

ABSTRACT
Influenza A virus (IAV) depends on cellular factors to complete its replication cycle; thus, investigation of the factors utilized by IAV may facilitate antiviral drug development. To this end, a cellular transcriptional repressor, DR1, was identified from a genome-wide RNA interference (RNAi) screen. Knockdown (KD) of DR1 resulted in reductions of viral RNA and protein production, demonstrating that DR1 acts as a positive host factor in IAV replication. Genome-wide transcriptomic analysis showed that there was a strong induction of interferon-stimulated gene (ISG) expression after prolonged DR1 KD. We found that beta interferon (IFN-β) was induced by DR1 KD, thereby activating the JAK-STAT pathway to turn on ISG expression, which led to a strong inhibition of IAV replication. This result suggests that DR1 in normal cells suppresses IFN induction, probably to prevent undesired cytokine production, but that this suppression may create a milieu that favors IAV replication once cells are infected. Furthermore, biochemical assays of viral RNA replication showed that DR1 KD suppressed viral RNA replication. We also showed that DR1 associated with all three subunits of the viral RNA-dependent RNA polymerase (RdRp) complex, indicating that DR1 may interact with individual components of the viral RdRp complex to enhance viral RNA replication. Thus, DR1 may be considered a novel host susceptibility gene for IAV replication via a dual mechanism, not only suppressing the host defense to indirectly favor IAV replication but also directly facilitating viral RNA replication.
IMPORTANCE Investigations of virus-host interactions involved in influenza A virus (IAV) replication are important for understanding viral pathogenesis and host defenses, which may manipulate influenza virus infection or prevent the emergence of drug resistance caused by a high error rate during viral RNA replication. For this purpose, a cellular transcriptional repressor, DR1, was identified from a genome-wide RNAi screen as a positive regulator in IAV replication. In the current studies, we showed that DR1 suppressed the gene expression of a large set of host innate immunity genes, which indirectly facilitated IAV replication in the event of IAV infection. Besides this scenario, DR1 also directly enhanced the viral RdRp activity, likely through associating with individual components of the viral RdRp complex. Thus, DR1 represents a novel host susceptibility gene for IAV replication via multiple functions, not only suppressing the host defense but also enhancing viral RNA replication. DR1 may be a potential target for drug development against influenza virus infection.
INTRODUCTION
Influenza A virus (IAV) infection results in serious respiratory illness and mortality. It is a highly contagious disease and has caused annual epidemics over the past century. Based on the properties of the viral surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA), IAVs are subtyped into different species (1, 2), among which H1N1, H2N2, and H3N2 cover the majority of human infections (3). Potential threats of IAV include consuming public health services and causing huge economic losses; thus, a better understanding of the regulatory mechanism of IAV replication is important for battling these threats.
IAV contains 8 segments of negative-sense, single-stranded viral RNAs (vRNAs), which are packaged into viral ribonucleoproteins (vRNPs), and the viral genome encodes up to 11 proteins. In the early stage of the IAV replication cycle, the virions are internalized through endocytosis into the endosomes of host cells. The low pH of endosomes triggers fusion of the viral membrane with the endosome membrane. In the meantime, M1 is proposed to be ubiquitinated by the ubiquitin ligase ITCH, resulting in the release of the interior viral components into the cytoplasm in a process called “uncoating” (4, 5). The vRNP is subsequently imported into the nucleus, facilitated by the nuclear localization signals of NP (6). vRNP is composed of NP and the viral RNA-dependent RNA polymerase (RdRp), including PB1, PB2, and PA, to carry out both the transcription and replication of the viral genome in the nuclei of infected host cells (7). In the late stage of the IAV replication cycle, the newly synthesized vRNP, M1, and viral envelope proteins assemble the virions, which are subsequently released from the host cell surface.
When host cells are invaded by viruses, the cells immediately respond by synthesizing and secreting alpha and/or beta interferon (IFN-α/β) (IFN pathway), which subsequently binds to its cognate receptors on the cell surface (8, 9). The engagement of IFN-α/β and its receptors activates JAK1 and TYK2 to phosphorylate the STAT1 and STAT2 signal transducers (JAK-STAT pathway). This process generates the ISGF3 transcriptional factor, a complex of STAT1, STAT2, and IRF9, which is transported into the nucleus and induces the expression of interferon-stimulated genes (ISGs), such as MxA, PKR, OAS, and many others (10). ISGs can inhibit IAV replication by using a variety of defense strategies, including translation inhibition, RNA degradation, apoptosis induction, etc. Despite the multiple defense mechanisms, viruses often prevail and cause disease, as viruses usually evolve a weapon(s) to counteract the host defense system. In the case of IAV infection, IFN-α/β synthesis can be inhibited by NS1, thus abolishing the effect of host innate immunity; indeed, IFN-α/β is highly induced when host cells are infected with a delNS1 mutant IAV lacking the NS1 gene (11). From this point of view, blockade of NS1 function by elevating host innate immunity is a promising approach to develop antiviral agents against IAV replication.
IAV transcription is a primer-dependent mechanism, which requires capped RNA primers snatched from the host pre-mRNAs transcribed by RNA polymerase II (Pol II). In eukaryotes, cellular transcription by Pol II involves a dynamic interplay of positive and negative factors (12, 13). At transcription initiation, the TATA box-binding protein (TBP), associated with the TATA box at the promoter, guides the assembly of the preinitiation complex (PIC). The PIC helps to place Pol II over gene transcription start sites, denatures the DNA, and positions the DNA into the active site of Pol II. Negative cofactor 2 (NC2), a repressor of basal transcription, functions through binding to TBP and inhibiting the assembly of the PIC (14). NC2 is composed of 2 subunits, NC2α and NC2β, also known as DRAP1 (DR1-asscociated protein 1) and DR1 (downregulator of transcription 1), respectively. DR1, first identified in HeLa cell nuclear extracts, binds to TBP and precludes the association of TFIIA and TFIIB with TBP to interfere with the assembly of the PIC, thus repressing Pol II transcription (15). DRAP1 is required for DR1-mediated repression of transcription (16). Given the fact that NC2 influences cellular pre-mRNA production, it might have potential to affect IAV RNA transcription.
IAV, like all other viruses, depends not only on viral proteins but also on host cellular factors to accomplish its replication cycle. Identifying novel host factors that participate in each step of the viral replication cycle may accelerate the development of antiviral therapy in the future (17–19). Previously, we performed a genome-wide RNA interference (RNAi) pooled screen to search for host cellular factors participating in the early stage of the IAV replication cycle (5). Based on this screen, we identified a cellular transcriptional repressor, DR1, involved in IAV replication. In this study, we show that knockdown (KD) of DR1 significantly correlates with decreases of viral RNA and protein production. Prolonged DR1 KD stimulates the expression of wide-ranging host innate immunity genes, thereby inhibiting IAV replication. Biochemical studies also revealed that DR1 associates with the three subunits of the viral RdRp complex and stimulates viral RNA replication. Hence, DR1 represents a novel host susceptibility gene which not only suppresses host innate immunity, indirectly providing an environment to facilitate IAV replication, but also directly participates in viral RNA replication. DR1 may be a potential target for antiviral development.
MATERIALS AND METHODS
Cell culture and virus.
HEK293T and MDCK cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; HyClone) and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin; Gibco). A549 cells were maintained in F-12K medium (Gibco) supplemented with 10% FBS (HyClone) and antibiotics (Gibco). All cells were cultured at 37°C in a 5% CO2 incubator. The A/WSN/33 strain of IAV was used in the study.
Primers.
All primers used in this study are listed in Table S2 in the supplemental material.
Generation of DR1 KD cells, wobble rescue, and IAV infection.
All plasmids required for lentivirus production were obtained from the National RNAi Core Facility, Academia Sinica, Taiwan (RNAi Core). The three pLKO.1-shRNA vectors used for knocking down DR1 or ITCH were as follows: TRCN0000013784 (shDR1-1), TRCN0000013787 (shDR1-2), and TRCN0000355778 (shITCH). The pLKO.1-shLacZ control plasmid was TRCN0000072240 (shLacZ). To generate the plasmids for wobble mutant rescue or overexpression experiments, the full-length coding region sequence of DR1, with or without wobble mutations (GCC AAT GAG ATT TGT AAC AAA to GCG AAC GAA ATC TGC AAT AAG) that cannot be targeted by shDR1-1 but do not change the amino acid sequence of DR1, was amplified, tagged with a Myc tag at the C terminus, and then inserted into pLAS2w.Pbsd, expressing a blasticidin S (BSD) selection marker (RNAi Core), at NheI and PmeI sites (pLAS2w-DR1-Myc or pLAS2w-DR1-Myc-wobble). Lentivirus production in HEK293T cells and determinations of virus titers in A549 cells followed the RNAi Core online protocols (http://rnai.genmed.sinica.edu.tw/webContent/web/protocols). Lentivirus transduction and IAV infection experiments mentioned throughout this paper generally followed the procedures described below. To generate DR1 or ITCH KD cells, lentivirus transduction followed the RNAi Core online protocols. Briefly, A549 or HEK293T cells were transduced with a lentivirus carrying shDR1s, shITCH, or the shLacZ control and subsequently selected with 3 μg/ml puromycin (Sigma) for 10 days to generate stable cell lines; for wobble mutant rescue experiments, after puromycin selection, the DR1 KD cells (shDR1-1) were additionally transduced with a lentivirus carrying the DR1 wobble mutant (pLAS2w-DR1-Myc-wobble) or the control (pLAS2w.Pbsd), selected with 10 μg/ml BSD (Invitrogen). For DR1 overexpression assay, A549 cells were transduced with a lentivirus carrying DR1 (pLAS2w-DR1-Myc) or the control (pLAS2w.Pbsd) and subsequently selected with 10 μg/ml BSD. All the resultant cells described above were washed twice with 1× phosphate-buffered saline (PBS) before being infected with IAV in minimal essential medium alpha (MEM-alpha) (Gibco) containing 0.5 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma) at the indicated multiplicity of infection (MOI) for 1 h. Afterwards, the cells were washed twice with 1× PBS to remove excess IAV and then replaced with complete growth medium. The samples were harvested at 6 h postinfection (hpi) for further analysis.
qRT-PCR.
Total RNA was extracted by using a High Pure RNA isolation kit (Roche) according to the manufacturer's protocol. cDNAs were synthesized by using the SuperScript III first-strand synthesis system (Invitrogen), an oligo(dT)20 primer, and an IAV-specific primer (uni-12). Quantitative reverse transcription-PCR (qRT-PCR) assays were performed by using the RealTime Ready universal probe library system (Roche), and the results were analyzed by use of LightCycler 480 software (Roche) according to the manufacturer's protocols. GAPDH mRNA was used as an internal control.
Genome-wide transcriptomic analysis.
shLacZ- or shDR1-1-transduced A549 cells were selected by use of puromycin for 9 days to generate stable cell lines. Afterwards, the cells were infected with IAV at an MOI of 50 to ensure 100% infection. Total RNA at 6 hpi was isolated by using a High Pure RNA isolation kit (Roche). Ten micrograms of RNA of each sample was reverse transcribed into cDNA and labeled with Alexa Fluor 555 by using the SuperScript Plus Indirect cDNA labeling system (Invitrogen), and the labeled cDNA was hybridized by using a SurePrint G3 human gene expression 8 × 60 v2 microarray kit (Agilent Technologies), according to the manufacturers' protocols. Expression analysis and Gene Ontology (GO) analysis were performed by using GeneSpring 12 software (Agilent Technologies).
IFN promoter-driven reporter assay.
shLacZ- or shDR1-1-transduced HEK293T cells at 4 or 10 days posttransfection (dpt) were cotransfected with pIFN-FLuc (containing an IFN promoter-driven firefly luciferase gene) and pRL-TK (containing a thymidine kinase promoter-driven Renilla luciferase gene). Total cell lysates were collected at 24 h posttransfection, and both luciferase activities were measured by using the Dual-Glo luciferase assay system (Promega) according to the manufacturer's protocol. Renilla luciferase activity was used as an internal control to normalize the transfection efficiencies.
IAV internalization assay.
shLacZ- or shDR1-1-transduced A549 cells at 4 or 10 dpt were infected with IAV at an MOI of 10 at 37°C for the indicated times and then washed twice with 1× PBS-HCl (pH 1.3 at 4°C) to remove the attached but not yet internalized virions. Afterwards, the cells were collected, and the total proteins were extracted by using M-PER reagent (Thermo Scientific) according to the manufacturer's protocol and then subjected to Western blotting.
Plaque assay.
MDCK cells were infected with serial 10-fold dilutions of IAV for 1 h, washed twice with 1× PBS, and then overlaid with 0.5% agarose (Lonza) containing MEM-alpha medium. After 2 days, the cells were fixed with 10% formaldehyde (Sigma) and stained with 0.1% crystal violet solution (Sigma).
Virus production assay.
shLacZ- or shDR1-1-transduced A549 cells at 4 or 10 dpt were infected with IAV at an MOI of 0.01 for 1 h, washed twice with 1× PBS, and then replaced with complete growth medium. The supernatants were harvested at the indicated times and titrated by plaque assay.
Suppression of both the JAK-STAT pathway and DR1.
To generate lenti-plasmids containing the indicated short hairpin RNAs (shRNAs), oligonucleotides for the shRNA sequences of TRCN0000231719 (shLuc) and TRCN0000280021 (shSTAT1) were synthesized, annealed, and constructed in pLKO-TRC16 according to RNAi Core online protocols, with pLKO-TRC16-shLuc used as a control. The fragment containing the U6P promoter and shSTAT2 was amplified from TRCN0000364400 and then inserted into pLKO-TRC16-shSTAT1 at the MluI site to generate pLKO-TRC16-shSTAT1/shSTAT2. Lentivirus production, titration, and transduction into A549 cells were performed according to RNAi Core online protocols. Briefly, A549 cells were transduced with the lentivirus carrying the shSTAT1/shSTAT2 clone to knock down the JAK-STAT pathway or with shLuc as a control and then selected by use of BSD for 7 days. Afterwards, the KD cells were transduced with the lentivirus carrying the shDR1-1 clone to knock down the endogenous DR1 or with shLacZ as a control and then selected by use of puromycin for an additional 9 days. The resultant KD cells were subjected to qRT-PCR assays or infected with IAV for Western blotting at 6 hpi.
Nuclear import of vRNP.
shLacZ- or shDR1-1-transduced A549 cells at 4 dpt were infected with IAV at an MOI of 5 at 37°C for the indicated times and then washed twice with 1× PBS-HCl (pH 5.5 at 4°C) to remove the attached but not internalized virions. Afterwards, the cells were harvested. The nucleus and cytoplasm were fractionated by use of NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific) according to the manufacturer's protocol, and their total RNAs were extracted by use of a High Pure RNA isolation kit (Roche). cDNA synthesis and qRT-PCR assays were performed as described above. The primers for reverse transcription were U6 snRNA-SYBR-Rv and oligo(dT)20 for the nuclear RNA and the cytoplasmic RNA, respectively, and uni-12 for both fractionated RNAs. The values for nuclear and cytoplasmic vRNAs were normalized with U6 small nuclear RNA (U6 snRNA) and GAPDH mRNA, respectively.
RNA-based minireplicon assay.
The DNA templates for an in vitro transcription system were prepared by the PCR method to incorporate a T7 promoter, a full-length coding sequence of the indicated gene, and an oligo(dA)30 tail into each PCR product. Capped PB1, PB2, PA, NP, and Renilla luciferase (RLuc) mRNAs were synthesized by using the Ribo m7G cap analog (Promega) and a MEGAscript kit (Invitrogen) according to the manufacturers' protocols. A virus-like negative-strand firefly luciferase RNA (FLuc-vRNA) in which the luciferase coding region was flanked by the packaging signals of the NS segment was synthesized by the same procedures as those described above.
To perform the RNA-based minireplicon assay, shLacZ- or shDR1-1-transduced HEK293T cells at 4 dpt were seeded for 24 h prior to transfections in 12-well plates. The cells in each well were cotransfected with 0.5 μg each of PB1, PB2, PA, and NP mRNAs and FLuc-vRNA and 0.01 μg RLuc-mRNA (3P/NP transfection) or with only FLuc-vRNA and RLuc-mRNA, as negative controls (mock transfection), by using DMRIE-C reagent (Invitrogen) according to the manufacturer's protocol. FLuc-vRNA was used as a template for the viral RdRp, including PB1, PB2, PA, and NP expressed from the corresponding mRNAs, to generate FLuc activity. RLuc activity expressed from RLuc-mRNA was used as an internal control to normalize the transfection efficiencies. Total cell lysates were collected at 24 h posttransfection, and both luciferase activities were measured by using the Dual-Glo luciferase assay system (Promega) according to the manufacturer's protocol.
Immunoprecipitation.
For the expression of DR1, the coding region sequence of DR1 was amplified, tagged with a Myc tag at the C terminus, and then inserted into pCAG.2 at NheI and NotI sites (pCAG.2-DR1-Myc; DR1-Myc). Plasmids expressing HA-tagged viral proteins were used as we described previously (20). For in vitro immunoprecipitation, HEK293T cells were cotransfected with one of the plasmids expressing viral proteins and DR1-Myc. Cells expressing only DR1-Myc or DR1-Myc with HA-tagged glutathione S-transferase (GST) were used as negative controls. Total cell lysates were extracted by using M-PER reagent (Thermo Scientific) at 48 hpt. The immunoprecipitation of HA-tagged proteins was carried out using a Profound Mammalian HA-Tag IP/co-IP kit (Thermo Scientific) according to the manufacturer's protocol, and the samples were analyzed by Western blotting. For in vivo immunoprecipitation, HEK293T cells were transfected with DR1-Myc for 48 h before infection with IAV at an MOI of 5 and then were harvested at 10 hpi. Cells only transiently expressing DR1-Myc, without IAV infection, or only infected with IAV were used as negative controls. Total cell lysates were extracted with M-PER reagent (Thermo Scientific) and immunoprecipitated with protein G-agarose (Roche) conjugated with the indicated antibodies according to the manufacturer's protocol. The final samples were analyzed by Western blotting.
Antibodies.
The antibodies used for immunoblotting were as follows: mouse anti-DR1 (ab88597) and mouse anti-NP (ab20343) antibodies were purchased from Abcam; mouse anti-actin antibody (MAB1501) was purchased from Millipore; mouse anti-NS1 (sc-130568), rabbit anti-HA (sc-805), and rabbit anti-Myc (sc-789) antibodies were purchased from Santa Cruz; mouse anti-M1 antibody (MCA401) was purchased from AbDSerotec; and rabbit anti-STAT1 (9712) and rabbit anti-P-STAT1(Tyr701) (9171) antibodies were purchased from Cell Signaling. For immunoprecipitation and immunoblotting, rabbit anti-PB1 (GTX125923), rabbit anti-PA (GTX118991), and rabbit anti-NS1 (GTX125990) antibodies were purchased from GeneTex, and goat anti-PB2 antibody (sc-17603) was purchased from Santa Cruz.
RESULTS
DR1 serves a positive role in IAV replication.
To identify the cellular factors involved in the early stage of the IAV replication cycle, we performed a high-throughput pooled RNAi library screen (5); the candidate genes were subsequently subjected to a secondary small-scale RNAi screen. In the secondary screen, the following two criteria were set for identifying candidate genes: (i) detecting a significant reduction of nuclear NP fluorescence signals in the shRNA-KD cells at 6 hpi compared to those of the control cells and (ii) having at least 2 hits of distinct shRNAs with such a phenotype. Based on these criteria, DR1 was identified as such a cellular factor and was selected for further studies.
To validate the role of DR1 in IAV replication, the endogenous DR1 in A549 cells was knocked down by transduction with a lentivirus carrying an shDR1 clone (shDR1-1 or shDR1-2) or an shLacZ control for 10 days to generate stable cell lines, followed by IAV infection. qRT-PCR assays and Western blotting of DR1 showed that both independent shDR1 clones had good KD efficiencies (Fig. 1A); correspondingly, the amounts of NP vRNA and viral protein (NP and NS1) production were substantially suppressed at 6 hpi (Fig. 1A and B). Conversely, overexpression of Myc-tagged DR1 in A549 cells was accompanied by increased NP production (Fig. 1C). Also, the virus titers of IAV produced from both DR1 KD cell lines were 2- to 3-log lower than those from the control cells at 24 or 48 hpi (Fig. 1D). In addition, there was no significant alteration of the DR1 protein level in A549 cells during IAV infection (data not shown), suggesting that DR1 expression is not tightly regulated by IAV infection. Taken together, these data showed that DR1 KD reduced IAV replication, with effects opposite to those of DR1 overexpression, suggesting that DR1 plays a positive role in IAV replication.
FIG 1.

DR1 acts as a positive regulator of IAV replication. A549 cells were transduced with the indicated lentiviruses for 10 days to generate stable cell lines and then infected with IAV for 6 h at an MOI of 1 for qRT-PCR assay or Western blotting. (A and B) The stable cells harboring shDR1-1, shDR1-2, or the shLacZ control were infected with IAV, harvested, and then subjected to qRT-PCR assays or Western blotting by using the indicated antibodies. Cellular DR1 mRNA and NP vRNA were measured and normalized to GAPDH mRNA, and the value for the control cells was set to 1. Data represent means ± standard deviations (SD) (n ≥ 3; *, P ≤ 0.05 by Student's t test). (C) For overexpression assay, overexpressed Myc-tagged DR1 (DR1-Myc OE) or control (vector only) cells at 8 dpt were infected with IAV, harvested, and then subjected to Western blotting. The band intensities of NP and actin were quantified, and the NP/actin ratios are shown below the blots. (D) The stable DR1 KD and shLacZ control A549 cells were infected with IAV at an MOI of 0.01. At 24 or 48 hpi, the supernatants were titrated by plaque assay to determine the virus titers. Data represent means ± SD (n ≥ 3; *, P ≤ 0.05 by Student's t test). (E) For the wobble mutant rescue experiment, the DR1 KD (shDR1-1) and shLacZ control cells were further transduced with a lentivirus overexpressing a DR1 wobble mutant (DR1-Myc-wobble) or the control (vector only) for an additional 8 days before being infected with IAV, and the cell lysates were subjected to Western blotting.
To rule out the possibility that the observed inhibition of IAV replication by DR1 KD was due to off-target effects, we performed a rescue experiment using a wobble DR1 mutant that expresses wild-type DR1 but cannot be targeted by shDR1-1 due to wobble mutations in the gene. As shown in Fig. 1E, endogenous DR1 was substantially knocked down by shDR1-1 (lanes 2 and 4). The expression of the Myc-tagged DR1 wobble mutant could be detected by Western blotting (Fig. 1E, lanes 3 and 4); correspondingly, the viral protein production was restored in the DR1 wobble mutant-expressing cells (compare lanes 2 and 4). These results demonstrated that shDR1-1 directly targeted the endogenous DR1 to inhibit IAV replication; therefore, we concluded that DR1 specifically enhances IAV replication. Based on the aforementioned results, shDR1-1 was chosen for further studies.
DR1 KD stimulates the expression of host innate immunity genes.
Given that DR1 functions as a general repressor in cellular transcription (15, 21), DR1 KD might have altered gene expression profiles of the host cells, thereby indirectly affecting IAV replication. To address this possible mechanism, the stable DR1 KD and control A549 cells were infected with IAV (shDR1-1-IAV or shLacZ-IAV) or mock infected (shDR1-1-Mock or shLacZ-Mock) for 6 h and then subjected to microarray analysis. Comparing shDR1-1-Mock with shLacZ-Mock control, the results showed that a total of only 360 genes were affected 3-fold or more in the DR1 KD cells in the absence of IAV infection; among them, 272 and 88 genes were up- and downregulated, respectively (Table 1). Analyses of genes affected by DR1 KD with a 2- or 5-fold change cutoff led to similar conclusions. These results showed that DR1 not only suppresses but also stimulates cellular transcription. Thus, in contrast to previous reports, DR1 is not a general repressor but regulates only a small subset of cellular genes. Furthermore, compared to shLacZ-Mock, IAV infection affected the expression of a large number of genes (shLacZ-IAV), but DR1 KD reduced the number of such genes by approximately 40% (shDR1-1-IAV) (Table 1), suggesting that DR1 KD alters the large-scale gene expression profiles of the host cells in IAV infection.
TABLE 1.
Comparison of expression profiles affected by DR1 KD with or without IAV infectiona
| Gene regulation | No. of genes (normalized to that for shLacZ-Mock) |
||
|---|---|---|---|
| shDR1-1-Mock | shLacZ-IAV | shDR1-1-IAV | |
| ≥2-fold up | 709 | 6,994 | 4,486 |
| ≤2-fold down | 367 | 3,589 | 1,497 |
| ≥3-fold up | 272 | 4,885 | 2,896 |
| ≤3-fold down | 88 | 1,335 | 353 |
| ≥5-fold up | 108 | 1,884 | 875 |
| ≤5-fold down | 12 | 251 | 48 |
Microarray analysis was performed on stable DR1 KD and control A549 cells (at 10 dpt), with or without IAV infection at an MOI of 50. At 6 hpi, the total RNA samples were subjected to microarray analysis. Each condition of the microarray data was analyzed by GeneSpring 12 software and normalized to the shLacZ-Mock sample, and the numbers of genes with different fold changes under different conditions were obtained by volcano plot analysis (P ≤ 0.05 by Student's t test).
To gain insight into the nature of the genes regulated by DR1 KD, the genes affected 3-fold or more in shDR1-1-Mock (a total of 360 genes) (Table 1) were subjected to GO analysis. The analysis showed that 60% of significant GO terms were involved in diverse categories not directly connected to virus; notably, the other 40% of significant GO terms were related to immune system regulation, and most were interferon- or cytokine-related terms (see Table S1 in the supplemental material), suggesting that a significant portion of DR1-regulated genes are involved in host innate immunity. Therefore, we further studied the gene expression profiles of host innate immunity for DR1 KD. The DR1 KD-associated signal transducers in the IFN pathway (RIG-I, MDA5, IRF1, and IRF7) or the JAK-STAT pathway (STAT1, STAT2, and IRF9), all of which have been reported to be involved in IAV infection (22), were first analyzed by qRT-PCR assays. All of the aforementioned genes in the DR1 KD cells were upregulated 3- to 70-fold in the absence of IAV infection (Fig. 2) and further enhanced in the presence of IAV infection (data not shown), confirming the microarray data and implying that downstream ISG expression may be induced by DR1 KD even without IAV infection. Therefore, we further analyzed ISG expression profiles from the microarray data. An ISG list of 371 genes, based on a published study (23), was used. Compared to the case in shLacZ-Mock cells (data not shown), 44 ISGs in the DR1 KD cells were upregulated 2-fold or more in the absence of IAV infection (shDR1-1-Mock). In the presence of IAV infection (shDR1-1-IAV or shLacZ-IAV), 107 and 57 ISGs in the DR1 KD and control cells, respectively, were upregulated. These data demonstrate that DR1 KD increases the expression of a large number of genes involved in host innate immunity, such as the signal transducers and the ISG family, suggesting that DR1 in normal cells may suppress host innate immunity, which, in turn, may create a beneficial milieu for IAV replication once the host cells are infected.
FIG 2.
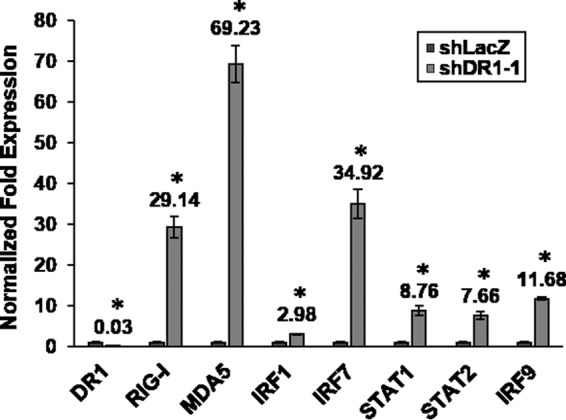
The expression levels of DR1 KD-induced signal transducers in host innate immunity were analyzed by qRT-PCR assays in the absence of IAV infection. Cellular signal transducer mRNAs were measured and normalized to GAPDH mRNA, with the value for each gene in the control cells set to 1. Data represent means ± SD (n ≥ 3; *, P ≤ 0.05 by Student's t test).
DR1 KD stimulates IFN induction in a time-dependent manner, thereby activating the JAK-STAT pathway to induce ISG expression.
To study the mechanism of the enhancement of host innate immunity by DR1 KD, we first studied the mechanism of IFN induction. Since host innate immunity genes are regulated by a cascade of signaling molecule expression, we studied the kinetics of gene expression following DR1 KD. The E3 ubiquitin ligase ITCH was included in these experiments to serve as a control, since it is not a transcriptional factor but enhances IAV replication by promoting IAV entry (5). For this purpose, DR1 or ITCH was knocked down by transduction of cells with a lentivirus carrying shDR1-1 or shITCH, and cells were harvested at 4, 7, or 10 dpt. The results showed that the DR1 or ITCH KD A549 cells had reached maximum KD efficiencies at 4 dpt and remained the same subsequently (Fig. 3A). The amounts of IFNB1 induction were about the same between the DR1 and control cells at 4 dpt; in contrast, IFNB1 induction in DR1 KD cells gradually increased until it reached a 3-fold higher level at 10 dpt (Fig. 3B). The induction of IFNB1 was confirmed by increased luciferase activities in IFN promoter-driven reporter assays (Fig. 3C). These data suggest that DR1 KD is associated with IFNB1 induction in a time-dependent manner and that IFNB1 induction may result from a cascade of gene regulation triggered by DR1 KD, thus explaining the delay between DR1 KD and IFNB1 induction.
FIG 3.

DR1 KD effects on IFNB1 induction and ISG expression at different postransduction times in the absence of IAV infection. DR1 KD, ITCH KD, and shLacZ control A549 cells at 4, 7, or 10 dpt were infected with IAV for 6 h at an MOI of 1 for qRT-PCR assays or Western blotting. (A and B) Cellular DR1, ITCH, or IFNB1 mRNA levels were determined and normalized to the GAPDH mRNA level. The value for shLacZ control cells under each condition was set to 1. Data represent means ± SD (n ≥ 3; *, P ≤ 0.05 by Student's t test). (C) DR1 KD and shLacZ control HEK293T cells at 4 or 10 dpt were cotransfected with pIFN-FLuc and pRL-TK. At 24 h posttransfection, the total cell lysates were subjected to luciferase activity assay. The value for the control cells was set to 1. Data represent means ± SD (n ≥ 3; *, P ≤ 0.05 by Student's t test). (D) Cell lysates obtained under each condition were subjected to Western blotting by using the indicated antibodies. (E) ISG mRNA levels under each condition were determined and normalized to those of GAPDH mRNA. The value for shLacZ control cells under each condition was set to 1. Data represent means ± SD (n ≥ 3; *, P ≤ 0.05 by Student's t test).
IFN-β induction is expected to activate the JAK-STAT pathway; we therefore analyzed the amounts of STAT1 and phosphorylated STAT1 (active form) in the DR1 KD cells. The results showed that the total amount of STAT1 at 10 dpt, but not 4 dpt, was significantly increased in DR1 KD cells (Fig. 3D), consistent with the qRT-PCR data (Fig. 2 and 3E). Also, phosphorylated STAT1 was detected in the DR1 KD cells at 10 dpt but not 4 dpt. These data suggest that the JAK-STAT pathway is activated by IFN-β induction in DR1 KD cells, but only after prolonged DR1 KD.
To address whether the activation of the JAK-STAT pathway by DR1 KD could induce ISG expression, we analyzed the expression levels of the signal transducers of the JAK-STAT signaling pathway and the representative ISGs, which have been reported to be involved in IAV infection (8, 9). The results showed that the various ISGs were not induced until 10 dpt, when there were at least 5-fold increases of all the ISGs (Fig. 3E). Above all, these data confirm the microarray data and demonstrate a good correlation between the activation of the JAK-STAT pathway and the enhancement of ISG expression after DR1 KD. Also, these findings demonstrate that there is a time lag between DR1 KD and the activation of IFN-β and ISGs, suggesting that the mechanism of activation of the IFN pathway is through a cascade of signal transduction and gene expression mediated by DR1, rather than the direct transcription-inhibitory effect of DR1.
To correlate the enhanced ISG expression with the possible inhibitory activity on IAV replication, we examined the DR1 or ITCH KD cells at 4 or 10 dpt in the presence of IAV infection. The qRT-PCR results showed that the amounts of NP vRNA in the DR1 KD cells were lower than those in the control cells, by about 50% at 4 dpt and over 93% at 10 dpt (Fig. 4A), consistent with the ISG expression levels in Fig. 3E. In contrast, the amount of NP vRNA in the ITCH KD cells was lower than that in the control cells, by 38% at 4 dpt, and remained at this level at 10 dpt. Western blotting of viral proteins after DR1 KD showed a pattern similar to that for the qRT-PCR data (Fig. 4B), demonstrating that IAV replication was only moderately suppressed (50%) at an early time point after DR1 KD (4 dpt) but dramatically suppressed (more than 99%) after prolonged DR1 KD (10 dpt).
FIG 4.
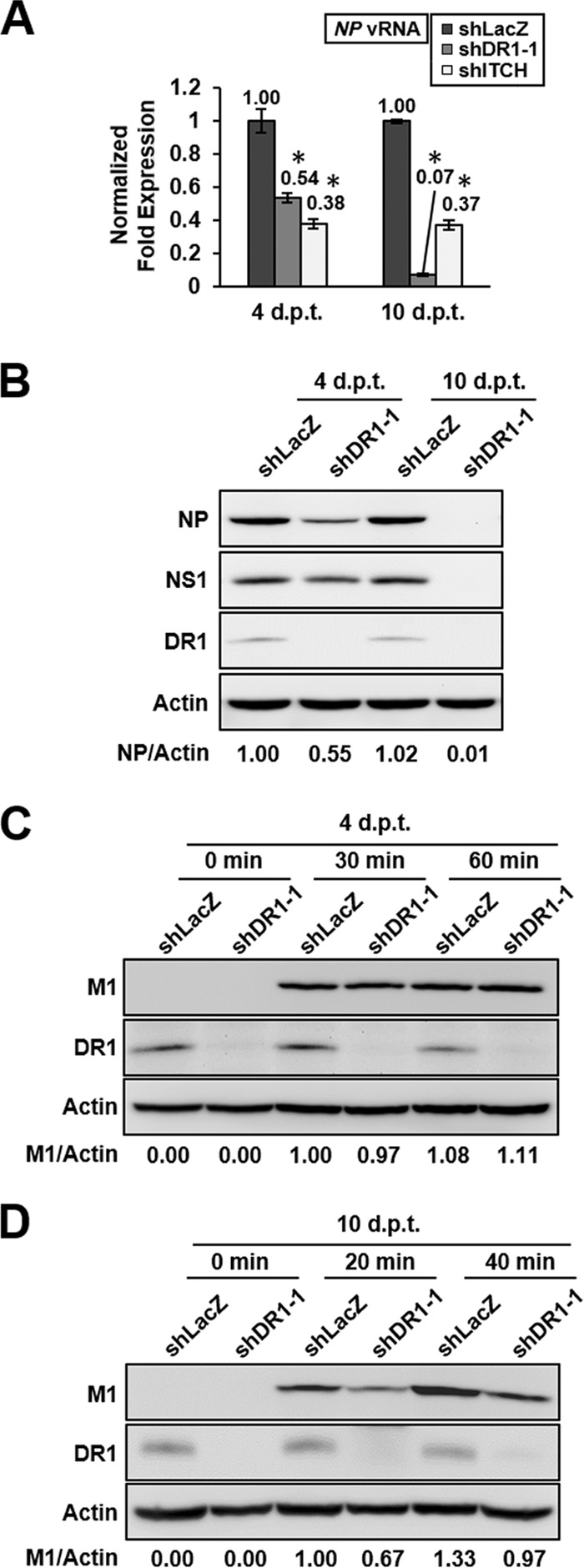
DR1 KD effects on IAV replication with early or prolonged DR1 KD. (A and B) DR1 KD and shLacZ control A549 cells at 4 or 10 dpt were infected with IAV for 6 h at an MOI of 1 for qRT-PCR assays or Western blotting by using the indicated antibodies. NP vRNA levels were determined and normalized to the GAPDH mRNA level. The value for shLacZ control cells under each condition was set to 1. Data represent means ± SD (n ≥ 3; *, P ≤ 0.05 by Student's t test). (C and D) DR1 KD and shLacZ control A549 cells at 4 or 10 dpt were infected with IAV at an MOI of 10 (at 37°C) for the indicated times, harvested, and then subjected to Western blotting using the indicated antibodies. The band intensities for M1 and actin were quantified, and the M1/actin ratios are shown below the blots.
Different ISGs inhibit different steps of the IAV replication cycle (8, 9, 23). Among these ISGs, IFITMs, which were enhanced by prolonged DR1 KD (10 dpt), have been reported to restrict IAV entry; thus, we examined whether IAV internalization was affected by DR1 KD. For this purpose, the DR1 KD and control A549 cells at 4 or 10 dpt were incubated with IAV at 37°C for the indicated times and washed extensively to remove the uninternalized virions. The internalized virions were detected by immunoblotting against M1, which has commonly been used as a marker of IAV internalization (24). The results showed that the amounts of internalized virions in the DR1 KD cells at 4 dpt were almost the same as those in the control cells (Fig. 4C), even though DR1 expression was almost completely shut off at this time point. In contrast, the amounts of internalized virions in the DR1 KD cells at 10 dpt were lower than those in the control cells, by about 33% to 36% (Fig. 4D), consistent with the kinetics of ISG induction in the DR1 KD cells (Fig. 3E), in which ISGs were enhanced by DR1 KD at 10 dpt but not at 4 dpt, suggesting that the enhanced ISG expression was associated with the inhibition of IAV internalization.
Overall, these data are consistent with, though they do not directly prove, the following sequence of events: prolonged DR1 KD stimulates IFN-β production, which consequently leads to the enhancement of ISG expression, thereby inhibiting IAV replication. It stands to reason that DR1 in normal cells may suppress IFN induction by its transcription-inhibitory activity; in the presence of IAV infection, this suppression may facilitate IAV replication. Thus, DR1 may represent a host susceptibility gene for IAV replication.
Abolishment of DR1 KD-associated ISG induction rescues IAV replication.
To support the above-mentioned sequence of events, we studied whether abolishment of DR1 KD-associated ISG induction could rescue IAV replication. For this purpose, the JAK-STAT pathway was disrupted in the DR1 KD cells, since this pathway mainly controls ISG induction (10) and also was activated by DR1 KD (Fig. 3D). Thus, both the JAK-STAT pathway and DR1 of A549 cells were knocked down by use of the respective shRNAs; the resultant STAT1/STAT2-KD cells, with or without DR1 KD, were used for IAV infection. We chose 4 ISGs (CXCL10, ISG15, MX1, and IFITM1) that have been reported to be involved in IAV infection (8, 9, 25) as representative ISGs. As shown in Table 2, both STAT1 and STAT2 were induced about 5-fold in the presence of shDR1, and the 4 ISGs were also enhanced, by as much as 400-fold. The induction of STAT1 and STAT2 by DR1 KD was suppressed by shSTAT1 and shSTAT2, as expected (compare both columns of Table 2); correspondingly, the induction of all ISGs by DR1 KD was almost completely abolished as well. Furthermore, DR1 KD caused an almost complete shutoff of viral protein production, but this inhibition was restored by shSTAT1 (Fig. 5). These results demonstrate that abolishment of DR1 KD-associated ISG induction can rescue IAV replication and suggest that the antiviral effect of DR1 KD on IAV replication may be mainly through activating the JAK-STAT pathway to induce ISG expression.
TABLE 2.
ISG expression with prolonged DR1 KD in the absence or presence of repression of STAT1 and STAT2a
| ISG | qRT-PCR result (normalized to that for shLuc/shLacZ) |
|
|---|---|---|
| shLuc/shDR1-1 | shSTAT1/shSTAT2/shDR1-1 | |
| DR1 | 0.03 ± 0.01 | 0.03 ± 0.01 |
| STAT1 | 5.94 ± 0.19 | 0.80 ± 0.08 |
| STAT2 | 5.20 ± 0.14 | 0.52 ± 0.08 |
| CXCL10 | 70.41 ± 4.56 | 2.54 ± 0.09 |
| ISG15 | 347.69 ± 4.65 | 3.46 ± 0.07 |
| MX1 | 402.93 ± 8.77 | 3.20 ± 0.13 |
| IFITM1 | 208.95 ± 5.21 | 4.06 ± 0.06 |
A549 cells were transduced with a lentivirus expressing both shSTAT1 and shSTAT2 to repress the JAK-STAT pathway or with the shLuc control and then were transduced with a lentivirus expressing shDR1-1 to knock down endogenous DR1 (or with the shLacZ control) (see Materials and Methods). All resultant KD cells were subjected to qRT-PCR assays. The value for each gene in the shLuc/shLacZ control cells was set to 1. Data represent means ± SD (n ≥ 3).
FIG 5.
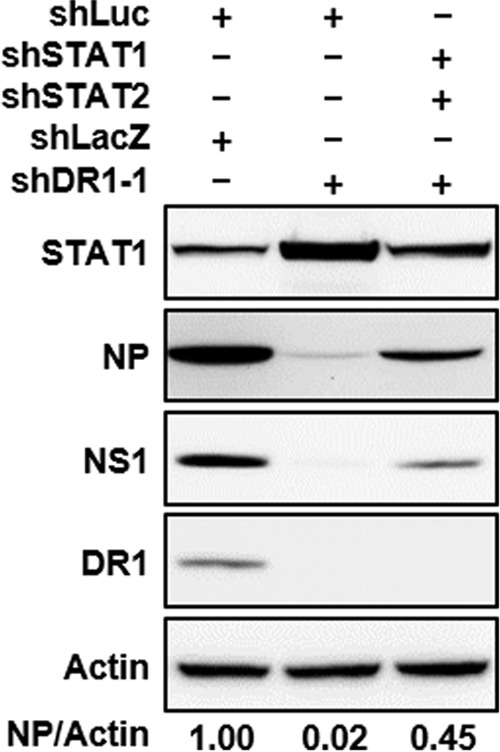
Abolishment of DR1 KD-associated ISG induction restored IAV replication. A549 cells were transduced with shRNAs as indicated at the top and then infected with IAV at an MOI of 1 for 6 h. Cells harboring both shLacZ and shLuc served as a negative control. The cell lysates were subjected to Western blotting using the indicated antibodies. The band intensities for NP and actin were quantified, and the NP/actin ratios are shown below the blots.
DR1 KD also inhibits viral RNA replication.
The aforementioned data showed that at an early stage of DR1 KD (4 dpt), there was a moderate reduction of IAV replication even though there was no significant induction of IFN-β and ISGs (Fig. 3 and 4); also, IAV internalization was not affected under these conditions (Fig. 4C), suggesting that the moderate reduction of IAV replication at early times of DR1 KD may not be contributed by ISGs but by other modes of action. In addition, the experimental design of the secondary screen (5), which transiently silenced cellular genes and detected NP production at 6 hpi, indicates that DR1 is likely involved in the early stage of the IAV replication cycle. To assess which step in the early stage of the IAV replication cycle is affected by early DR1 KD, we also examined nuclear import of vRNP and the viral RdRp activity.
To examine nuclear import of vRNP, the DR1 KD A549 cells at 4 dpt were infected with IAV at 37°C for 15 or 25 min and then harvested for subcellular fractionation. The amounts of vRNA in the nucleus and in the cytoplasm were analyzed by qRT-PCR assay. The results showed that the percentage of nuclear vRNA in the DR1 KD cells was slightly lower than that in the control cells, indicating that the nuclear import of vRNA was slightly impaired in the DR1 KD cells (Fig. 6A). However, this minor variation (<9%) is likely not sufficient to account for the observed 40 to 50% reduction in IAV replication at early DR1 KD (4 dpt) (Fig. 4).
FIG 6.
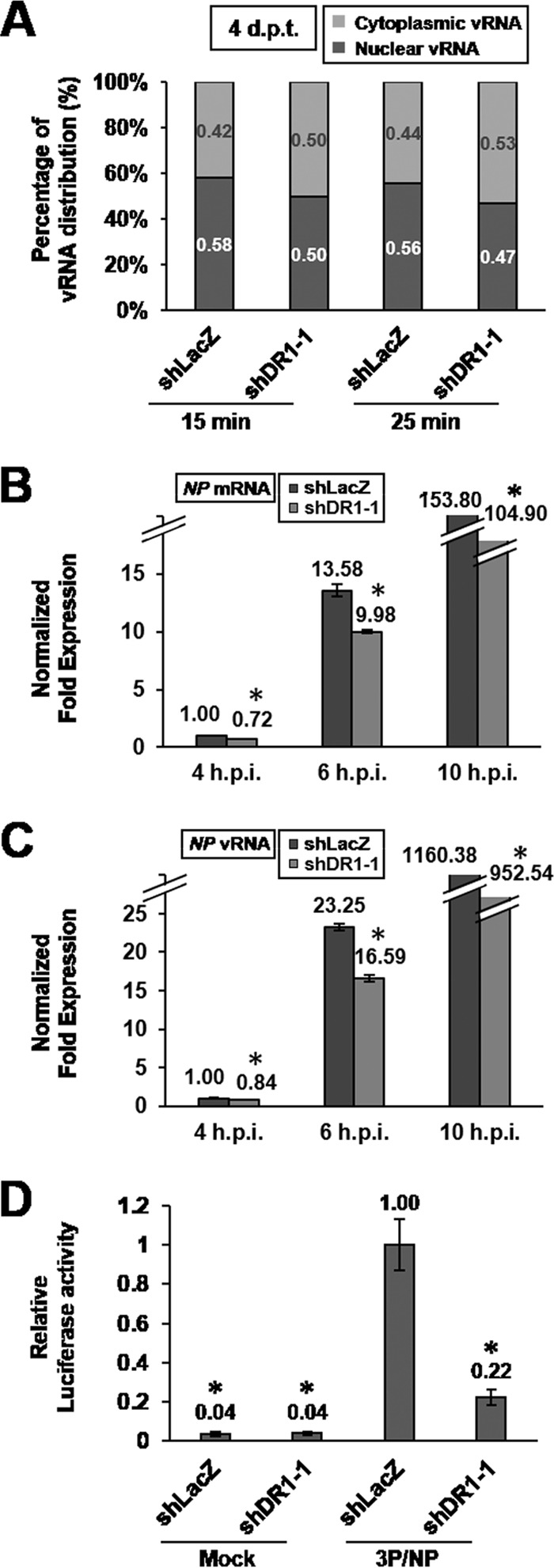
Early DR1 KD affects viral RNA transcription and replication. (A) DR1 KD and shLacZ control A549 cells at 4 dpt were infected with IAV at an MOI of 5 (at 37°C) for 15 or 25 min, harvested, and then subjected to subcellular fractionation of total RNA. The NP vRNA in different fractions was measured by qRT-PCR and normalized to U6 snRNA or GAPDH mRNA for the nuclear fraction or the cytoplasmic fraction, respectively. The percentage of vRNA was the ratio of the amount of vRNA from each fraction to the amount of vRNA from whole cells. Data represent the means for two independent experiments. (B and C) DR1 KD and shLacZ control A549 cells at 4 dpt were infected with IAV at an MOI of 1 for the indicated times, harvested, and then subjected to qRT-PCR assays. Data represent means ± SD (n ≥ 3; *, P ≤ 0.05 by Student's t test). (D) DR1 KD and shLacZ control HEK293T cells at 4 dpt were transfected with PB1, PB2, PA, and NP mRNAs, RLuc-mRNA, and FLuc-vRNA (3P/NP), or only with FLuc-vRNA and RLuc-mRNA, as negative controls (mock). At 24 h posttransfection, the total cell lysates were subjected to luciferase activity assay. The value for the control cells transfected with 3P/NP was set to 1. Data represent means ± SD (n ≥ 3; *, P ≤ 0.05 bt Student's t test).
We next monitored the time course of viral RNA synthesis by cells at an early time of DR1 KD (4 dpt). The results showed that the amounts of both NP mRNA and vRNA were reduced in the DR1 KD cells at all time points after infection compared to those in the control cells (Fig. 6B and C), suggesting that the viral RdRp activity was reduced by DR1 KD. We also independently examined the effects of DR1 KD on viral transcription and replication by using a modified influenza virus minireplicon assay. The conventional minireplicon assay (26) is based on the expression of the viral RdRp, NP, and a reporter RNA from the respective expression plasmids. However, because DR1 functions as a repressor of cellular transcription, the conventional plasmid-based influenza virus minireplicon assay may be biased by different transcription efficiencies of the input plasmids between the DR1 KD and control cells. To circumvent this potential problem, we developed an RNA-based minireplicon assay to bypass the effect of DR1 KD on the transcription of the input plasmids. For this purpose, 6 RNAs, including the mRNAs for PB1, PB2, PA, NP, and RLuc and the reporter RNA FLuc-vRNA, were generated by in vitro transcription from the appropriate PCR fragments. These RNAs were cotransfected into the DR1 KD HEK293T cells at 4 dpt, and the luciferase activity was then determined at 24 h posttransfection. The results showed that the amounts of the viral RdRp (PB1, PB2, and PA; 3P) and NP in both DR1 KD and control cells were almost the same at 24 h posttransfection (data not shown). Also, luciferase activity could be detected only in those cells transfected with 3P/NP (Fig. 6D), indicating the authenticity of the RNA-based minireplicon assay. Significantly, the luciferase assay showed that the viral RdRp activity in the DR1 KD cells was lower than that in the control cells, by about 80% (Fig. 6D). Taken together, these data demonstrate that the viral RdRp activity is reduced at early times of DR1 KD, suggesting that DR1 facilitates viral RNA replication in IAV infection.
DR1 specifically interacts with all three subunits of the viral RdRp complex in vivo.
To investigate whether DR1 is present in the viral RdRp complex, thereby affecting viral RdRp activity, we performed an in vitro coimmunoprecipitation assay between DR1 and individual components of the viral RdRp. HEK293T cells were transfected with plasmids expressing Myc-tagged DR1 (DR1-Myc) and one of the HA-tagged viral or control proteins, and then the total cell lysates were immunoprecipitated with anti-HA agarose. As shown in Fig. 7A, DR1-Myc was coprecipitated with PB1-, PB2-, and PA-HA (lanes 8 to 10, respectively) but not with NP-HA or NS1-HA (lanes 11 and 12). These results indicate that DR1 interacts with the three subunits of the viral RdRp complex. Moreover, we performed an in vivo coimmunoprecipitation assay to independently verify the in vitro results for the IAV-infected cells. HEK293T cells were first transfected with a DR1-Myc-expressing plasmid for 48 h before IAV infection, and the total lysates at 10 hpi were immunoprecipitated with the indicated protein G-agarose-conjugated antibodies, including anti-PB1, anti-PB2, anti-PA, anti-NP, and anti-NS1 antibodies. As shown in Fig. 7B, DR1-Myc was specifically coprecipitated in vivo with all three subunits of the viral RdRp complex. Interestingly, in this in vivo system, DR1-Myc was also coprecipitated with NP, which was not seen in the in vitro assay. This result suggests that DR1 might be present in the viral RdRp complex, so NP may indirectly coprecipitate DR1-Myc. In contrast, DR1-Myc could not be coprecipitated with NS1, consistent with the in vitro result in Fig. 7A. In summary, these data suggest that DR1 enhances viral RNA replication, likely through direct association with the three subunits of the viral RdRp complex in IAV-infected cells.
FIG 7.

In vitro and in vivo pulldown assays of DR1 with distinct components of the viral RdRp complex. (A) HEK293T cells were cotransfected with the indicated plasmids expressing individual HA-tagged viral proteins and Myc-tagged DR1. Cell lysates were harvested at 48 h posttransfection and immunoprecipitated (IP) by using anti-HA agarose. The resultant products were subjected to Western blotting. The input cell lysates are shown in the left panel. (B) HEK293T cells were transfected with or without Myc-tagged DR1 (DR1-Myc) for 48 h before infection with or without IAV at an MOI of 5 for 10 h. The cell lysates were immunoprecipitated by using protein G-agarose conjugated with the indicated antibodies. The resultant products were subjected to Western blotting by using the indicated antibodies. The input cell lysates are shown in the left panel.
DISCUSSION
Virus infection relies on complex interplays between viruses and their hosts (27). In our previous study, we identified a set of host factors with the potential to be involved in the early stage of the IAV replication cycle (5). Among them, a cellular transcriptional repressor, DR1, was identified. Our current studies demonstrate that DR1 serves as a positive regulator facilitating IAV replication by suppressing host innate immunity and is also involved in viral RNA replication. A genome-wide transcriptomic analysis revealed that DR1 KD affected only a small subset of genes, among which are wide-ranging host innate immunity genes, including RIG-I, MDA5, IRF7, IFNB1, STAT1, STAT2, IRF9, ISG15, MX1, IFITM genes, etc., most of which have been reported to function in the IAV replication cycle (8, 9). For example, a ubiquitin-like protein, ISG15, conjugates with NS1 and inhibits NS1 function to reduce IAV replication (28); MX1, a GTPase, associates with PB2 and prevents viral transcription (29); and the IFITM family inhibits the viral entry of IAV (30). Indeed, in DR1 KD cells, the enhanced ISGs inhibited IAV internalization (Fig. 4D). Taken together, these data suggest that in uninfected cells, DR1 per se suppresses host innate immunity genes by its transcription-inhibitory activity on IFN induction (Fig. 3B and C), thereby preventing untimely release and unwanted deleterious effects of downstream ISGs. But in IAV-infected cells, this suppression of ISGs by DR1 might help to create a beneficial environment to indirectly facilitate IAV replication. Thus, DR1 may be considered a host susceptibility gene for IAV replication. If this is true, one may predict that DR1 should also facilitate the infection and replication of other viruses. Our preliminary data indeed showed that DR1 also helped the replication of Japanese encephalitis virus, dengue virus, and vaccinia virus (data not shown). In this scenario, the expression level of DR1 might be tightly regulated in IAV-infected cells. However, in either microarray analysis or Western blotting studies of DR1 at different postinfection times, we did not detect any change in the expression pattern during the viral life cycle (data not shown). Further studies are required to unlock the mechanism of association between DR1 and immune-related genes.
DR1 works in coordination with DRAP1 to form a heterodimer called NC2, which possesses inhibitory activity on Pol II transcription (16). We also examined the effects of DRAP1 KD by use of shDRAP1 in IAV replication. The results demonstrated that DRAP1 also functions as a positive regulator of IAV replication, suggesting that DRAP1 may have functional redundancy with DR1 in IAV replication (data not shown). This finding may explain why the enhancement of ISGs requires such prolonged DR1 KD, because endogenous DRAP1 may also suppress IFN induction to supplement the effects of DR1 KD. On the other hand, RIG-I and MDA5 were not induced by early DR1 KD but were highly stimulated by prolonged DR1 KD (Fig. 3E, data for 4 and 10 dpt). These data may suggest that DR1 does not directly affect the expression of these signal transducers to induce IFN production; perhaps it regulates the expression of other cellular factors through a cascade of transcriptional regulation to indirectly mediate IFN induction and its downstream signaling pathway. Regardless, DR1 appears to be an important regulator of immune-related genes and may serve as a guardian against self-destruction of cells by innate immune molecules. As such, DR1 may be considered a host susceptibility gene for viral infection. It will be interesting to know why and how DR1 regulates and links to a large number of immune-related genes. All these issues need to be investigated further.
We note that while the KD efficiencies of shDR1 were very high, the reduction of IAV replication was only moderate during early DR1 KD (4 dpt); however, there was a dramatic increase in antiviral activity later (10 dpt). Also, there was no significant IFN induction or enhanced ISG expression at an early time of DR1 KD (4 dpt) (Fig. 3), and IAV internalization was not affected (Fig. 3C), thus ruling out the roles of ISGs at early times of DR1 KD. In this study, we showed that DR1 directly interacts with all three subunits of the viral RdRp complex and enhances the viral RdRp activity; thus, DR1 directly plays a positive role in IAV RNA replication. So far, IAV RNA replication has been shown to be coordinated by several host factors; for instance, (i) hCLE and cyclin T1/CDK9 interact with Pol II and the viral RdRp, thus enhancing Pol II activity to promote viral transcription (31, 32); and (ii) the MCM complex and Tat-SF1 associate with the viral RdRp and NP, respectively, to promote viral replication (33, 34). Intriguingly, unlike cyclin T1/CDK9 and Tat-SF1, which are positive cellular transcription elongation factors, DR1 is thought to be a cellular transcription repressor by precluding the assembly of the PIC to inhibit Pol II initiation (15). Yet all these factors promote IAV transcription. This contradiction might be explained by the finding that DR1 associates with the individual components of the viral RdRp complex (Fig. 7); this interaction might reduce the interaction of DR1 with TBP, thereby abolishing the inhibitory activity of DR1 on Pol II and consequently enhancing Pol II activity to promote the pre-mRNA production which is required for viral transcription. This scenario also applies to adenovirus. In adenovirus, E1A can compete away the binding ability of TBP for DR1 and allow the productive interaction of TBP with TFIIA to initiate cellular transcription (35). Our microarray data also indicated that DR1 KD caused not only up- but also downregulation of some cellular genes (Table 1); thus, DR1 has both transcriptional repressor and enhancer functions for various genes, representing a novel class of cellular factors for IAV replication.
Besides the well-established function of NC2 as a transcriptional repressor, NC2 also has been shown to have a positive activity on cellular transcription by the downstream promoter element (DPE) in Drosophila (12, 36, 37). The DPE is an element in the Pol II core promoter that is conserved from Drosophila to humans, and its function provides a TFIID recognition site to initiate cellular transcription. NC2 has also been reported to associate with the hyperphosphorylated carboxy-terminal domain (CTD) of Pol II (active form) (38). These findings suggest that DR1 might directly enhance Pol II activity by DPE as well, which is beneficial to viral transcription. Furthermore, during viral transcription, PB2 recognizes cellular capped pre-mRNAs, and PA cuts 10 to 13 nucleotides downstream from the caps, allowing the capped oligonucleotides to serve as primers for PB1-catalyzed transcription initiation (7). Since DR1 associates with all 3 components of the viral RdRp complex, it is likely that DR1 affects the vRNA-binding ability, cap-snatching process, or catalytic activity of PB1 to enhance viral RNA replication. Taken together, the data indicate that this RdRp-DR1 complex may become part of an active RNA transcription machinery and move to the site of a cellular gene promoter, allowing the viral RdRp to be adjacent to the site of pre-mRNA synthesis, which generates primers for viral RNA transcription. On the other hand, the viral polymerase may use DR1 to modulate DR1-associated gene expression, such as that of host innate immunity genes. All these issues need to be studied further.
Based on our findings, we present a model (Fig. 8) proposing that DR1 may simultaneously perform the following dual functions in IAV replication: (i) DR1 suppresses IFN induction to block a large set of ISG functions, providing a friendly environment favoring IAV replication; and (ii) DR1 facilitates viral RNA replication by interacting with the viral RdRp complex. Therefore, DR1 represents a new class of host susceptibility genes for IAV replication. It should be noted that this dual mode of action is unique to DR1. The ubiquitin ligase ITCH, which inhibits IAV uncoating (5), does not have effects on production and action; thus, the antiviral effects of ITCH KD are considerably weaker than those of DR1 KD.
FIG 8.
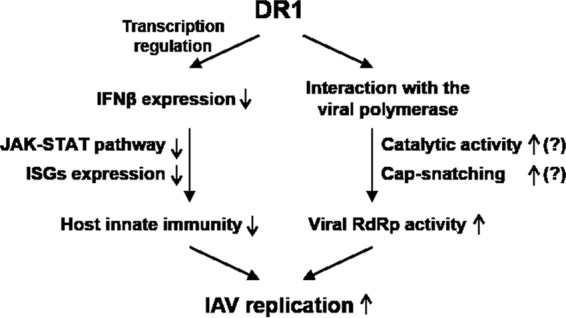
Proposed model of DR1 functions in IAV replication. DR1 may involve the following dual mechanism in IAV replication: (i) DR1 modulates the expression of cellular genes involved in IFN production and/or action to suppress host innate immunity, providing a beneficial environment to favor IAV replication; and (ii) DR1 facilitates the viral RdRp activity through interacting with the viral RdRp complex and enhancing either the catalytic activity of the viral polymerase or the supply of precursor pre-mRNA for cap-snatching function. As such, DR1 represents a new class of host susceptibility genes for IAV replication. The question marks indicate issues yet to be investigated.
The development of effective antiviral drugs is important for combating virus invasion. To date, commercial drugs that target IAV proteins readily acquire drug resistance. For example, oseltamivir and zanamivir, which are neuraminidase inhibitors, target the active site of the highly conserved NA protein of influenza A and B virus subtypes (39). However, the high error rate of viral polymerases during virus replication, especially in RNA viruses such as HIV and influenza virus, may speed up the emergence of drug resistance. A recent study indicated that the vast majority of mutations conferring drug resistance are single amino acid residue substitutions (H274Y in N1) in NA (40). Using cellular factors as drug targets may be a good choice to reduce the problem of viral resistance. Our microarray analysis showed that only a small subset of host genes were affected by DR1 KD (Table 1, shDR1-1-Mock), similar to the microarray data obtained from the thermally inactivated NC2 cofactor in yeast (41). It also showed that DR1 KD resulted in 107 upregulated ISGs with a 2-fold change in the presence of IAV infection. Taken together, our results show that the effects of DR1 KD on reducing viral RNA replication and simultaneously enhancing the expression of host innate immunity genes are attractive reasons to select DR1 as a drug target.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grant MOST 103-2320-B-039-013-MY2 from the Ministry of Science and Technology, Taiwan.
We thank the National RNAi Core Facility, Academia Sinica, Taiwan, for providing the lentivirus vectors and for technical support. We also thank the Genomic Center, Institute of Molecular Biology, Academia Sinica, Taiwan, for the analysis of the microarray data and for technical support.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03610-14.
REFERENCES
- 1.Fouchier RA, Munster V, Wallensten A, Bestebroer TM, Herfst S, Smith D, Rimmelzwaan GF, Olsen B, Osterhaus AD. 2005. Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J Virol 79:2814–2822. doi: 10.1128/JVI.79.5.2814-2822.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morens DM, Taubenberger JK, Fauci AS. 2009. The persistent legacy of the 1918 influenza virus. N Engl J Med 361:225–229. doi: 10.1056/NEJMp0904819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carr CM, Kim PS. 1994. Flu virus invasion: halfway there. Science 266:234–236. doi: 10.1126/science.7939658. [DOI] [PubMed] [Google Scholar]
- 5.Su WC, Chen YC, Tseng CH, Hsu PW, Tung KF, Jeng KS, Lai MM. 2013. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc Natl Acad Sci U S A 110:17516–17521. doi: 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Neill RE, Jaskunas R, Blobel G, Palese P, Moroianu J. 1995. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J Biol Chem 270:22701–22704. doi: 10.1074/jbc.270.39.22701. [DOI] [PubMed] [Google Scholar]
- 7.Lamb RA, Krug RM. 2001. Orthomyxoviridae: the viruses and their replication, p 1487–1530 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 8.García-Sastre A. 2011. Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu SY, Sanchez DJ, Cheng G. 2011. New developments in the induction and antiviral effectors of type I interferon. Curr Opin Immunol 23:57–64. doi: 10.1016/j.coi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stark GR, Kerr IM, Williams BRG, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu Rev Biochem 67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 11.Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, Garcia-Sastre A. 2000. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 74:7989–7996. doi: 10.1128/JVI.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butler JE, Kadonaga JT. 2002. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev 16:2583–2592. doi: 10.1101/gad.1026202. [DOI] [PubMed] [Google Scholar]
- 13.Smale ST, Kadonaga JT. 2003. The RNA polymerase II core promoter. Annu Rev Biochem 72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- 14.Goppelt A, Stelzer G, Lottspeich F, Meisterernst M. 1996. A mechanism for repression of class II gene transcription through specific binding of NC2 to TBP-promoter complexes via heterodimeric histone fold domains. EMBO J 15:3105–3116. [PMC free article] [PubMed] [Google Scholar]
- 15.Inostroza JA, Mermelstein FH, Ha I, Lane WS, Reinberg D. 1992. Dr1, a TATA-binding protein-associated phosphoprotein and inhibitor of class II gene transcription. Cell 70:477–489. doi: 10.1016/0092-8674(92)90172-9. [DOI] [PubMed] [Google Scholar]
- 16.Mermelstein F, Yeung K, Cao J, Inostroza JA, Erdjument-Bromage H, Eagelson K, Landsman D, Levitt P, Tempst P, Reinberg D. 1996. Requirement of a corepressor for Dr1-mediated repression of transcription. Genes Dev 10:1033–1048. doi: 10.1101/gad.10.8.1033. [DOI] [PubMed] [Google Scholar]
- 17.Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. 2008. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, Mäurer AP, Müller E, Wolff T, Rudel T, Meyer TF. 2010. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 19.König R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, García-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. 2010. Human host factors required for influenza virus replication. Nature 463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao TL, Wu CY, Su WC, Jeng KS, Lai MM. 2010. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J 29:3879–3890. doi: 10.1038/emboj.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeung KC, Inostroza JA, Mermelstein FH, Kannabiran C, Reinberg D. 1994. Structure-function analysis of the TBP-binding protein Dr1 reveals a mechanism for repression of class II gene transcription. Genes Dev 8:2097–2109. doi: 10.1101/gad.8.17.2097. [DOI] [PubMed] [Google Scholar]
- 22.Ehrhardt C, Seyer R, Hrincius ER, Eierhoff T, Wolff T, Ludwig S. 2010. Interplay between influenza A virus and the innate immune signaling. Microbes Infect 12:81–87. doi: 10.1016/j.micinf.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eierhoff T, Ludwig S, Ehrhardt C. 2009. The influenza A virus matrix protein as a marker to monitor initial virus internalisation. Biol Chem 390:509–515. doi: 10.1515/BC.2009.053. [DOI] [PubMed] [Google Scholar]
- 25.Law AH, Lee DC, Yuen KY, Peiris M, Lau AS. 2010. Cellular response to influenza virus infection: a potential role for autophagy in CXCL10 and interferon-alpha induction. Cell Mol Immunol 7:263–270. doi: 10.1038/cmi.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Watanabe T, Hatta M, Watanabe S, Nanbo A, Ozawa M, Kakugawa S, Shimojima M, Yamada S, Neumann G, Kawaoka Y. 2009. Mutational analysis of conserved amino acids in the influenza A virus nucleoprotein. J Virol 83:4153–4162. doi: 10.1128/JVI.02642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Müller KH, Kakkola L, Nagaraj AS, Cheltsov AV, Anastasina M, Kainov DE. 2012. Emerging cellular targets for influenza antiviral agents. Trends Pharmacol Sci 33:89–99. doi: 10.1016/j.tips.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Zhao C, Hsiang TY, Kuo RL, Krug RM. 2010. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc Natl Acad Sci U S A 107:2253–2258. doi: 10.1073/pnas.0909144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haller O, Stertz S, Kochs G. 2007. The Mx GTPase family of interferon-induced antiviral proteins. Microbes Infect 9:1636–1643. doi: 10.1016/j.micinf.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pérez-González A, Rodriguez A, Huarte M, Salanueva IJ, Nieto A. 2006. hCLE/CGI-99, a human protein that interacts with the influenza virus polymerase, is a mRNA transcription modulator. J Mol Biol 362:887–900. doi: 10.1016/j.jmb.2006.07.085. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Li G, Ye X. 2010. Cyclin T1/CDK9 interacts with influenza A virus polymerase and facilitates its association with cellular RNA polymerase II. J Virol 84:12619–12627. doi: 10.1128/JVI.01696-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawaguchi A, Nagata K. 2007. De novo replication of the influenza virus RNA genome is regulated by DNA replicative helicase, MCM. EMBO J 26:4566–4575. doi: 10.1038/sj.emboj.7601881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naito T, Kiyasu Y, Sugiyama K, Kimura A, Nakano R, Matsukage A, Nagata K. 2007. An influenza virus replicon system in yeast identified Tat-SF1 as a stimulatory host factor for viral RNA synthesis. Proc Natl Acad Sci U S A 104:18235–18240. doi: 10.1073/pnas.0705856104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraus VB, Inostroza JA, Yeung K, Reinberg D, Nevins JR. 1994. Interaction of the Dr1 inhibitory factor with the TATA binding protein is disrupted by adenovirus E1A. Proc Natl Acad Sci U S A 91:6279–6282. doi: 10.1073/pnas.91.14.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willy PJ, Kobayashi R, Kadonaga JT. 2000. A basal transcription factor that activates or represses transcription. Science 290:982–985. doi: 10.1126/science.290.5493.982. [DOI] [PubMed] [Google Scholar]
- 37.Burke TW, Kadonaga JT. 1997. The downstream core promoter element, DPE, is conserved from Drosophila to humans and is recognized by TAFII60 of Drosophila. Genes Dev 11:3020–3031. doi: 10.1101/gad.11.22.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castaño E, Gross P, Wang Z, Roeder RG, Oelgeschläger T. 2000. The C-terminal domain-phosphorylated IIO form of RNA polymerase II is associated with the transcription repressor NC2 (Dr1/DRAP1) and is required for transcription activation in human nuclear extracts. Proc Natl Acad Sci U S A 97:7184–7189. doi: 10.1073/pnas.140202297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burch J, Corbett M, Stock C, Nicholson K, Elliot AJ, Duffy S, Westwood M, Palmer S, Stewart L. 2009. Prescription of anti-influenza drugs for healthy adults: a systematic review and meta-analysis. Lancet Infect Dis 9:537–545. doi: 10.1016/S1473-3099(09)70199-9. [DOI] [PubMed] [Google Scholar]
- 40.Ward P, Small I, Smith J, Suter P, Dutkowski R. 2005. Oseltamivir (Tamiflu) and its potential for use in the event of an influenza pandemic. J Antimicrob Chemother 55(Suppl 1):i5–i21. doi: 10.1093/jac/dki018. [DOI] [PubMed] [Google Scholar]
- 41.Geisberg JV, Holstege FC, Young RA, Struhl K. 2001. Yeast NC2 associates with the RNA polymerase II preinitiation complex and selectively affects transcription in vivo. Mol Cell Biol 21:2736–2742. doi: 10.1128/MCB.21.8.2736-2742.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.