

Death receptor 6 (DR6) was recently shown to bind amyloid precursor protein (APP) via the protein extracellular regions, stimulate axonal pruning, and inhibit synapse formation. Xu et al. report the crystal structure of the DR6 ectodomain in complex with the E2 domain of APP and show that it supports a model for APP-induced dimerization and activation of cell surface DR6.
Keywords: amyloid precursor protein, crystal structure, death receptor 6, dimerization, signal activation
Abstract
The amyloid precursor protein (APP) has garnered considerable attention due to its genetic links to Alzheimer's disease. Death receptor 6 (DR6) was recently shown to bind APP via the protein extracellular regions, stimulate axonal pruning, and inhibit synapse formation. Here, we report the crystal structure of the DR6 ectodomain in complex with the E2 domain of APP and show that it supports a model for APP-induced dimerization and activation of cell surface DR6.
The amyloid precursor protein (APP) is a single-pass transmembrane protein best known for its genetic association with Alzheimer's disease (AD). APP serves as the proteolytic precursor to Aβ, the major peptide component of amyloid plaques in the brains of Alzheimer's patients (Selkoe 2011). Therapeutic strategies to modify APP processing or clear Aβ from plaques in Alzheimer's patients have not yet proven effective (Gandy and DeKosky 2013), encouraging the search for additional approaches for treating AD. This has stimulated interest in understanding the normal physiological role of APP in the developing and adult nervous system in the hope that this will provide new insights into how perturbed APP function may contribute to disease onset and/or progression. These studies have implicated APP in several physiological processes, including axon pruning, synapse formation, axonal transport, and cellular adhesion (Kamal et al. 2001; Priller et al. 2006; Bittner et al. 2009; Nikolaev et al. 2009; Wang et al. 2009). The extracellular domain of APP is comprised of an N-terminal domain termed E1 and a more C-terminal domain termed E2 that are linked via an acidic unstructured sequence and followed by a transmembrane helix and a cytoplasmic region. Previous studies have elucidated the structures of the unbound APP extracellular domains (Wang and Ha 2004; Dahms et al. 2010, 2012; Hoopes et al. 2010; Lee et al. 2011; Xue et al. 2011a,b; Coburger et al. 2013, 2014). High-affinity binding of APP to death receptor 6 (DR6, also known as TNFRSF21), a death domain-containing member of the extended TNF receptor superfamily (Pan et al. 1998), was recently documented (Nikolaev et al. 2009) and shown to be mediated by the E2 domain of APP (Olsen et al. 2014). DR6 has four cysteine-rich domain (CRD) modules in its extracellular region, followed by a transmembrane domain and death domain in its cytoplasmic region. The structure of the unbound DR6 ectodomain (ECD) (Kuester et al. 2011; Ru et al. 2012) revealed an elongated architecture with a kink between CRD modules 2 and 3. Genetic analysis of DR6 and APP mutant mice showed that the animals share several coincident phenotypes, including behavioral deficits, altered synapse formation, and delayed axon pruning during development or following sensory deprivation (Kallop et al. 2014; Olsen et al. 2014). These and other data (Kallop et al. 2014; Olsen et al. 2014) indicate that APP and DR6 function cell-autonomously and in the same pathway to stimulate axon pruning and synapse elimination. However, how APP and DR6 interact to mediate their actions is poorly understood.
Results and Discussion
Structure of the APP/DR6 complex
To advance our understanding of how APP and DR6 function together to initiate downstream signaling controlling axon pruning and synapse elimination, we determined the crystal structure of the APP/DR6 complex. The complex between the APP E2 domain and the ECD of DR6 was crystallized, and its structure was determined at 2.2 Å resolution, as described in the Materials and Methods. Consistent with previous studies, DR6 contains four CRD modules composed mostly of strands and loops of various lengths, with either two or three pairs of disulfide bonds stabilizing each CRD module. The DR6 ECD adopts an elongated shape with a kink between CRD modules 2 and 3. N-linked glycosylation was predicted for residues N467 and N496 of APP and residues N82 and N141 of DR6. However, clear electron density was observed and modeled only for the carbohydrate moieties attached to N467 of APP and N82 of DR6 (Fig. 1A, gray spheres). A magnesium ion stabilizes the third CRD of DR6 and is coordinated by six oxygen-containing ligands, including the side chain of S177; the main chain carbonyls of C150, P151, W154, and V179; and a water molecule (Fig. 1A, inset).
Figure 1.

Crystal structure of the DR6/APP-E2 complex. (A) Two 90°-rotated views of the complex. The E2 domain of APP is colored in green, while DR6 is colored in magenta. Disulfide bonds are colored in yellow, and the carbohydrate glycosylation moieties are colored in gray. The N and C termini of the molecules are labeled. The bound magnesium ion is shown as a blue sphere, and a schematic representation of its coordination by DR6 residues and a water molecule is shown in the middle. (B) Structure of the APP-E2/DR6 complex with zoom-ins of the two interface regions (insets). The E2 domain of APP is colored in green, while DR6 is colored in magenta. Key interface residues are labeled.
APP/DR6 interaction interface
The DR6/APP interface is relatively small, burying ∼680 Å2 in each molecule (Fig. 1B). The majority of the interactions are between helices H1 and H2 of APP-E2 and the first CRD module of DR6, with the angle between these two interacting regions being ∼30°. The interface is arranged as a sandwich of hydrophilic and hydrophobic interactions. The top hydrophobic portion is defined by the insertion of L85DR6 into a pocket formed by K350, K353, and K354 of APP; the middle hydrophilic portion is defined by two salt bridges formed between E342APP and R86DR6 and between K354APP and D68DR6; and the bottom hydrophobic portion is centered on a patch formed by residues M335APP, W338APP, P71DR6, and A72DR6. The interface is further strengthened by additional van der Waals contacts and a few hydrogen bonds, including between Q361APP and E100DR6 as well as between Q361APP and the main chain carbonyl of G52DR6. Further away from this major interface, there is a second, minor DR6/APP interface, specifically a salt bridge formed between E310APP, located at the N terminus of E2 helix H1, and R157DR6, located at the third CRD module of DR6. A list of all APP/DR6 contacts is shown in Supplemental Figure S2. It should be noted that all contact residues in both APP (Supplemental Fig. S3A) and DR6 (Supplemental Fig. S3B) are highly conserved across species.
Conformational rearrangements in APP and DR6 upon complex formation
Slight differences are found in the orientation of the E2 helices between the bound and unbound structures of this domain (Supplemental Fig. S4A). These are facilitated by the flexibility of the helix–turn–helix assembly, especially in the “turn” as well as the unusually long H1 and H2 helices. Specifically, APP-E2 in the DR6 complex can be superimposed with the unbound APP-E2 (3NYL), with an RMSD of 1.7 Å over 161 Cα atoms, and with the metal-bound APP-E2 (3UMH), with an RMSD of 2.5 Å over 175 Cα atoms.
In DR6, the linker between CRD modules 2 and 3 is flexible, as previously predicted (Kuester et al. 2011). This flexibility allows for a conformational change upon APP binding, resulting in the movement of the third and fourth DR6 CRD modules closer to APP (∼20° shift). This enables it to interact more intimately with APP via both the major interface involving CRD module 1 and the minor interface involving CRD module 3 (Fig. 1B; Supplemental Fig. S4B).
Structure-based mutagenesis confirms the functional significance of the complex
To confirm the functional significance of the observed DR6/APP interface, we mutated key residues in APP and evaluated their effects on binding to DR6. Specifically, we selected four APP residues that are located at the main APP/DR6 interface: M335 and W338, which form the hydrophobic interface patch discussed above, were mutated to aspartic acid and alanine, respectively; E342, which is part of the middle hydrophilic portion of the interface, was mutated to alanine to eliminate the salt bridge that this residue forms with R86DR6; and R328, which is located on the edge of the main interface and forms a hydrogen bond with R98 of DR6, was mutated to alanine. The wild-type and mutant APP proteins were expressed as either secreted N-terminal alkaline phosphatase (AP) fusion constructs of the E2 domain-containing region of the APP ECD (AP-APP-E2) Fig. 2, legend; Materials and Methods) or full-length proteins on the surface of COS-7 cells; DR6 was expressed as a secreted C-terminal AP fusion construct of its ECD (DR6-AP) or a full-length protein in COS-7 cells. We first used a cell-based assay to assess binding of soluble AP-APP-E2 fusions (mutant and wild type) to cell surface DR6 and soluble DR6-AP fusion binding to full-length cell surface APP (mutant and wild type). As previously described (Olsen et al. 2014), wild-type AP-APP-E2 binds to DR6 on cells, and DR6-AP binds to APP on cells (Fig. 2A). We found that two of the mutations mentioned above (M335D/W338A and E342A) completely blocked the DR6/APP interaction in both directions (Fig. 2A), confirming the interface observed in the crystal structure. On the other hand, mutation of R328A, a residue located at the periphery of the interface, did not significantly decrease binding (Fig. 2A). We also made the E310A substitution, altering the much smaller secondary DR6/APP interface, but this substitution also did not significantly decrease the APP affinity to DR6 in this assay (data not shown).
Figure 2.

Mutations of amino acids at the APP/DR6 interaction interface disrupt binding, and a single amino acid substitution in APLP2 is sufficient to confer DR6 binding. (A) COS-7 cells were transfected with constructs that encode full-length DR6 or APP or APP mutants with single or double amino acid substitutions at the APP/DR6 interaction interface. Two days after transfection, cells were incubated with DR6-AP (amino acids 1–349), AP-APP-E2 (amino acids 287–506), or AP-APP-E2 mutants. (Top left) COS-7 cells expressing DR6 interacted with AP-APP-E2. (Bottom left) Similarly, COS-7 cells expressing APP specifically bound DR6-AP. An APP double mutation (M335D, W338A; these residues are adjacent and together form a single hydrophobic patch at the interface) or a single substitution (E342A) disrupted the interaction, whereas the R328A mutation did not affect DR6 binding. (B) Partial alignment of amino acid sequences of APP and APLP2. Shading denotes the amino acids in APP that contribute to the APP/DR6 interface. Shade color specifies the extent of conservation of analogous amino acids in APLP2: (green) conservative substitution; (orange) semiconservative; (red) nonconservative. COS-7 cells were transfected with constructs that encode full-length DR6, APLP2, or APP and APLP2 mutants. Two days after transfection, cells were incubated with DR6-AP (amino acids 1–349), AP-APLP2-E2 (amino acids 306–523), AP-APLP2-E2 mutants, or an APP point mutant (M335K). APLP2 did not bind DR6, but the single K353M substitution was sufficient to confer DR6 binding. The converse change in APP (M335K), which mimics the APLP2 sequence, disrupted APP binding to DR6. The Q372K mutation in APLP2 did not confer DR6 binding. (C) Binding affinities between DR6-AP and wild-type or mutated AP-APP-E2 and AP-APLP2-E2 measured using biolayer interferometry (Materials and Methods).
We next verified the results of the cell-based assays by assessing direct binding of purified soluble fusion constructs to one another using biolayer interferometry (see the Materials and Methods). As shown in Figure 2C, in this assay, the KD of the interaction between wild-type AP-APP-E2 (in solution) and a DR6-Fc fusion (immobilized on the sensor chip) was 85 nM; this is somewhat higher than the EC50 of ∼10 nM observed by ELISA for binding to a DR6-Fc fusion to APP-E2-His immobilized on plastic (Olsen et al. 2014), perhaps reflecting differences in which protein was adsorbed, the degree of clustering on the different adsorption surfaces, or protein tags. Importantly, binding of roughly similar affinity was observed by interferometry for the AP-APP-E2 construct carrying the R328A mutation, but binding was not detectable with constructs carrying the E324A or M335D/W338A mutations (Fig. 2C). These changes parallel the effects of the mutations on binding in the cell-based assay, thus cross-validating those results.
Switching the death receptor-binding specificities of APP and APLP2
APP has a close homolog called APLP2, but, in contrast to APP, it does not interact with DR6, as assessed by binding of DR6-AP to APLP2-expressing cells or binding of soluble AP-APLP2-E2 to DR6-expressing cells (Fig. 2B). Analysis of the APP/DR6 interface residues reveal that there are only two nonconserved substitutions in the APLP2 E2 domain that are likely to account for the difference in DR6 binding: M335 and K354 in APP are replaced by lysine and glutamine, respectively, in APLP2 (here we use APP residue numbering to label corresponding APLP2 residues to facilitate cross-comparisons). Therefore, we decided next to evaluate whether we could confer DR6 binding to APLP2 by making the substitutions K335M or Q354K in APLP2 and, conversely, abolish DR6 binding to APP by making the substitution M335K in APP. Remarkably, the single K335M mutation in APLP2 is sufficient to transform APLP2 into a DR6-binding protein, whereas the converse M335K mutation in APP is sufficient to abolish APP binding to DR6, as assessed by both binding of soluble APLP2/APP-E2 fusions to DR6 on cells and binding of soluble DR6-AP to APLP2/APP on cells (Fig. 2B). The single K335M mutation was similarly sufficient to confer binding of soluble AP-APLP2-E2 to DR6-Fc in solution, as assessed by biolayer interferometry (Fig. 2C). In contrast, the Q354K substitution, affecting a residue that, based on the structure, is not expected to be as key a player at the interface, did not significantly alter the DR6-binding properties of APLP2. Our ability to confer DR6 binding to APLP2 via a single point mutation that was predicted by the structure further validates our model for the binding interface.
Implication for death receptor activation and signaling
Death receptor activation and signaling initiation are thought to be effected through receptor oligomerization, with either dimerization or trimerization having been proposed as the activation mechanism for various family members (Idriss and Naismith 2000; Chan 2007). In our crystal structure, the APP-E2 binds DR6 with 1:1 stoichiometry. Size exclusion chromatography (SEC) experiments suggest that the while the isolated E1 and E2 domains of APP are monomeric in solution, the full APP ECD (residues 18–596) is dimeric (Supplemental Fig. S1A). This observation is consistent with previous reports (Wang and Ha 2004; Lee et al. 2011; Xue et al. 2011a,b; Hoefgen et al. 2014). Thus, the studies described here suggest that DR6 activation could be triggered by dimerization induced by APP binding. Interestingly, it was also suggested that the dimeric state of APP can be stabilized by heparin and, specifically, that heparin induces APP dimerization via interactions with either the E1 or E2 domain (Hoopes et al. 2010; Lee et al. 2011; Xue et al. 2011a,b; Coburger et al. 2013; Hoefgen et al. 2014). Based on the experiments reported here and the previously published data, we propose a simple model for DR6 activation by APP-induced dimerization that ultimately leads to axon pruning and synapse elimination (Fig. 3). Specifically, in our model, two DR6 molecules are brought together by binding to a dimeric APP molecule at the neuronal surface, which positions the C-terminal death domains of adjacent DR6 molecules in close proximity and triggers the activation of downstream signaling. Whether and how heparin or other molecules modulate the oligomeric state of APP in vivo are matters for further investigation, but it should be noted that (1) our data (Supplemental Fig. S1A) indicate that the APP ECD exists as a preformed dimer even in the absence of heparin, but (2), since both the E1 and E2 domains of APP are positively charged, a highly negatively charged agent, such as heparin, is likely to facilitate APP dimerization or oligomerization, as supported by our SEC analysis (Supplemental Fig. S1B). Our “activation via dimerization” model is further supported by the observation that a bivalent monoclonal antibody can induce DR6 activation (Hu et al. 2014).
Figure 3.
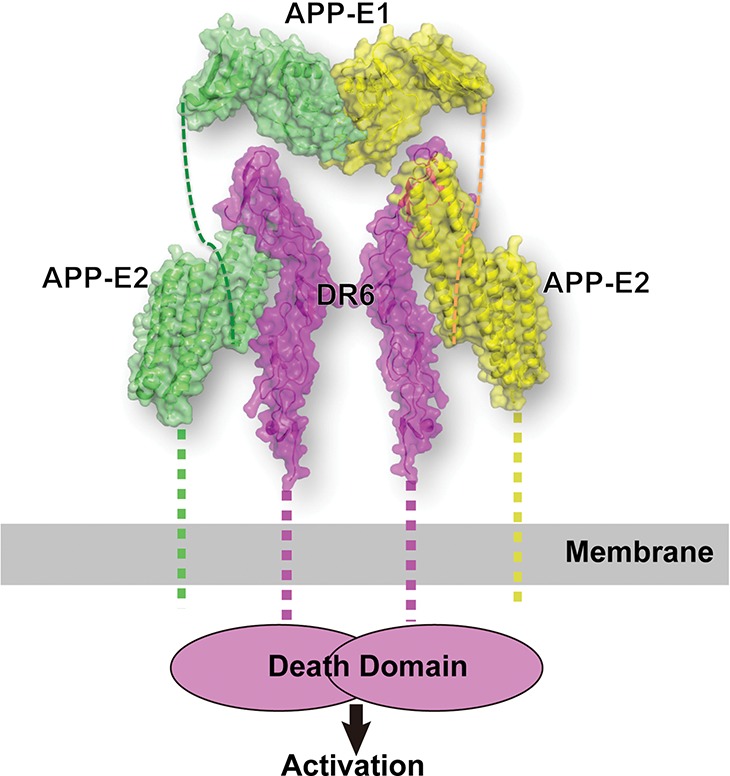
Model of the DR6/APP signaling complex at the neuronal surface. APP binding induces dimerization and activation of DR6. The model was generated by combining the structure reported here of the APP-E2/DR6 complex with the dimeric APP-E1 structure (Protein Data Bank ID3KTM) (Dahms et al. 2010). The two molecules in the APP dimer are colored in yellow and green, while the two DR6 molecules are colored in magenta. The linker regions in the structure were depicted as dashed lines.
Previous work implied that APP and DR6 function in the same genetic pathway that activates a downstream caspase cascade to regulate axon pruning and synapse density in vivo. It was initially suggested that DR6 activation involves proteolytic cleavage of APP (Nikolaev et al. 2009), but subsequent work showed that APP cleavage may not be required (Kallop et al. 2014; Olsen et al. 2014). Our structure confirms a direct interaction of DR6 with APP, and we present a model that is compatible with a membrane-delimited interaction between APP and DR6 at the cell surface (Fig. 3). This model would most easily explain the cell-autonomous function of both molecules in axon pruning and synapse formation. However, we cannot rule out alternative modes of pathway activation. For example, our structure is also compatible with DR6 being activated by the cleaved APP ECD as well as with APP and DR6 forming a coreceptor complex for another ligand—perhaps one that facilitates APP dimerization. Finally, it is also noteworthy that an APP/DR6 complex at the membrane would potentially allow for an interaction between their cytoplasmic tails. This is an interesting possibility considering that the DR6 cytoplasmic tail contains a death domain (Pan et al. 1998) and that peptide fragments of the APP cytoplasmic tail have been linked to neuronal death (Lu et al. 2000). Further in vivo studies on the APP/DR6 complex will be essential for clarifying these possibilities and understanding how their interaction and APP dimerization are regulated.
Materials and methods
Protein expression and purification
All protein constructs, including the E1 domain (residues 18–199), E2 domain (residues 295–516), and ECD (residues 18–624) of mouse APP (accession ID NP_031497) as well as the ECD (residues 42–220) of mouse DR6 (accession ID NP_848704), were expressed in HEK293 cells. Recombinant protein purification was facilitated by fusion of all expression constructs to a thrombin-cleavable Fc region of human IgG. Gel filtration in SD-200 column (GE Healthcare) was used as the final purification step after Fc tag removal by thrombin protease.
Crystallization and structure determination
APP-E2 and DR6-ECD did not form a stable complex in the gel filtration column (Supplemental Fig. S1B). For complex formation, purified APP-E2 and DR6-ECD were mixed in a 1:1 molar ratio in 5 mM Hepes (pH 7.2) and 500 mM NaCl buffer. Crystallization trials were conducted with a “mosquito” robot (TTL Technology) using the sitting drop vapor diffusion method by mixing 100 nL of protein solution with 100 nL of well solution. Crystals (in the P61 space group with one 1:1 complex in an asymmetric unit) grew when the crystallization drops were set against wells containing 100 mM Tris (pH 8.6) and 18% PEG 8000 and reached optimal size in a week. Well solution supplemented by 25% glycerol was used as cryoprotectant for flash-freezing the crystals in liquid nitrogen. Diffraction data sets were collected remotely in beamline ID-24 of the Advance Photon Source. The best crystals diffract to ∼2.2 Å resolution. The data sets were processed with HKL2000 (Otwinowski and Minor 1997). Phases were calculated by molecular replacement in Phaser (McCoy 2007) using human APP-E2 (3UMH) and human DR6 (3QO4) as search models. Model building and refinement were performed by using Coot (Emsley and Cowtan 2004) and Phenix refine (Adams et al. 2010). The Rfree of the refined model was 24.5%. Crystallographic statistics (Supplemental Table S1) and complex packing within the crystal lattice (Supplemental Fig. S6) are presented as Supplemental Material. The APP/DR6 complex structure has been deposited in the Protein Data Bank under ID 4YN0.
Cell-binding experiments
Cell-binding experiments were performed as described previously (Olsen et al. 2014). Briefly, AP fusion proteins were produced in HEK293 cells. The concentration of the AP fusion protein in supernatants was determined by measuring AP activity and diluting to 250 nM in AP-binding buffer (Hank's balanced salt solution [HBSS], 20 mM HEPES, 0.2% BSA, 0.1% NaN3, 5 mM CaCl2, 1 mM MgCl2). To measure binding, transfected COS-7 cells were rinsed twice with AP-binding buffer and incubated with diluted AP proteins for 2 h on ice. Cells were then rinsed five times using AP-binding buffer and fixed (3% paraformaldehyde in PBS) for 12 min at room temperature. Following fixation, cells were equilibrated three times using HBS buffer, and endogenous AP activity was quenched for 30 min at 65°C. Cells were then rinsed three times using AP developing buffer (100 mM Tris at pH 9.5, 100 mM NaCl, 50 mM MgCl2), and AP protein binding was observed by incubating cells overnight at 4°C in AP developing buffer containing NBT/BCIP (Roche). Mutations in APP and APLP2 were generated by site-directed mutagenesis. Single or double point mutants at the APP/DR6 complex interface were designed using the crystal structure of the complex.
Affinity measurements
The affinities between DR6 and wild-type and mutant AP fusion APP and APLP2 constructs were measured using the biolayer interferometry method with a BLITZ instrument (ForteBio, Inc.). Specifically, DR6-Fc was immobilized on a protein-A sensor chip and used to measure the binding of the APP/APLP2 constructs. KD values were determined by fitting the binding curve with the program provided with the BLITZ instrument. All proteins were purified by affinity chromatography and gel filtration chromatography, and their purity was confirmed using SDS-PAGE.
SEC analysis
SEC analysis was performed by running the samples on a Superdex-200 column (GE Healthcare) with 5 mM Hepes (pH 7.2) and 500 mM NaCl buffer. Complexes (APP-E2/DR6, APP-E2/heparin, and APP-E1/heparin) were prepared by incubating the two components for 2 h at 4°C.
Illustrations
All molecular representations were generated with PyMOL (Delano Scientific LLC). Figures were prepared using Adobe Illustrator and Adobe Photoshop.
Supplementary Material
Acknowledgments
We thank Dr. Zhuhao Wu (Rockefeller University) for valuable discussions, Dr. Nicholas Hertz (Rockefeller University) for assistance with protein purification, and Dr. Yehuda Goldgur (Memorial Sloan Kettering Cancer Center) for help with data collection. This work was funded by National Institutes of Health grant 1R01NS089786 to M.T.-L. X-ray diffraction studies were conducted remotely at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines ID24, which are supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under contract number DE-AC02-06CH11357.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.257675.114.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Jung CK, Volbracht C, Steiner H, Mitteregger G, Kretzschmar HA, Haass C, Herms J. 2009. γ-Secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci 29: 10405–10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK. 2007. Three is better than one: pre-ligand receptor assembly in the regulation of TNF receptor signaling. Cytokine 37: 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburger I, Dahms SO, Roeser D, Guhrs KH, Hortschansky P, Than ME. 2013. Analysis of the overall structure of the multi-domain amyloid precursor protein (APP). PLoS One 8: e81926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburger I, Hoefgen S, Than ME. 2014. The structural biology of the amyloid precursor protein APP—a complex puzzle reveals its multi-domain architecture. Biol Chem 395: 485–498. [DOI] [PubMed] [Google Scholar]
- Dahms SO, Hoefgen S, Roeser D, Schlott B, Guhrs KH, Than ME. 2010. Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc Natl Acad Sci 107: 5381–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahms SO, Konnig I, Roeser D, Guhrs KH, Mayer MC, Kaden D, Multhaup G, Than ME. 2012. Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J Mol Biol 416: 438–452. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Gandy S, DeKosky ST. 2013. Toward the treatment and prevention of Alzheimer's disease: rational strategies and recent progress. Annu Rev Med 64: 367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefgen S, Coburger I, Roeser D, Schaub Y, Dahms SO, Than ME. 2014. Heparin induced dimerization of APP is primarily mediated by E1 and regulated by its acidic domain. J Struct Biol 187: 30–37. [DOI] [PubMed] [Google Scholar]
- Hoopes JT, Liu X, Xu X, Demeler B, Folta-Stogniew E, Li C, Ha Y. 2010. Structural characterization of the E2 domain of APL-1, a Caenorhabditis elegans homolog of human amyloid precursor protein, and its heparin binding site. J Biol Chem 285: 2165–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Du Q, Yin X, Li J, Wang T, Zhang L. 2014. Agonist antibody activates death receptor 6 downstream signaling involving TRADD recruitment. FEBS Lett 588: 401–407. [DOI] [PubMed] [Google Scholar]
- Idriss HT, Naismith JH. 2000. TNFα and the TNF receptor superfamily: structure–function relationship(s). Microsc Res Tech 50: 184–195. [DOI] [PubMed] [Google Scholar]
- Kallop DY, Meilandt WJ, Gogineni A, Easley-Neal C, Wu T, Jubb AM, Yaylaoglu M, Shamloo M, Tessier-Lavigne M, Scearce-Levie K, et al. 2014. A death receptor 6-amyloid precursor protein pathway regulates synapse density in the mature CNS but does not contribute to Alzheimer's disease-related pathophysiology in murine models. J Neurosci 34: 6425–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. 2001. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature 414: 643–648. [DOI] [PubMed] [Google Scholar]
- Kuester M, Kemmerzehl S, Dahms SO, Roeser D, Than ME. 2011. The crystal structure of death receptor 6 (DR6): a potential receptor of the amyloid precursor protein (APP). J Mol Biol 409: 189–201. [DOI] [PubMed] [Google Scholar]
- Lee S, Xue Y, Hu J, Wang Y, Liu X, Demeler B, Ha Y. 2011. The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry 50: 5453–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. 2000. A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat Med 6: 397–404. [DOI] [PubMed] [Google Scholar]
- McCoy AJ. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 63: 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. 2009. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457: 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Olsen O, Kallop DY, McLaughlin T, Huntwork-Rodriguez S, Wu Z, Duggan CD, Simon DJ, Lu Y, Easley-Neal C, Takeda K, et al. 2014. Genetic analysis reveals that amyloid precursor protein and death receptor 6 function in the same pathway to control axonal pruning independent of β-secretase. J Neurosci 34: 6438–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymology 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pan G, Bauer JH, Haridas V, Wang S, Liu D, Yu G, Vincenz C, Aggarwal BB, Ni J, Dixit VM. 1998. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett 431: 351–356. [DOI] [PubMed] [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. 2006. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci 26: 7212–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru H, Zhao L, Ding W, Jiao L, Shaw N, Liang W, Zhang L, Hung LW, Matsugaki N, Wakatsuki S, et al. 2012. S-SAD phasing study of death receptor 6 and its solution conformation revealed by SAXS. Acta Crystallogr D Biol Crystallogr 68: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. 2011. Alzheimer's disease. Cold Spring Harb Perspect Biol 3: a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ha Y. 2004. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell 15: 343–353. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang B, Yang L, Guo Q, Aithmitti N, Songyang Z, Zheng H. 2009. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J Neurosci 29: 10788–10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Lee S, Ha Y. 2011a. Crystal structure of amyloid precursor-like protein 1 and heparin complex suggests a dual role of heparin in E2 dimerization. Proc Natl Acad Sci 108: 16229–16234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Lee S, Wang Y, Ha Y. 2011b. Crystal structure of the E2 domain of amyloid precursor protein-like protein 1 in complex with sucrose octasulfate. J Biol Chem 286: 29748–29757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.