

Biran et al. show that senescent cells directly transfer proteins to neighboring cells and that this process facilitates immune surveillance of senescent cells by NK cells. The transfer is strictly dependent on cell–cell contact and CDC42-regulated actin polymerization and is mediated at least partially by cytoplasmic bridges. These findings reveal a novel mode of intercellular communication by which senescent cells regulate their immune surveillance and might impact tumorigenesis and tissue aging.
Keywords: cellular senescence, cytoplasmic bridges, actin polymerization, natural killer cells
Abstract
Mammalian cells mostly rely on extracellular molecules to transfer signals to other cells. However, in stress conditions, more robust mechanisms might be necessary to facilitate cell–cell communications. Cellular senescence, a stress response associated with permanent exit from the cell cycle and the development of an immunogenic phenotype, limits both tumorigenesis and tissue damage. Paradoxically, the long-term presence of senescent cells can promote tissue damage and aging within their microenvironment. Soluble factors secreted from senescent cells mediate some of these cell-nonautonomous effects. However, it is unknown whether senescent cells impact neighboring cells by other mechanisms. Here we show that senescent cells directly transfer proteins to neighboring cells and that this process facilitates immune surveillance of senescent cells by natural killer (NK) cells. We found that transfer of proteins to NK and T cells is increased in the murine preneoplastic pancreas, a site where senescent cells are present in vivo. Proteomic analysis and functional studies of the transferred proteins revealed that the transfer is strictly dependent on cell–cell contact and CDC42-regulated actin polymerization and is mediated at least partially by cytoplasmic bridges. These findings reveal a novel mode of intercellular communication by which senescent cells regulate their immune surveillance and might impact tumorigenesis and tissue aging.
Effective communication between cells and their microenvironments is essential for tissue homeostasis. Senescent cells can impact their microenvironment and influence tissue homeostasis with both positive and negative outcomes, depending on physiological context (Acosta et al. 2008; Coppe et al. 2008; Kuilman et al. 2008; Wajapeyee et al. 2008; Calabrese et al. 2009). Senescent cells are present in premalignant lesions and sites of wound healing, where they limit tumorigenesis and tissue damage (Krizhanovsky et al. 2008; Jun and Lau 2010; Kuilman et al. 2010; Salama et al. 2014). These cells also contribute to morphogenesis in embryos (Munoz-Espin et al. 2013; Storer et al. 2013) and possibly maternal–embryonic transport in the placenta (Chuprin et al. 2013). However, senescent cells also accumulate in tissues with age, apparently contributing to tissue impairment and the development and progression of age-related diseases (Dimri et al. 1995; Herbig et al. 2006; Baker et al. 2011).
The entry into senescence is typically a cell-autonomous response to genomic stress. In particular, cells can become senescent in response to persistent DNA damage triggered by telomere shortening, exposure to a DNA-damaging agent, or expression of activated oncogenes (Serrano et al. 1997; Vaziri et al. 1997; Bartkova et al. 2006; Di Micco et al. 2006; Campisi and d'Adda di Fagagna 2007; Mallette et al. 2007). In response to these stimuli, cells activate p53 and pRb pathways to permanently arrest cell proliferation and thereby block tumorigenesis (Collado and Serrano 2010; Freund et al. 2010; Kuilman et al. 2010; Salama et al. 2014).
Despite the terminal shutdown of proliferative pathways, senescent cells are metabolically active and influence surrounding cells. In response to DNA damage, NF-κB and p38 pathways become activated, resulting in the secretion of proinflammatory molecules, growth factors, and proteases (Acosta et al. 2008; Kuilman et al. 2008; Freund et al. 2010; Chien et al. 2011). This secretory phenotype functions in communicating with immune cells, such as natural killer (NK) cells and T cells, attracting them to the location of the senescent cells and enabling their removal (Xue et al. 2007; Krizhanovsky et al. 2008; Kang et al. 2011; Sagiv and Krizhanovsky 2013). However, if senescent cells persist in tissues, the secretory phenotype can contribute to local inflammation and attenuate tissue repair, features associated with aging (Freund et al. 2010; Yoshimoto et al. 2013; Ovadya and Krizhanovsky 2014). In this manner, senescent cells communicate with and influence the behavior of neighboring cells through paracrine signaling.
For many cell types, secreted factors constitute a predominant, but not the sole, mechanism for intercellular transfer of information. Nevertheless, cells can also transfer proteins from different cellular compartments and even entire organelles to other cells without secretion (Davis and Sowinski 2008; Abounit and Zurzolo 2012; Naphade et al. 2015). Cytoplasmic bridges (CBs), which are membrane protrusions connecting two cells over a long distance, are such a way of cellular communication that allows direct protein transfer between cells. Different forms of CBs that vary in both diameter and length have been documented; examples include tunneling nanotubes (TNTs), filopodial bridges, and epithelial bridges (Rustom et al. 2004; Davis and Sowinski 2008; Abounit and Zurzolo 2012). CBs have been observed in a variety of cell types, including neurons, cancer cells, and immune cells. For instance, NK cells were shown to interact with target cells via CBs, leading to increased lysis (Chauveau et al. 2010). Therefore, CBs provide a functional route for communication between cells.
Intercellular communication of senescent cells is essential for both negative and positive effects of senescent cells on their microenvironment. Proteins secreted from senescent cells can account only partially for these effects. Here we demonstrate that senescent cells can also influence neighboring cells by direct intercellular protein transfer (IPT). Senescent cells transfer proteins to NK cells in vitro and in vivo, leading to increased cytotoxicity. We identified the proteins transferred from senescent cells by the trans-SILAC high-throughput proteomic method. The proteomic analysis and functional studies of the transferred proteins revealed that the transfer is strictly dependent on cell–cell contact and CDC42-regulated actin polymerization and is mediated at least partially by CBs. Taken together, our data suggest that senescent cells communicate with neighboring cells by IPT, which regulates their immune surveillance and their impact on the microenvironment.
Results
Senescent fibroblasts transfer protein to NK cells
NK cells specifically recognize and efficiently eliminate senescent cells in vitro and in vivo (Xue et al. 2007; Krizhanovsky et al. 2008; Sagiv et al. 2013). We examined whether proteins can transfer from senescent cells to NK cells during their interaction. To induce senescence and monitor protein transfer, normal human fibroblasts (IMR-90) were transduced with a vector encoding the mCherry-H-Ras12V fusion protein. As expected, this procedure resulted in oncogene-induced senescence (OIS) of these cells (Supplemental Fig. S1A,B). NK92 cells, similarly to primary NK cells, can specifically interact with senescent cells (Sagiv et al. 2013). NK92 cells were cocultured with OIS cells, fixed, stained for the NK-specific marker CD56, and analyzed by confocal microscopy. After the coculture, some of the NK cells were positive for the mCherry-H-Ras12V fusion protein, indicating that protein was transferred from senescent cells to NK cells (Fig. 1A). To further study this phenomenon, we designed a quantitative transfer assay in which NK92 or primary human NK cells were cocultured with senescent cells and then collected, stained with anti CD56 antibodies and DAPI, and analyzed by fluorescence-activated cell sorting (FACS). To ensure specific identification, NK cells were gated according to FSC-A/SSC-A, live cells (DAPI-negative), CD56-positive, and doublet discrimination (Supplemental Fig. S2A). Following the transfer assay, intercellular transfer of the mCherry-H-Ras12V protein to NK cells was detected (27.2% ± 1% and 40.1% ± 0.8% of primary NK and NK92 cells, respectively) (Fig. 1B–D). Importantly, the transferred mCherry-H-Ras12V was localized to the plasma membrane in NK cells, as identified by the ImageStream system, which combines FACS and microscopy analysis (Fig. 1E). This suggests that H-Ras12V transferred to the NK cells maintains normal subcellular localization. Further analysis revealed no differences in cell size and CD56 membrane distribution between the transfer-positive (T+) and transfer-negative (T−) populations (Fig. 1B; Supplemental Fig. S2B,C), thus excluding fusion of senescent and NK cells as a mechanism of protein transfer. To evaluate the presence of mCherry-H-Ras12V in the NK cells by an alternative approach, we sorted NK cells after the transfer assay into T+ and T− populations (Fig. 1B), lysed the cells, and performed a Western blot analysis for mCherry-H-Ras12V. Higher levels of mCherry-H-Ras12V protein were detected in the T+ population compared with the T− population (Fig. 1F). mCherry-H-Ras12V was not detected in the control NK cell population. Moreover, mCherry-H-Ras12V band size in the T+ population is similar to mCherry-H-Ras12V band size in the OIS population (Fig. 1F; Supplemental Fig. S3A), indicating that the full-length mCherry-H-Ras12V had transferred to NK92 cells and not a fragmented form. Interestingly, transfer of mCherry-H-Ras12V was detected as early as 15 min after coculture was started (Supplemental Fig. S3B), suggesting the transfer of the protein itself and not mRNA or DNA.
Figure 1.

mCherry is preferentially transferred from senescent cells to NK cells by a contact-dependent mechanism. Primary human NK cells or NK92 cells were cocultured with senescent IMR-90 cells expressing the mCherry-H-Ras12V fusion protein (OIS cells) for 2 h. (A) The cells were fixed and stained for CD56 (green). Bar, 10 µM. (B) NK92 cells were collected, stained for CD56 and with DAPI, and analyzed by FACS compared with NK92 cells without coculture (control [C]). NK92 cells that received mCherry were designated as T+. (C) The percentage of T+ NK92 cells was determined following 2 h of coculture with OIS cells. (D) The percentage of T+ human primary NK cells was determined following 1 h of coculture with OIS cells. (E) NK92 cells were analyzed by ImageStream following coculture with OIS cells. (F) Following coculture with OIS cells, NK92 cells were FACS-sorted for T+ and T− populations and analyzed by Western blot for the presence of mCherry-H-Ras12V (using anti-pan-Ras antibodies). (G) IPT to NK92 cells from OIS, DNA damage-induced senescence (DIS), or growing IMR-90 cells was determined by FACS analysis after 2 h of coculture. (H) Representative analysis of IPT from growing, quiescent, apoptotic, or DIS cells, all expressing mCherry, to NK92 cells after 2 h of coculture. (I) NK92 cells were cocultured with growing, OIS, or DIS cells or separated within the same well by a transwell filter (0.4-µm pore) and then analyzed by FACS. (J) Primary human NK cells were cocultured for 1 h with OIS cells or separated by a transwell filter and analyzed for IPT. All graphs are expressed as means ± SEM from at least three independent experiments. (***) P < 0.001.
To evaluate whether senescent cells preferentially transfer proteins to NK cells, we compared IPT from control growing, OIS, and DNA damage-induced senescence (DIS) cells. Senescent and growing cells expressed comparable levels of mCherry (Supplemental Fig. S3A), thus allowing direct comparison between them. IPT was significantly higher from both OIS and DIS cells compared with growing cells (P < 0.001) (Fig. 1G). Senescent cells also showed higher IPT levels compared with quiescent cells or apoptotic cells (Fig. 1H). Therefore, senescent cells preferentially participate in IPT with NK cells.
Senescent cells influence their surroundings via their secretory response. To determine whether secreted factors contribute to IPT, OIS, DIS, or growing cells were cocultured with NK cells in a transwell chamber that prevents direct contact between the cells but enables them to share the same medium. In addition, NK cells were cultured in medium collected from growing, DIS, or OIS cells. Coculture in the chamber led to a complete ablation of protein transfer to NK92 cells and primary NK cells (Fig. 1I,J). No transfer was observed when NK92 cells were cultured with medium collected from growing or senescent cells (Supplemental Fig. S3C). These results indicate that cell–cell contact is essential for the observed IPT.
Identification of transferred proteins by SILAC-mediated proteomic analysis
To obtain a global view of the proteins transferred from senescent cells to NK cells, a trans-SILAC approach (Rechavi et al. 2010) followed by mass spectrometry analysis of the transferred proteins approach was used (see Fig. 2A for schematic description). IMR-90 cells were grown in “heavy” medium containing [13C615N4] arginine and [13C615N2] lysine for eight population doublings. Cells were then treated with etoposide to induce senescence or with vehicle control. We confirmed that the SILAC labeling procedure did not affect the induction of senescence (Supplemental Fig. S4A). The “heavy” senescent and “heavy” growing, vehicle-treated cells were cocultured with NK92 cells containing unlabeled, “light” amino acids. After 2 h of coculture, NK cells were isolated by sorting, lysed, and analyzed by mass spectrometry. Identification of the labeled proteins in the NK cells indicates that these proteins were transferred from the IMR-90 cells. We performed two independent experiments; each experiment included three repeats of NK cells cocultured with growing cells and three repeats of NK cells cocultured with DIS cells. NK cells alone were used as a control. We identified the proteins that were significantly higher in the NK cells incubated with IMR-90 compared with the control samples and found, overall, 47 proteins that were transferred to NK cells (Fig. 2B). A distance matrix analysis of the samples, based on the transferred proteins, indicated that the samples of each experimental setting from both experiments form distinct homogeneous groups, indicating high consistency of our assay (Supplemental Fig. S4B). The identified transferred proteins were ordered in the expression matrix using a SPIN algorithm (Fig. 2B; Tsafrir et al. 2005). A clear distinction was seen between NK cells cocultured with growing and DIS cells, with 90% of the proteins being transferred exclusively from the senescent cells. These data support our finding that senescent cells preferentially initiate IPT to NK cells. Analysis of these proteins by molecular weight demonstrated a wide distribution of protein sizes from 12 kDa to 475 kDa (Fig. 2C). In addition, the transferred proteins were analyzed according to subcellular localization using the cellular component branch of gene ontology (GO). This analysis revealed that most of the transferred proteins are derived from the cytoplasm but may also be localized to intracytosolic organelles and the cell membrane (Fig. 2D).
Figure 2.

Transferred proteins identified by proteomic analysis lead to NK activation and cytotoxicity. (A) Diagram of the trans-SILAC experiment. (B) SPIN-ordered expression matrix. Colors depict relative levels of transfer (centered and normalized), where red denotes high levels of transfer, and blue denotes low levels of transfer. Rows represent proteins, and columns represent samples: NK92 cells without coculture (Ctrl) or cocultured with growing control (Grow) or DIS cells. (C) Size distribution (in kilodaltons) of the identified transferred proteins. (D) Subcellular localization of the identified transferred proteins. (E) Quantitative proteomic analysis of DIS cells. Proteins from senescent cells were ranked according to their relative intensity levels, reflecting protein abundance. Transferred proteins from DIS to NK92 cells are marked in red. (F) A matrix of protein ratios R1/R2, where the transferred protein abundance is denoted by R1 and protein abundance in DIS cells is denoted by R2. (G) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis identified the pathways affected by the transferred proteins. (H–K) Primary human NK cells were cocultured for 1 h with OIS or DIS cells, collected, and gated by FACS as T− or T+ compared with control (C) NK cells. (H) Representative FACS analysis of CD107a levels in control, T−, and T+ populations following IPT from DIS cells. (I) Average percentage of CD107a-positive cells in T− and T+ populations. (J) Average percentage of CD69-positive cells in T− and T+ populations. (K) Average percentage of phosphorylated AKT (p-AKT)-positive cells between T− and T+ populations. Results are expressed as means ± SEM from at least three independent experiments. (***) P < 0.001.
In order to determine whether IPT is an actively regulated process, we combined the proteomic data of the transferred proteins with a quantitative proteomic analysis of senescent cells. This analysis revealed that the transferred proteins were highly abundant in senescent cells (Fig. 2E). Analysis of protein expression ratios for each of the two SILAC data sets (the transferred proteins and protein abundance in senescent cells) showed that protein transfer is highly correlated with high protein abundance (Fig. 2F). However, 12.5% of the identified proteins transferred independently of protein abundance, indicating that, in some instances, IPT may be a regulated process (Fig. 2F). GO analysis of the transferred proteins identified enrichment in processes related to glycolysis, regulation of the actin cytoskeleton, and antigen processing and presentation (Fig. 2G). These results suggest that IPT might lead to alterations in cytoskeleton organization of the recipient cells, thereby leading to functional changes.
IPT leads to increased NK activation and cytotoxicity
Actin polymerization and cytoskeletal reorganization are required for NK cell-mediated cytotoxicity (Topham and Hewitt 2009). Since transfer of proteins from senescent cells might influence actin reorganization in NK cells, we asked whether IPT correlates with elimination of senescent cells by NK cells. Following coculture of senescent cells (OIS or DIS) with primary human NK cells, NK cells were gated for T− and T+ populations, as determined by the levels of mCherry transferred from senescent cells (Fig. 1B). Each of these NK cell populations was then analyzed for the presence of an activation marker (CD69) and a degranulation marker (CD107a), which indicates cytotoxicity. In primary NK cells, both the activation and degranulation markers were elevated twofold in T+ cells compared with T− cells (P < 0.001) (Fig. 2H–J). Phosphorylation of AKT is one of the initiating molecular events in lymphocyte activation (Lanier 2008). We therefore cocultured NK cells with senescent cells and stained NK cells for phosphorylated AKT (p-AKT). In line with the increase in the activation level, T+ primary NK cells displayed higher levels of p-AKT (P < 0.001) (Fig. 2K). Overall, these results demonstrate that NK cells that received proteins from senescent cells became more activated and cytotoxic.
IPT is dependent on cell contact and actin polymerization
Since protein transfer is contact-dependent and because we observed the transfer of cytoskeleton-related proteins, we aimed to evaluate the functional role of the cytoskeleton components in protein transfer from senescent cells. Additionally, actin polymerization was reported to play a role in IPT in other cell systems (Rustom et al. 2004). To determine the role of actin polymerization in our model, we performed a transfer assay in the presence of Latrunculin A (LatA), a reversible actin polymerization inhibitor (Supplemental Fig. S5A). NK92 cells were cocultured with growing, DIS, or OIS cells in the presence of LatA or a vehicle. LatA caused a fourfold reduction in IPT from senescent cells (P < 0.001) (Fig. 3A). To evaluate whether IPT to primary NK cells is also dependent on actin polymerization, we cocultured these cells with OIS cells in the presence of LatA. Similarly to NK92 cells, the IPT to the primary NK cells was significantly inhibited by LatA (P < 0.001) (Fig. 3B). Thus, IPT between senescent cells and NK cells is facilitated by actin polymerization.
Figure 3.
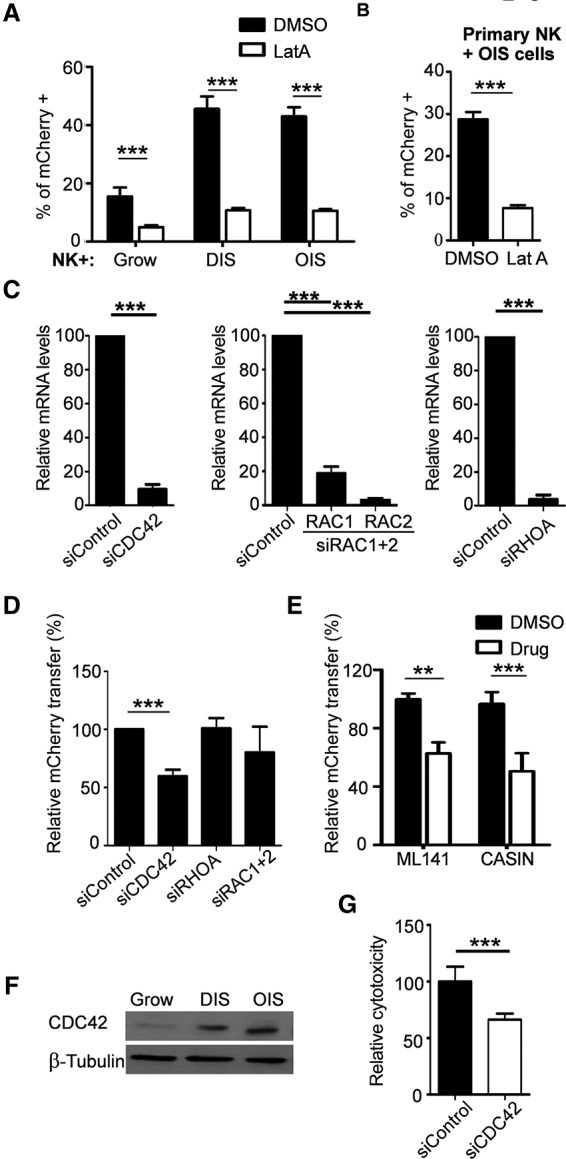
IPT is dependent on actin polymerization. NK cells were cocultured with growing, DIS, or OIS cells. NK cells were then collected, stained for CD56 and with DAPI, and analyzed by FACS for the presence of mCherry. (A) The percentage of T+ NK92 cells following 2 h of coculture in the presence of 200 nM LatA or vehicle (DMSO). (B) The percentage of T+ primary human NK cells following 1 h of coculture in the presence of 200 nM LatA or vehicle. (C) CDC42, RAC1, RAC2, and RHOA siRNAs induce efficient knockdown 3 d after transfection. (D) DIS cells with knockdown of CDC42, RHOA, or RAC1 and RAC2 were cocultured for 2 h with NK92 cells. (E) DIS cells were treated with ML141, CASIN (CDC42 inhibitors), or DMSO (control); washed; and then cocultured with NK92 cells. (F) Western blot analysis of CDC42 levels in growing, DIS, and OIS IMR-90 cells. (G) NK-mediated cytotoxicity toward DIS cells following CDC42 knockdown (siCDC42). Results are expressed as means ± SEM from at least three independent experiments. (***) P < 0.001.
CDC42, RHOA, RAC1, and RAC2 differentially regulate actin polymerization (Heasman and Ridley 2008). To determine the molecular mechanisms of actin polymerization that control IPT from senescent cells, we knocked down CDC42, RHOA, or RAC1/2 (combined) in DIS cells using siRNAs. The knockdown efficiency was validated 3 d after transfection (Fig. 3C), and protein transfer was evaluated. CDC42 knockdown in DIS cells led to 40.3% ± 2.6% (P < 0.001) reduction in protein transfer to NK cells, whereas knockdown of RHOA or RAC1/2 did not cause a significant reduction (Fig. 3D). To evaluate the role of CDC42 by an independent approach, we used ML141 and CASIN, two specific inhibitors of CDC42 (Sakamori et al. 2012; Hong et al. 2013). DIS cells were cultured with ML141 or CASIN overnight, washed, and cocultured for 2 h with NK cells. Inhibition of CDC42 by ML141 or CASIN significantly reduced protein transfer (by 37% ± 3.8%, P < 0.01; and 50.5% ± 4.6%, P < 0.001 for ML141 and CASIN, respectively) (Fig. 3E). Overall, our findings indicate that CDC42 is an important molecular component mediating IPT from senescent cells.
IPT is increased from senescent cells compared with growing cells, and we hypothesized that elevated levels of CDC42 in these cells may explain this phenomenon. Thus, we evaluated CDC42 mRNA and protein levels in growing and senescent cells. Western blot analysis demonstrated that CDC42 protein levels were elevated in DIS and OIS IMR-90 cells compared with control growing cells, whereas no significant change was observed in the mRNA levels (Fig. 3F; Supplemental Fig. S5B). This suggests that post-transcriptional mechanisms are responsible for the increased abundance of CDC42 protein in senescent cells. CDC42 is a small GTPase protein and is activated when bound to GTP. To evaluate CDC42 activation in growing and DIS cell, a CDC42-GTP pull-down assay was performed. The amount of active CDC42-GTP is elevated in DIS cells as compared with growing control cells (Supplemental Fig. S5C). Overall, these findings suggest that up-regulation of CDC42 protein and its activity in senescent cells are responsible for increased IPT from these cells. Thus, protein transfer is dependent on cell contact mediated by actin polymerization and regulated primarily by CDC42.
We demonstrated that IPT from senescent cells leads to NK activation and cytotoxicity (Fig. 2I,J). As such, we wanted to determine whether inhibition of CDC42 and hence reduction in IPT impact killing of senescent cells by the NK cells. DIS cells were transfected with siRNA targeting CDC42 or nontargeting control siRNA. Of note, CDC42 knockdown did not significantly affect cell viability or senescent state (Supplemental Fig. S5D,E). Three days post-transfection, NK cytotoxicity toward these cells was evaluated using a Real-Time Cell Analyzer (RTCA). Down-regulation of CDC42 in DIS cells reduced the cytotoxicity by NK cells by 33.6% ± 5.3% (P < 0.001) (Fig. 3G). Thus, IPT from senescent target cells facilitates killing of these cells by NK cells.
IPT is not limited to NK cells
Senescent cells can alter the functional behavior of cells in their microenvironment, and this activity has been associated with the enhanced tumorigenic capacity of adjacent cancer cells (Freund et al. 2010). To determine whether protein transfer is a general mechanism of communication used by senescent cells as opposed to a specific mode of interaction with NK cells, we investigated protein transfer to epithelial cells. We cocultured mCherry-expressing senescent or growing cells with GFP-expressing MCF10A (epithelial normal [EpN]) cells and analyzed IPT by FACS (Supplemental Fig. S6A). A significant increase in IPT from DIS or OIS to EpN (24.6% ± 1.6% and 46% ± 10%, respectively) (Fig. 4A,B) was observed, whereas IPT from growing cells was only 11.8% ± 3%. No IPT was detected when EpN cells were incubated with the medium from growing or senescent cells (Fig. 4C). When mCherry-expressing senescent or growing cells were cocultured with H1299 cells (epithelial cancer [EpC]), significant and contact-dependent IPT was observed (Fig. 4D–F). The presence of LatA during coculture reduced IPT by threefold in both DIS and OIS cells (P < 0.001) (Fig. 4F). These results indicate that IPT from senescent cells, dependent on cell–cell contact and actin polymerization, is not limited to NK cells and may therefore represent a general feature of cell senescence. To study the role of CDC42 in IPT from senescent cells to epithelial cells, EpN cells were cocultured with DIS cells in the presence of the CDC42 inhibitor CASIN or DMSO as a control. Treatment with CASIN markedly inhibited IPT to EpN cells (44.5% ± 6.6% reduction, P < 0.001) (Supplemental Fig. S6B). Next, we examined p-AKT levels in T− and T+ EpN cells following coculture with senescent cells. p-AKT levels were more than twofold elevated in T+ cells compared with T− cells (P < 0.001) (Supplemental Fig. S6C). This result indicates that transfer of proteins from senescent cells to epithelial cells might have a functional impact.
Figure 4.

Senescent cells transfer proteins to epithelial cells. (A–C) MCF10A cells expressing GFP (EpN) were cocultured with growing, DIS, or OIS cell for 24 h. Transfer of mCherry to MCF10A was analyzed by FACS. (A) Representative FACS analysis of mCherry levels in EpN cells compared with MCF10A alone (Ep only). (B) The percentage of mCherry-positive EpN cells following the coculture. (C) EpN cells were cultured with medium collected from growing, DIS, or OIS cells. (D–F) H1299 cells expressing GFP (EpC) were cocultured for 24 h with growing, DIS, and OIS cells and analyzed by FACS. (D) Representative FACS analysis of mCherry levels in EpC cells. (E) EpC cells were separated by a transwell chamber from growing, DIS, or OIS cells. (F) The percentage of mCherry-positive EpC cells following coculture in the presence of LatA or in a transwell chamber. Results are expressed as means ± SEM from at least three independent experiments. (***) P < 0.001.
One of the structures that mediate IPT and also depend on actin polymerization and cell–cell contact is the CB (Davis and Sowinski 2008; Abounit and Zurzolo 2012). Thus, we hypothesized that CBs may mediate protein transfer from senescent cells. CB structures were indeed detected in cocultures of senescent and EpC cells (Fig. 5A). In order to facilitate visualization of even short bridges, EpC cells cocultured with senescent cells were stained with a red cell tracker, CMTPX (Fig. 5B). CBs were very abundant and had a diverse length ranging from 7 to 226 µm. To evaluate the presence of actin cytoskeleton in the CBs, F-actin was labeled with phalloidin-TRITC. F-actin was detected within all of the CBs observed (Fig. 5C). Moreover, electron microscopy pictures of CBs from senescent cells revealed that vesicles are located within the bridge, supporting the hypothesis that organelles can transfer from senescent cells (Supplemental Fig. S7A). Next, we wanted to examine whether CB structures can mediate transfer between senescent cells and EpC cells. EpC cells were cocultured with DIS cells, and the green fluorescent dye calcein was injected into a DIS cell with a CB to an EpC cell. Transfer of the dye was monitored over time using time-lapse microscopy. Transfer of the dye from the senescent cells to the epithelial cells was observed a few seconds following injection (Fig. 5D), indicating that the CBs are open at both ends and may represent a functional communication channel between cells. CBs containing actin were also observed between senescent cells and NK cells (Supplemental Fig. S7B; Supplemental Movie S1). Overall, CBs formed between senescent cells and other cells can transfer solutes and proteins.
Figure 5.
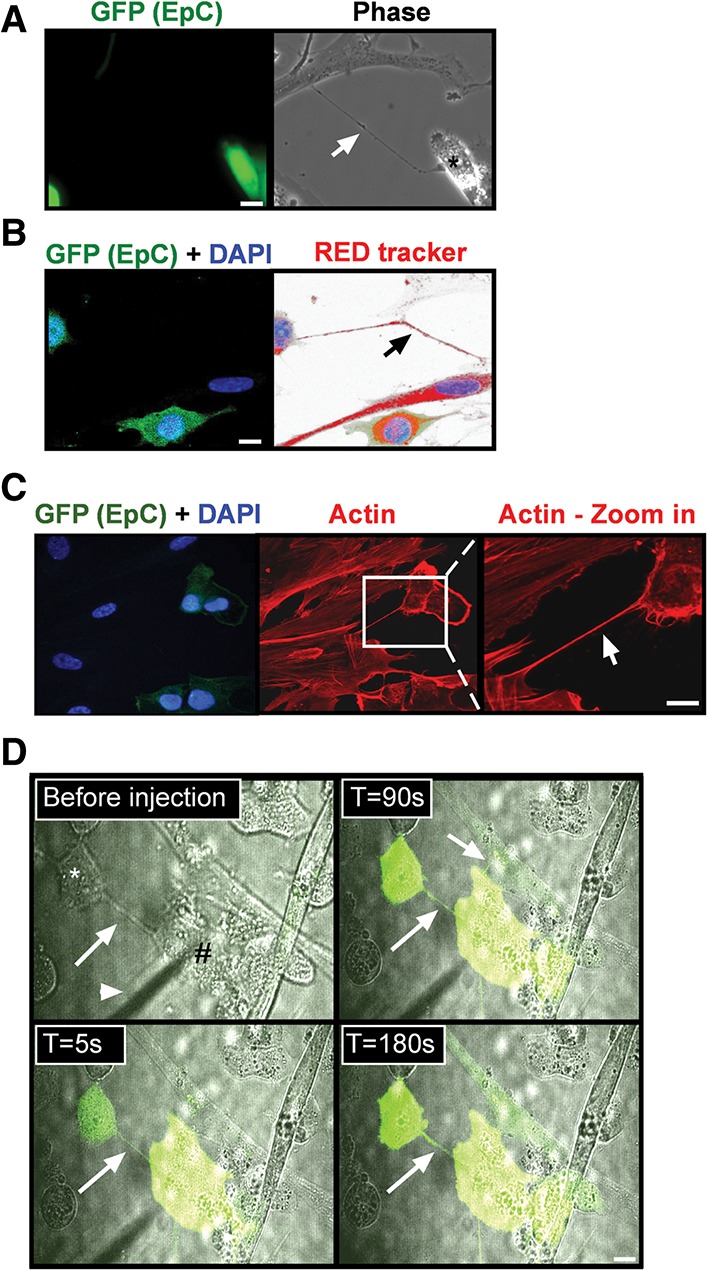
Senescent cells form CBs with cancer cells. (A–C) GFP-expressing EpC cells were cocultured with DIS cells, stained, and analyzed by confocal microscope. (A) The CB (arrow) was detected in the coculture. (B) A CB (arrow) was detected when the cells were stained with RedTracker dye and DAPI. (C) Cells were stained for F-actin (red) and DAPI. (D) Using a microinjection needle (arrowhead), a DIS fibroblast (#) was injected with calcein fluorescent dye. Transfer of the dye via a CB (arrow) from the senescent cell to the cancer cells (asterisk) was monitored over time. Bars, 10 µM.
IPT occurs in vivo
While IPT was previously observed in several in vitro and ex vivo models (Rustom et al. 2004; Rechavi et al. 2007; Stern-Ginossar et al. 2007; Davis and Sowinski 2008; Abounit and Zurzolo 2012), evidence of transfer in vivo in mammals is limited. We therefore aimed to monitor IPT in vivo. For this study, we focused on the premalignant pancreas, in which OIS cells are present in both human patients and mouse models (Caldwell et al. 2011; Guerra et al. 2011; Guerra and Barbacid 2013). We used the Pdx1-Cre;Lox-Stop-Lox(LSL)-K-Ras12V model to express mutant K-Ras in the pancreas (Caldwell et al. 2011; Guerra et al. 2011). In this model, expression of the mutant K-Ras oncogene is turned on during the early stages of embryonic pancreatic development by expressing a Cre recombinase under the control of the Pdx1 promoter (Fig. 6A). Expression of the oncogene K-Ras triggers pancreatic intraepithelial neoplasia lesions (PanINs), which can progress into invasive adenocarcinoma if tumor suppressor pathways are inactivated (Guerra and Barbacid 2013). We then crossed the Pdx1-Cre;LSL-K-Ras12V (designated as Ras) mice with LSL-mRFP mice (Luche et al. 2007). The resulting Pdx1-Cre;LSL-K-Ras12V;LSL-mRFP mice (designated as Ras;mRFP mice) express mRFP in all cells that express the oncogenic K-Ras (Supplemental Fig. S8A), thus allowing the monitoring of mRFP in these mice. In concordance with previous studies, we detected senescent cells at the PanIN stage 1 in the pancreas of the Ras;mRFP mice at the age of 10 wk (Fig. 6B). Pancreata of these mice were dissociated to single-cell suspension, analyzed by FACS, and compared with the blood of the same mice. mRFP was detected in 18% ± 3.2% of the NK cells extracted from pancreata samples of Ras;mRFP mice and in 11.5% ± 4.3% of the NK cells extracted from pancreata samples of Pdx1-Cre;LSL-mRFP mice (designated as mRFP mice) (P < 0.001) (Fig. 6C). mRFP was not detected in NK cells extracted from the blood of the same mice or in the pancreas or blood of the mice that express only K-RasV12 in the pancreas (designated as Ras mice) (Supplemental Fig. S8B). Moreover, we observed mRFP-positive NK cells in sections of pancreata from Ras;mRFP mice but not control Ras mice (Fig. 6D). These results demonstrate that mRFP can be transferred from pancreatic cells to NK cells in a premalignant pancreas.
Figure 6.

mRFP is transferred to immune cells in the premalignant pancreas in vivo. (A) Schematic representation of the Ras;mRFP mouse model. (B) H&E- and SA-β-gal-stained sections from Ras;mRFP mice. Bar, 50 µm. (C) FACS quantification of mRFP-positive NK cells extracted from the pancreata. n ≥ 5 mice for each genotype; (***) P < 0.001; (**) P < 0.01. (D) Sections of a pancreas from Ras;mRFP mice were stained for NK1.1 (yellow) and with DAPI (blue) and imaged by confocal microscope. Bar, 5 µm. (E) Sections of a pancreas from Ras;mRFP mice were stained for CD45 (yellow) and CD3 (green) and with DAPI (blue). Bar, 5 µm. (F,G) ImageStream analysis of cells derived from the blood or pancreata of mice of the indicated genotype. (F) T cells (CD45+;CD3+) and pancreatic resident cells have different characteristics and are clearly distinguishable by ImageStream analysis. Bar, 20 μm. (G) Representative images of mRFP-positive and mRFP-negative T cells acquired using ImageStream. Bar, 10 µm.
In addition to NK cells, T cells also specifically interact with OIS cells (Kang et al. 2011). mRFP-positive T cells were detected in sections of pancreata from Ras;mRFP mice but not control Ras mice (Fig. 6E). To further visualize mRFP-positive T cells, we analyzed blood and pancreatic samples of Ras;mRFP and Ras mice using ImageStream. Differences in both cell size and labeling of the immune markers CD45 and CD3 were used to identify T cells and exclude pancreatic cells (Fig. 6F). Analysis of these samples confirmed the presence of mRFP exclusively in T cells derived from pancreata of Ras:mRFP mice (Fig. 6G). Overall, these findings demonstrate that proteins can be transferred from K-Ras12V-expressing cells to immune cells in vivo.
Discussion
The prevailing view of how senescent cells facilitate changes within their microenvironment has primarily been focused on secreted factors acting in a paracrine manner (Freund et al. 2010; Kuilman et al. 2010; Acosta et al. 2013; Burton and Krizhanovsky 2014). However, in this study, we found that IPT serves as an additional mechanism of such communication both in vitro and in vivo. As compared with normal cells, senescent cells preferentially use IPT to regulate self-elimination by immune cells and communicate with epithelial cells.
The physiological significance of IPT from senescent cells is yet to be understood. It is conceivable that IPT is a general feature of cells undergoing stress and that IPT extends the stress to the neighboring cells. However, the physiological response to IPT is likely to be dependent on the nature of transferred proteins as well as the cell type of the recipient cell. Recent studies have demonstrated a physiological role of IPT in other systems, including initiation and modulation of immune responses and supporting cell survival (Roda-Navarro et al. 2006; McCann et al. 2007; Stern-Ginossar et al. 2007; Yasuda et al. 2011).
The identification of CBs from senescent cells to neighboring cells suggests that this may be an efficient mode of intercellular communication by which senescent cells can execute their cell-nonautonomous effects. Since CBs, such as TNTs, are predominantly made up of F-actin, a number of studies have shown that inhibition of actin polymerization can inhibit CB formation (Rustom et al. 2004; Sowinski et al. 2008). Although in this study, we demonstrated a decrease in IPT following treatment with actin polymerization inhibitors, it is uncertain whether this is a result of a reduction in CB formation or in the transport rate within the CBs or both. Our study further identified CDC42, a known regulator of actin organization, as a novel mediator of IPT in senescent cells. In line with our results, inhibition of CDC42 was shown to block TNT formation in T cells (Arkwright et al. 2010), suggesting that the reduction in protein transfer following CDC42 knockdown or inhibition may be due to lack of CB formation. Identifying CDC42 activity as a key regulator of CB formation in senescent cells provides molecular insight that could be exploited to prevent IPT in pathological conditions. An increase in CDC42 activity in senescent cells might result from the Ras signaling in OIS cells or activation of downstream signaling pathways in DIS cells.
The biological significance of senescence-mediated IPT via CBs may be relevant in many areas of biology, including normal embryonic development, disease development, and cancer resistance. During embryonic development, CBs may function to transfer molecular, electrical, and mechanical signals between cells (Gerdes et al. 2013). Intriguingly, three recent studies have demonstrated that senescent cells play a role in embryonic development (Chuprin et al. 2013; Munoz-Espin et al. 2013; Storer et al. 2013). In light of these findings, it could be suggested that senescent cells at least in part aid embryonic development by facilitating intercellular communication via CB formation.
In contrast to the beneficial roles of IPT in normal physiological conditions, IPT could function in a pathological manner to promote disease progression, allowing the transfer of infectious pathogens such as prions, viruses, and bacteria, thereby facilitating the spread of disease (Sowinski et al. 2008; Abounit and Zurzolo 2012). Senescent cells accumulate in tissues with age and promote tissue aging in their microenvironment (Dimri et al. 1995; Baker et al. 2011). IPT might, in principle, be one of the ways that senescent cells communicate and therefore extend their stress to other cells. However, whether the cell type of the recipient cells influences the components transferred from senescent cells has yet to be determined. We found that IPT to NK cells resulted in NK cell activation and increased cytotoxicity. This observation was further supported by analysis of the transferred proteins, including enrichment in proteins such as HSPA5 and CALR, which are associated with regulation of NK cell activity. Alternatively, IPT may function to support neighboring cells and promote their survival, thereby acting as a protective mechanism. Cancer cells could take functional advantage of such a mechanism. The transport of cell organelles and proteins from senescent cells to cancer cells could facilitate their survival and tumorigenesis by providing them with additional resources. Therefore, limiting IPT from senescent cells, possibly by inhibition of CDC42, may reduce their proaging and procancer effects. However, such an approach may also limit the surveillance of senescent cells by the immune system. Understanding how senescent cells influence their microenvironment will provide critical insight into the mechanisms governing aging, cancer, and disease development and progression.
In a variety of organisms, IPT by CBs is part of a stress response that functions to either gain valuable resources to help with cell survival or provide resources to neighboring cells for protection against ongoing stress (Dubey and Ben-Yehuda 2011; Abounit and Zurzolo 2012; Pasquier et al. 2013). Cellular senescence might represent a cellular stress response that uses cell–cell communication via IPT to support the adaptation of tissues to stress.
Materials and methods
Cell culture
IMR-90, NK92, MCF10A, and H1299 cells (American Type Culture Collection) were grown in standard conditions. Primary human NK cells were a gift from O. Mandelboim, The Hebrew University Hadassah Medical School, Jerusalem, Israel. DIS was induced by etoposide (Sigma) treatment as described previously (Krizhanovsky et al. 2008). OIS was induced by infection of pLPC mCherry-H-Ras12V plasmid into IMR-90 cells.
Transfer assay
To evaluate transfer of mCherry from IMR-90 cells to target cells, IMR-90 cells expressing mCherry were plated in 12-well plates (5 × 104 cells per well). NK cells or epithelial cells were added for the indicated time (1:2 ratio). Cells were then centrifuged, washed with PBS, incubated for 30 min on ice in 5 mM EDTA/PBS, washed, stained with AlexaFluor488 anti-CD56 antibodies for 30 min (for NK cells), costained with DAPI, and analyzed by FACS. For blocking of actin polymerization, the cells were cocultured in the presence of the 200 nM LatA (Sigma). For induction of cell quiescence, growing IMR-90 cells expressing mCherry were serum-starved for 3 d (0.1% FCS). Quiescent cells were than washed and incubated with NK92 cells for 2 h in full medium. To induce apoptosis, DIS cherry cells were incubated for 24 h with 1 µM staurosporine. Cell debris was collected, washed, and incubated with NK92 cells for 2 h. For the transwell assay, IMR-90 and NK cells or epithelial cells were plated in different chambers. NK cells or epithelial cells were then collected and analyzed for mCherry transfer. For evaluation of CDC42 involvement in protein transfer from DIS to NK cells, IMR-90 cells were treated with ML141 (Sigma), CASIN (Xcessbio Biosciences, Inc.), or DMSO (control) overnight; cells were then washed and cocultured with NK cells. For evaluation of CDC42 involvement in protein transfer from DIS to epithelial cells, epithelial cells were cultured with DIS cells for 5 h, and then CASIN or DMSO was added for 24 h. For evaluation of CD107a-positive cells, NK cells were treated with 1:1000 monensin (Biolegend) for 1 h. NK cells were then cocultured with DIS IMR-90 in the presence of AF647 anti-CD107a antibodies (Biolegend). For measuring CD69 and p-AKT levels, NK cells or epithelial cells were collected after the transfer assay and labeled with AlexaFluor488 anti-CD56 (for NK), AlexaFluor 647 anti-CD69 (Biolegend), or AlexaFluor 647 anti-pAKT (BD Biosciences) antibodies.
Combined microscopy and FACS analysis
NK92 cells or epithelial cells were gated by their size (FSC/SSC), DAPI-negative (live cells), CD56-positive (NK cells), or GFP-positive (epithelial cells). Data were collected from at least 10,000 single-cell events. The percentage of gated cells positive for mCherry was determined. To acquire cell images, we used ImageStream technology (Amnis Corporation).
Modification and evaluation of gene expression
Detection of protein expression by immunoblotting was performed using anti-CDC42 (Cell Signaling Technology), anti-β-tubulin, anti-pan-Ras, and anti-p16 (Santa Cruz Biotechnology).
ON-TARGETplus SMARTpool siRNA targeting RHOA, RAC1, RAC2, CDC42, and the nontargeting (control) pool were transfected into senescent IMR-90 cells with Dharmafect 1 reagent (all from Dharmacon). Cells were washed 24 h after transfection; transfer assay, cell viability assay, and evaluation of knockdown were performed 48 h later.
For retroviral transductions, the following plasmids were used: pLPC mCherry or pLPC EGFP (mCherry or GFP fused to the gene of interest from an internal CMV promoter with a puromycin resistance gene driven by the LTR). Cells were infected with mCherry-H-Ras12V, GFP, or mCherry alone. Retrovirus-mediated gene transfer was performed as previously described.
Total RNA was isolated using a NucleoSpin kit (Macherey Nagel) and reverse-transcribed using the RevertAid H Minus first strand cDNA synthesis kit (Fermentas). The cDNA samples were used for real-time PCR (Fast SYBR Green master mix, Applied Biosystems). PCR amplifications were carried out using StepOnePlus real-time PCR systems (Applied Biosystems). The relative expression of each gene was normalized using the expression levels of GAPDH.
Immunofluorescence and confocal microscopy
IMR-90 cells were plated as described for the transfer assay. After 24 h NK92 or epithelial cells were added for 2 or 48 h, respectively. Cells were then fixed and stained with Alexa488 anti-CD56 antibodies, Phalloidin-TRITC (Sigma), Red mito-tracker (Cell Signaling), or CellTracker Red CMTPX (Life Technologies). The cells were imaged on Zeiss LSM710 confocal microscopes.
Trans-SILAC analysis
IMR-90 cells were cultured for eight doublings in SILAC medium in which the natural lysine and arginine were replaced by [13C615N4] arginine and [13C615N2] lysine and supplemented with 10% (v/v) dialyzed serum. IMR-90 cells were treated for 48 h with etoposide to induce senescence. After 9 d, the cells were cocultured with NK92 cells for 2 h. NK92 cells were collected, sorted to ensure maximal purity, and lysed with a buffer containing 2 M thiourea, 6 M urea, 0.1 M Tris (pH 8.5), and protease. Proteins were reduced with 1 mM DTT and alkylated with 5 mM iodoacetamide followed by 3 h of digestion with lysC (1:100 enzyme to protein ratio) and an overnight trypsin digestion (1:50 enzyme to protein ratio). Peptides were then purified on C18 StageTips. Mass spectrometric analysis was performed on the EASY-nLC1000 HPLC system coupled to the Q-Exactive mass spectrometer. Data analysis was performed with MaxQuant software and the SPIN algorithm. Transferred proteins were analyzed according to subcellular localization using the cellular component branch of GO. For SILAC analysis of growing and senescent cells, growing cells were labeled as described, whereas senescent cells were unlabeled. Cells were than lysed and analyzed as described above.
Monitoring NK cytotoxicity using a RTCA
IMR-90 cells were transfected with siRNAs and then seeded in triplicates on RTCA (Roche) E-plates (7000 cells per well). The cell index, indicating the presence of live cells, was monitored before the addition of NK cells. NK92 cells were added at a 1:2 ratio in a volume of 100 µL per well. Treatment with 1% Triton was used as an indicator of maximal elimination of target cells. Cocultures were assessed by the RTCA system every 5 min for up to 24 h.
Mice
All experiments were done with approval from the Weizmann Institute Animal Care and Use Committee. Animals were sacrificed at the age of 10 wk. Blood and pancreata samples were stained with AlexaFluor488 anti-CD45, AlexaFluor647 anti-NK1.1, and AlexaFluor647 anti-CD3 antibodies (Biolegend) and analyzed by FACS or ImageStram. The SA-β-gal and H&E stainings were performed as described (Krizhanovsky et al. 2008). To detect immune cells in tissue sections from the pancreas, the mice were subjected to a series of seven hourly intraperitoneal injections of Caerulein (50 ng/g per injection) for 2 d in a row to induce pancreatitis. Twenty-four hours after the last injection, mice were sacrificed, and pancreata were taken, fixed in 4% PFA for 5 h at room temperature, cryoprotected in 30% sucrose, and snap-frozen in OCT. Cryosections (10 µm) were then stained with the same antibodies as for FACS.
CDC42-GTP pull-down assay
GST (glutathione S-transferase)-PBD (Rac/Cdc42-binding domain of p21-activated kinase 1 protein) pull-down assays were used to detect cellular GTP-bound Cdc42. In brief, cells were lysed in a buffer containing 50 mm Tris-HCl (pH 7.2), 1% (v/v) Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mm NaCl, 10 mm MgCl2, 1 mm phenylmethanesulphonyl fluoride (PMSF), and protease inhibitor cocktail (Sigma). The supernatants of the lysates were incubated for 1.5 h at 4°C with GST-PBD-coupled glutathione-sepharose beads. The beads were washed three times with buffer containing 50 mm Tris-HCl (pH 7.2), 1% (v/v) Triton X-100, 150 mm NaCl, 10 mm MgCl2, 0.1 mm PMSF, and protease inhibitor cocktail. The amount of GTP-bound CDC42 was detected by Western blotting with an antibody against CDC42.
Statistical analysis
Statistical analysis of the results was performed using one-sided two-sample t-tests or one-way ANOVA followed by a Tukey post-hoc test. Data are presented as means ± SEM, a P-value of <0.05 was considered significant.
Supplementary Material
Acknowledgments
We are grateful to D. Fass and I. Ben-Porath for critical reading of the manuscript and insightful suggestions, O. Mandelboim (Hadassah Medical Center) for providing primary human NK cells, M. Narita (Cancer Research UK) for pLPC vectors for mCherry fusion constructs, M. Segal and E. Korkotian (Weizmann Institute) for Calcein injection and microscopy, Z. Porat (Weizmann Institute FACS unit) for ImageStream analysis, and K. Rechav and E. Shimoni (Weizmann Institute electron microscopy unit) and all the members of the Krizhanovsky laboratory for insightful discussions. T.G. is supported by the Israel Cancer Research Fund. This work was supported by grants to V.K. from the European Research Council under the European Union’s FP7; the Ministry of Science, Technology, and Space of the State of Israel; the German Cancer Research Center (DKFZ); and the Marie Curie International Reintegration Grant. V.K. is an incumbent of the Karl and Frances Korn Career Development Chair in Life Sciences.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.259341.115.
References
- Abounit S, Zurzolo C. 2012. Wiring through tunneling nanotubes—from electrical signals to organelle transfer. J Cell Sci 125: 1089–1098. [DOI] [PubMed] [Google Scholar]
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. 2008. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018. [DOI] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, et al. 2013. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15: 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkwright PD, Luchetti F, Tour J, Roberts C, Ayub R, Morales AP, Rodriguez JJ, Gilmore A, Canonico B, Papa S, et al. 2010. Fas stimulation of T lymphocytes promotes rapid intercellular exchange of death signals via membrane nanotubes. Cell Res 20: 72–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637. [DOI] [PubMed] [Google Scholar]
- Burton DG, Krizhanovsky V. 2014. Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci 71: 4373–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, Mallette FA, Deschenes-Simard X, Ramanathan S, Gagnon J, Moores A, Ilangumaran S, Ferbeyre G. 2009. SOCS1 links cytokine signaling to p53 and senescence. Mol Cell 36: 754–767. [DOI] [PubMed] [Google Scholar]
- Caldwell ME, Denicola GM, Martins CP, Jacobetz MA, Maitra A, Hruban RH, Tuveson DA. 2011. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene 33: 1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F. 2007. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740. [DOI] [PubMed] [Google Scholar]
- Chauveau A, Aucher A, Eissmann P, Vivier E, Davis DM. 2010. Membrane nanotubes facilitate long-distance interactions between natural killer cells and target cells. Proc Natl Acad Sci 107: 5545–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, et al. 2011. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev 25: 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V. 2013. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 27: 2356–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Serrano M. 2010. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10: 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DM, Sowinski S. 2008. Membrane nanotubes: dynamic long-distance connections between animal cells. Nat Rev Mol Cell Biol 9: 431–436. [DOI] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444: 638–642. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci 92: 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey GP, Ben-Yehuda S. 2011. Intercellular nanotubes mediate bacterial communication. Cell 144: 590–600. [DOI] [PubMed] [Google Scholar]
- Freund A, Orjalo AV, Desprez PY, Campisi J. 2010. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16: 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes HH, Rustom A, Wang X. 2013. Tunneling nanotubes, an emerging intercellular communication route in development. Mech Dev 130: 381–387. [DOI] [PubMed] [Google Scholar]
- Guerra C, Barbacid M. 2013. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol Oncol 7: 232–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, Rodriguez-Justo M, Serrano M, Barbacid M. 2011. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 19: 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701. [DOI] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. 2006. Cellular senescence in aging primates. Science 311: 1257. [DOI] [PubMed] [Google Scholar]
- Hong L, Kenney SR, Phillips GK, Simpson D, Schroeder CE, Noth J, Romero E, Swanson S, Waller A, Strouse JJ, et al. 2013. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem 288: 8531–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JI, Lau LF. 2010. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12: 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–551. [DOI] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. 2008. Senescence of activated stellate cells limits liver fibrosis. Cell 134: 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133: 1019–1031. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. 2010. The essence of senescence. Genes Dev 24: 2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL. 2008. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. 2007. Faithful activation of an extra-bright red fluorescent protein in ‘knock-in’ Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol 37: 43–53. [DOI] [PubMed] [Google Scholar]
- Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. 2007. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 21: 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann FE, Eissmann P, Onfelt B, Leung R, Davis DM. 2007. The activating NKG2D ligand MHC class I-related chain A transfers from target cells to NK cells in a manner that allows functional consequences. J Immunol 178: 3418–3426. [DOI] [PubMed] [Google Scholar]
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. 2013. Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118. [DOI] [PubMed] [Google Scholar]
- Naphade S, Sharma J, Gaide Chevronnay HP, Shook MA, Yeagy BA, Rocca CJ, Ur SN, Lau AJ, Courtoy PJ, Cherqui S. 2015. Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 33: 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovadya Y, Krizhanovsky V. 2014. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 15: 627–642. [DOI] [PubMed] [Google Scholar]
- Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, et al. 2013. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi O, Goldstein I, Vernitsky H, Rotblat B, Kloog Y. 2007. Intercellular transfer of oncogenic H-Ras at the immunological synapse. PLoS One 2: e1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi O, Kalman M, Fang Y, Vernitsky H, Jacob-Hirsch J, Foster LJ, Kloog Y, Goldstein I. 2010. Trans-SILAC: sorting out the non-cell-autonomous proteome. Nat Methods 7: 923–927. [DOI] [PubMed] [Google Scholar]
- Roda-Navarro P, Vales-Gomez M, Chisholm SE, Reyburn HT. 2006. Transfer of NKG2D and MICB at the cytotoxic NK cell immune synapse correlates with a reduction in NK cell cytotoxic function. Proc Natl Acad Sci 103: 11258–11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. 2004. Nanotubular highways for intercellular organelle transport. Science 303: 1007–1010. [DOI] [PubMed] [Google Scholar]
- Sagiv A, Krizhanovsky V. 2013. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology 14: 617–628. [DOI] [PubMed] [Google Scholar]
- Sagiv A, Biran A, Yon M, Simon J, Lowe SW, Krizhanovsky V. 2013. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 32: 1971–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamori R, Das S, Yu S, Feng S, Stypulkowski E, Guan Y, Douard V, Tang W, Ferraris RP, Harada A, et al. 2012. Cdc42 and Rab8a are critical for intestinal stem cell division, survival, and differentiation in mice. J Clin Invest 122: 1052–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama R, Sadaie M, Hoare M, Narita M. 2014. Cellular senescence and its effector programs. Genes Dev 28: 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602. [DOI] [PubMed] [Google Scholar]
- Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Kohler K, Oddos S, Eissmann P, Brodsky FM, Hopkins C, et al. 2008. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol 10: 211–219. [DOI] [PubMed] [Google Scholar]
- Stern-Ginossar N, Nedvetzki S, Markel G, Gazit R, Betser-Cohen G, Achdout H, Aker M, Blumberg RS, Davis DM, Appelmelk B, et al. 2007. Intercellular transfer of carcinoembryonic antigen from tumor cells to NK cells. J Immunol 179: 4424–4434. [DOI] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. 2013. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119–1130. [DOI] [PubMed] [Google Scholar]
- Topham NJ, Hewitt EW. 2009. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology 128: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsafrir D, Tsafrir I, Ein-Dor L, Zuk O, Notterman DA, Domany E. 2005. Sorting points into neighborhoods (SPIN): data analysis and visualization by ordering distance matrices. Bioinformatics 21: 2301–2308. [DOI] [PubMed] [Google Scholar]
- Vaziri H, West MD, Allsopp RC, Davison TS, Wu YS, Arrowsmith CH, Poirier GG, Benchimol S. 1997. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J 16: 6018–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. 2008. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 132: 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Khandare A, Burianovskyy L, Maruyama S, Zhang F, Nasjletti A, Goligorsky MS. 2011. Tunneling nanotubes mediate rescue of prematurely senescent endothelial cells by endothelial progenitors: exchange of lysosomal pool. Aging 3: 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. 2013. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.