

ABSTRACT
Lethal mutagenesis is a broad-spectrum antiviral strategy that exploits the high mutation rate and low mutational tolerance of many RNA viruses. This approach uses mutagenic drugs to increase viral mutation rates and burden viral populations with mutations that reduce the number of infectious progeny. We investigated the effectiveness of lethal mutagenesis as a strategy against influenza virus using three nucleoside analogs, ribavirin, 5-azacytidine, and 5-fluorouracil. All three drugs were active against a panel of seasonal H3N2 and laboratory-adapted H1N1 strains. We found that each drug increased the frequency of mutations in influenza virus populations and decreased the virus' specific infectivity, indicating a mutagenic mode of action. We were able to drive viral populations to extinction by passaging influenza virus in the presence of each drug, indicating that complete lethal mutagenesis of influenza virus populations can be achieved when a sufficient mutational burden is applied. Population-wide resistance to these mutagenic agents did not arise after serial passage of influenza virus populations in sublethal concentrations of drug. Sequencing of these drug-passaged viral populations revealed genome-wide accumulation of mutations at low frequency. The replicative capacity of drug-passaged populations was reduced at higher multiplicities of infection, suggesting the presence of defective interfering particles and a possible barrier to the evolution of resistance. Together, our data suggest that lethal mutagenesis may be a particularly effective therapeutic approach with a high genetic barrier to resistance for influenza virus.
IMPORTANCE Influenza virus is an RNA virus that causes significant morbidity and mortality during annual epidemics. Novel therapies for RNA viruses are needed due to the ease with which these viruses evolve resistance to existing therapeutics. Lethal mutagenesis is a broad-spectrum strategy that exploits the high mutation rate and the low mutational tolerance of most RNA viruses. It is thought to possess a higher barrier to resistance than conventional antiviral strategies. We investigated the effectiveness of lethal mutagenesis against influenza virus using three different drugs. We showed that influenza virus was sensitive to lethal mutagenesis by demonstrating that all three drugs induced mutations and led to an increase in the generation of defective viral particles. We also found that it may be difficult for resistance to these drugs to arise at a population-wide level. Our data suggest that lethal mutagenesis may be an attractive anti-influenza strategy that warrants further investigation.
INTRODUCTION
Influenza virus is a single-stranded, negative-sense RNA virus with a genome consisting of 8 segments (1). Like other RNA viruses, influenza virus replicates with extremely low fidelity. Its RNA-dependent RNA polymerase (RdRp) complex, which includes the viral proteins PB1, PB2, PA, and NP (2, 3), has a mutation rate of approximately 2.3 × 10−5 substitutions per nucleotide per cell infection (4). This high mutation rate limits the effectiveness of seasonal vaccines and antivirals, as it allows the virus to generate mutations that mediate escape from neutralizing antibodies and resistance to antiviral drugs (5–9). While a high mutation rate allows RNA viruses to rapidly adapt to new selective pressures, most newly generated mutations are deleterious (10–12). RNA viruses, therefore, exist at a threshold of viability, where even small increases in mutational load can cause population extinction (13, 14).
Lethal mutagenesis is a broad-spectrum antiviral strategy that exploits the high mutation rate and low mutational tolerance of many RNA viruses. This approach utilizes mutagenic drugs to increase the virus' mutation rate, thereby burdening the population with a large number of mutations that are either lethal or highly detrimental to ongoing replication. Extinction of the population will occur when the number of infectious progeny generated by each infectious particle drops to less than one (13). Lethal mutagenesis has been applied to a number of RNA viruses, most commonly with nucleoside (e.g., ribavirin and 5-azacytidine) and base (e.g., 5-fluorouracil) analogs. Ribavirin is a broad-spectrum antiviral that has been demonstrated to cause lethal mutagenesis of poliovirus, Hantaan virus, lymphocytic choriomeningitis virus (LCMV), GB virus, and West Nile virus in vitro (15–19). While ribavirin is used clinically for hepatitis C virus and respiratory syncytial virus, its mode of action against these viruses in vivo is less clear (20–22). Lethal mutagenesis with 5-azacytidine has been demonstrated in HIV-1 and foot-and-mouth disease virus (FMDV) in vitro (23, 24). The base analog 5-fluorouracil is processed intracellularly into a nucleoside analog and has demonstrated activity as a lethal mutagen against LCMV, exonuclease-deficient coronaviruses, and FMDV in vitro (24–26). For simplicity, we refer to all three drugs as nucleoside analogs.
In most cases, the mutagenic activity of nucleoside analogs is attributable to the misincorporation of their triphosphate forms into replicating genomes by the viral RdRp. The structure of the nucleoside analog, its base-pairing properties, and the sense of the RNA strand determine the classes of mutations observed (27). Ribavirin, a guanosine analog, causes an increase in both C-to-U and G-to-A transitions (15), and 5-fluorouracil, which mimics uridine, leads to the accumulation of A-to-G and U-to-C transitions (25). Interestingly, 5-azacytidine, a cytidine analog, is able to induce both C-to-G and G-to-C transversions by virtue of a pyrimidine ring-opening mechanism that allows it to base pair with cytosine (23, 28). Ribavirin also has additional mechanisms that may play a role in its antiviral activity. Within host cells, it alters GTP pool concentrations by inhibiting IMP dehydrogenase (IMPDH) (29, 30). Other modes of action may include direct inhibition of the influenza virus RdRp (31, 32) and interference with capping of viral RNA (33). Some data suggest that ribavirin affects inflammatory and T-cell responses in vivo (34–36).
Initially, lethal mutagenesis was believed to be a “resistance-proof” strategy, since a newly arising resistance mutation would, in many cases, be linked to a lethal one on the same genome (37). However, in poliovirus, FMDV, and Chikungunya virus, mutagen-resistant variants have been recovered after fewer than 14 passages in sublethal concentrations of drug (38–40). The RdRps of these resistant viruses have replication fidelity phenotypes that make them less sensitive to mutagenesis by nucleoside analogs. Population genetic theory suggests that high mutation rates will also select for viruses that are more tolerant of mutation, or mutationally robust (14, 41). This mechanism of mutagenic drug tolerance has recently been identified in vesicular stomatitis virus populations passaged in 5-fluorouracil and in coxsackievirus populations passaged in ribavirin (42, 43).
The viability of lethal mutagenesis as a therapeutic approach to influenza virus infection has yet to be systematically explored. While the anti-influenza virus activity of ribavirin has long been recognized, its mechanism of action is unclear (31, 44). Recent data suggest that it may function as a lethal mutagen for influenza A virus and that a high-fidelity polymerase variant is less sensitive to its antiviral activity (45). Similarly, a new broad-spectrum antiviral, favipiravir, has been shown to be mutagenic to influenza virus in vitro and norovirus in vivo (46, 47).
We performed a systematic investigation of lethal mutagenesis as a therapeutic strategy to target influenza virus. We utilized ribavirin, 5-azacytidine, and 5-fluorouracil, which are three structurally distinct nucleoside analogs that are known to increase the frequency of specific mutations in a range of RNA viruses. We set four criteria for demonstrating lethal mutagenesis: (i) a concentration-dependent decrease in infectious viral titer, (ii) an increase in viral mutation frequency, (iii) a concentration-dependent decrease in the specific infectivity of the viral population, and (iv) the ability to extinguish the viral population upon multiple rounds of replication in the presence of drug. In addition to demonstrating their mutagenic action, we investigated the ability of influenza virus populations passaged in sublethal concentrations to acquire resistance to each of these nucleoside analogs.
MATERIALS AND METHODS
Cells, viruses, and drugs.
Madin-Darby canine kidney (MDCK) cells were provided by Arnold S. Monto (University of Michigan School of Public Health) and were maintained in Dulbecco's modified Eagle medium (Gibco 11965) supplemented with 10% fetal bovine serum (Gibco 10437) and 25 mM HEPES (Gibco 15630). Cells were maintained at 37°C and 5% CO2 in a humidified incubator.
A biological clone of influenza A/Puerto Rico/8/1934(H1N1) virus was obtained from ATCC (VR-1469). Influenza A/WSN/33(H1N1) virus was rescued following transfection of 8 plasmids that express the viral RNA and proteins from each genome segment (48). The plasmids were provided by Robert G. Webster (St. Jude Children's Research Hospital). Biological clones of influenza A/Panama/2007/1999(H3N2) and A/Wyoming/03/2003(H3N2) viruses were provided by Arnold S. Monto (University of Michigan School of Public Health). Unless otherwise indicated, infections were performed in viral infection medium consisting of Dulbecco's modified Eagle medium supplemented with 25 mM HEPES, 0.18% bovine serum albumin (Gibco 15260), and 2 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Worthington Biochemical 3740).
Ribavirin {1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-1,2,4-triazole-3-carboxamide} (Sigma-Aldrich R9644) was dissolved in phosphate-buffered saline (PBS) to make a 100 mM stock. 5-Azacytidine [4-amino-1-(β-d-ribofuranosyl)-1,3,5-triazin-2(1H)-one] (Sigma-Aldrich A2385), guanosine (Sigma-Aldrich G6752), and mycophenolic acid (Sigma-Aldrich M5255) were dissolved in dimethyl sulfoxide (DMSO) at 100 mM. 5-Fluorouracil (2,4-dihydroxy-5-fluoropyrimidine) (Sigma-Aldrich F6627) was dissolved in DMSO at 384 mM. Aliquots of all stocks were stored at −20°C.
Cellular toxicity assays.
The viability of MDCK cells after drug treatment was measured using an 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyl tetrazolium bromide (MTT) assay (49). Briefly, 24-well plates were seeded with 20,000 MDCK cells in 500 μl of medium. The following day, the culture medium was replaced with viral infection medium containing drug. After 24 h of incubation in drug, 50 μl of 5 mg/ml MTT (Sigma-Aldrich M5655) was added, and the cells were incubated at 37°C. After 2 h, 550 μl of 10% Triton X-100 (Acros Organics 327372500) and 0.1 N aqueous HCl in isopropanol (Fisher BP2610) were added. One hour later, the resulting precipitates were dissolved by repeated pipetting. One additional hour later, the absorbance at 595 nm was measured using a Synergy HT microplate reader (Bio-Tek).
Cytotoxicity was measured using the CytoTox-Glo cytotoxicity assay (Promega G9290), according the manufacturer's protocol. Briefly, 3,200 MDCK cells per well were seeded in a white, flat-bottom, 96-well plate (Fisher 353296). Drugs were diluted in viral infection medium and added to the cells as described above. Luminescence was measured after 24 h using a Synergy HT microplate reader both before and after digitonin permeabilization of the cell membrane.
Drug treatment of viruses.
MDCK cells were seeded in 24-well cell culture plates at a density of 6.5 × 104 cells per well in 500 μl medium. The next day, cells were washed with PBS and treated for 3 h with drug diluted in viral infection medium. Cells were infected with influenza virus at a multiplicity of infection (MOI) of 0.1 50% tissue culture infectious dose (TCID50)/cell in 200 μl of drug medium. The inoculum was removed after 1 h, and the cells were washed with PBS. Infected cells were incubated in 500 μl of fresh drug medium for an additional 24 h. Viral supernatants were clarified by centrifugation for 4 min at 1400 × g and stored at −80°C with glycerol at a final concentration of 0.5%. Viral titers were determined using either plaque assay (50) or TCID50. For TCID50 assays, 4 × 103 MDCK cells per well were seeded into a 96-well tissue culture plate in 100 μl of viral infection medium lacking TPCK-treated trypsin. The next day, serial 10-fold dilutions of viral supernatants were added to each row on the plate in 100 μl of viral infection medium with 4 μg/ml of TPCK-treated trypsin. After 4 days, the wells were scored for cytopathic effect (CPE), and the titers were calculated using the method of Reed and Muench (51).
RNA extraction, reverse transcription-PCR (RT-PCR), and quantitative PCR (qPCR).
RNA was extracted from clarified supernatants using either TRIzol reagent (Ambion 15596026), Purelink Pro 96 viral RNA/DNA kits (Invitrogen 12280), or QIAamp viral RNA minikits (Qiagen 52904) according to the manufacturers' instructions. cDNA was generated using random hexamer priming and the SuperScript III first-strand synthesis system (Invitrogen 18080). Quantitative PCR was performed on a 7500 fast real-time PCR system (Applied Biosystems) using FastStart universal SYBR green master mix (Roche 04913850001) with primers PB2for (5′-GTTGGGAGAAGAGCAACAGC-3′) and PB2rev (5′-GATTCGCCCTATTGACGAAA-3′). Serial 10-fold dilutions of plasmid containing the PB2 gene of A/WSN/33(H1N1) were used to generate a standard curve for quantification of cDNA copy number based on cycle threshold (CT) values.
Measurement of viral mutation frequency.
cDNAs corresponding to the eight viral RNAs were generated with primer uni12 (5′-AGCAAAAGCAGG-3′) using Superscript III as described above. A 957-base fragment of the hemagglutinin (HA) gene was amplified using Taq DNA polymerase (Invitrogen 18038) with primers HAfor (5′-GAAGGCAAACCTACTGGTCC-3′) and HArev (5′-GCACTCTCCTATTGTGACTGG-3′). PCR products were purified using the GeneJET PCR purification kit (Thermo K0701) and terminally adenylated by incubation with 500 μM dATP and Taq DNA polymerase for 10 min at 72°C. PCR products were then cloned into the pCR4-TOPO-TA vector using the TOPO-TA cloning kit for sequencing (Invitrogen 45-0071). Individual clones were sequenced at the University of Michigan DNA sequencing core using both T3 and T7 primers. Sequences were aligned over an 859-bp region that had adequate-quality sequencing reads for all clones, and mutations were identified using SeqMan Pro version 10.1.1 (DNASTAR). Only mutations present in both the forward and reverse reads of a clone were counted, and mutations found in multiple clones were counted once.
Drug passages.
To evolve influenza virus mutants that are resistant to the detrimental effects of mutagens, virus was passaged in low concentrations of drug. Three passage lineages for the mock-treated control and each drug were generated in the following way. Three million MDCK cells were seeded into 75-cm2 flasks. The next day, cells were washed with PBS and treated with 10 ml of viral infection medium containing either 7.5 μM ribavirin, 7.5 μM 5-azacytidine, 30 μM 5-fluorouracil, or an equivalent volume of DMSO for 3 h. The medium was then removed and replaced with 7.5 ml of drug-containing medium with 5 × 104 PFU of influenza A/Puerto Rico/8/1934(H1N1) virus (MOI, 0.01). After 1 h, the virus was removed, the cells were washed with PBS, and 18.5 ml of drug-containing medium was added. Culture supernatants were harvested at 24 h postinfection, and titers were determined by plaque assay. This procedure was performed iteratively, with 5 × 104 PFU from the previous passage being used to infect the next. If titers dropped below the level that allowed for this MOI, 1 ml of undiluted culture supernatant was used. Passaged viral populations were tested for their sensitivity to drugs at an MOI of 0.01 and 0.1 using the above-described drug treatment protocol.
To demonstrate lethal mutagenesis and population extinction through serial drug passage, virus was passaged in high concentrations of drug. Three lineages in 40 μM ribavirin, 25 μM 5-azacytidine, 100 μM 5-fluorouracil, or an equivalent volume of DMSO were passaged as described above, except at an MOI of 0.1 and scaled to 25-cm2 flasks. If titers dropped below 1.6 × 105 PFU, 500 μl of undiluted culture supernatant from the previous passage were used. If titers dropped below detectable levels, we attempted to recover any remaining virus by adding 800 μl of undiluted culture supernatant to MDCK cells in the absence of drug. Supernatants of recovery passages were harvested at 4 days, and titers were determined by plaque assay. If titers were still undetectable, the viral population was considered extinct.
Next-generation sequencing.
Multiplex reverse transcription-PCR amplification of all 8 influenza virus genome segments was performed on RNA samples using Superscript III with HiFi platinum Taq (Invitrogen 12574) with the primers Uni12/Inf1 (5′-GGGGGGAGCAAAAGCAGG-3′), Uni12/Inf3 (5′-GGGGGAGCGAAAGCAGG-3′), and Uni13/Inf1 (5′-CGGGTTATTAGTAGAAACAAGG-3′) (52). Seven hundred fifty nanograms of the each amplified cDNA was sheared to an average size of 300 to 400 bp using a Covaris S220 focused ultrasonicator. Sequencing libraries were prepared using the NEBNext Ultra DNA library prep kit (NEB E7370L), Agencourt AMPure XP beads (Beckman Coulter A63881), and NEBNext multiplex oligonucleotides for Illumina (NEB E7600S). Indexed samples were pooled in equal volumes and sequenced on an Illumina MiSeq instrument with 2 × 250-base paired end reads.
Sequencing reads that passed standard Illumina quality control filters were binned by index, culled of low-quality bases (phred, <25), and aligned to the reference genome using bowtie (53). Single-nucleotide variants (SNV) were identified and analyzed using DeepSNV (54). The DeepSNV algorithm relies on a clonal control to estimate the local error rate within a given sequence context and to identify strand bias in base calling. It then applies a hierarchical binomial model based on mutation calls for test and control at each base and position to identify true-positive SNV. The clonal control was a library prepared in an identical fashion from 8 plasmids containing the A/Puerto Rico/8/1934 genome and sequenced in the same flow cell. The optimal P value and frequency cutoffs for variant base calls were determined empirically from MiSeq data in which a mutant virus was spiked in at known frequencies. We calculated our sensitivity and specificity for variant detection using DeepSNV based on a P value of 0.01. For a mutation at 5% frequency, sensitivity was 0.9916 and specificity was 0.9926. For a mutation at 2.5% frequency, sensitivity was 0.9875 and specificity was 0.9933. For a mutation at 1.25% frequency, sensitivity was 0.9562 and specificity was 0.9934. For a mutation at 0.63% frequency, sensitivity was 0.9102 and specificity was 0.9928. Based on these data, only SNV with a Benjamini-Hochberg-corrected P value of <0.01 and present at a frequency of ≥1% were used in downstream analyses.
Statistical analysis.
Mutation frequencies were compared to those in the mock-treated control using the chi-square test. The one-tailed Mann-Whitney U test was used to analyze the number of mutations per clone compared to those for the mock-treated control. Viral titers and specific infectivities were compared to those for the no-drug control using the Kruskal-Wallis test with Dunn's multiple-comparison test. All statistical tests were performed using either R or GraphPad Prism 6.
RESULTS
Anti-influenza virus effects of nucleoside analogs.
In our study of lethal mutagenesis, we used ribavirin, 5-azacytidine, and 5-fluorouracil, each of which is a known mutagen of other RNA viruses (15, 23, 25, 55). These three drugs were selected, in part, due to their activity against a range of RNA viruses as well as differences in the types of mutations that they are known to induce. We tested the activity of each drug against two laboratory-adapted H1N1 strains (A/PR8/34 and A/WSN/33) and two seasonal H3N2 strains (A/Panama/2007/1999 and A/Wyoming/03/2003). All three drugs were active against this panel of influenza viruses, reducing the viral titer in a concentration-dependent manner (Fig. 1). Ribavirin and 5-azacytidine exhibited comparable activity in this assay, with 2 to 3 log10 reductions in influenza virus titer at 10 μM. 5-Fluorouracil was less potent, with similar reductions at 80 μM. The A/Panama/2007/1999 (H3N2) strain appeared to be less sensitive to both ribavirin and 5-fluorouracil than the other three strains. This strain is not known to be inherently resistant to these drugs and does not contain either PB1 D27N or PB1 V43I, two mutations that are known to confer ribavirin resistance (32, 45). The reduced sensitivity could be due to the reduced replicative capacity of A/Panama/2007/1999 in MDCK cells, where its average titer of 3.2 × 105 TCID50/ml in the absence of drug was at least 1.5 log10 lower than those of the other strains. This reduced replicative capacity may decrease the number of replication cycles over which an antiviral can act. Given the general similarity in drug effect on the other three influenza virus strains, we used A/PR8/34 for all subsequent experiments.
FIG 1.
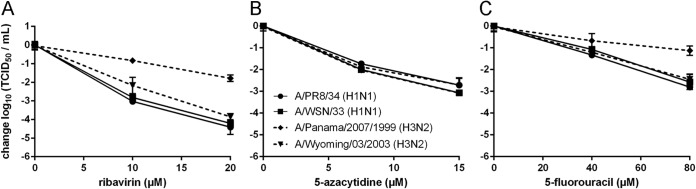
Sensitivity of influenza virus to nucleoside analogs. MDCK cells were infected with influenza A/PR8/34 (●), A/WSN/33 (◼), A/Panama/2007/1999 (◆), or A/Wyoming/03/2003 (▼) virus at an MOI of 0.1 in the presence of nucleoside analogs at the indicated concentrations (x axis). Cells were treated with ribavirin (A), 5-azacytidine (B), or 5-fluorouracil (C). Titers in supernatants were determined at 24 h and are shown relative to the 0 μM drug control. Solid lines, H1N1 strains; dashed lines, H3N2 strains. Points are plotted as mean ± standard deviation for 3 replicates.
Effects of nucleoside analogs on cultured cells.
Nucleoside analogs have structural similarity to cellular nucleosides and may reduce viral titer through pleiotropic effects on cellular polymerases and metabolic pathways. We investigated this possibility by quantifying both direct cytotoxicity and the effect of each drug on cellular viability. Cellular viability is distinct from direct cytotoxicity, as it reflects both cell proliferation and cell death over the assay period. We assayed relative cell viability using the MTT assay, which measures mitochondrial succinate dehydrogenase activity. The CytoTox-Glo assay, which quantifies the release of cellular proteases, was used to assay drug-induced cytotoxicity.
Using the MTT assay, we found a modest decrease in cellular viability in both 120 μM ribavirin and 480 μM 5-fluorouracil and a 50% decrease in cellular viability in 20 μM 5-azacytidine (Fig. 2A). Using the protease release assay, we determined that direct cytotoxicity was minimal for both ribavirin and 5-fluorouracil up to the maximal concentrations tested (60 μM and 200 μM, respectively). We found approximately 50% cytotoxicity with 5-azacytidine at 25 μM (Fig. 2B). We also assessed the health of our cell cultures by light microscopy after incubation in drug for 24 h. At the highest drug concentrations used in any of our assays involving influenza virus, the drug-treated cells were less overgrown than the mock-treated control cells and did not display signs of cell death, such as rounding or detachment. (Fig. 2C). Taken together, these data suggest that the observed decreases in cellular viability are due to reduced proliferation rather than direct cytotoxicity, as is expected based upon the effects of these drugs on cellular physiology (56, 57). Given the relatively small reductions in cellular viability at the doses used, the large decreases in viral titer observed with each of the three nucleosides (Fig. 1) are unlikely to be due to drug-associated cytotoxicity.
FIG 2.

Effect of nucleoside analogs on MDCK cells. (A) Number of viable cells relative to mock-treated controls after 24 h of drug treatment at the indicated concentrations (x axis) as analyzed by MTT assay. Each point represents the mean ± standard deviation for 3 replicates. (B) Cytotoxicity was measured using the CytoTox-Glo protease release assay on cells plated at low density and treated for 24 h with nucleoside analogs. Percent cytotoxicity (y axis) is expressed relative to untreated cells. (C) Images of cells after treatment with the indicated nucleoside analogs for 24 h. The drug concentrations shown are the highest used in any of the experiments involving influenza virus that are described in the text. Magnification, ×20.
Mutagenic effects of nucleoside analogs on influenza virus.
We used clonal sequencing to determine whether the three drugs are mutagenic to influenza virus over a single passage in MDCK cells (58). In this mutation frequency assay, we passaged influenza virus on cells in the presence of drug for 24 h at concentrations that caused a 2-log10 decrease in viral titers. We then amplified a 957-base HA gene fragment from RNA in the culture supernatant. These cDNAs, which include sequences from both viable and nonviable progeny, were cloned and sequenced. Using this assay, we observed a statistically significant increase in overall mutation frequency for viruses passaged in 10 μM 5-azacytidine (P = 0.013 by chi-square test) and 50 μM 5-fluorouracil (P = 0.027) (Fig. 3A). Viruses treated with 5-azacytidine also exhibited an increase in C-to-G transversions (P = 0.00023) and a strong statistical trend of increased A-to-G mutations (P = 0.13). Viruses recovered from cells treated with 50 μM 5-fluorouracil exhibited a trend of increased A-to-G (P = 0.15) and U-to-C (P = 0.063) mutations. In this analysis, we noted that some mutations were present in multiple clones. Since we could not exclude their presence as stable polymorphisms in the population prior to drug treatment, we reanalyzed the data set excluding these mutations. In all but one case, the significance level of the P values remained unchanged. In this more conservative analysis, the change in A-to-G transitions in viruses exposed to 10 μM 5-azacytidine achieved statistical significance (P = 0.043).
FIG 3.

Mutation frequency in influenza virus populations treated with nucleoside analogs. MDCK cells were infected with influenza A/PR8/34 (H1N1) virus at an MOI of 0.1 in drug-containing medium. Supernatants were harvested at 24 h postinfection. A 957-base fragment of the HA gene was amplified and cloned. Between 51 and 110 clones from each sample were sequenced, and mutations were identified. Overall mutation frequencies are expressed per 104 bases sequenced. Wild-type bases are on the left in each table. Specific mutation types are expressed per 104 wild-type bases sequenced. Mutations identified in multiple clones were counted once. A chi-square test was used to determine the statistical significance of the differences in total mutation frequency for each mutation type relative to the no-drug control. *, P < 0.05; **, P < 0.005. Statistically significant increases are highlighted by shading. (A) Each nucleoside analog compared to a no-drug control. (B) Treatment with multiple concentrations of ribavirin.
In our initial mutation frequency assay, virus exposed to 10 μM ribavirin exhibited an increase in C-to-U transitions (P = 0.0025) compared to the mock-treated control, but the change in the overall mutation frequency did not achieve statistical significance (Fig. 3A). In an independent experiment, we measured mutation frequency over a range of ribavirin concentrations. Here, we used ribavirin at 5 and 40 μM, as these concentrations caused moderate and large reductions in specific infectivity, respectively (see the next section). In this experiment, we identified a significant increase in overall mutation frequency at both drug concentrations (5 μM, P = 0.014; 40 μM, P = 0.0091) relative to the control population (Fig. 3B). The frequency of C-to-U transitions in the 40 μM ribavirin sample was also significantly higher than those in the 5 μM ribavirin sample (P = 0.038) and the no-drug control (P = 0.00074), suggesting a concentration dependence. We noted a relationship between drug concentration and the frequency of G-to-A mutations, which has been reported for other viruses treated with ribavirin (15). Importantly, the statistical significance of these results was not affected by exclusion of mutations present in multiple clones. The fact that the overall mutation frequency in viral populations treated with 5 μM ribavirin was significantly higher than that in mock-treated populations, but the overall frequency in the viral populations treated with 10 μM ribavirin was not, may be due to differences in the background mutation frequency in the untreated control, experiment-to-experiment variability, and issues of statistical power.
We also assessed the mutagenic activity of each drug by comparing the number of mutations present per clone. We found that the 5 μM ribavirin (1.74 mutations per clone; P = 0.018 by 1-tailed Mann-Whitney U test), 40 μM ribavirin (1.66 mutations per clone; P = 0.031), and 10 μM 5-azacytidine (1.35 mutations per clone; P = 0.025) samples had significantly more mutations per clone than the mock-treated controls (1.13 for the ribavirin control and 0.93 for the 5-azacytidine and 5-fluorouracil controls). There was a strong statistical trend in the 50 μM 5-fluorouracil-treated sample (1.27 mutations per clone; P = 0.057). Together, these results demonstrate the mutagenic activity of ribavirin, 5-azacytidine, and 5-fluorouracil.
Effect of nucleoside drugs on specific infectivity.
A hallmark of lethal mutagenesis is a reduction in the specific infectivity of a viral population. As mutations induced by the drugs accumulate in progeny genomes, fewer of the corresponding virions maintain infectivity. We calculated the specific infectivity of drug-treated viral populations relative to mock-treated control samples based on the titer and genome copy number in cell-free supernatants. All three drugs caused a concentration-dependent decrease in specific infectivity (Kruskal-Wallis test: ribavirin, P = 0.0036; 5-azacytidine, P = 0.0029; and 5-fluorouracil, P = 0.0071) (Fig. 4). When virus was treated with 20 μM ribavirin, there was a greater-than-5-fold reduction in specific infectivity, which persisted at higher concentrations (Fig. 4A). We found that treatment with 12.5 μM 5-azacytidine was sufficient to cause a greater-than-10-fold decrease in specific infectivity, with larger reductions at higher drug concentrations (Fig. 4B). Similar reductions in specific infectivity were achieved at 100 μM 5-fluorouracil (Fig. 4C). These reductions in specific infectivity are consistent with a mutagenic mode of action for each of the nucleoside analogs.
FIG 4.
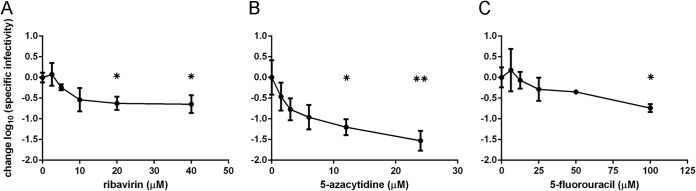
Specific infectivity of influenza virus populations treated with nucleoside analogs. MDCK cells were infected with influenza A/PR8/34 (H1N1) virus at an MOI of 0.1 and treated with ribavirin (A), 5-azacytidine (B), or 5-fluorouracil (C). Supernatants were harvested at 24 h postinfection, and titers of infectious virus were determined by TCID50 assay. Quantitative reverse transcription-PCR was used to determine the genome copy number in the samples. This was used to calculate the specific infectivity (TCID50/genome copy), which is shown relative to the 0 μM drug sample. Statistical significance was determined using the Kruskal-Wallis test with a Dunn correction. *, P < 0.05; **, P < 0.005. Points are plotted as mean ± standard deviation for 4 replicates.
Alternative mechanisms of ribavirin activity.
Ribavirin is known to have both mutagenic and nonmutagenic effects on viral replication. A well-characterized antiviral mechanism is its inhibition of IMPDH, a cellular enzyme that catalyzes the rate-limiting step in de novo guanine nucleotide synthesis (44, 59). This inhibition typically causes a reduction in intracellular GTP pools (60), which can be reversed with guanosine supplementation (29). To determine the potential contribution of IMPDH inhibition to the antiviral action of ribavirin, we measured the effect of mycophenolic acid, a potent and specific IMPDH inhibitor, on influenza virus titer and specific infectivity. Mycophenolic acid has been reported to have 100-fold-greater inhibitory activity against IMPDH in MDCK cells than ribavirin (59). Using the MTT cell viability assay, we determined that in 10 μM mycophenolic acid, there was a 50% reduction in the number of viable MDCK cells (data not shown). We found that treatment with mycophenolic acid caused a decrease in viral titer (Fig. 5A) but not in specific infectivity (Fig. 5B), suggesting a nonmutagenic effect of IMPDH inhibition on viral replication. We determined that there was no additional decrease in viral titer at concentrations above 10 μM mycophenolic acid (data not shown). This effect was completely reversed by guanosine supplementation, demonstrating the importance of adequate GTP pools to influenza virus replication.
FIG 5.

Effect of IMPDH inhibition on influenza virus. MDCK cells were treated with ribavirin or mycophenolic acid either with or without 40 μM guanosine and infected with influenza A/PR8/34 (H1N1) virus at an MOI of 0.1. At 24 h postinfection, culture supernatants were harvested and used for both determination of infectious virus titers by TCID50 assay and quantitative reverse transcription-PCR. Infectious titer (A) and specific infectivity (TCID50/genome copy) (B) data are shown normalized to 0 μM drug. Specific infectivities were compared to the 0 μM drug samples using the Kruskal-Wallis test with a Dunn correction. *, P < 0.05. Solid lines, samples treated with drug only; dashed lines, samples with drug plus 40 μM guanosine. Points are plotted as mean ± standard deviation for 4 replicates.
In contrast, we found that ribavirin significantly reduced the specific infectivity of the viral population (Fig. 4A and 5B) and was able to cause titer reductions 100-fold greater than those maximally achieved by mycophenolic acid (Fig. 5A). While the effect of ribavirin on infectious titer was greater at an MOI of 0.01 than at an MOI of 5, its effect on specific infectivity was not (data not shown). Guanosine supplementation completely reversed the antiviral effect and reduced the decrease in specific infectivity caused by ribavirin. Together, these results suggest that ribavirin's inhibition of IMPDH is important for its activity against influenza virus and that the resulting changes in GTP pools may augment the drug's mutagenic activity.
Lethal mutagenesis of influenza virus.
We next determined whether the mutagenic action of each nucleoside analog was sufficient to drive influenza virus populations to extinction. Influenza virus populations were passaged in nucleoside analog concentrations that were sufficient to cause significant decreases in specific infectivity and 3- to 4-log10 reductions in viral titer (Fig. 6). At 40 μM ribavirin, virus was undetectable by passage 3, and we were unable to recover any infectious virus after blind passage in the absence of drug. Using an identical approach, we observed extinction at passages 4 and 5 with 100 μM 5-fluorouracil and 25 μM 5-azacytidine, respectively. Together with the results above, these data suggest that lethal mutagenesis of influenza virus can be achieved provided that the viral population accumulates a significant mutational load.
FIG 6.
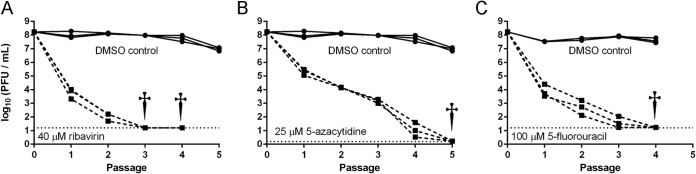
Lethal mutagenesis of influenza virus. Influenza A/PR8/34 (H1N1) virus was passaged on MDCK cells in ribavirin (A), 5-azacytidine (B), or 5-fluorouracil (C). Cells were infected at each passage with an MOI of ≤0.1 as described in Materials and Methods, and progeny were harvested at 24 h postinfection. Three viral lineages were passaged for each condition. Solid lines, 3 mock-treated control lineages; dashed lines, 3 drug-treated lineages. The horizontal dotted lines indicate the limit of detection for the last passage of each experiment. When titers dropped below the limit of detection, 0.8 ml of supernatant was added to fresh MDCK cells in the absence of drug, and titers were determined at 4 days postinfection. Daggers indicate that no virus was recovered from any of the three lineages at that passage.
Antiviral susceptibility after serial passage in drug.
While lethal mutagenesis was initially believed to have a high genetic barrier to resistance, serial passage in sublethal concentrations of drug may select for drug-resistant viruses that express variant, high-fidelity polymerases (38, 40, 45). To evaluate the potential for evolved resistance in influenza virus, we passaged virus in each nucleoside analog at concentrations that cause 1- to 2-log10 reductions in viral titer. These sublethal concentrations were chosen because they impose a significant selective pressure for evolved resistance without driving the viral population into extinction. As in our single-passage experiments, we initially passaged viral populations at an MOI of 0.1. We found that titers of both drug-treated and mock-treated populations initially declined, which was followed by a rapid rebound. As this cyclical effect was independent of drug treatment, we hypothesized that it was related to the generation and purging of defective interfering (DI) particles (61, 62). To control for this effect, we repeated the experiment at an MOI of 0.01, which should limit the accumulation of DI particles. Under these conditions, titers for the control lineages (D1 to D3) remained stable over 16 passages (Fig. 7A). For the three lineages passaged in the presence of 7.5 μM ribavirin (R1 to R3), infectious titers initially dropped for the first several passages and then reached a new equilibrium by passage 12. By passage 16, the titers of the ribavirin-treated populations were higher than those observed after a single drug passage but lower than those for mock-treated samples. Lineages A1 to A3, which were passaged in 7.5 μM 5-azacytidine, exhibited significant fluctuations in their titers across the 16 passages, but the titers remained lower than those of mock-treated samples. Viruses passaged in 30 μM 5-fluorouracil (F1 to F3) maintained their titer after the initial 10-fold drop at passage 1.
FIG 7.

Serial passage of influenza virus in sublethal concentrations of nucleoside analogs. Influenza A/PR8/34 (H1N1) virus was passaged on MDCK cells in the presence of nucleoside analogs. Passages were performed at an MOI of 0.01 using supernatant from the previous passage, and cells were harvested at 24 h postinfection. (A) Infectious titers for 16 passages at the indicated drug concentrations. (B) Infectious titers of passage 16 drug-treated and mock-treated (D2) populations after a single passage at an MOI of 0.1 (black bars) or 0.01 (gray bars) over a 24-h period in the absence of nucleoside analogs. (C) Sensitivity of passage 16 populations to the drugs in which they had been passaged. Infectious titers of viruses after a single passage at an MOI of 0.01 in drug for 24 h are shown. Titers are relative to those from virus passaged in the absence of drug. Solid lines with solid symbols, unpassaged and DMSO-passaged controls. Dashed lines with open symbols, viruses that had been passaged in drug. The horizontal dotted line indicates the limit of detection. Points are plotted as mean ± standard deviation for 3 replicates.
Given the stabilization in titer across all 9 drug-treated lineages by passage 16, we assessed the drug sensitivity of these populations using the same drug concentrations as for Fig. 1. We tested the sensitivity of each passage 16 population to the drug in which it had been passaged at the MOI used in single-passage (0.1) and serial-passage (0.01) drug treatments. In both cases, we found no significant differences in the sensitivity of drug-passaged populations relative to either the unpassaged stock or the control lineages at the tested concentrations (Fig. 7C). The only exception was the F1 population, which exhibited a statistically significant decrease in drug sensitivity at 80 μM 5-fluorouracil relative to that of the unpassaged stock (P < 0.05). The biological significance of this finding is unclear, as the lineage was just as sensitive to drug as the mock-treated passage control (see D2 passage 16).
We also noticed that in the absence of drug, the titers of drug-passaged viral lineages were at least 10-fold lower when infections were carried out at an MOI of 0.1 as opposed to 0.01 (Fig. 7B). The mock-treated control passages replicated to equivalent titers at both multiplicities of infection. The sensitivity of the drug-treated populations to the multiplicity of infection suggests that drug treatment accelerates the accumulation of highly mutated defective particles that interfere with the replication of other, less mutated progeny through lethal defection. Together, these data indicate the importance of lethal defection in drug-passaged populations, which may limit the emergence of population-level resistance to mutagenic nucleoside analogs (63).
Drug-induced shifts in the viral mutant spectrum.
The stabilization of viral titers in each drug-passaged population suggested that a new equilibrium had been reached by passage 16. We performed deep sequencing of the passage 16 populations to determine the consensus sequences, predominant mutation types, and minority variants found within each viral population. Using a conservative, empirically determined cutoff for variant detection (see Materials and Methods), we identified relatively few nonsynonymous mutations at a frequency of >50% in passaged populations (Table 1). Many of the consensus mutations (>50% frequency) within the drug-passaged lineages were also identified at lower levels in the mock-treated lineages or in the unpassaged population. Since the drug-passaged and the control populations were equally sensitive to drug (Fig. 7B), it is unlikely that any of these high-level mutations confer resistance to the antiviral effects of the nucleoside analogs.
TABLE 1.
Frequency of nonsynonymous, consensus mutations within passaged viral lineages
| Mutation | Amino acid change | Frequency (% of population) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Passage 0 | Passage 16 |
|||||||||||||
| D1 | D2 | D3 | R1 | R2 | R3 | A1 | A2 | A3 | F1 | F2 | F3 | |||
| HA G483T | A151S | 3.6 | 0.4 | 0.7 | —a | 47.3 | 81.4 | 54.9 | 3.3 | 0.8 | 5.2 | 3.7 | 6.2 | 2.3 |
| HA T627C | S199P | 11.5 | 1.9 | 1.1 | 2.2 | 50.1 | 81.6 | 53.1 | 2.9 | 0.6 | 5.1 | 3.6 | 5.6 | 2.6 |
| HA G634A | S201N | — | — | — | — | 1.2 | 1.2 | 53.0 | — | 0.1 | — | 0.7 | 0.7 | 0.7 |
| HA A641C | E203D | — | 96.5 | 96.8 | 96.5 | — | 0.8 | — | 87.4 | 94.7 | 86.4 | 93.0 | 92.2 | 95.0 |
| HA A1089G | I353V | 6.7 | 0.7 | 0.8 | 0.6 | 47.1 | 81.7 | 55.7 | 3.0 | 0.5 | 5.4 | 4.1 | 6.5 | 4.5 |
| HA A1404G | M458V | 24.0 | 1.8 | 0.9 | 2.4 | 62.9 | 84.4 | 59.9 | 7.5 | 0.4 | 7.7 | 3.6 | 5.6 | 2.7 |
| M A792G | T27A M2 | 5.8 | 37.0 | 38.3 | 38.1 | 56.7 | 29.3 | 8.1 | 66.3 | 74.0 | 39.7 | 58.1 | 62.4 | 55.3 |
| M G434A | A137T M1 | 0.5 | 24.5 | 31.3 | 28.1 | 8.9 | 30.2 | 45.8 | 21.3 | 15.7 | 53.1 | 15.6 | 14.1 | 7.8 |
| NA G412A | S131N | 4.6 | 4.9 | 7.0 | 7.6 | 7.1 | 10.6 | 29.9 | 60.5 | 46.7 | 46.0 | 11.5 | 14.8 | 22.0 |
| NP T906A | S287R | — | — | — | — | — | — | — | 2.2 | 63.7 | — | 0.5 | — | — |
| NS A575G | E26G NS2 | 35.3 | 33.8 | 30.2 | 25.5 | 17.0 | 36.5 | 31.9 | 64.5 | 55.4 | 57.6 | 61.5 | 71.2 | 63.3 |
| PA G292A | V90I | 5.9 | 60.6 | 64.8 | 65.9 | 4.9 | 31.9 | 27.6 | 51.9 | 47.5 | 46.8 | 67.7 | 81.1 | 80.9 |
| PA C314T | T97I | — | — | — | — | 88.3 | 10.9 | 55.2 | — | — | — | 0.7 | 0.9 | — |
| PB1 G1959A | M645I | — | — | — | — | 90.3 | 44.6 | 14.5 | — | — | — | 0.3 | — | — |
| PB2 G2128A | D701N | — | 45.6 | 64.5 | 63.1 | 1.5 | 1.9 | 3.2 | 19.4 | — | 20.8 | 65.8 | 60.4 | 67.1 |
—, mutation was not found.
We determined the type, location, and frequency of all mutations present at a frequency of greater than 1%. In each case, the drug-passaged populations exhibited a large number of mutations across the genome (Fig. 8). In the ribavirin lineages, we observed an increase in both C-to-U and G-to-A transitions. Viruses passaged in 5-azacytidine had an excess of C-to-G and G-to-C transversions, and the 5-fluorouracil-passaged lineages had an accumulation of A-to-G and U-to-C transitions. All of these mutations are characteristic of the drug used. While the drug-treated populations had a higher mutational load than the control lineages, nearly all of the mutations were present at a frequency of less than 5%. In one of the ribavirin-passaged populations (R2), we identified PB1 D27N, a mutation that has been reported to confer ribavirin resistance, at a frequency of 2.5% (32). A second previously described ribavirin resistance mutation, PB1 V43I, was not observed in any of the lineages (45). Our comprehensive analysis of drug-passaged populations confirms the mutagenic activity of each drug and suggests that influenza virus populations do not readily acquire resistance to nucleoside analogs.
FIG 8.

Mutation accumulation within viral populations after serial passage in nucleoside analogs. Influenza A/PR8/34 (H1N1) virus was serially passaged in 7.5 μM ribavirin (R1 to R3), 7.5 μM 5-azacytidine (A1 to A3), or 30 μM 5-fluorouracil (F1 to F3), or without drug (D1 to D3). At passage 16, viral populations were sequenced to a high depth of coverage using the Illumina platform. The location, frequency, and type of all mutations above 1% frequency and with a P value of below 0.01 are shown. The influenza virus genome segments are concatenated with positions 1 to 2341 representing PB2, 2342 to 4682 representing PB1, 4683 to 6915 representing PA, 6916 to 8693 representing HA, 8694 to 10258 representing NP, 10259 to 11671 representing NA, 11672 to 12698 representing M, and 12699 to 13588 representing NS. Mutations above the dashed line (frequency of 0.5) are consensus mutations within the population. Red dots, C-to-U and G-to-A transition mutations; blue dots, C-to-G and G-to-C transversion mutations; black dots, A-to-G and U-to-C transition mutations; white dots, all other mutation types, including deletions.
DISCUSSION
We used three structurally distinct nucleoside analogs to systematically explore the potential of lethal mutagenesis as a therapeutic strategy for influenza virus infection. Our studies of viruses exposed to ribavirin have established mutagenic and nonmutagenic modes of action for this drug against influenza virus. We also found that two other broad-spectrum antivirals, 5-azacytidine and 5-fluorouracil, are effective lethal mutagens for influenza virus in vitro. Additionally, we found that populations subjected to extended passage in sublethal concentrations of these mutagens did not readily acquire resistance. These results support the utility of lethal mutagenesis as an approach for the treatment of influenza virus.
Ribavirin has broad-spectrum antiviral activity with documented mutagenic and nonmutagenic modes of action (31, 33, 36, 44) Previous work suggests that ribavirin's mode(s) of action may be specific to certain viruses or taxa. For influenza virus, we found that its mode of action is more complex than previously realized. We determined that ribavirin was mutagenic to influenza virus by assessing mutation frequency and specific infectivity. While ribavirin is known to inhibit IMPDH in MDCK cells, this mechanism is unlikely to directly account for mutagenesis, as a more potent IMPDH inhibitor, mycophenolic acid, did not affect the virus' specific infectivity (44, 59). Guanosine supplementation reversed ribavirin's effect on both titer and specific infectivity, suggesting that GTP pool concentrations are an important factor in the mutagenic activity of ribavirin. The simplest explanation for these data is that ribavirin's inhibition of IMDPH creates a cellular environment that allows for mutagenesis. Since ribavirin is a guanosine analog, reduced levels of GTP within the cell may increase the probability of ribavirin triphosphate incorporation into replicating viral RNA. The concentrations of ribavirin that are effective against influenza virus are lower than those reported for several other RNA viruses (15, 18), which may reflect increased sensitivity to this drug. Alternatively, differences in host cell transport and metabolism of a given nucleoside analog may affect its antiviral activity.
The results from our mutation frequency assay and deep sequencing of serially passaged populations support a mutagenic mode of action for ribavirin, 5-azacytidine, and 5-fluorouracil. After 16 passages in drug, we observed an increase in two mutation types for each drug that are consistent with their chemical structures and previous work in other viral systems (15, 23, 25, 28). In contrast, only one of the two expected mutation types was observed in the mutation frequency assay after a single passage in ribavirin or 5-azacytidine. The mutation frequency assay may have insufficient power to detect increases in transition mutations, which are already present at high frequencies in the control populations due to the inherent transition bias of the influenza virus RdRp. This issue of statistical power could explain the lack of significant increases in G-to-A mutations in ribavirin-treated populations and both A-to-G and U-to-C mutations in 5-fluorouracil-treated populations using this assay. It is also possible that the sense of the RNA strand into which the nucleotide analog is incorporated may bias the observed mutation types in the single-passage experiment. During influenza virus replication, negative-sense viral RNA is known to be transcribed at levels 10-fold to 100-fold higher than for positive-sense cRNA (64). This suggests that for a given nucleotide position, there would be a greater likelihood of nucleoside analog incorporation into the negative-sense strand. Indeed, our data support this model, as we observed an increase in mutations predicted from 5-azacytidne incorporation into the negative-sense strand (C to G), but not the positive-sense strand (G to C), in our mutation frequency assay.
The mutational space explored by a virus may be an important aspect of its genetics that influences the effectiveness of a lethal mutagen. We determined that the influenza virus RdRp has a strong bias toward transition mutations, especially A to G and U to C. This mutational bias suggests that influenza virus populations may more thoroughly explore the sequence space accessible through these mutation types. This natural exploration could confer a certain level of genetic robustness to the detrimental effects of A-to-G and U-to-C transitions (65, 66). Therefore, mutagens that induce the same types of mutations as influenza virus's normal bias may be less effective at inducing lethal mutagenesis. In support of this model, we observed that the least potent mutagen, 5-fluorouracil, templates the two mutation types that are most commonly made by the influenza virus RdRp. We also found that 5-azacytidine, which induces transversion mutations rarely made by the influenza virus RdRp, was able to cause the largest reductions in specific infectivity. These results suggest that for maximal effectiveness, a lethal mutagen should force a viral population to explore regions of sequence space that are not normally accessed under normal replication conditions.
As opposed to other viral systems, where resistant variants quickly rose to prominence within the viral population, we did not observe emergence of high-level resistance after serial passage in sublethal concentrations of nucleoside analog (38–40). This lack of resistance is despite the fact that viral populations persisted through 16 passages and achieved a new equilibrium with titers higher than those observed at passage 1. These observations suggest that population-wide resistance, through either the evolution of resistance mutations or genetic robustness, is not readily acquired by influenza virus. Since the lack of a resistance phenotype suggests that a more mutationally robust population has not evolved, the mechanism by which the viral population persists may be through the maintenance of lightly mutated genomes. At sublethal concentrations of mutagen, a small percentage of genomes may remain unmutated. Together with genomes containing neutral mutations, these viruses will have a selective advantage over their highly, or lethally, mutated brethren and will be maintained within the population. We also found that concentrations of drug 3- to 5-fold higher than those used for the serial passage experiment were able to completely extinguish influenza virus populations. Together, these results suggest that for these three nucleoside analogs, there is a narrow window between the concentrations that allow for persistence and those that quickly cause extinction. The utility of lethal mutagenesis as an anti-influenza strategy is further supported by these results, because they suggest that even if the drug dose is insufficient to cause extinction of the viral population, it may be unlikely to lead to the evolution of a population resistant to the drug.
Our data suggest that DI particles, by interfering with the replication of less-mutated progeny through lethal defection, contribute to the antiviral effect of lethal mutagens on influenza virus populations. Consistent with this model, we found that virus passaged in mutagenic nucleoside analogs replicated to lower titers when infected at a high MOI. The dependency on MOI suggests that this effect is the result of DI particles within the viral population (61, 63). Our deep sequencing data show that hundreds of mutations accumulate at frequencies of greater than 1% when influenza virus is passaged in drug. There are likely hundreds or even thousands more mutations that accumulate at lower frequencies. Thus, under conditions in which coinfection of cells is more likely, mutagenized genomes can effectively interfere with the replication of their less-mutated brethren. The effect of lethal defection may be more pronounced in a segmented, negative-stranded RNA virus due to its mode of replication. In support of this model, treatment of LCMV, another segmented, negative-stranded RNA virus, with 5-fluorouracil was shown to generate defective particles capable of interfering with the replication of the viral population (63). Lethal defection may also explain why we did not observe population-level resistance. Even though single mutations are known to mediate mutagen resistance, defective interfering particles may keep those mutations from increasing in frequency within the population. In addition to causing lethal defection, the mutational burden induced by the drugs also would increase the likelihood of a beneficial mutation arising within a defective genome.
We note that other investigators have failed to identify population-wide resistance in influenza virus populations exposed to lethal mutagens. In a recent study, a ribavirin resistance variant (PB1 V43I in influenza A/Wuhan/359/95 virus [H3N2]) was identified only in a plaque reduction assay and subsequent screening of 182 plaques (45). Additionally, passaging influenza virus in the mutagen favipiravir did not cause the viral population to become resistant (46). Ribavirin has also been demonstrated to suppress resistance to conventional anti-influenza drugs when used in combination (67). Each of these examples, along with our results, suggests that an increased mutational burden reduces the potential for high-level resistance within an influenza virus population.
Our study reinforces the attractiveness of lethal mutagenesis as an antiviral strategy. We have demonstrated that three nucleoside analogs, which each induce different types of mutations, can function as lethal mutagens of influenza virus. Our data suggest that segmented, negative-stranded RNA viruses may have a higher barrier to resistance to this class of antiviral due to the impact of lethal defection. Influenza virus is now one of several viruses that have been shown to be sensitive to drugs that are capable of inducing lethal mutagenesis. The fact that favipiravir, which is currently in clinical trials as an influenza therapy, also functions as a lethal mutagen suggests the clinical relevance of this strategy. Current drawbacks to using nucleoside analogs clinically as antivirals are off-target effects and their relatively low potency (27, 56, 57). If novel mutagenic nucleoside analogs that overcome these hurdles can be identified, lethal mutagenesis may emerge as an effective strategy for treating a broad spectrum of RNA viruses, including influenza virus.
ACKNOWLEDGMENTS
This project was supported in part by a Clinician Scientist Development Award from the Doris Duke Charitable Foundation (to A.L.). M.P. was supported by the Michigan Predoctoral Training Program in Genetics (T32GM007544).
We thank Judy Opp from the microbial sequencing core of the University of Michigan Host Microbiome Initiative for assistance with next-generation sequencing, J. T. McCrone for assistance with sequence analysis, and Mike Imperiale and David Miller for helpful discussions.
REFERENCES
- 1.Steinhauer DA, Skehel JJ. 2002. Genetics of influenza virus. Annu Rev Genet 36:305–332. doi: 10.1146/annurev.genet.36.052402.152757. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal S, Bradel-Tretheway B, Takimoto T, Dewhurst S, Kim B. 2010. Biochemical characterization of enzyme fidelity of influenza A virus RNA polymerase complex. PLoS One 5:e10372. doi: 10.1371/journal.pone.0010372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruigrok RW, Crépin T, Hart DJ, Cusack S. 2010. Towards an atomic resolution understanding of the influenza virus replication machinery. Curr Opin Struct Biol 20:104–113. doi: 10.1016/j.sbi.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Sanjuan R, Nebot MR, Chirico N, Mansky LM, Belshaw R. 2010. Viral mutation rates. J Virol 84:9733–9748. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiraishi K, Mitamura K, Sakai-Tagawa Y, Goto H, Sugaya N, Kawaoka Y. 2003. High frequency of resistant viruses harboring different mutations in amantadine-treated children with influenza. J Infect Dis 188:57–61. doi: 10.1086/375799. [DOI] [PubMed] [Google Scholar]
- 6.Ghedin E, Holmes EC, DePasse JV, Pinilla LT, Fitch A, Hamelin M-E, Papenburg J, Boivin G. 2012. Presence of oseltamivir-resistant pandemic A/H1N1 minor variants before drug therapy with subsequent selection and transmission. J Infect Dis 206:1504–1511. doi: 10.1093/infdis/jis571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das SR, Hensley SE, Ince WL, Brooke CB, Subba A, Delboy MG, Russ G, Gibbs JS, Bennink JR, Yewdell JW. 2013. Defining influenza A virus hemagglutinin antigenic drift by sequential monoclonal antibody selection. Cell Host Microbe 13:314–323. doi: 10.1016/j.chom.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park AW, Daly JM, Lewis NS, Smith DJ, Wood JLN, Grenfell BT. 2009. Quantifying the impact of immune escape on transmission dynamics of influenza. Science 326:726–728. doi: 10.1126/science.1175980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheu TG, Fry AM, Garten RJ, Deyde VM, Shwe T, Bullion L, Peebles PJ, Li Y, Klimov AI, Gubareva LV. 2011. Dual resistance to adamantanes and oseltamivir among seasonal influenza A(H1N1) viruses: 2008-2010. J Infect Dis 203:13–17. doi: 10.1093/infdis/jiq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domingo-Calap P, Cuevas JM, Sanjuán R. 2009. The fitness effects of random mutations in single-stranded DNA and RNA bacteriophages. PLoS Genet 5:e1000742. doi: 10.1371/journal.pgen.1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrasco P, de la Iglesia F, Elena SF. 2007. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in tobacco etch virus. J Virol 81:12979–12984. doi: 10.1128/JVI.00524-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanjuán R, Moya A, Elena SF. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc Natl Acad Sci U S A 101:8396–8401. doi: 10.1073/pnas.0400146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bull JJ, Sanjuan R, Wilke CO. 2007. Theory of lethal mutagenesis for viruses. J Virol 81:2930–2939. doi: 10.1128/JVI.01624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauring AS, Andino R. 2010. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 6:e1001005. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crotty S, Cameron CE, Andino R. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci U S A 98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Severson WE, Schmaljohn CS, Javadian A, Jonsson CB. 2003. Ribavirin causes error catastrophe during Hantaan virus replication. J Virol 77:481–488. doi: 10.1128/JVI.77.1.481-488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moreno H, Gallego I, Sevilla N, de la Torre JC, Domingo E, Martin V. 2011. Ribavirin can be mutagenic for arenaviruses. J Virol 85:7246–7255. doi: 10.1128/JVI.00614-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanford RE, Chavez D, Guerra B, Lau JYN, Hong Z, Brasky KM, Beames B. 2001. Ribavirin induces error-prone Replication of GB virus B in primary tamarin hepatocytes. J Virol 75:8074–8081. doi: 10.1128/JVI.75.17.8074-8081.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Day C, Smee D, Julander J, Yamshchikov V, Sidwell R, Morrey J. 2005. Error-prone replication of West Nile virus caused by ribavirin. Antiviral Res 67:38–45. doi: 10.1016/j.antiviral.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Shah NR, Sunderland A, Grdzelishvili VZ. 2010. Cell type mediated resistance of vesicular stomatitis virus and Sendai virus to ribavirin. PLoS One 5:e11265. doi: 10.1371/journal.pone.0011265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dietz J, Schelhorn S-E, Fitting D, Mihm U, Susser S, Welker M-W, Fuller C, Daumer M, Teuber G, Wedemeyer H, Berg T, Lengauer T, Zeuzem S, Herrmann E, Sarrazin C. 2013. Deep sequencing reveals mutagenic effects of ribavirin during monotherapy of hepatitis C virus genotype 1-infected patients. J Virol 87:6172–6181. doi: 10.1128/JVI.02778-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brillanti S, Mazzella G, Roda E. 2011. Ribavirin for chronic hepatitis C: and the mystery goes on. Dig Liver Dis 43:425–430. doi: 10.1016/j.dld.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Dapp MJ, Clouser CL, Patterson S, Mansky LM. 2009. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J Virol 83:11950–11958. doi: 10.1128/JVI.01406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sierra S, Davila M, Lowenstein PR, Domingo E. 2000. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J Virol 74:8316–8323. doi: 10.1128/JVI.74.18.8316-8323.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruiz-Jarabo CM, Ly C, Domingo E, de la Torre JC. 2003. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 308:37–47. doi: 10.1016/S0042-6822(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 26.Smith EC, Blanc H, Vignuzzi M, Denison MR. 2013. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog 9:e1003565. doi: 10.1371/journal.ppat.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graci JD, Cameron CE. 2008. Therapeutically targeting RNA viruses via lethal mutagenesis. Future Virol 3:553–566. doi: 10.2217/17460794.3.6.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson-Grusby L, Laird PW, Magge SN, Moeller BJ, Jaenisch R. 1997. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc Natl Acad Sci U S A 94:4681–4685. doi: 10.1073/pnas.94.9.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee H-J, Pawlak K, Nguyen BT, Robins RK, Sadée W. 1985. Biochemical differences among four inosinate dehydrogenase inhibitors, mycophenolic acid, ribavirin, tiazofurin, and selenazofurin, studied in mouse lymphoma cell culture. Cancer Res 45:5512–5520. [PubMed] [Google Scholar]
- 30.Leyssen P, Balzarini J, De Clercq E, Neyts J. 2005. The Predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J Virol 79:1943–1947. doi: 10.1128/JVI.79.3.1943-1947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eriksson B, Helgstrand E, Johansson NG, Larsson A, Misiorny A, Noren JO, Philipson L, Stenberg K, Stening G, Stridh S, Oberg B. 1977. Inhibition of Influenza virus ribonucleic acid polymerase by ribavirin triphosphate. Antimicrob Agents Chemother 11:964–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Binh NT, Wakai C, Kawaguchi A, Nagata K. 2014. Involvement of the N-terminal portion of influenza virus RNA polymerase subunit PB1 in nucleotide recognition. Biochem Biophys Res Commun 443:975–979. doi: 10.1016/j.bbrc.2013.12.071. [DOI] [PubMed] [Google Scholar]
- 33.Bougie I, Bisaillon M. 2004. The broad spectrum antiviral nucleoside ribavirin as a substrate for a viral RNA capping enzyme. J Biol Chem 279:22124–22130. doi: 10.1074/jbc.M400908200. [DOI] [PubMed] [Google Scholar]
- 34.Ogbomo H, Michaelis M, Altenbrandt B, Doerr HW, Cinatl J. 2010. A novel immunomodulatory mechanism of ribavirin in suppressing natural killer cell function. Biochem Pharmacol 79:188–197. doi: 10.1016/j.bcp.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 35.Rigopoulou E, Abbott W, Williams R, Naoumov N. 2007. Direct evidence for immunomodulatory properties of ribavirin on T-cell reactivity to hepatitis C virus. Antiviral Res 75:36–42. doi: 10.1016/j.antiviral.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Mondelli MU. 2014. The multifaceted functions of ribavirin: antiviral, immunomodulator, or both? Hepatology 60:1126–1129. doi: 10.1002/hep.27186. [DOI] [PubMed] [Google Scholar]
- 37.Freistadt M. 2004. Lethal mutagens: broad-spectrum antivirals with limited potential for development of resistance? Drug Resist Updat 7:19–24. doi: 10.1016/j.drup.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Pfeiffer JK, Kirkegaard K. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci U S A 100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng J, Wang H, Xie X, Yang D, Zhou G, Yu L. 2013. An increased replication fidelity mutant of foot-and-mouth disease virus retains fitness in vitro and virulence in vivo. Antiviral Res 100:1–7. doi: 10.1016/j.antiviral.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 40.Coffey LL, Beeharry Y, Borderia AV, Blanc H, Vignuzzi M. 2011. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc Natl Acad Sci U S A 108:16038–16043. doi: 10.1073/pnas.1111650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visser J, Hermisson J, Wagner GP, Meyers LA, Bagheri-Chaichian H, Blanchard JL, Chao L, Cheverud JM, Elena SF, Fontana W, et al. . 2003. Perspective: evolution and detection of genetic robustness. Evolution 57:1959–1972. doi: 10.1554/02-750R. [DOI] [PubMed] [Google Scholar]
- 42.Sanjuán R, Cuevas JM, Furió V, Holmes EC, Moya A. 2007. Selection for robustness in mutagenized RNA viruses. PLoS Genet 3:e93. doi: 10.1371/journal.pgen.0030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graci JD, Gnadig NF, Galarraga JE, Castro C, Vignuzzi M, Cameron CE. 2012. Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J Virol 86:2869–2873. doi: 10.1128/JVI.05712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wray S, Gilbert B, Noall M, Knight V. 1985. Mode of action of ribavirin: effect of nucleotide pool alterations on influenza virus ribonucleoprotein synthesis. Antiviral Res 5:29–37. [DOI] [PubMed] [Google Scholar]
- 45.Cheung PPH, Watson SJ, Choy K-T, Fun Sia S, Wong DDY, Poon LLM, Kellam P, Guan Y, Malik Peiris JS, Yen H-L. 2014. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat Commun 5:4794. doi: 10.1038/ncomms5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baranovich T, Wong S-S, Armstrong J, Marjuki H, Webby RJ, Webster RG, Govorkova EA. 2013. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J Virol 87:3741–3751. doi: 10.1128/JVI.02346-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arias A, Thorne L, Goodfellow I. 2014. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. eLife 3:e03679. doi: 10.7554/eLife.03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 50.Balish AL, Katz JM, Klimov AI. 2013. Influenza: propagation, quantification, and storage. Curr Protoc Microbiol 15G.1. doi: 10.1002/9780471729259.mc15g01s29. [DOI] [PubMed] [Google Scholar]
- 51.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. [Google Scholar]
- 52.Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J Virol 83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B, Trapnell C, Pop M, Salzberg SL, others 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, Moch H, Beerenwinkel N. 2012. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun 3:811. doi: 10.1038/ncomms1814. [DOI] [PubMed] [Google Scholar]
- 55.Anderson JP, Daifuku R, Loeb LA. 2004. Viral error catastrophe by mutagenic nucleosides. Annu Rev Microbiol 58:183–205. doi: 10.1146/annurev.micro.58.030603.123649. [DOI] [PubMed] [Google Scholar]
- 56.Li M-H, Ito D, Sanada M, Odani T, Hatori M, Iwase M, Nagumo M. 2004. Effect of 5-fluorouracil on G1 phase cell cycle regulation in oral cancer cell lines. Oral Oncol 40:63–70. doi: 10.1016/S1368-8375(03)00136-2. [DOI] [PubMed] [Google Scholar]
- 57.Krečmerová M, Otmar M. 2012. 5-Azacytosine compounds in medicinal chemistry: current stage and future perspectives. Future Med Chem 4:991–1005. doi: 10.4155/fmc.12.36. [DOI] [PubMed] [Google Scholar]
- 58.Beaucourt S, Bordería AV, Coffey LL, Gnädig NF, Sanz-Ramos M, Beeharry Y, Vignuzzi M. 2011. Isolation of fidelity variants of RNA viruses and characterization of virus mutation frequency. J Vis Exp 16:2953. doi: 10.3791/2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K. 2005. Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother 49:981–986. doi: 10.1128/AAC.49.3.981-986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carr S, Papp E, Wu J, Natsumeda Y. 1993. Characterization of human type I and type I1 IMP dehydrogenases. J Biol Chem 268:27286–27290. [PubMed] [Google Scholar]
- 61.Nayak DP. 1980. Defective interfering influenza viruses. Annu Rev Microbiol 34:619–644. doi: 10.1146/annurev.mi.34.100180.003155. [DOI] [PubMed] [Google Scholar]
- 62.Thompson KA, Yin J. 2010. Population dynamics of an RNA virus and its defective interfering particles in passage cultures. Virol J 7:257. doi: 10.1186/1743-422X-7-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grande-Pérez A, Lázaro E, Lowenstein P, Domingo E, Manrubia SC. 2005. Suppression of viral infectivity through lethal defection. Proc Natl Acad Sci U S A 102:4448–4452. doi: 10.1073/pnas.0408871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J Virol Methods 173:1–6. doi: 10.1016/j.jviromet.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lauring AS, Acevedo A, Cooper SB, Andino R. 2012. Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe 12:623–632. doi: 10.1016/j.chom.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Plotkin JB, Dushoff J. 2003. Codon bias and frequency-dependent selection on the hemagglutinin epitopes of influenza A virus. Proc Natl Acad Sci U S A 100:7152–7157. doi: 10.1073/pnas.1132114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoopes JD, Driebe EM, Kelley E, Engelthaler DM, Keim PS, Perelson AS, Rong L, Went GT, Nguyen JT. 2011. Triple combination antiviral drug (TCAD) composed of amantadine, oseltamivir, and ribavirin impedes the selection of drug-resistant influenza A Virus. PLoS One 6:e29778. doi: 10.1371/journal.pone.0029778. [DOI] [PMC free article] [PubMed] [Google Scholar]