

ABSTRACT
The roles of host genetics versus exposure and contact frequency in driving cross-species transmission remain the subject of debate. Here, we used a multitaxon lemur collection at the Saint Louis Zoo in the United States as a model to gain insight into viral transmission in a setting of high interspecies contact. Lemurs are a diverse and understudied group of primates that are highly endangered. The speciation of lemurs, which are endemic to the island of Madagascar, occurred in geographic isolation apart from that of continental African primates. Although evidence of endogenized viruses in lemur genomes exists, no exogenous viruses of lemurs have been described to date. Here we identified two novel picornaviruses in fecal specimens of ring-tailed lemurs (Lemur catta) and black-and-white ruffed lemurs (Varecia variegata). We found that the viruses were transmitted in a species-specific manner (lesavirus 1 was detected only in ring-tailed lemurs, while lesavirus 2 was detected only in black-and-white ruffed lemurs). Longitudinal sampling over a 1-year interval demonstrated ongoing infection in the collection. This was supported by evidence of viral clearance in some animals and new infections in previously uninfected animals, including a set of newly born triplets that acquired the infection. While the two virus strains were found to be cocirculating in a mixed-species exhibit of ring-tailed lemurs, black-and-white ruffed lemurs, and black lemurs, there was no evidence of cross-species transmission. This suggests that despite high-intensity contact, host species barriers can prevent cross-species transmissions of these viruses.
IMPORTANCE Up to 75% of emerging infectious diseases in humans today are the result of zoonotic transmission. However, a challenge in understanding transmission dynamics has been the limited models of cross-species transmission. Zoos provide a unique opportunity to explore parameters defining viral transmission. We demonstrated that ongoing virus transmission in a mixed lemur species exhibit was species specific. This suggests that despite high contact intensity, host species barriers contribute to protection from cross-species transmission of these viruses. While the combinations of species might differ, most zoological parks worldwide commonly feature mixed-species exhibits. Collectively, this report demonstrates a widely applicable approach toward understanding infectious disease transmission.
INTRODUCTION
The origin of many emerging infectious diseases can be traced to transmissions between humans and nonhuman animals. For example, the severe acute respiratory syndrome (SARS) outbreak resulted from the transmission of SARS coronavirus from civets to humans, and the ongoing human immunodeficiency virus (HIV)-AIDS pandemic originated from cross-species transmissions of simian immunodeficiency virus (SIV) from chimpanzees and related primates (1, 2). Host genetic factors, such as cellular receptors and immunity genes, can act as species barriers to viral transmission (3–5). For RNA viruses, it has been proposed that host barriers that share closer genetic similarities between species correspond to the flattened fitness valley that viruses can traverse in their adaptation to new hosts (4, 6). Consequently, species-specific barriers can be overcome by virus evolution through adaptive mutations and neofunctionalization (7–10). Alternatively, it has been argued that high contact rate is the key driver of virus emergence (11–13). However, a major challenge to studying the dynamics of cross-species transmission has been the lack of models in relevant settings. Hence, most studies have relied on prospective inference and reconstruction.
Zoological parks feature collections that house different animal species within an enclosure (i.e., mixed-species exhibits). Mixed-species exhibits benefit both the animals and public visitors by providing a more enriched environment and improving the educational experience (14, 15). Mixed-species exhibits also provide a practical solution to the problem of the limited space available at most zoos. However, this creates an environment where interspecies interactions may occur through physical contact (16).
Lemurs, endemic to Madagascar, are prosimians that diverged from other primates on the African mainland approximately 62 million years ago (17). Lemurs are highly diverse, in part because, unlike African and Asian prosimians that are strictly nocturnal, they evolved in the absence of anthropoid primates (monkeys and apes), branching out to occupy the diurnal and nocturnal niches of the island's different ecosystems. There are only limited data regarding viruses that infect lemurs. Serological studies suggest that lemurs have been exposed to pathogens similar to West Nile virus and lentiviruses (18). Moreover, endogenous gammaherpesvirus, lentivirus, and spumavirus sequences have been identified in lemur genomes (19–23). However, there has been no direct evidence to date of extant exogenous viruses in lemurs.
One Health has been defined as an initiative that aims to merge animal and human health sciences to benefit both (24). Emerging infectious diseases of animals and humans, along with the continued anthropogenic environmental stressors that challenge wildlife and human health, have been the catalyst for the growing One Health approach in the veterinary, medical, and environmental fields (25). Within this framework, mixed-species exhibits provide a unique opportunity to examine viral transmission in a setting of high interspecies contact. In this study, we demonstrated the species-specific transmission of two novel picornaviruses in lemurs housed in single-species and mixed-species exhibits at the Saint Louis Zoo.
MATERIALS AND METHODS
Specimens.
The study was approved by the Saint Louis Zoo's Institutional Animal Care and Use Committee. A total of 35 fecal specimens were collected during September to October 2012 from ring-tailed lemurs (Lemur catta), black lemurs (Eulemur macaco macaco), a blue-eyed black lemur (Eulemur macaco flavifrons), mongoose lemurs (Eulemur mongoz), black-and-white ruffed lemurs (Varecia variegata), and Coquerel's sifakas (Propithecus coquereli). Details of the individual species in the collection are listed in Table S1 in the supplemental material. A fecal specimen from 1 Coquerel's sifaka was not available at the time of collection. A second set of 33 fecal specimens was collected in September 2013. One ring-tailed lemur and 3 black-and-white ruffed lemurs died since the 2012 collection. Samples from 1 black-and-white ruffed lemur (transferred to another zoo) and 1 black lemur were not available at the time of the 2013 collection.
Sequencing.
A subset of specimens from the 2012 collection was subjected to unbiased next-generation sequencing. Fecal specimens were diluted in 6:1 in phosphate-buffered saline (PBS) and filtered through a 0.45-μm-pore-size membrane to minimize recovery of intact bacteria. Total nucleic acid was extracted from the filtrate. The sequencing library for the specimen from lemur Mis101308 was prepared using ScriptSeq (Epicentre, Madison, WI, USA). Total nucleic acid extracted from the specimen from lemur Nai108015 was subjected to random-priming cDNA synthesis and amplification, and the sequencing library was generated using a standard TruSeq (Illumina, San Diego, CA, USA) protocol. Libraries were sequenced on an Illumina MiSeq instrument. High-quality reads with no detectable similarity to the reference human genome or NCBI nucleotide database by BLASTn were analyzed by BLASTx alignment against the NCBI nonredundant (nr) protein database as previously described (26), in order to identify divergent viral sequences. Contigs were assembled from viral sequences using Newbler (27).
Amplification of complete genome.
PCR primers were designed from contigs assembled from Illumina sequences. The complete genome of lesavirus 1 (the name we propose) was amplified by reverse transcription-PCR (RT-PCR) in five overlapping fragments using a SuperScript III reverse transcriptase kit (Invitrogen, Grand Island, NY, USA), cloned using a TOPO cloning kit (Invitrogen, Grand Island, NY, USA), and Sanger sequenced as previously described (28). The following primers were used: LV1-1F (5′-TCACATTAAGCCATGTTGCCTGCG-3′) with LV1-1r (5′-CATCACCTGGGCTGAAGAATTGGTC-3′); LV1-2F (5′-CAAGTACAAGTGAACGCAACACGC-3′) with LV1-2r (5′-GGAGGTGGTTCAGTCTTCATAAGC-3′); LV1-3F (5′-TAGTTCAGATCCGTCTCTGGCTGC-3′) with LV1-3r (5′-TGCAGCTACTTTCCTGGCTCAGAC-3′); LV1-4F (5′-ACAGGTTCCTGGTTGTAGCCATCC-3′) with LV1-4r (5′-AACTCCATGGGCACCAGCGCAATG-3′); and LV1-5F (5′-CTGCACCAGGCTTCTGTGGTTCAC-3′) with LV1-5r (5′-TGGAATGGTTCCGTTGTCAAAGTGG-3′). 5′ Rapid amplification of cDNA ends (RACE) was performed with LV1-5RACE1r (5′-CCATGAAGGGGCTGCTAACCCG-3′); 3′ RACE was performed with LV1-3RACE1F (5′-ATGACGAGGAGTACACGCTGACTG-3′).
The complete genome of the lesavirus 2 was amplified by RT-PCR in four overlapping fragments. The following primers were used: LV2-1F (5′-GGAATTCCAGGGAGCCGGAGC-3′) with LV2-1r (5′-CATTTCGTGGTCCAGTTGCACCTG-3′); LV2-2F (5′-CAGGTGCAACTGGACCACGAAATG-3′) with LV2-2r (5′-GCTGCCAGCATAGGGTCTGAAGC-3′); LV2-3F (5′-TGACTCTCAGAGCAGCTTCAGACC-3′) with LV2-3r (5′-GACATCCGTCGGGATTCTTGAACG-3′); and LV2-4F (5′-CAGCTCTTAGCTGCAGAGACCCA-3′) with LV2-4r (5′-ACTGGCCCACTGTGTACAGCCAG-3′). 5′ RACE was performed with LV2-5RACE1r (5′-ACCAAGCCATACTCATTCTGTAC-3′); 3′ RACE was performed with LV2-3RACE1F (5′-CACCTGCCCAGAAGGATGGAGATC-3′).
The VP1 sequence for LV1 was amplified from nucleic acid extracted from fecal specimens collected in 2012 and 2013 from a ring-tailed lemur, Mis101308, using primer set LV1-VP1F (5′-CAGGTGCTACAACACCCACTGATG-3′) and LV1-VP1r (5′-TGAACCACCAAGCAGAAACACTGC-3′). LV2 VP1 was amplified from nucleic acid extracted from fecal specimens collected in 2012 and 2013 from a black-and-white ruffed lemur, Mah951211, using primer set LV2-2F and LV2-2r.
PCR amplification of cytochrome B.
Part of the mitochondrial cytochrome B gene sequence was PCR amplified from total nucleic acid extracted from fecal specimens using an AccuPrime Taq DNA polymerase kit (Invitrogen). The following primer set was used: LemurCytB400F (5′-CCATGAGGACAAATATCMTTCTGAG-3′) and LemurCytB1032r (5′-TTCRACGGGTTGVCCTCCRATTC-3′). PCR products were cloned and sequenced.
Diversity analyses and phylogenetic methods.
Amino acid sequences of the full-length polyprotein from lesavirus 1 (KM396707), lesavirus 2 (KM396708), hunnivirus A1 (NC_018668), hunnivirus A2 (HM153767), and porcine teschovirus 1 (NC_003985) were aligned by MUSCLE (29). Diversity plots were generated with Simplot (30), employing sliding windows of 250 amino acids in length and a step size of 10 amino acids, with Kimura (2-parameter) correction.
Phylogenetic trees were constructed from alignments of the concatenated 2C3CD and P1 (VP4231) regions from the following picornaviruses: enterovirus A (NC_001612), simian sapelovirus (NC_004451), foot-and-mouth disease virus (NC_004004), cosavirus A (NC_012800), equine rhinitis B virus (NC_003983), encephalomyocarditis virus (NC_001479), Seneca valley virus (NC_011349), porcine teschovirus 1 (NC_003985), hunnivirus A1 (NC_018668), hunnivirus A2 (HM153767), Aichi virus (NC_001918), salivirus A (GQ253930), cadicivirus A (JN819202), melegrivirius A (HM751199), human parechovirus (FM178558), duck hepatitis A virus (NC_008250), hepatitis A virus (NC_001489), aquamavirus A (EU142040), avian encephalomyelitis virus (NC_003990), mosavirus A (JF973687), mischivirus A (JQ814851), gallivirus A (JQ691613), passerivirus A (GU182406), oscivirus A (GU182408), rosavirus A (JF973686), avisivirus A (KC465954), and pasivirus A (KM259923). Phylogenies were constructed with PhyML v3.0 (31) by the maximum-likelihood (ML) method using the LG substitution model. A discrete γ distribution of 4 rate categories was used to model heterogeneity among sites. Analyses were performed at least twice, and support for ML trees was assessed by 1,000 nonparametric bootstraps. The best-fit model of protein evolution was determined by ProtTest v 2.4 (32). Bayesian Markov chain Monte Carlo (MCMC) inference (WAG + I + G + F) was performed with BEAST v1.7.5 (33). A total of 10,000,000 MCMC states were run with a 25% burn-in period under a lognormal relaxed clock and Yule prior. Convergence and mixing were assessed with Tracer v1.5 and AWTY (34, 35). The two methods yielded trees with similar topologies.
For the phylogenetic analysis of cytochrome B genes and lesavirus sequences obtained through the screening assay, nucleotide sequences were aligned by Muscle (29) and primer sequences were trimmed from the alignment. A phylogeny was constructed by the neighbor-joining method using the Jukes-Cantor model of nucleotide substitution and maximum-likelihood method. The two methods yielded similar phylogenies.
Diagnostic RT-PCR amplification.
Standard precautions to avoid end product contamination were taken for all PCR assays, including the use of PCR hoods and maintaining separate areas for PCR setup and analysis. Seven no-template negative controls were interspersed between the actual samples. OneStep RT-PCR (Qiagen, Valencia, CA, USA) was used to amplify 5 μl of extracted samples using the following PCR program: 50°C for 35 min, 95°C for 15 min, and 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 21 s, followed by 72°C for 10 min. A consensus-degenerate primer pair, LVScreenF (5′-TTGTMACCTTYCTCAARGATGAGAC-3′) and LVScreenr (5′-GTGTAYTCCTCRTCATCCCAGATRTG-3′), used to screen samples for the presence of lesavirus 1 and lesavirus 2, generated a 388-nucleotide (nt) amplicon from the 3D polymerase (3Dpol) region, one of the most highly conserved regions of the genomes. Products were visualized following electrophoresis on 1.25% agarose gels. Amplicons were cloned and sequences verified.
Nucleotide sequence accession numbers.
The sequences of the complete genomes of lesavirus 1 and lesavirus 2, VP1 sequences, amplicons obtained through screening, and cytochrome B amplicon sequences have been entered into the GenBank database under accession numbers KM396707 to KM396752.
RESULTS
Two novel picornaviruses in lemurs.
Fecal specimens (35 from 6 taxa of lemurs at the Saint Louis Zoo) were collected for this study in 2012. The lemurs were housed in 9 single-species exhibits (ring-tailed lemurs, black-and-white ruffed lemurs, Coquerel's sifakas, mongoose lemurs, a blue-eyed black lemur, and black lemurs) and a mixed-species exhibit (4 ring-tailed lemurs with 4 black-and-white ruffed lemurs and 2 black lemurs in one exhibit) (Fig. 1A). As there have been no known exogenous viruses of lemurs described to date, we first sought to identify viruses associated with lemurs by performing unbiased deep sequencing on total nucleic acid samples extracted from fecal specimens from a subset of lemurs.
FIG 1.
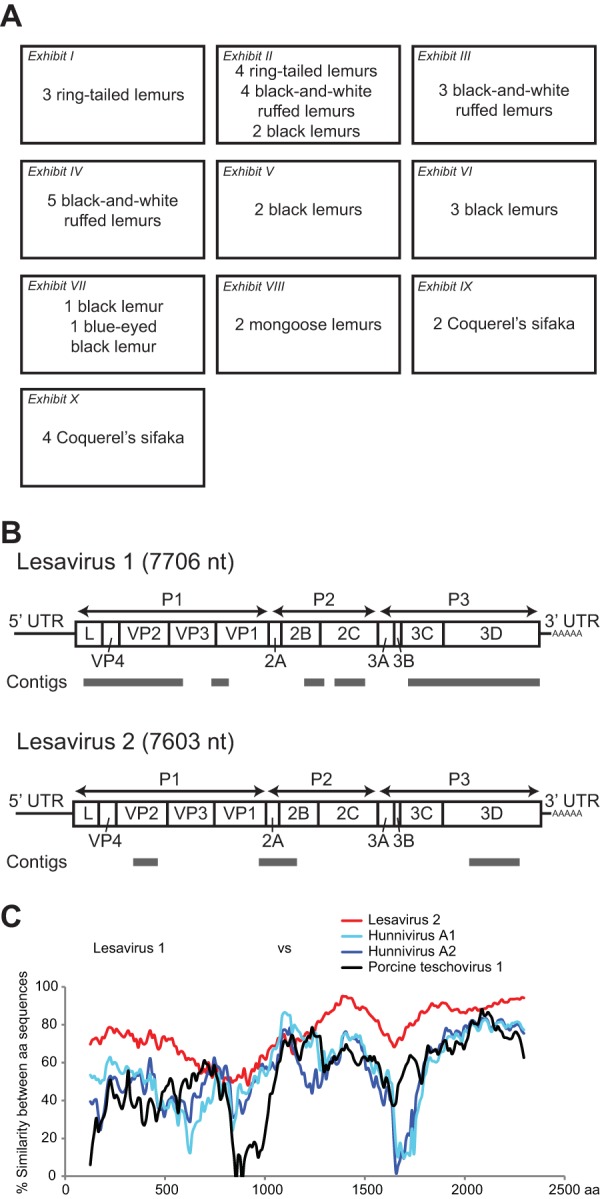
Identification of 2 novel picornaviruses in lemurs. (A) The species allocation of the lemur exhibits at the time of the 2012 collection is shown. (B) The diagram shows the complete genomes of lesavirus 1 (above) and lesavirus 2 (below) confirmed by RACE/RT-PCR. Contigs assembled from the Illumina sequencing reads are indicated in gray. UTR, untranscribed region. (C) Diversity plots of amino acid sequences are shown, comparing the lesavirus 1 polyprotein to lesavirus 2 (red), hunniviruses (light blue and dark blue), and porcine teschovirus 1 (black).
From 3,349,958 total sequencing reads in a ring-tailed lemur (Mis101308), we identified 20 reads that had limited sequence identity to known picornaviruses. De novo assembly of the picornavirus-like sequence reads yielded five contiguous sequences (contigs) that shared between 37% and 65% amino acid identity to hunniviruses, picornaviruses previously identified from cattle and sheep (36). Picornaviruses are single-stranded RNA viruses. The genome of typical picornaviruses includes a single open reading frame, flanked by untranslated regions at the 5′ and 3′ ends. Using a combination of RT-PCR and RACE methods, the complete genome of 7,687 nt was obtained and was verified to more than 3× coverage by Sanger sequencing (Fig. 1B). This virus was named lesavirus 1 (LV1; lemur stool-associated picornavirus 1).
Analyses of 579,108 reads from a black-and-white ruffed lemur (Nai108015) identified 341 reads that assembled into 3 contigs with limited sequence identity to picornaviruses (Fig. 1C). Sequence comparison demonstrated that the 3 contigs shared only 64.1% nucleotide identity with LV1, suggesting that the viral sequences in each specimen were distinct. Therefore, we sequenced the complete genome (7,593 nt) of the virus and named it lesavirus 2 (LV2; lemur stool-associated picornavirus 2). Sliding window analysis demonstrated that LV1 and LV2 were indeed distinct viruses, and limited similarity to the next most closely related hunnivirus and porcine teschovirus 1 species was observed throughout the genome (Fig. 1C).
We then examined the genomes for molecular features characteristic of picornaviruses. The NPGP cleavage motif shown in the 2A protein was conserved in LV1 (N970PGP) and LV2 (N949PGP). The putative 2C proteins had both the GXXGXGKS NTP binding motif (LV1, G1250RPGQGKS; LV2, G1231KPGQGKS) and the DDLXQ helicase activity motif (LV1, D1299DLGQ; LV2, D1280DLGQ). Additionally, the GXCG cysteine active site in the 3C protein was also conserved (LV1, G1725FCG; LV2, G1698YCG). Finally, the putative 3D protein maintains the YGDD active site (LV1, Y2101GDD; LV2, Y2074GDD) and the KDELR (LV1, K1936DETR; LV2, K1909DETR), FLKR (LV1, F2149LKR; LV2, F2122LKR), and GGLPSG (LV1, G2063GLPSG; LV2, G2036GLPSG) motifs. Thus, LV1 and LV2 encode conserved molecular hallmarks of picornaviruses.
Lesavirus 1 and lesavirus 2 define a novel genus in the family Picornaviridae.
The Picornaviridae family consists of 26 genera (37). We examined the evolutionary relationship of LV1 and LV2 in the family Picornaviridae. Phylogenetic trees were constructed with Bayesian and maximum-likelihood methods using a concatenated amino acid alignment of 2C and 3CD genes that included representative members from 26 picornavirus genera. Identical topologies were obtained when reconstructed with Bayesian and maximum-likelihood methods. The phylogenetic analyses strongly supported the idea that LV1 and LV2 formed a monophyletic clade and that they be placed sister to hunniviruses (Fig. 2A). These findings were also well supported by phylogenetic reconstruction using the P1 (VP4321) region (Fig. 2B). This indicated that LV1 and LV2 have an evolutionary history distinct from those of other picornaviruses.
FIG 2.

Lesavirus 1 and lesavirus 2 form a monophyletic clade in the Picornaviridae family. Phylogenetic relationships of representative members of the Picornaviridae family were inferred from the concatenated 2C3CD (A) and P1 (B) amino acid alignment, generated by the Bayesian MCMC method. The P1 phylogeny was consistent with phylogenetic analyses of the VP1 region (data not shown). Internal branch labels indicate the posterior probability. The maximum-likelihood method yielded trees with similar topologies. aa, amino acid.
International Committee on Taxonomy of Viruses (ICTV) guidelines for picornavirus species demarcation specify <70% amino acid identities in the P1 and 2C3CD regions and within-genus criteria as >40% in the P1 region, >40% in the P2 region, and >50% in the P3 region (37, 38). The pairwise amino acid identities of LV1 to LV2 in the P1, 2C3CD, P2, and P3 regions were 54.8%, 75.2%, 68.9%, and 72.8%, respectively (Table 1). While the region 2C3CD data support the idea of LV1 and LV2 being the same species, the P1 region had <70% identity, suggesting that they are of different species. Nonetheless, this indicated that LV1 and LV2 should be placed within the same genus. We next performed pairwise comparisons to hunniviruses and porcine teschovirus 1, which were most similar to LV1 and LV2. Comparison of LV1 and LV2 to hunniviruses in the P1, P2, and P3 regions gave results that ranged from 39.0% to 42%, 35.0% to 36.2%, and 46.2% to 46.6%, respectively. Results of sequence comparison to porcine teschovirus 1 ranged from 30.3% to 31.3%, from 36.5% to 37.9%, and from 40.5% to 40.9% in the P1, P2, and P3 regions, respectively. Taking the data together, this indicated that LV1 and LV2 define a novel picornavirus genus.
TABLE 1.
Pairwise amino acid comparison between lesavirus 1 and lesavirus 2
| Virus type | Genus | GenBank accession no. | Size (bp) | % G+C content | Comparison to lesavirus 1 (% aa identity) |
Comparison to lesavirus 2 (% aa identity) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | 2C | 3CD | 2C3CD | P1 | P2 | P3 | 2C | 3CD | 2C3CD | |||||
| Lesavirus 1 | “Lesavirus” | KM396707 | 7,687 | 46.0 | 54.8 | 68.9 | 72.8 | 76.1 | 74.7 | 75.2 | ||||||
| Lesavirus 2 | “Lesavirus” | KM396708 | 7,593 | 45.0 | 54.8 | 68.9 | 72.8 | 76.1 | 74.7 | 75.2 | ||||||
| Hunnivirus A1 | Hunnivirus | JQ941880 | 7,583 | 46.0 | 39.7 | 36.2 | 46.2 | 45.3 | 51.4 | 49.4 | 41.0 | 35.4 | 46.6 | 46.1 | 51.6 | 49.8 |
| Hunnivirus A2 | Hunnivirus | HM153767 | 7,588 | 46.0 | 39.0 | 35.9 | 46.2 | 46.6 | 51.4 | 49.8 | 42.0 | 35 | 46.5 | 45.8 | 51.3 | 49.5 |
| Porcine teschovirus 1 | Teschovirus | NC_003985 | 7,117 | 45.0 | 30.3 | 37.9 | 40.5 | 40.7 | 44.0 | 42.9 | 31.3 | 36.5 | 39.0 | 40.9 | 42.7 | 42.1 |
Lemur picornaviruses are highly prevalent and species specific.
We examined the epidemiology of the two novel picornaviruses in the lemur collection using longitudinally collected fecal specimens. We designed and validated a consensus-degenerate RT-PCR assay to amplify a 388-nt product from the 3Dpol region of LV1 and LV2 (Fig. 3A). Thirty-five fecal specimens representing 6 lemur taxa (ring-tailed lemurs, black lemurs, a blue-eyed black lemur, mongoose lemurs, black-and-white ruffed lemurs, and Coquerel's sifakas) collected from September and October 2012 were screened by the RT-PCR assay. Additionally, 33 fecal specimens collected approximately a year later (September 2013) were evaluated. In the period between the two samplings, 4 lemurs (3 black-and-white ruffed lemurs and 1 ring-tailed lemur) had died, a triplet of black-and-white ruffed lemurs was born, and a black-and-white ruffed lemur was transferred to a different zoo. To verify the species origin of the specimens, we sequenced the mitochondrial cytochrome B gene from nucleic acid extracted from the fecal specimens. Mitochondrial gene sequences from lemur species clustered into well-supported clades that matched the generally accepted phylogeny of lemurs (Fig. 3B).
FIG 3.

Species-specific prevalence of lesavirus 1 and lesavirus 2. (A) RT-PCR analysis of lesavirus is shown for water (control) or representative specimens found to be negative (Neg) for lesavirus 1 and lesavirus 2 (Gay88020) or positive for lesavirus 1 (Giz107097) or lesavirus 2 (Mah951211). The band corresponds to a 388-nt PCR product. (B) A maximum-likelihood phylogenetic tree constructed from partial cytochrome B sequences is shown. The alignment was based on sequences from samples found to be positive for either lesavirus 1 (LV1) or lesavirus 2 (LV2) and from samples from representative individuals of other species. Reference sequences from GenBank were included for L. catta (LCU53575), V. variegata (AB371089), and P. coquereli (AF285528). The phylogeny was outgrouped to an aye-aye sequence (DMU53569). Branch labels indicate bootstrap proportion. Individuals in the mixed-species exhibit are marked with an asterisk. E.m.macaco, Eulemur macaco macaco; E.m. flavifrons, Eulemur macaco flavifrons; E. mongoz, Eulemur mongoz; D. madagascariensis, Daubentonia madagascariensis. (C) The prevalences of LV1 and LV2 in specimens collected in 2012 and 2013 are shown. (D) A phylogeny inferred from the nucleotide sequences of all LV1 and LV2 strains that were screened in the experiments represented in panel C using the neighbor-joining method is shown. Virus sequences are highlighted with the host species origin as either ring-tailed lemur (open boxes) or black-and-white ruffed lemur (gray boxes) as determined by the cytochrome b genotype (see panel B). Virus sequences from individuals in the mixed-species exhibit are marked with an asterisk. (E) The prevalences of lemurs in the mixed-species exhibit in 2012 and 2013 are shown. The exhibit consisted of 4 ring-tailed lemurs (circles), 4 black-and-white ruffed lemurs (squares), and 2 black lemurs (triangles). Boxes and squares representing individuals infected by LV1 (gray) and LV2 (black) are shaded. The black-and-white ruffed lemurs were housed separately (Exhibit XI) in 2013. Two previously LV2-infected black-and-white ruffed lemurs died prior to the 2013 sampling (crossed squares).
LV1 was detected in 5 of 7 (71.4%) ring-tailed lemur fecal specimens collected in 2012 and in 5 of 6 (83.3%) ring-tailed lemur specimens in the 2013 collection (Fig. 3C). LV1 was not detected in black lemurs, a blue-eyed black lemur, mongoose lemurs, black-and-white ruffed lemurs, or Coquerel's sifakas. We detected LV2 in 6 of 12 (50%) black-and-white ruffed lemurs in the 2012 collection and in 7 of 11 (63.6%) black-and-white ruffed lemurs in 2013 (Fig. 3C). We did not detect LV2 in ring-tailed lemurs, black lemurs, a blue-eyed black lemur, mongoose lemurs, or Coquerel's sifakas. A phylogenetic tree constructed with all LV1 and LV2 sequences overlaid with each host species origin confirmed that all 10 specimens that were positive for LV1 were from ring-tailed lemurs and that all 13 LV2-positive specimens were from black-and-white ruffed lemurs (Fig. 3D). Picornaviruses evolve rapidly due to the presence of the error-prone RNA-dependent RNA polymerase. Therefore, we compared the VP1 sequences of LV1 from fecal specimens collected in 2012 and 2013 from the same ring-tailed lemur (Mis101308). A similar analysis was done for the VP1 of LV2 from a black-and-white ruffed lemur (Mah951211) whose results were positive at both time points. The estimated mean rates of LV1 and LV2 VP1 evolution were approximately 9.22 × 10−3 and 8.26 × 10−3 nucleotide substitutions per site per year, respectively, within the range of previous estimates for enteroviruses (39, 40).
We next examined the virus prevalence in the context of their single-species or mixed-species housing. Examples of both new infection and viral clearance were observed. In one single-species exhibit, a previously LV2-positive black-and-white ruffed lemur tested negative in 2013 (exhibit 3; see Table S1 in the supplemental material). A set of black-and-white ruffed lemur triplets born after the 2012 sampling and kept in a single-species exhibit were all positive for LV2 at the 2013 testing. In the mixed-species exhibit that housed 4 ring-tailed lemurs, 4 black-and-white ruffed lemurs, and 2 black lemurs together for approximately 5 months, both LV1 and LV2 were detected (Fig. 3E). Initially, in 2012, two ring-tailed lemurs were positive for LV1 and one black-and-white ruffed lemur was positive for LV2. In 2013, both LV1-positive ring-tailed lemurs remained positive whereas an additional ring-tailed lemur became infected with LV1. Both black lemurs remained negative for LV1 and LV2. Approximately 2 months after the first sampling, the four black-and-white ruffed lemurs were transferred to a separate exhibit, after which one lemur was found to have acquired LV2 in 2013 and the initially LV2-positive individual died. These observations demonstrated that, even in a high-contact mixed-species exhibit, the viruses were transmitted in a species-specific manner.
A ring-tailed lemur (Geo101895) that was positive for LV1 in 2012 subsequently died prior to the 2013 sampling. Two of the three black-and-white ruffed lemurs (Man105690 and Bon101605) that died prior to the 2013 sampling were positive for LV2 in 2012. An additional positive black-and-white ruffed lemur (And113831) died after the 2013 evaluation. However, the causes of death were different among the 5 lemurs—malignant neoplasia (Geo101895), progressive neurological disease (Man105690, Bon101605, and Jir105691), and suppurative meningoencephalitis (And113831) (see Table S1 in the supplemental material).
DISCUSSION
It is widely accepted that many emerging infectious diseases in humans are the result of zoonotic transmissions. However, the conditions that facilitate or prevent these transmissions are less well understood. This poses an urgent challenge in predicting disease emergence. Here, we investigated the transmission of two previously undescribed picornaviruses in lemurs at the Saint Louis Zoo. We chose lemurs because their species diverged on comparable evolutionary timescales to continental African primates, thus providing a parallel model for primate host genetic divergence. For example, black-and-white ruffed lemurs and ring-tailed lemurs diverged from their common ancestor approximately 26 and 21 million years ago, respectively (41). This evolutionary time scale is comparable to the divergence of the Catarrhini parvorder of primates that includes humans, gibbons, great apes, and Old World monkeys. Using the housing organization at the Saint Louis Zoo, we studied viral transmission in a setting of high interspecies contact (mixed-species exhibit) and a setting of minimal interspecies contact (single-species exhibit). Mixed-species exhibits are common in most zoological parks worldwide, and this report illustrates an approach that can be widely applied to other zoo settings to study viral transmission.
The role of host genetic barriers in helping to prevent cross-species transmission and viral adaptation and in determining whether transmission between species is primarily driven by contact intensity remains the subject of debate (4, 6, 13). In this study, we found that both lemur picornaviruses were highly prevalent and species specific in the lemur collection (Fig. 3C and D). LV1 was detected only in ring-tailed lemurs and LV2 only in black-and-white ruffed lemurs; neither virus was detected in black lemurs, mongoose lemurs, or Coquerel's sifakas. In other studies, enteroviruses and parechoviruses have been found cocirculating between humans and nonhuman primates (42, 43). For example, rhesus macaques and baboons in a multispecies cage at the Dhaka Zoo harbored human enterovirus 112 (43). In contrast, we found that the two lemur viruses were species specific despite cocirculating in an environment of high physical exposure and contact within a mixed-species exhibit (Fig. 3E). A ring-tailed lemur and a black-and white ruffed lemur in the mixed-species exhibit that were previously negative were positive at the second sampling time, demonstrating that infection could be newly acquired in the exhibit during this time frame. Together, these observations suggest that the lemur species have evolved host barriers to prevent cross-species transmission of these viruses, possibly shaped by selection to survive past pathogenic pressures (44). In addition to understanding the host genetic determinants of species specificity, future work can track the adaptive evolutionary trajectory in the event of cross-species transmission and establishment in the new host species.
All three lemur species in the mixed-species exhibit were fed the same diet, suggesting that the presence of the viruses was not simply the result of dietary ingestion. We are unable to exclude the possibility that the viruses originated from other host sources at the zoo, such as mice and insects that may be commonly encountered despite efforts to control their environment. Nonetheless, regardless of the prior host origin, we have demonstrated that both viruses can be detected in sequential samples in lemurs. Samples collected in 2012 and 2013 showed that the majority of the lemurs that tested positive in 2012 remained positive 1 year later, with viral clearance observed in only a minority of them. It is possible that the viruses cause persistent infection or, alternatively, that there may be clearance followed by reinfection. Additionally, black-and-white ruffed lemur triplets born after the initial sampling were found positive for LV2 in 2013, suggesting that de novo virus infection occurred. The test results from the dam (Lul105694) of the triplets were negative at both time points, suggesting that the infections were the result of horizontal transmission. The host range of these picornaviruses remains to be experimentally determined. This might be difficult to address in vivo, as many lemur species, including the critically endangered black-and-white ruffed lemur and endangered ring-tailed lemur, are threatened with extinction and are the focus of multifaceted conservation efforts (45). However, our studies are noninvasive (fecal collection) and could help assess the potential risk of viral infections on lemur survival.
No exogenous viruses had been described in lemurs prior to this study. The discovery of the lemur picornaviruses raises important questions about infectious causes of morbidity and mortality in lemurs. While we have detected viral nucleic acid in fecal specimens, this finding may not reflect the site of disease or provide clues to the pathogenicity of the virus. For example, poliovirus (a picornavirus) is shed in feces but causes neurologic disease. While four of the five lemurs that died also tested positive for the viruses at the prior evaluation, the causes of death were different among the individuals. Further studies are necessary to determine the potential pathogenicity of these lemur picornaviruses and to better define the epidemiology of infection in captive and wild lemurs.
According to ICTV guidelines for picornavirus taxonomy (37), the criteria for species demarcation are <70% amino acid identity in the P1 and 2C3CD regions and genera defined as sharing greater than 40%, 40%, and 50% identity in the P1, P2, and P3 regions, respectively. While the 2C3CD comparison of LPV1 and LPV2 falls within the species guidelines, indicating that they belong to the same species, the P1 region identity is lower than the 70% cutoff and indicates that LPV1 and LPV2 should be considered separate species. Based on the P1 divergence and the observed species specificity of infection, we propose that the two viruses are distinct species. Regardless, there is consistent agreement in the broader comparison of the P1, P2, and P3 regions, supporting the suggestion that LV1 and LV2 should be classified within a novel genus in the Picornaviridae family. Thus, we propose the name “Lesavirus” for the novel genus.
Supplementary Material
ACKNOWLEDGMENTS
D.W. holds an investigatorship in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund. E.S.L. is an Eli & Edythe Broad Fellow of the Life Sciences Research Foundation.
We thank the Department of Animal Health and the primate keeper staff at the Saint Louis Zoo. We also thank Henry Huang for comments on the manuscript and Tolison Fowler for technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03342-14.
REFERENCES
- 1.Bailes E, Gao F, Bibollet-Ruche F, Courgnaud V, Peeters M, Marx PA, Hahn BH, Sharp PM. 2003. Hybrid origin of SIV in chimpanzees. Science 300:1713. doi: 10.1126/science.1080657. [DOI] [PubMed] [Google Scholar]
- 2.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JSM, Poon LLM. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 3.Demogines A, Abraham J, Choe H, Farzan M, Sawyer SL. 2013. Dual host-virus arms races shape an essential housekeeping protein. PLoS Biol 11:e1001571. doi: 10.1371/journal.pbio.1001571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Streicker DG, Turmelle AS, Vonhof MJ, Kuzmin IV, McCracken GF, Rupprecht CE. 2010. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 329:676–679. doi: 10.1126/science.1188836. [DOI] [PubMed] [Google Scholar]
- 5.Malim MH, Bieniasz PD. 2012. HIV restriction factors and mechanisms of evasion. Cold Spring Harb Perspect Med 2:a006940. doi: 10.1101/cshperspect.a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuiken T, Holmes EC, McCauley J, Rimmelzwaan GF, Williams CS, Grenfell BT. 2006. Host species barriers to influenza virus infections. Science 312:394–397. doi: 10.1126/science.1122818. [DOI] [PubMed] [Google Scholar]
- 7.Etienne L, Hahn BH, Sharp PM, Matsen FA, Emerman M. 2013. Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe 14:85–92. doi: 10.1016/j.chom.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim ES, Malik HS, Emerman M. 2010. Ancient adaptive evolution of tetherin shaped the functions of Vpu and Nef in human immunodeficiency virus and primate lentiviruses. J Virol 84:7124–7134. doi: 10.1128/JVI.00468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim ES, Fregoso OI, McCoy CO, Matsen FA, Malik HS, Emerman M. 2012. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe 11:194–204. doi: 10.1016/j.chom.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dortmans JCFM, Dekkers J, Wickramasinghe INA, Verheije MH, Rottier PJM, van Kuppeveld FJM, de Vries E, de Haan CAM. 2013. Adaptation of novel H7N9 influenza A virus to human receptors. Sci Rep 3:3058. doi: 10.1038/srep03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anishchenko M, Bowen RA, Paessler S, Austgen L, Greene IP, Weaver SC. 2006. Venezuelan encephalitis emergence mediated by a phylogenetically predicted viral mutation. Proc Natl Acad Sci U S A 103:4994–4999. doi: 10.1073/pnas.0509961103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song H-D, Tu C-C, Zhang G-W, Wang S-Y, Zheng K, Lei L-C, Chen Q-X, Gao Y-W, Zhou H-Q, Xiang H, Zheng H-J, Chern S-WW, Cheng F, Pan C-M, Xuan H, Chen S-J, Luo H-M, Zhou D-H, Liu Y-F, He J-F, Qin P-Z, Li L-H, Ren Y-Q, Liang W-J, Yu Y-D, Anderson L, Wang M, Xu R-H, Wu X-W, Zheng H-Y, Chen J-D, Liang G, Gao Y, Liao M, Fang L, Jiang L-Y, Li H, Chen F, Di B, He L-J, Lin J-Y, Tong S, Kong X, Du L, Hao P, Tang H, Bernini A, Yu X-J, Spiga O, Guo Z-M, et al. 2005. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci U S A 102:2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parrish CR, Holmes EC, Morens DM, Park E-C, Burke DS, Calisher CH, Laughlin CA, Saif LJ, Daszak P. 2008. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev 72:457–470. doi: 10.1128/MMBR.00004-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonardi R, Buchanan-Smith HM, Dufour V, MacDonald C, Whiten A. 2010. Living together: behavior and welfare in single and mixed species groups of capuchin (Cebus apella) and squirrel monkeys (Saimiri sciureus). Am J Primatol 72:33–47. doi: 10.1002/ajp.20748. [DOI] [PubMed] [Google Scholar]
- 15.Pearson EL, Davis JM, Litchfield CA. 2010. A case study of orangutan and siamang behavior within a mixed-species zoo exhibit. J Appl Anim Welf Sci 13:330–346. doi: 10.1080/10888705.2010.507125. [DOI] [PubMed] [Google Scholar]
- 16.Mullin SJ. 1998. Inter- and intraspecific interaction rates of three species of lemurs (subfamily Lemurinae) in an enclosure at the Memphis Zoo and Aquarium. J Tenn Acad Sci 73:77–81. [Google Scholar]
- 17.Yoder AD, Yang Z. 2004. Divergence dates for Malagasy lemurs estimated from multiple gene loci: geological and evolutionary context. Mol Ecol 13:757–773. doi: 10.1046/j.1365-294X.2004.02106.x. [DOI] [PubMed] [Google Scholar]
- 18.Sondgeroth K, Blitvich B, Blair C, Terwee J, Junge R, Sauther M, VandeWoude S. 2007. Assessing flavivirus, lentivirus, and herpesvirus exposure in free-ranging ring-tailed lemurs in southwestern Madagascar. J Wildl Dis 43:40–47. doi: 10.7589/0090-3558-43.1.40. [DOI] [PubMed] [Google Scholar]
- 19.Gilbert C, Maxfield DG, Goodman SM, Feschotte C. 2009. Parallel germline infiltration of a lentivirus in two Malagasy lemurs. PLoS Genet 5:e1000425. doi: 10.1371/journal.pgen.1000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gifford RJ, Katzourakis A, Tristem M, Pybus OG, Winters M, Shafer RW. 2008. A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc Natl Acad Sci U S A 105:20362–20367. doi: 10.1073/pnas.0807873105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aswad A, Katzourakis A. 2014. The first endogenous herpesvirus, identified in the tarsier genome, and novel sequences from primate rhadinoviruses and lymphocryptoviruses. PLoS Genet 10:e1004332. doi: 10.1371/journal.pgen.1004332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han GZ, Worobey M. 2012. An endogenous foamy virus in the aye-aye (Daubentonia madagascariensis). J Virol 86:7696–7698. doi: 10.1128/JVI.00650-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katzourakis A, Aiewsakun P, Jia H, Wolfe ND, LeBreton M, Yoder AD, Switzer WM. 2014. Discovery of prosimian and afrotherian foamy viruses and potential cross species transmissions amidst stable and ancient mammalian co-evolution. Retrovirology 11:61. doi: 10.1186/1742-4690-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enserink M. 2007. Medicine. Initiative aims to merge animal and human health science to benefit both. Science 316:1553. doi: 10.1126/science.316.5831.1553a. [DOI] [PubMed] [Google Scholar]
- 25.Deem SL. 2015. Conservation medicine to one health: the role of zoologic veterinarians, p 698–703 InMiller RE, Fowler ME (ed), Fowler's zoo and wild animal medicine, vol 8 Saunders Elsevier, Saint Louis, MO. [Google Scholar]
- 26.Zhao G, Krishnamurthy S, Cai Z, Popov VL, Travassos da Rosa AP, Guzman H, Cao S, Virgin HW, Tesh RB, Wang D. 2013. Identification of novel viruses using VirusHunter – an automated data analysis pipeline. PLoS One 8:e78470. doi: 10.1371/journal.pone.0078470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim J-B, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim ES, Cao S, Holtz LR, Antonio M, Stine OC, Wang D. 2014. Discovery of rosavirus 2, a novel variant of a rodent-associated picornavirus, in children from The Gambia. Virology 454–455:25–33. doi: 10.1016/j.virol.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 32.Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 33.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drummond AJ, Andrew R. 2009. Tracer v1.5. http://tree.bio.ed.ac.uk/software/tracer/.
- 35.Nylander JA, Wilgenbusch JC, Warren DL, Swofford DL. 2008. AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 24:581–583. doi: 10.1093/bioinformatics/btm388. [DOI] [PubMed] [Google Scholar]
- 36.Reuter G, Pankovics P, Knowles NJ, Boros A. 2012. Two closely related novel picornaviruses in cattle and sheep in Hungary from 2008 to 2009, proposed as members of a new genus in the family Picornaviridae. J Virol 86:13295–13302. doi: 10.1128/JVI.01142-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knowles NJ, Hovi T, Hyypiä T, King AMQ, Lindberg AM, Pallansch MA, Palmenberg AC, Simmonds P, Skern T, Stanway G, Yamashita T, Zell R. 2011. Picornaviridae, p 855–880 InVirus taxonomy: classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London, United Kingdom. [Google Scholar]
- 38.Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. 2005. Virus taxonomy: eighth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 39.Hicks AL, Duffy S. 2011. Genus-specific substitution rate variability among picornaviruses. J Virol 85:7942–7947. doi: 10.1128/JVI.02535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McWilliam Leitch EC, Cabrerizo M, Cardosa J, Harvala H, Ivanova OE, Kroes AC, Lukashev A, Muir P, Odoom J, Roivainen M, Susi P, Trallero G, Evans DJ, Simmonds P. 2010. Evolutionary dynamics and temporal/geographical correlates of recombination in the human enterovirus echovirus types 9, 11, and 30. J Virol 84:9292–9300. doi: 10.1128/JVI.00783-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MAM, Kessing B, Pontius J, Roelke M, Rumpler Y, Schneider MPC, Silva A, O'Brien SJ, Pecon-Slattery J. 2011. A molecular phylogeny of living primates. PLoS Genet 7:e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harvala H, Van Nguyen D, McIntyre C, Ahuka-Mundeke S, Ngole EM, Delaporte E, Peeters M, Simmonds P. 2014. Co-circulation of enteroviruses between apes and humans. J Gen Virol 95:403–407. doi: 10.1099/vir.0.059048-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oberste MS, Feeroz MM, Maher K, Nix WA, Engel GA, Begum S, Hasan KM, Oh G, Pallansch MA, Jones-Engel L. 2013. Naturally acquired picornavirus infections in primates at the Dhaka Zoo. J Virol 87:572–580. doi: 10.1128/JVI.00838-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emerman M, Malik HS. 2010. Paleovirology—modern consequences of ancient viruses. PLoS Biol 8:e1000301. doi: 10.1371/journal.pbio.1000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwitzer C, Mittermeier RA, Johnson SE, Donati G, Irwin M, Peacock H, Ratsimbazafy J, Razafindramanana J, Louis EE Jr, Chikhi L, Colquhoun IC, Tinsman J, Dolch R, LaFleur M, Nash S, Patel E, Randrianambinina B, Rasolofoharivelo T, Wright PC. 2014. Conservation. Averting lemur extinctions amid Madagascar's political crisis. Science 343:842–843. doi: 10.1126/science.1245783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.