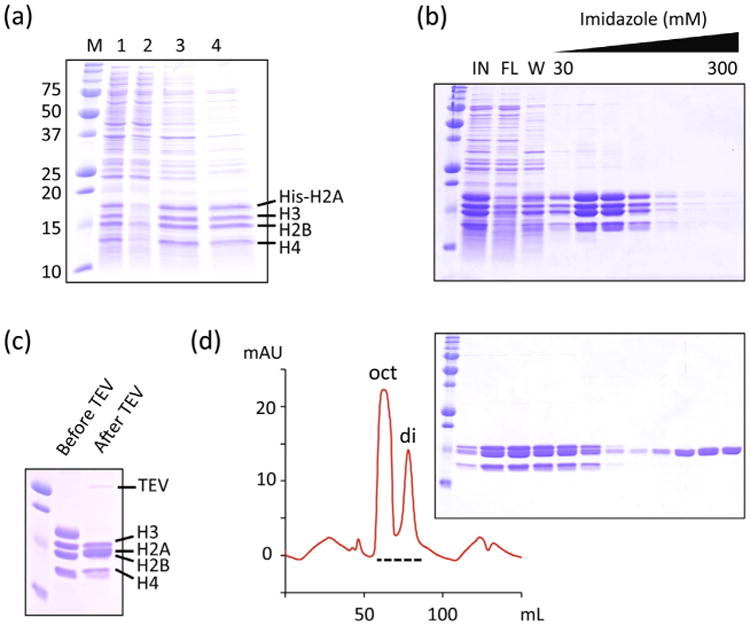
Histone octamer can be rapidly reconstituted by one-pot refolding. (a) Histones are insoluble after bacterial overexpression (lane 1; total cell lysate, lane 2; soluble fraction), but solubilized after one-pot refolding (lane 3; total fractions, lane 4; soluble fraction). M; size markers (kDa). (b) Metal-affinity purification of histones after one-pot refolding. In; input refolded histones, FL; flowthrough, W; wash. (c) Polyhistidine-tag was cleaved from H2A by TEV protease. (d) Final purification step on a Superdex 200 PG column. The fractions on the dashed line were analyzed by SDS-PAGE.
Fig. 3.
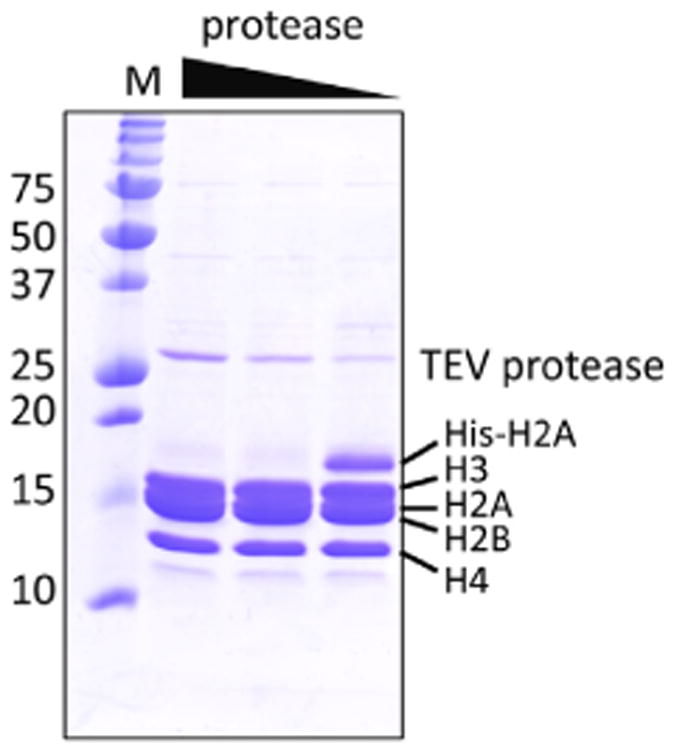
Optimization of TEV protease cleavage reaction. Two-fold serially diluted TEV protease was added to the histone octamer purified from the metal-affinity chromatography. Cleavage reaction was carried out at 4 °C for overnight. M; size markers (kDa).
Mononucleosomes were reconstituted by salt gradient dialysis and migrated as a single band on a native PAGE (Fig. 4A), confirming high purity of histone octamer. Reconstituted nucleosomes were further tested as a substrate for histone methyltransferases using an in vitro assay to determine if they are a suitable enzymatic assay substrate. Using histone methyltransferase DOT1L, the recombinant nucleosomes prepared using the methods described in this report demonstrated good activity as a methyltransferase substrate and were comparable to nucleosomes extracted from HeLa cells. Next, we tested whether DOT1L inhibition by the generic methyltransferase inhibitor, S-adenosine homocysteine (SAH), was maintained with recombinant nucleosomes. Indeed, the inhibition of DOT1L with SAH was unaffected by the source of substrate nucleosomes and was the same for the reconstituted and extracted nucleosomes from HeLa cells, IC50 = 375 ± 120 and 433 ± 116 nM, respectively (Fig. 4B). In addition to DOT1L, MLL1 methyltransferase activity was also comparable in two parallel reactions using HeLa-cell extracted nucleosome and the reconstituted nucleosomes (Fig. 4C). These data show that the reconstituted nucleosomes prepared by the current method are comparable to HeLa nucleosomes as enzyme substrates for histone methyltransferases.
Fig. 4.
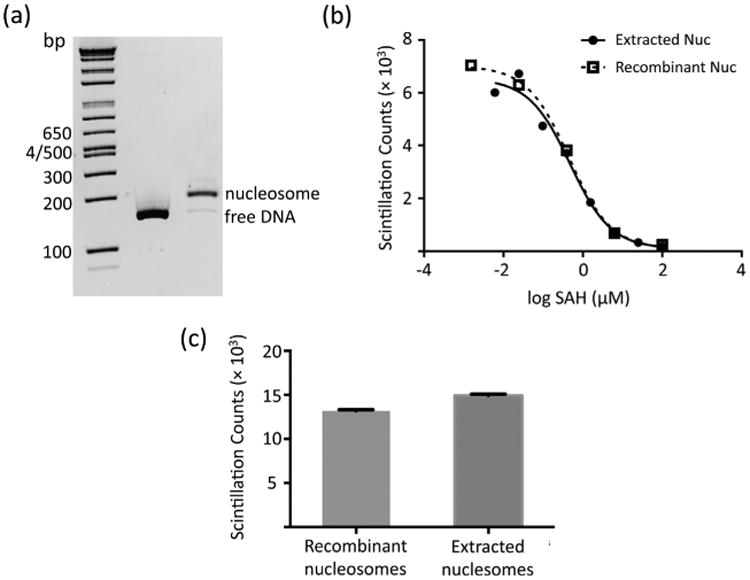
Reconstituted nucleosome from the purified histone octamer serves as a valid substrate to histone methyltransferases. (a) 4-12% native PAGE of the reconstituted nucleosome. (b) DOT1L HMTase assay demonstrating inhibition of DOT1L by SAH with recombinant and extracted nucleosomes. (c) Human MLL1 HMT activity assay using reconstituted nucleosomes exhibits similar methyltransferase activity to HeLa cell extracted nucleosomes. Each data was performed in duplicate.
We also collected small angle X-ray data to further confirm that we have obtained recombinant nucleosomes that are properly reconstituted. The overall scattering profile and the pair distance distribution function of the reconstituted nucleosome in this study was similar to the data from the traditionally reconstituted nucleosome (Fig. 5A, B) [19]. Dmax in the pair distance distribution function was around 120 Å, close to the diameter of the X-ray crystal structure of nucleosome [16], which was well fit to the envelope of the reconstructed ab initio model of nucleosome (Fig. 5C). These results show that the histone octamer and DNA prepared in this study were correctly integrated into nucleosome.
Fig. 5.
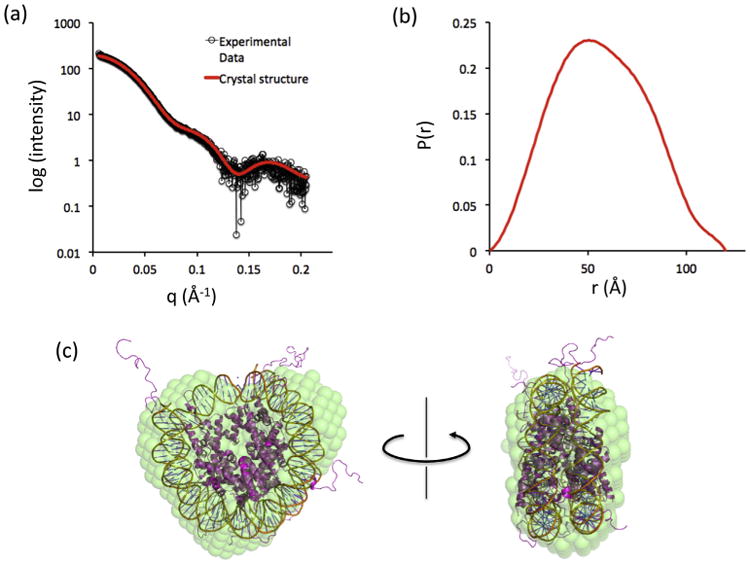
SAXS measurements demonstrate correct integration of the histone octamer and the 147 bp Widom DNA into the nucleosomes. (a) Superimposed SAXS profiles of experimental scattering data and theoretical scattering data from the crystal structure of nucleosome (χ = 1.4, Rg = 403). (b) The pair distance distribution function of the experimental scattering data. (c) Superimposed structures of the X-ray crystal structure and the averaged ab initio model of nucleosome.
Discussion
In this report, we described an optimized method for rapid reconstitution of histone octamer. Our strategy is unique in that all histones were combined together before denaturation from inclusion bodies and refolded into soluble histone octamer. This one-pot refolding of all histones allowed us to bypass the lengthy purification of individual histones under denaturing conditions that are the most time-consuming steps. This method greatly reduces the amount of column purification of individual histones, while providing histone octamer with high purity.
In order to employ nickel-affinity chromatography, we added a polyhistidine-tag and a TEV recognition sequence at the N-terminus of H2A. Shim et al. previously employed nickel-affinity chromatography to purify soluble histone octamer coexpressed polycistronically in bacteria [6]. In this case, polyhistidine-tags were added both on H2A and H4. After cleaving off the tags, residual residues remaining on histones did not interfere reconstitution into nucleosome [6]. Therefore, we employed a similar purification strategy, however we added a polyhistidine-tag only to H2A. A single polyhistidine-tag on H2A facilitates the subsequent purification that removes excess histone tetramer of H3/H4. As histone tetramer elutes very close to octamer on a size exclusion column [3], removal of any extra tetramer beforehand permits complete separation of histone octamer from smaller units.
We routinely reconstituted nucleosomes up to 300 μg by stepwise salt dialysis or by linear salt gradient dialysis for a larger scale reconstitution. The quality of nucleosome was virtually the same by both methods when evaluated by native PAGE analysis. Depending on the scale and individual researcher's needs, reconstitution can be carried out by either method. On a larger scale employing a linear gradient dialysis, 5 mg of nucleosomes was successfully reconstituted for structural and biophysical studies. SAXS measurements indeed demonstrated that the physical integrity of the reconstituted nucleosome was compatible to the previously determined X-ray crystal structure of nucleosome. All of these results show that the histone octamer purified in this study were correctly integrated into nucleosome, which should be suitable for both biochemical and biophysical experiments.
In conclusion, we described a new method for preparation of histone octamer by one-pot refolding of histones. Our approach ensures fast and robust reconstitution of histone octamer and could be easily adopted by laboratories that need nucleosomes for studying histone-modifying enzymes but have limited experiences and resources in protein purifications. Furthermore, the overall procedure is highly efficient and purification of octamers with histone variants or mutants can be easily pursued in parallel.
Highlights.
We report a rapid method to reconstitute histone octamer by one-pot refolding of all histones from inclusion bodies.
The optimized protocol eliminates time-consuming steps of individual histone purification but ensures high purity of reconstituted octamer.
Nucleosomes reconstituted from the purified octamer serve as valid substrates for the histone methyltransferases, DOT1L and MLL1.
Reconstituted nucleosomes show correct structural integration of histone octamer and DNA when evaluated by small angle X-ray scattering.
This simplified protocol is expected to facilitate research on histone modifying enzymes.
Acknowledgments
We thank Dr. Weifeng Sheng and Dr. Srinivas Chakravarthy at the BioCAT for help with SAXS data collection.
Footnotes
Abbreviations used: His6, hexahistidine tag; HMTase, histone methyltransferase; SAH, S-adenosyl homocysteine.
Contributor Information
Young-Tae Lee, Email: lyoungta@med.umich.edu.
Yali Dou, Email: yalid@med.umich.edu.
References
- 1.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 2.Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 3.Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 4.Klinker H, Haas C, Harrer N, Becker PB, Mueller-Planitz F. Rapid purification of recombinant histones. PLoS One. 2014;9:e104029. doi: 10.1371/journal.pone.0104029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson M, Huh JH, Ngo T, Lee A, Hernandez G, Pang J, Perkins J, Dutnall RN. Co-expression as a convenient method for the production and purification of core histones in bacteria. Protein Expr Purif. 2010;72:194–204. doi: 10.1016/j.pep.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Shim Y, Duan MR, Chen X, Smerdon MJ, Min JH. Polycistronic coexpression and nondenaturing purification of histone octamers. Anal Biochem. 2012;427:190–192. doi: 10.1016/j.ab.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown WC, DelProposto J, Rubin JR, Lamiman K, Carless J, Smith JL. New ligation-independent cloning vectors compatible with a high-throughput platform for parallel construct expression evaluation using baculovirus-infected insect cells. Protein Expr Purif. 2011;77:34–45. doi: 10.1016/j.pep.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 9.Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, Hess JL, Atadja P, Lei M, Qin ZS, Malek S, Wang S, Dou Y. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53:247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 11.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 12.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 13.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Crystallogr. 2001;34:33–41. [Google Scholar]
- 16.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 17.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–W544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grossman TH, Kawasaki ES, Punreddy SR, Osburne MS. Spontaneous cAMP-dependent derepression of gene expression in stationary phase plays a role in recombinant expression instability. Gene. 1998;209:95–103. doi: 10.1016/s0378-1119(98)00020-1. [DOI] [PubMed] [Google Scholar]
- 19.Yang C, van der Woerd MJ, Muthurajan UM, Hansen JC, Luger K. Biophysical analysis and small-angle X-ray scattering-derived structures of MeCP2-nucleosome complexes. Nucleic Acids Res. 2011;39:4122–4135. doi: 10.1093/nar/gkr005. [DOI] [PMC free article] [PubMed] [Google Scholar]