

ABSTRACT
Human immunodeficiency virus (HIV) transmission typically results from infection by a single transmitted/founder (T/F) variant. Are T/F variants chosen uniformly at random from the donor pool, or are they selected based on advantageous traits facilitating transmission? Finding evidence for selection during transmission is of particular interest, because it would indicate that phenotypic and/or genetic properties of the viruses might be harnessed as potential vaccine targets or immunotherapies. Here, we systematically evaluated the differences between the Env proteins of simian immunodeficiency virus/simian HIV (SIV/SHIV) stock and T/F variants in search of “signature” sites of transmission. We also surveyed residue preferences in HIV at the SIV/SHIV signature sites. Four sites of gp120 showed significant selection, and an additional two sites showed a similar trend. Therefore, the six sites clearly differentiate T/F viruses from the majority of circulating variants in the stocks. The selection of SIV/SHIV could be inferred reasonably across both vaccinated and unvaccinated subjects, with infections resulting from vaginal, rectal, and intravenous routes of transmission and regardless of viral dosage. The evidence for selection in SIV and SHIV T/F variants is strong and plentiful, and in HIV the evidence is suggestive though commensurate with the availability of suitable data for analysis. Two of the signature residues are completely conserved across the SIV, SHIV, and HIV variants we examined. Five of the signature residues map to the C1 region of gp120 and one to the signal peptide. Our data raise the possibility that C1, while governing the association between gp120 and gp41, modulates transmission efficiency, replicative fitness, and/or host cell tropism at the level of virus-cell attachment and entry.
IMPORTANCE The present study finds significant evidence of selection on gp120 molecules of SIV/SHIV T/F viruses. The data provide ancillary evidence suggesting the same sites are under selection in HIV. Our findings suggest that the signature residues are involved in increasing the transmissibility of infecting viruses; therefore, they are potential targets for developing a vaccine or other protective measures. A recent study identified the same T/F signature motif but interpreted it as an effect of neutralization resistance. Here, we show that the T/F motif has broader functional significance beyond neutralization sensitivity, because it is present in nonimmune subjects. Also, a vaccine regimen popular in animal trials might have increased the transmission of variants with otherwise low transmission fitness. Our observations might explain why many animal vaccine trials have not faithfully predicted outcomes in human vaccine trials and suggest that current practices in vaccine design need to be reexamined accordingly.
INTRODUCTION
For over 2 decades, it has been clear that human immunodeficiency virus (HIV) transmission and the inception of a productive infection involves a population bottleneck (1, 2). In about 80% of heterosexual HIV transmissions, a single transmitted/founder (T/F) variant establishes infection (3). To date, it is unclear whether any infectious HIV in the donor pool is equally likely to initiate transmission or whether HIV variants undergo selection during transmission (4). The population bottleneck in the transmission of T/F viruses suggests that selection plays a role in HIV transmission, but quantitative evidence supporting this intuition has yet to come forward. Finding evidence for selection during HIV transmission is of particular interest, because it would indicate that phenotypic and/or genetic properties of the T/F viruses can be harnessed as vaccine targets.
Phenotypically, transmitted viruses are known to be preferentially CCR5-tropic viruses (5). However, the specific molecular features increasing risk of T/F transmission, if selection applies, have not been clearly defined. On the other hand, T/F viruses might be chosen uniformly at random and not as a result of selective pressures. Thus, establishing whether HIV transmission is under selection or a uniform random process is central to designing effective HIV vaccines and other protective measures.
The present study couples logically simple statistical methods to computational sequence analyses to evaluate the properties of T/F variants from six simian immunodeficiency virus (SIV), two hybrid SIV-HIV (SHIV), and four HIV-1 infection cohorts. The datasets contain examples of primate lentiviral infection in the presence (6–8) and absence (9) of a humoral response through vaginal (10), rectal, or intravenous (6, 9, 11) routes of transmission, as well as from multiple low-dose, single high-dose, and natural human infection dosages.
Our findings indicate that the gp120 molecules of primate lentiviruses are under selection during transmission. Furthermore, we identify at least four (and up to six) sites on gp120 under selection. The T/F “signature” motif is highly conserved regardless of vaccination status, viral dosage, or transmission route. Here, we pose several hypotheses regarding the role of the infection signatures in facilitating enhanced infectiousness of T/F viruses, which make the C1 signatures likely targets for developing an effective vaccine or treatment. We also find evidence to indicate that the transmission signatures have broader functional significance than previously reported.
MATERIALS AND METHODS
Datasets.
The present study placed specific requirements on its datasets (as detailed in Results), so the following datasets were available for entry into the study. Note that the requirements stipulated that single-genome amplification (SGA) generate all sequences.
Unvaccinated Keele-SIVsmE660 and Keele-SIVmac251 datasets.
The present study analyzed data from the 18 adult Indian rhesus macaques (Macaca mulatta) in the study by Keele et al., which were inoculated with low-dose intrarectal and intravenous SIVsmE660 and SIVmac251 (termed Keele-SIVsmE660 and Keele-SIVmac251, respectively, or Keele-SIV datasets) (9). The animals had no preexisting humoral response to Env at the time of exposure. The SIV inocula contained a viral quasispecies with env diversity comparable to that observed in humans 1 to 2 years after infection by HIV-1 (9, 12). Since the present study focuses on the properties of T/F viruses, we identified the animals with data at ramp-up viremia (5 SIVsmE660 and 8 SIVmac251 animals). Under experimental conditions, the transmitted viruses are unknown and must be inferred. Thus, examining viruses early in the infection improves the accuracy of phylogenetic inference of the T/F virus. We dropped one SIVsmE660-infected animal (CP37) from the present study because a discrete transmitted lineage could not be clearly identified (see Fig. S1 in the supplemental material). The 12 remaining animals (4 SIVsmE660 and 8 SIVmac251) showed one or more transmitted lineages on neighbor-joining (NJ) trees of the stock and animal sequences (see Fig. S1A and S2 in the supplemental material).
Vaccinated SIV smE660 (Roederer-SIV) datasets.
We analyzed the env sequences of viruses in the inoculum and in the infected rhesus macaques from the four treatment arms of the Roederer et al. study (termed the Roederer-SIV datasets) (6). All animals received a DNA prime, recombinant adenovirus type 5 (rAd5) vaccine regimen. The control group received vectors with no inserts, while the three other groups received Gag mosaic, mosaic Env, and SIVmac239 Env immunogens. The env sequences in the Roederer et al. study come from the earliest time point with detectable plasma viral load. We used the T/F env sequences deduced by Roederer et al. and deposited in GenBank.
SHIV-BaL (Klein-SHIV) dataset.
Four female rhesus macaques in the Klein et al. study (10) were inoculated vaginally with a single high dose of the SHIV-BaL strain (termed the Klein-SHIV dataset). The four SHIV-BaL control animals received no antibodies. Klein et al. collected blood samples 6 h after infusion (day 0) and at 7, 14, and 21 days postinfusion. The env sequences we analyzed came from the first viral RNA-positive plasma sample. Figure S3 in the supplemental material shows SHIV-BaL stock and animal trees and transmitted lineages.
pSF257-SHIV clone.
pSF257 is a clade B SHIV we made based on an infectious molecular clone of a T/F HIV-1 AD17 virus (11). The AD17 virus came from an acutely infected subject in a cohort of acutely infected men who have sex with men (MSM).
Step-HIV datasets.
We analyzed the T/F viruses on the vaccine and placebo arms of the Step vaccine trial (termed Step-HIV datasets). Subjects in the vaccine group received Merck adenovirus 5 (MRKAd5) HIV-1 subtype B Gag/Pol/Nef immunogens (7, 8). The Step trial tested for HIV-1 infection at day 1 and weeks 12, 30, and 52 and every 6 months for 4 years. Sequencing was performed on plasma specimens collected at the time of HIV-1 diagnosis.
Gnanakaran-HIV datasets.
Gnanakaran et al. collected sequence variants from HIV-1, subtype B, sexually transmitted infections in individuals from the United States and Trinidad (termed the Gnanakaran-HIV datasets) (13). Gnanakaran et al. classified infections according to Fiebig stages and divided the data into two sets, original and holdout; the latter set was created for hypothesis testing. Sequences from Fiebig stages 2 to 5 were grouped into the Gnanakaran-HIV original/holdout acute group, and chronic sequences were put in the Gnanakaran-HIV original/holdout chronic group. Sequences from Fiebig stage 1 were not available. By following the protocol of Gnanakaran et al., we derived a consensus sequence for each patient. The consensus sequences in the acute subsets represent the T/F variant population. In the case of a single transmitted variant, the consensus sequence generally models the transmitted virus. Using a consensus in the case of multiple transmitted variants will generate a chimeric T/F variant, which is the best approximation we can derive given the type of data available. Since donor data were not available for the Gnanakaran datasets, we could not perform the type of phylogenetic analysis described below to enumerate the T/F variants. Thus, in contrast to the experimental datasets, we could not select a single predominant transmitted lineage for the Gnanakaran datasets. The consensus sequence for the chronic patients captures the predominant residues at each site within a subject (i.e., the major variants).
Sequence analysis.
We translated the env genes of stock and subject variants using HIValign and generated consensus sequences using Consensus maker v.2.0.1, which are tools developed at Los Alamos National Laboratory (14). To deduce T/F viruses, we implemented a modified version of the protocol by Keele et al. (9). First, HIValign using MAFFT (16) was used to create a multiple-sequence alignment of stock and recipient Env sequences for each animal (Fig. 1). PHYLIP's protdist and neighbor methods, v. 3.69 (17), then generated a distance matrix and a resulting NJ tree. To ensure independent samples, our statistical tests required us to represent the transmission to each subject with exactly one T/F virus, so when multiple T/F viruses were transmitted, only sequences from the predominant transmitted lineage were used, as shown in Fig. 1 and in Fig. S1 to S3 in the supplemental material. For each animal, we created phylogenetic trees of stock and animal variants to visualize the number of transmitted lineages (see Fig. S1 to S3). In the majority of animals, a single transmitted lineage was evident (see Fig. S1 to S3). In cases of multiple transmitted lineages, we chose the most heavily populated animal lineage as the predominant transmitted lineage. In addition, to delimit the number of animal variants belonging to the predominant lineage, we required each predominant-lineage animal variant to have no more than 10% stock sequences as its closest variants. Lastly, the Env sequence of the T/F virus was deduced by generating a consensus sequence from the multiple-sequence alignment of the predominant transmitted lineage.
FIG 1.
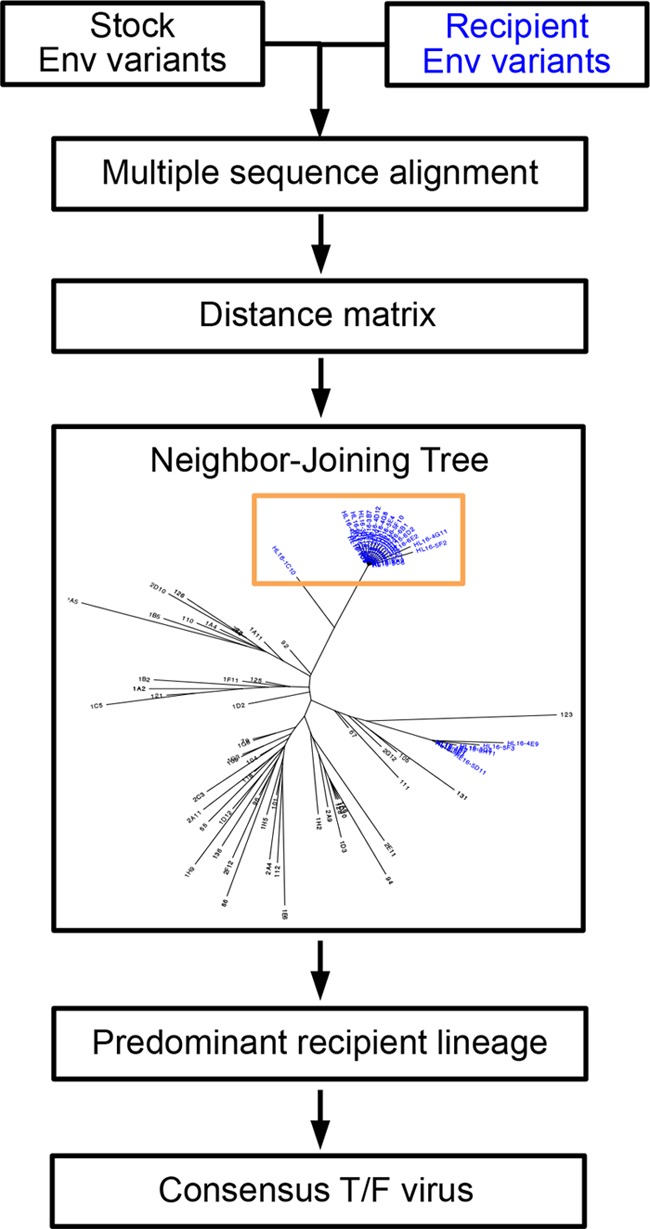
Deducing transmitted/founder (T/F) viruses. For each dataset, we generated a multiple-sequence alignment of stock and animal Env sequences. A distance matrix computed from the alignment guided the construction of a neighbor-joining (NJ) phylogenetic tree. To identify a single T/F virus per transmission event, we selected the predominant recipient lineage from the NJ tree. The predominant lineage (orange box) was defined as the set of recipient (blue) sequences whose closest neighborhood consisted of 90% or more recipient sequences. The T/F virus was deduced as the consensus of the sequences in the predominant recipient/transmitted lineage.
Statistical analysis.
To ensure a conservative statistical analysis, all P values reported are two sided. To facilitate reproduction of our calculations, we reported P values directly; they are reported as significant only if they survive a statistically conservative procedure, namely, Tarone's modification (18) of the Bonferroni correction for multiple testing (19). Fisher exact tests (20) were used for signature scanning, Mann-Whitney-Wilcoxon tests (20) were performed on glycosylation and loop length analyses, and Fisher inverse chi-square tests (21) were used to perform the metadata analyses.
RESULTS
Criteria to evaluate selection during lentiviral transmission.
Often, only one HIV variant from the donor pool establishes a productive clinical infection in the exposed subject (3), leading to the following question: are HIV T/F viruses chosen uniformly at random from the donor pool, or are they selected because they have advantageous traits facilitating transmission? To reflect transmission events faithfully, relevant data must satisfy several stringent criteria. We required datasets involving infections (i) in vivo; (ii) sampled during the earliest stages of the acute phase; (iii) with known genetic compositions of donor/stock and recipient pools; and (iv) with sequences generated through SGA methods.
Given the uncertainty of host or virus factors affecting the success of viral transmission, in vivo infections are preferable. However, deciphering early events of in vivo infections is complicated by the viral “eclipse” period. During viral eclipse, the single or a few successful transmitted variant(s) establishing infection replicate in local tissues. However, current nucleic acid tests cannot quantitatively detect viral variants until the swarm size surpasses the detection threshold (i.e., ∼50 copies/ml) (reviewed in reference 4). Thus, the best chance to deduce T/F viruses accurately is to use datasets with viral sequence data from the earliest stages of the infection.
We searched specifically for datasets with viral sequences sampled from the seroconverted recipients at the earliest phases of acute infection (i.e., the equivalent of Fiebig stages 1 to 4). We required early acute variants in the recipient because T/F variants cannot be explicitly determined but can be inferred only by phylogenetic modeling. T/F viruses are deduced routinely by phylogeny as the ancestral sequence of a mostly homogenous swarm of early viruses (3, 9, 11, 22).
In addition, our study used sequence data only for infections where the genetic composition of both donor (stock or seropositive donor) and recipient (animal or seroconverted recipient) variants were known. Knowing the genetic composition of donor and recipient variants was necessary to meet the main objective of the present study, i.e., to determine whether T/F viruses differ significantly from the remaining circulating variants in the donor pool, and if so, to identify any advantageous traits facilitating their increased transmissibility.
We also decided to omit any dataset with sequences generated prior to the advent of SGA methods. By omitting non-SGA datasets (e.g., the mother-to-child transmission [MTCT] pairs from references 15 and 23), we hoped to avoid drawing conclusions subject to possible biases by Taq polymerase or recombination errors (3).
Given the need to meet the above-described stringent criteria, our study evaluated selection at transmission mainly in experimental infections with SIV and SHIV. Although HIV data obviously are relevant to the eventual aim of developing AIDS therapies, the data associated with natural HIV infections generally come from long sampling intervals (e.g., ∼six months in the MTCT pairs from references 15 and 23), which impairs our ability to capture viruses early enough to ascertain the properties of initial transmitted viruses accurately. Several HIV datasets we considered had nonhomogeneous viral populations in the seroconverted recipients, suggesting infection had progressed past ramp-up acute viremia; thus, the viral population had begun to diversify. Unfortunately, the long intervals between actual transmission and blood sampling make capturing a diversified HIV population in recent seroconverted recipients of human trials common.
Some other HIV datasets included data from early stages of infection (e.g., the Step trial [7, 8] and the acute datasets of Gnanakaran et al. [13]), permitting us to deduce the corresponding T/F variants reasonably well, but donor data were unavailable. Thus, we used the datasets from the experimental infections in Table 1 to evaluate the role of selection and used the datasets from natural infections to provide ancillary evidence of residue preferences in HIV at the SIV/SHIV signature sites.
TABLE 1.
Datasets of SIV, SHIV, and HIV-1 transmission examined in the present study
| Source of infection | Dataseta | Viral doseb | Vaccination status | Route of transmissionc | Reference |
|---|---|---|---|---|---|
| Experimental | Keele-SIVsmE660 | Multiple low | Unvaccinated | i.r., i.v. | 9 |
| Experimental | Keele-SIVmac251 | Multiple low | Unvaccinated | i.r., i.v. | 9 |
| Experimental | Roederer-SIV control | Multiple low | DNA/Ad5 | i.r. | 6 |
| Experimental | Roederer-SIV mosaic gag | Multiple low | DNA/Ad5 Gag | i.r. | 6 |
| Experimental | Roederer-SIV mosaic Env | Multiple low | DNA/Ad5 mosaic Env | i.r. | 6 |
| Experimental | Roederer-SIV mac239 Env | Multiple low | DNA/Ad5 mac239Env | i.r. | 6 |
| Experimental | Klein-SHIV | Single high | Unvaccinated | Vaginal | 10 |
| Natural | pSF257-SHIV | NA | Unvaccinated | MSM | |
| Natural | Step-placebo-HIV | NA | Diluent vaccinated (no Ad5) | Unspecified | 7, 8 |
| Natural | Step-vaccine-HIV | NA | Ad5 gag-nef-pol vaccine | Unspecified | 7, 8 |
| Natural | Gnanakaran-HIV original FS2-FSC | NA | Unvaccinated | Unspecified | 15 |
| Natural | Gnanakaran-HIV holdout FS2-FSC | NA | Unvaccinated | Unspecified | 15 |
FS, Fiebig stage.
NA, not applicable.
i.r., intrarectal. i.v., intravenous.
The experimental infections also afforded the additional benefit of observing multiple subjects infected with the same stock. The Keele-SIV study infected four animals with SIVsmE660 and eight with SIVmac251, the Roederer-SIV study infected 20 animals in each treatment group (a total of 80 animals), and the Klein-SHIV study infected four with SHIV-BaL. In our statistical treatment, each animal inoculation becomes a different, independent sample from the same population, contributing to the statistical power of our study. Matching the same conditions in a natural human infection would require the same donor subject infecting multiple recipients. We are not aware of HIV transmission data linked epidemiologically by a common donor also fitting our other criteria. Thus, at present, the SIV/SHIV animal infections proved to be the best models to address the present question of selection during HIV transmission.
Our statistical tests require only that samples from a viral population be probabilistically independent, not that the phylogeny of the samples within the population be independent. Note, therefore, that although the two usages of “independent” can create linguistic illusions, our statistical methods remain valid.
Signature sites under selection in SIV transmission.
To evaluate the role of selection in SIV transmission, we analyzed Env variants from seven sets of macaques infected experimentally with stocks of SIVsmE660 or SIVmac251. The experimental datasets covered vaccinated as well as unvaccinated subjects, with infections resulting from rectal or intravenous routes of transmission through low-dose inocula (Table 1). The sequences of T/F viruses were inferred analytically by applying the standard protocol of Keele et al. and others (3, 9, 11, 22) as described in Materials and Methods. Phylogenetic trees of stock and animal variants showed low-diversity transmitted lineages (see Fig. S1 and S2 in the supplemental material), reflecting diversification from discrete T/F viral genomes.
We analyzed multiple sequence alignments of stock and T/F Env proteins in search of signature sites of transmission within each dataset (Fig. 2; also see Fig. S4 in the supplemental material). Signature residues were defined as sites where a Fisher exact test determined that the amino acid preference in the T/F variants was significantly different from the corresponding residue in the stock variants. Signature scanning was performed by dataset; i.e., we aligned each stock to the T/F sequences in the given dataset and identified sites with differential residue frequencies between the two groups (stock versus T/F viruses). For concise illustration, Fig. 2A shows a multiple-sequence alignment of the consensus of each dataset along the Env region containing the signature sites. The supplemental material shows extensive alignments of all variants in the present study (see Fig. S4). Figure 2B zooms in on the signature columns where the residue difference between stock and T/F variant was statistically significant.
FIG 2.

Alignment of sequence variants in the present study showing the signature sites. For concise illustration, the figure uses a single consensus sequence to represent either the stock or T/F variants in the given dataset. Our statistical analyses were not based on consensus sequences except to deduce the T/F variants, i.e., we examined all variants in each stock and one T/F variant per infected animal, as described in Materials and Methods. (A) An alignment showing the signal peptide (where available) and the C1 region of gp120 for stock and T/F variants in the various datasets examined by this study. Signature sites were defined as sites where the residue preference of T/F variants was significantly different (based on a Fisher exact test) from the corresponding residue in the stock. The alignment shows signature sites at positions 9, 33, 55, 64, and 88 in SIV and site 65 in SHIV. HXB2 was added to the alignment for reference purposes, standardizing the coordinates across datasets. Residues in blue mark variants carrying the predominant stock residue, whereas yellow highlighting shows residues preferred by T/F viruses. The sites in gray show Gnanakaran et al.'s H12 site (H9 in our coordinates) proposed as a signature of acute viruses. Abbreviations and symbols: spaces represent gaps; f, T/F viruses; *, sites conserved across all datasets; ·, residues identical to those in the SIV or HIV variant at the top, i.e., the Keele-SIV stock or the HXB2 sequence. For visual clarity, residues at the signature sites are shown explicitly rather than using dots. (B) An abbreviated alignment showing only the columns for the six signature sites as seen in the experimental datasets. The alignment is grouped by dataset and depicts stock residues in blue and T/F residues in purple.
We examined the residue preferences in full-length Env sequences, including the signal peptides (where available), gp120, and gp41 proteins of stock and T/F variants. Nonetheless, all signature sites consistently mapped to the signal peptide and the C1 region of gp120 (Fig. 2A). The Keele-SIVsmE660 dataset showed five signature sites: one site on the Env signal peptide and four in the C1 region of gp120. The Roederer-SIV datasets showed the same C1 signatures. Detailed results are outlined below, listing individual P values by site. To evaluate whether the individual P values from the Keele SIV datasets are significant on their own, a Bonferroni multiple-test correction factor of five may be applied (to account for five sites showing significant variation according to recommendations in reference 18). A correction factor of four is appropriate for the Roederer-SIV datasets, considering that only four sites show significant variation. Minimal variation across SIV variants was evident elsewhere on Env, but no other sites proved statistically significant.
The Keele-SIVsmE660 dataset sequenced 42 env genes from the stock and 108 ramp-up viremia variants from four challenges in Indian rhesus macaques. The Env proteins of the Keele-SIVsmE660 stock and T/F variants were virtually identical throughout, except at five sites with starkly different amino acid preferences (Fig. 2; also see Fig. S4 in the supplemental material). A Fisher exact test showed strong residue differences at positions 9 (P = 4.99e−3), 33 (P = 4.99e−3), 55 (P = 2.14e−4), 64 (P = 9.19e−5), and 88 (P = 1.35e−2) in the Keele-SIV dataset (Fig. 2A). The RIAK Keele-SIVsmE660 motif survives the modified Bonferroni adjustment at α = 0.05 (see Fig. S5 in the supplemental material). The N T/F signature at position 88 on the Keele-SIVsmE660 set showed a trend toward selection with a corrected P value significant at α = 0.06 (see Fig. S5). The alignment positions in Fig. 2 correspond to HXB2 coordinates; the HIV-1 HXB2 protein was added to the alignment for reference purposes. Residues KVTRS predominated in the Keele-SIVsmE660 stock pool, while T/F viruses featured an RIAKN signature at the corresponding positions (Fig. 2A).
Keele et al. also inoculated eight additional animals with a stock of SIVmac251. The Keele-SIVmac251 dataset reported 61 env genes from the stock and 260 ramp-up (acute) viremia variants from eight animals. We could not detect any statistically significant differences between the SIVmac251 stock and the T/F variants (see Fig. S5 in the supplemental material). However, three of the SIVsmE660 sites were completely conserved in the mac251 T/F viruses. Positions 55, 64, and 88 featured AKN, but so did the stock viruses, which indicates that the SIVmac251 stock does not present a diverse viral challenge. The SIVmac251 sequences showed KT in both stock and transmitted lineages at sites 9 and 33.
The animal trial by Roederer et al. (the Roederer-SIV dataset) also challenged a set of rhesus macaques with the SIVsmE660 strain (6). Roederer and colleagues immunized 80 rhesus macaques with one of four DNA-prime/rAD5 vaccine regimens prior to viral challenge. The authors obtained 34 env sequences from the SIVsmE660 challenge inoculum and 1,496 from the infected animals. Roederer et al. reported the same IAKN signature for T/F viruses in the mosaic Env and mac239 Env arms of their study at positions 33, 55, 64, and 88. Unfortunately, the Roederer et al. study did not sequence upstream enough of gp120 to allow us to test for the presence of the signature at position 9 of the signal peptide. We confirmed the IAKN signature in the mac239 Env treatment with the following Fisher exact test P values: I, 6.49e−6; A, 1.55e−3; K, 3.15e−3; N, 5.72e−4. All four Roederer-SIV mac239Env sites remain highly significant after multiplicity correction (see Fig. S5 in the supplemental material). T/F viruses in the mosaic Env treatment showed a strong preference for I(A|T)KN (P values: I, 4.11e−2; K, 3.02e−2; N, 5.02e−3) (Fig. 2B). Preference for A over T at position 45 is not significant (P = 0.124); half of the mosaic Env T/F variants carried A, while the other half carried the donor signature T at position 45. Only the N site in T/Fs of Roederer-SIV mosaic Env group survives at α = 0.05 after the stringent multiplicity adjustment (see Fig. S5). The majority of T/F viruses in the Roederer-SIV control and gag treatment groups carried the same signature residues as the stock viruses (i.e., VTRS) (Fig. 2B).
We found a variant in the Keele-SIVsmE660 stock with all four residues of the IAKN T/F signature: 1/42 stock sequences carried IAKN at sites 33, 55, 64, and 88. Three stock variants featured R at position 9, but a stock variant carrying the full RIAKN motif could not be identified. Current sequencing techniques have the limitation that they cannot sequence deeply enough to provide a full account of all variants present in the stock or the animal. Thus, our inferences of predominance in the stock and transmitted lineages are close, yet imperfect, approximations of actual viral swarms. The IAKN variants also represented a minor fraction of the Roederer-SIVsmE660 stock. Only 2/34 stock variants featured the IAKN signature motif, whereas 24/34 featured VTRS at the same sites. The genetic compositions of stock variants sequenced by Keele et al. and Roederer et al. suggest that IAKN was indeed a minor variant in the SIVsmE660 stock. Despite its low frequency in the stock viral swarm, IAKN was the only lineage transmitted in 38 independent instances: i.e., in 4 Keele-SIV and 34 Roederer-SIV animals (19/20 mosaic Env and 15/20 mac239Env animals, since 6 animals in the Roederer et al. study never got infected [M. Roederer, personal communication]).
On the one hand, we have applied (Tarone's variant of) the Bonferroni multiple-test correction to evaluate whether individual P values from the Keele-SIV and Roederer-SIV datasets are significant on their own. On the other hand, weighing evidence from all six SIV datasets together through meta-data analyses permits generating broader scientific conclusions. Fisher inverse chi-square P values were calculated based on two-sided, Bonferroni-corrected, Fisher exact test P values (see Fig. S5 in the supplemental material). Metadata analyses applied to the six datasets indicated that sites 33, 55, 64, and 88 on the C1 region of gp120 are under selection during SIV transmission. The application of the Fisher inverse chi-square test to the SIV datasets yielded overall P values for the signature residues (I, 1.33e−3; A, 1.37e−2; K, 4.92e−3; N, 8.45e−3). However, the conservation of signature site R9 was not statistically significant (P = 0.117) over the two SIV datasets with signal peptide sequences. All four C1 region combined P values remain highly significant despite our conservative statistical treatment, indicating strongly that the IAKN residues provide signatures for SIV infectivity.
Signature sites under selection in SHIV transmission.
We also searched for transmission signatures in the SHIV study by Klein et al. (10). T/F viruses were inferred analytically as described in Materials and Methods. The Klein-SHIV infections also showed diversification from discrete T/F viral genomes evidenced by low-diversity transmitted lineages (see Fig. S3A to D in the supplemental material).
Klein et al. challenged four macaques vaginally with SHIV-BaL in the absence of a vaccination regimen (10). The Klein-SHIV study sequenced 51 stock and 197 animal env variants. None of the SIV signature sites showed a significant difference in Klein-SHIV stock versus T/F variants. The predominant residues in the SHIV variants at positions 9, 33, 55, 64, and 88 were HKAEN, respectively. We found similar residue preferences on pSF257-SHIV, a clade B SHIV clone we made based on an infectious molecular clone of a T/F HIV-1 AD17 virus. pSF257-SHIV featured HQAEN at the SIV signature positions (Fig. 2A). Thus, sites A55 and N88 were completely conserved across T/F viruses in all experimental infections of SIV and SHIV. The Klein-SHIV dataset showed one signature site different from those in SIV. The majority of variants in the Klein-SHIV stock preferred V, while T/F viruses preferred E at position 65 (P = 0.012; correction factor, 2).
The signature site at position 65 is exclusive to the Klein-SHIV dataset, but incidentally, it also maps to the C1 region of gp120 and is next to one of the SIVsmE660 signature sites (position 64). The location of the SHIV signature in the vicinity of the other SIV transmission signatures strongly suggests that the C1 region of gp120 is of particular importance to the transmissibility and infectivity of primate lentiviruses.
SIV/SHIV transmission signatures are conserved in HIV.
Various studies have collected data from linked HIV transmission pairs; however, the lack of donor data or late sampling of transmitted lineages make them unsuitable datasets to evaluate selection during transmission. The literature provides HIV transmission data for infections at various stages, including early-, mid-, late-acute, and chronic stages. However, matched donor-recipient data (i.e., chronic data representing the parent population of a known early-acute transmission) also meeting the other criteria deemed essential to this study were unavailable. Thus, we used the signature sites identified in the experimental models to survey residue preferences at those sites in various datasets from natural HIV infections.
The Step trial identified 21 T/F viruses in the placebo and 30 T/F viruses in the vaccine group (7, 8). T/F variants from the placebo and vaccine groups of the Step trial showed (K|Q)AEN at the SIV signature locations. Similarly, the original and holdout sets of the Gnanakaran-HIV acute and chronic sets also featured QAEN at the signature sites.
Despite sequence differences between SIV and HIV (24), positions A55 and N88 were completely conserved across the primate lentiviruses sampled in all 12 datasets from SIV/SHIV/HIV infections (Table 1 and Fig. 2). Additionally, the N-Glycosite package (14) identified the signature residue at position 88 as part of a potential N-linked glycosylation site with the NXT motif.
Identifying a new SIV signature or confirming a previous one on the HIV signal peptide.
Gnanakaran et al. reported position H12 (H9 according to HXB2 numbering) in the signal peptide as a transmission signature (13). Because of alignment ambiguities caused by gaps, Gnanakaran et al.'s H9 aligned exactly with our R9 signature on the SIV signal peptide (Fig. 2) or aligned three residues upstream (see Fig. S6 in the supplemental material). Figure 2 grouped variants from Fiebig stages 2 to 5 into a single acute set, whereas Fig. S6 aligned the Gnanakaran-HIV variants separately by Fiebig stage.
Thus, the alignment ambiguity implies that we are either confirming Gnanakaran et al.'s HIV signature in SIV or uncovering a new, independent signature site on the SIV signal peptide. Additional experimental work needs to be done before a SIV T/F preference for R can be assigned to HXB2 site 6 or 9 and its role in the preferential transmission of SIV and HIV T/F viruses can be deciphered.
Glycosylation motifs and length of hypervariable loops in T/F viruses.
Previous studies have reported signature patterns on the hypervariable loops of T/F viruses. The hypervariable loops of T/F variants have been described as shorter and carrying fewer potential N-linked glycosylation (PNLG) sites than donor variants (24–27). We delimited the V1-V2 and V4 hypervariable regions of the SIV variants according to the coordinates in reference 28. We found no consensus in terms of hypervariable loop lengths across the experimental datasets. According to a Mann-Whitney-Wilcoxon test, the V1-V2 region of T/F viruses was shorter in the Klein-SHIV dataset (P = 8.08e−4), longer in all of the Roederer-SIV datasets (control P = 3.19e−8; gag P = 3.42e−8; mosaic Env P = 1.79e−6; mac239 Env P = 1.33e−6), and not significantly different from the stock in the Keele-SIV founders. The V4 region was significantly shorter in Keele-SIV (P = 1.2e−10) and Klein-SIV (P = 2.62e−13) T/F viruses and equal in length in the stock and T/F variants of all four Roederer-SIV datasets. We also searched for PNLG sites on the hypervariable regions of T/F variants using the N-Glycosite tool. We found no significant difference between the number of PNLGs in T/F versus donor viruses in any of the SIV datasets. We found similar results when analyzing all V1 to V5 regions.
DISCUSSION
HIV transmission typically results from infection by a single variant. However, the question of whether transmitted variants are chosen uniformly at random from the donor pool or whether they are selected based on a set of advantageous traits continues to be unresolved. Even with no quantitative evidence of selection to date, the general consensus in the scientific community has been that selective pressures during transmission must drive the choice of T/F variants. Phenotypically, transmitted viruses are known to be preferentially CCR5-tropic viruses (5). However, the specific molecular features increasing risk of T/F transmission have not been defined. Identifying the distinguishing properties of transmitted viruses is of special interest, since it could reveal potential vulnerabilities of the infecting virus, which in turn could guide the design of an effective vaccine or treatment.
The present study systematically evaluates the differences between the Env proteins of stock and T/F variants from seven sets of experimental SIV and SHIV infections. Our results demonstrate that transmission in SIV and SHIV, and most likely in HIV, involves selection on at least four (and up to six) sites of gp120. Selection of SIV/SHIV could be inferred reasonably across both vaccinated and unvaccinated subjects, with infections resulting from vaginal, rectal, and intravenous routes of transmission and regardless of viral dosage. The evidence for selection in SIV and SHIV T/F variants is strong and plentiful, and in HIV it is suggestive though commensurate with the availability of suitable data for analysis. Unfortunately, trials involving human subjects often produce sparse data scattered in time. The lack of early-acute T/F HIV data with matched donor sequence populations precludes the type of rigorous statistical analysis possible in the animal models.
T/F viruses always possess the properties required for viral transmission, but knowledge of the donor/stock viruses that were not transmitted is required to define the selection pressures on the T/F viruses and to decide whether the transmitted viruses are chosen uniformly at random or result from selection. Thus, the donor/stock population limits the detection of selection by our protocol or any other. Our protocol can test only for selection on the sites where the donor/stock population shows variation.
For instance, let us assume sufficiently strong selection, so that only viruses with I at site 33 (Ile 33) in gp120 can infect. If the recipient is challenged with variants carrying Val 33, Ile 33, or any other residue, only variants with Ile 33 will successfully transmit. However, if the challenging stock has no diversity at site 33, with all its variants having Ile 33, the T/F lineage will carry Ile 33. Note that in the latter case, selection indeed required Ile 33, but neither our protocol nor any other can detect the sieving effect of selection. Therefore, experimenters might well be able to elucidate more signature sites than we present here by diversifying their challenge stocks. In fact, the low diversity of the stock suffices to explain our findings in the Keele-SIVmac251 experiment. The SIVmac251 T/F viruses have IAKN at the signature sites, but since all stock variants also have IAKN, we cannot detect whether selection directs the choice of residues at those sites.
Natural HIV infections might also be displaying similar phenomena due to limited diversity in donor viruses. The residue preferences of all HIV variants we examined bore striking similarities to those seen at the signature sites of SIV/SHIV T/F viruses. The (K|Q)AEN motif in HIV variants was highly conserved regardless of Fiebig stage, geographic location, or transmission route. Given the high mutability of HIV, such conserved residue preferences throughout the course of the infection suggest strong selection to keep those sites fixed. Given that those sites remain fixed, it is likely that any mutations away from the motif are not viable for long, since they seem to be eliminated from the set of fit circulating variants. Since natural HIV circulating variants do not seem to offer a complete combinatorial set of point mutations at the SIV/SHIV signature sites, the best way to gather stronger evidence of selection during HIV transmission might be to examine SHIV and SIV models. Experiments could examine the necessity of particular motifs to transmission by using synthetic viral swarms in the stocks with a diverse set of point mutants to test for the sieving effect of selection.
Although the viral stocks we examined contained variants with only part of the IAKN signature motif, the only variants establishing successful infections with T/F lineages carried the full signature motif. The T/F lineages in 38 animals showed the complete signature motif, even though the signature motif represented only a minor variant in the infecting stock. Singleton point mutants were present in viral stocks but absent from the T/F viruses, strongly suggesting that selection requires all four mutations within a single virus to permit transmissibility.
The present study applies logically simple, conservative statistical tests to yield robust evidence of selection on the gp120 molecules of T/F viruses. While one may evaluate selection at the individual dataset level, metadata analyses increase the statistical power to evaluate selection over all of the SIV datasets. Regardless of which approach the reader finds more appealing, both individual and metadata analyses showed strong support for selection at several signature sites. In addition, evaluating the same hypotheses on different datasets under different treatment regimens revealed signatures conserved over differences in viral species, viral dosages, and routes of transmission. A priori, one might expect different transmission routes to display different selective pressures. However, the sites of amino acid signatures associated with transmission were consistent in mucosal, intravenous, and breastfeeding infections. We also observed the same T/F motif in the mother-to-child transmission (MTCT) pairs from references 15 and 23: those data were excluded from this study only because the MTCT sequences were obtained prior to the advent of SGA. Different transmission routes might lead to selection pressures at sites other than the ones found in this study, but as we have pointed out, experiments using challenge stocks with a variety of point mutants per site could elucidate other sites of selection.
Our main aim here was to discover strong signatures that would survive the rigor of conservative statistical approaches. Therefore, we have presented only two-sided P values of nonparametric Fisher exact and Fisher inverse chi-square tests and indicated multiple-test corrections with Tarone's modification of the Bonferroni correction. In the present context, two-sided nonparametric tests with Bonferroni corrections are exceedingly conservative statistical procedures. It is reassuring indeed that even after our conservative statistical treatment, evidence for selection on the signature sites remains statistically significant.
Previous studies have reported shorter and less glycosylated hypervariable loops in subtypes A and C, but not in B, as potential signatures of transmitted viruses (25, 26). In contrast, a more recent study reports acute subtype B viruses with shorter and less glycosylated loops than their chronic counterparts (13). Our findings in the SIV/SHIV datasets display a similar inconsistency. T/F viruses from some of the datasets had shorter, while others had longer, hypervariable loops or loops no different from those in the donor viruses. Likewise, we could detect no donor versus T/F differences in loop glycosylation patterns for any of the SIV/SHIV datasets. Therefore, we could not confirm length and glycosylation motifs of hypervariable loops as defining characteristics of T/F viruses.
The four strongest signature sites of infection mapped to the C1 region of gp120, indicating that C1 is involved in transmission fitness. Moreover, finding all signature sites in close proximity to one another suggests a cooperative effect among the signature residues to enhance transmissibility.
The C1 region is important in three aspects of Env biology. First, C1 is involved critically in the association of gp120 with gp41 in soluble Env trimers (27–29). Second, it harbors an epitope region recognized by nonneutralizing antibodies mediating potent antibody-dependent cellular cytotoxicity (ADCC) (30–33). Epitopes in this region are sites of ADCC-mediated escape mutations in HIV-1-infected people (30), and they have been implicated as targets of potentially protective antibodies in the RV144 vaccine trial (34, 35); the first atomic structures of this epitope region were published recently (36). Further, there is emerging evidence that HIV-1 has evolved a global escape mechanism involving Nef and Vpu to keep these epitopes from being exposed on nascently infected cells (33, 37, 38), suggesting their importance in protective immunity against HIV-1. Third, the signature sites in the C1 region affect sensitivity to neutralizing antibodies where the T/F signatures confer neutralization resistance (6).
Recent structural studies of soluble Env timer analogs (39–41) permits the generation of a hypothesis as to why the signature residues A55, E64, and N88 are selectively transmitted (Fig. 3). This region is located within mobile layers that undergo conformational changes after binding to CD4 and the coreceptor, exposing the hydrophobic N terminus of gp41 and leading to fusion of the virus and cell membranes (28, 29). Both signature sites A55 and E64 map to mobile layer 1 (Fig. 3). Signature site A55 is in the β2 strand, whereas signature site E64 is at the N terminus of the α0 helix (39–41). This is significant because the β2 strand contacts gp41 at F53 and the α0 helix contacts gp41 at H72 and A73 in the Env trimer analog (41). Further, mutation of an F, two residues N-terminal to signature site E64, affects the association of gp120 and gp41 (28, 29), as does mutation of a V, one position C terminal to E64 (28). Signature site N88 is an N-linked glycosylation site located between mobile layers 1 and 2 (Fig. 3). Mutations of this site and flanking residues also affect the association between gp120 and gp41 (28). Collectively, these data raise the possibility that C1, governing the association between gp120 and gp41, modulate transmission efficiency, replicative fitness, and/or host cell tropism (3, 22) at the level of virus-cell attachment and entry. It is known that T/F viruses are more infectious for memory CD4+ T cells, the principal early targets of transmission, than for monocytes (3, 22). The effect of the signature residues in increasing transmissibility could be proximal (e.g., lowering the activation energy required to trigger gp120-gp41 dissociation) or distal (e.g., affecting Env density on virions) (42). Thus, it is possible that the specific combination of T/F signature residues tips the structural mechanics of the envelope trimer in favor of supporting the first steps in primary infection.
FIG 3.
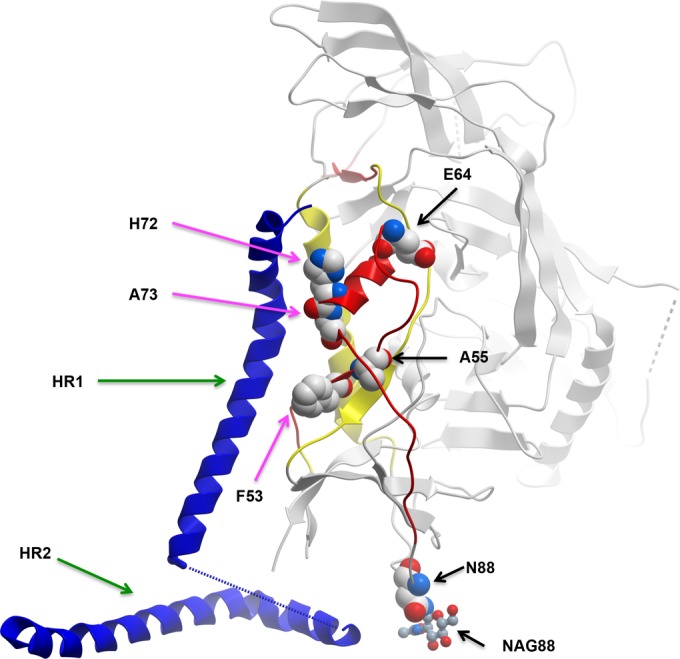
Location of the T/F signature residues on the gp120-gp41 interactive region. The figure depicts one monomer of gp120 (gray) and gp41 (dark blue helix) from 4NCO (39). Mobile layer 1 is shown in red, and mobile layer 2 is shown in yellow. The mobile layers were identified in references 3 and 40. The black arrows point toward the T/F residues, and the magenta arrows point toward the gp41 contact residues identified (39). The numbers are HXB2 coordinates. The green arrows point toward heptad-repeat 1 (HR1) and heptad-repeat 2 (HR2) of gp41.
Another puzzling aspect of our findings is that should the gp120 signature residues modulate transmission by their interaction with gp41, one might expect to also identify sites under selection in gp41; however, the present study did not find evidence of significant selection in gp41. Still, the lack of evidence of selection on gp41 or on other sites in other genes does not preclude selection there. As explained in the third paragraph of the Discussion, low-diversity stocks can prevent our ability to detect selection. We expect that the sieving effect of selection will be apparent only in experiments infecting with diverse viral swarms and only at sites where variants in the stock present a variety of residues.
The transmitted signature residues also could directly and indirectly affect the antigenicity of the A32-epitope subregion of gp120, where it is a potent ADCC target. Signature site 55 is flanked by contact residues in the β2 sheet for A32-like monoclonal antibodies, which mediate potent ADCC (28, 36, 43, 44). It is not known whether the T/F signature motif affects the antigenicity of this region, but it could favor poor recognition by such antibodies promoting the transmission of variants that are ADCC resistant. Similarly, signature site 64 at the top of the α0 helix is adjacent to key contact residues for A32-like antibodies (36). Signature site 88 is located between the β0 and β1 sheets connecting mobile layers 1 and 2. Layer 2 also contains key contact residues for A32-like antibodies in the α1 helix. Thus, the T/F motif might disfavor the conformation required for A32-like antibodies to recognize the cognate epitopes and mediate ADCC.
At this point, we have raised three main hypotheses, all speculative, to explain the mechanisms of increased transmissibility by the T/F residues: gp120-gp41 dissociation, Env density on virions, and recognition by A32-like antibodies. A definitive determination of whether and how the T/F signature residues modulate transmissibility requires further experimentation.
In addition to the T/F signatures, however, this study has other major findings: it reinterprets the conclusions of a recent animal vaccine trial, in turn raising some serious unanticipated possibilities in HIV vaccine trials.
In our study, only the T/F variants of the control and gag arms of the Roederer-SIV study did not carry the standard IAKN signature motif; control and gag T/F viruses carried the same residues, VTRS, as the majority of viruses in the stock. Roederer et al. interpreted the presence of the IAKN signature in the two Env treatments as purely an effect of their vaccine, accounting for neutralization resistance (6). It is most likely that this effect is indirect, as the region where the signature residues reside is internal to the epitopes recognized by the monoclonal antibodies for which resistance was increased by the mutations. Most likely, neutralization resistance and increased infectivity for CD4+ memory cells of T/F viruses are related phenomena. However, the IAKN residues at positions 33, 55, 64, and 88 likely have broader functional significance beyond just neutralization sensitivity, because the animals in the Keele-SIV dataset had no preexisting humoral response to Env at the time of exposure, yet they also displayed the IAKN signature, showing that the IAKN motif has a broader functional significance beyond just neutralization sensitivity.
Still, if in the Keele-SIV dataset the IAKN T/F signature provided a disproportionate selective advantage increasing transmissibility, why did Roederer-SIV's control and gag T/F viruses carry the anomalous VTRS signature predominant in the stock, which has a lower transmission fitness in all other datasets? To answer this question, we note that in the Keele-SIV dataset, the controls received only diluent, whereas in Roederer et al.'s vaccine regimens, all groups, including the control group, received Ad5 vectors. Thus, the only stated difference we found between the Keele- and Roederer-SIV controls was that the Roederer-SIV control animals received empty vectors (sham vaccination). Although other, unstated differences might exist between the Keele- and Roederer-SIV controls, we found ourselves forced to entertain the hypothesis that T/F viruses in Roederer et al.'s control and gag groups contained an anomalous VTRS signature because of some unintended effect of sham vaccination with Ad5 vectors.
To test the possible effects of sham vaccination, we analyzed the T/F viruses of the Step trial. The Step trial inoculated vaccine subjects with an Ad5 vector and placebo subjects with a vaccine diluent only without sham vaccinations. Our analysis of the Step trial showed that both placebo and vaccine T/F viruses featured the standard (K|Q)AEN signature we found in all of the other HIV datasets. Therefore, based on the data from the Step trial alone, the use of Ad5 vectors in the Roederer-SIV study does not appear to have influenced the control and gag VTRS preference. However, in the Los Alamos HIV database (in particular, in the Gnanakaran-HIV datasets), most HIV-1 variants in circulation have the standard (K|Q)AEN signature. Thus, even if sham vaccination were to increase the relative fitness of an HIV homolog of the anomalous VTRS variant, we might not see any anomalous transmission in humans, because circulating HIV populations present low diversity at the SIV signature sites. Thus, HIV homologs of the anomalous VTRS variant probably could not have been observed in the Step trial, because they are not common enough in the human donor pool. Thus, analyzing the Ad5-vaccinated subjects of the Step trial did not allow us to rule out whether the anomalous control and gag transmission patterns in the Roederer-SIV dataset result from sham vaccination.
Although we recognize that the hypothesis is counterintuitive, we could not rule out the possibility that the unusual transmission in the Roederer-SIV control and gag datasets is specifically linked to the DNA/Adeno vaccine regimen as applied to macaques. Thus, the vaccine regimen in the Roeder et al. study, which is a common strategy in animal trials, might have increased the transmission of variants with usually low transmission fitness (i.e., the VTRS variants), and the Env vaccination might merely have brought transmission down to include only the usual IAKN variants seen in completely naive animals, possibly leading to misinterpretation of data as a neutralizing antibody effect. Although we again emphasize that other differences might exist between the Keele- and Roederer-SIV controls, experiments with a different type of control group, one inoculated with vaccine diluent only and no sham vaccination, as in the placebo group in the Step trial, appear necessary. All our findings from various SIV trials suggest VTRS variants have low transmission fitness in naive subjects. Thus, whether due to Ad5 vector or not, the successful transmission of low-fit VTRS variants suggests the disturbing possibility that some aspect of the vaccination protocol is boosting variant transmissibility in control animals. Further testing is paramount to establishing whether our observations partly explain the failure of HIV/AIDS vaccine trials using regimens of DNA priming and/or sham Env vaccination. On the other hand, our findings may be revealing important differences among lentiviral infections of nonhuman versus human primates, which is perhaps why many animal vaccine trials have not faithfully predicted outcomes in human vaccine trials.
Our findings indicate that the gp120 molecules of primate lentiviruses are under selection during transmission. Given that the main four T/F signatures map to the C1 region of gp120, it is likely that the structural mechanics of the gp120-gp41 association take part in facilitating the preferential transmission of T/F viruses. The role of the C1 region in supporting the first steps in primary infection may further come into play in the face of vaccination if it also alters neutralizing antibody susceptibility. Here, we have posed several hypotheses regarding the role of the infection signatures in facilitating enhanced infectiousness of transmitted viruses, which make the C1 signatures likely targets for developing an effective vaccine or treatment.
Supplementary Material
ACKNOWLEDGMENTS
M.W.G. and J.L.S. were supported by the Intramural Research Program of the NIH, National Library of Medicine. A.L.D. and G.K.L. were funded by grants from The Bill and Melinda Gates Foundation and NIAID, NIH.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03235-14.
REFERENCES
- 1.Wolfs TF, Zwart G, Bakker M, Goudsmit J. 1992. HIV-1 genomic RNA diversification following sexual and parenteral virus transmission. Virology 189:103–110. doi: 10.1016/0042-6822(92)90685-I. [DOI] [PubMed] [Google Scholar]
- 2.Zhang LQ, MacKenzie P, Cleland A, Holmes EC, Brown AJ, Simmonds P. 1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J Virol 67:3345–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw GM, Hunter E. 2012. HIV transmission. Cold Spring Harb Perspect Med 2:a006965. doi: 10.1101/cshperspect.a006965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger EA, Murphy PM, Farber JM. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 6.Roederer M, Keele BF, Schmidt SD, Mason RD, Welles HC, Fischer W, Labranche C, Foulds KE, Louder MK, Yang ZY, Todd JP, Buzby AP, Mach LV, Shen L, Seaton KE, Ward BM, Bailer RT, Gottardo R, Gu W, Ferrari G, Alam SM, Denny TN, Montefiori DC, Tomaras GD, Korber BT, Nason MC, Seder RA, Koup RA, Letvin NL, Rao SS, Nabel GJ, Mascola JR. 2013. Immunological and virological mechanisms of vaccine-mediated protection against SIV and HIV. Nature 505:502–508. doi: 10.1038/nature12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rolland M, Tovanabutra S, deCamp AC, Frahm N, Gilbert PB, Sanders-Buell E, Heath L, Magaret CA, Bose M, Bradfield A, O'Sullivan A, Crossler J, Jones T, Nau M, Wong K, Zhao H, Raugi DN, Sorensen S, Stoddard JN, Maust BS, Deng WJ, Hural J, Dubey S, Michael NL, Shiver J, Corey L, Li FS, Self SG, Kim J, Buchbinder S, Casimiro DR, Robertson MN, Duerr A, McElrath MJ, McCutchan FE, Mullins JI. 2011. Genetic impact of vaccination on breakthrough HIV-1 sequences from the STEP trial. Nat Med 17:366–371. doi: 10.1038/nm.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, del Rio C, McElrath MJ, Casimiro DR, Gottesdiener KM, Chodakewitz JA, Corey L, Robertson MN, Step Study Protocol Team . 2008. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keele BF, Li H, Learn GH, Hraber P, Giorgi EE, Grayson T, Sun C, Chen Y, Yeh WW, Letvin NL, Mascola JR, Nabel GJ, Haynes BF, Bhattacharya T, Perelson AS, Korber BT, Hahn BH, Shaw GM. 2009. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J Exp Med 206:1117–1134. doi: 10.1084/jem.20082831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein K, Veazey RS, Warrier R, Hraber P, Doyle-Meyers LA, Buffa V, Liao HX, Haynes BF, Shaw GM, Shattock RJ. 2013. Neutralizing IgG at the portal of infection mediates protection against vaginal simian/human immunodeficiency virus challenge. J Virol 87:11604–11616. doi: 10.1128/JVI.01361-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Bar KJ, Wang S, Decker JM, Chen Y, Sun C, Salazar-Gonzalez JF, Salazar MG, Learn GH, Morgan CJ, Schumacher JE, Hraber P, Giorgi EE, Bhattacharya T, Korber BT, Perelson AS, Eron JJ, Cohen MS, Hicks CB, Haynes BF, Markowitz M, Keele BF, Hahn BH, Shaw GM. 2010. High multiplicity infection by HIV-1 in men who have sex with men. PLoS Pathog 6:e1000890. doi: 10.1371/journal.ppat.1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol 73:10489–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gnanakaran S, Bhattacharya T, Daniels M, Keele BF, Hraber PT, Lapedes AS, Shen T, Gaschen B, Krishnamoorthy M, Li H, Decker JM, Salazar-Gonzalez JF, Wang S, Jiang C, Gao F, Swanstrom R, Anderson JA, Ping LH, Cohen MS, Markowitz M, Goepfert PA, Saag MS, Eron JJ, Hicks CB, Blattner WA, Tomaras GD, Asmal M, Letvin NL, Gilbert PB, Decamp AC, Magaret CA, Schief WR, Ban YE, Zhang M, Soderberg KA, Sodroski JG, Haynes BF, Shaw GM, Hahn BH, Korber B. 2011. Recurrent signature patterns in HIV-1 B clade envelope glycoproteins associated with either early or chronic infections. PLoS Pathog 7:e1002209. doi: 10.1371/journal.ppat.1002209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaschen B, Kuiken C, Korber B, Foley B. 2001. Retrieval and on-the-fly alignment of sequence fragments from the HIV database. Bioinformatics 17:415–418. doi: 10.1093/bioinformatics/17.5.415. [DOI] [PubMed] [Google Scholar]
- 15.Nduati R, John G, Mbori-Ngacha D, Richardson B, Overbaugh J, Mwatha A, Ndinya-Achola J, Bwayo J, Onyango FE, Hughes J, Kreiss J. 2000. Effect of breastfeeding and formula feeding on transmission of HIV-1: a randomized clinical trial. JAMA 283:1167–1174. doi: 10.1001/jama.283.9.1167. [DOI] [PubMed] [Google Scholar]
- 16.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felsenstein J. 1989. PHYLIP–phylogeny inference package (version 3.2). Cladistics 5:164–166. [Google Scholar]
- 18.Tarone RE. 1990. A modified Bonferroni method for discrete data. Biometrics 46:515–522. doi: 10.2307/2531456. [DOI] [PubMed] [Google Scholar]
- 19.Miller RG. 1981. Simultaneous statistical inference. Springer-Verlag, New York, NY. [Google Scholar]
- 20.Siegel S, Castellan NJJ. 1988. Nonparametric statistics for the behavioral sciences, 2nd ed McGraw-Hill, New York, NY. [Google Scholar]
- 21.Fisher RA. 1970. Statistical methods for research workers, 14th ed Hafner Press, New York, NY. [Google Scholar]
- 22.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med 206:1273–1289. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X, Parast AB, Richardson BA, Nduati R, John-Stewart G, Mbori-Ngacha D, Rainwater SM, Overbaugh J. 2006. Neutralization escape variants of human immunodeficiency virus type 1 are transmitted from mother to infant. J Virol 80:835–844. doi: 10.1128/JVI.80.2.835-844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finzi A, Pacheco B, Xiang SH, Pancera M, Herschhorn A, Wang L, Zeng X, Desormeaux A, Kwong PD, Sodroski J. 2012. Lineage-specific differences between human and simian immunodeficiency virus regulation of gp120 trimer association and CD4 binding. J Virol 86:8974–8986. doi: 10.1128/JVI.01076-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chohan B, Lang D, Sagar M, Korber B, Lavreys L, Richardson B, Overbaugh J. 2005. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J Virol 79:6528–6531. doi: 10.1128/JVI.79.10.6528-6531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frost SD, Liu Y, Pond SL, Chappey C, Wrin T, Petropoulos CJ, Little SJ, Richman DD. 2005. Characterization of human immunodeficiency virus type 1 (HIV-1) envelope variation and neutralizing antibody responses during transmission of HIV-1 subtype B. J Virol 79:6523–6527. doi: 10.1128/JVI.79.10.6523-6527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helseth E, Olshevsky U, Furman C, Sodroski J. 1991. Human immunodeficiency virus type 1 gp120 envelope glycoprotein regions important for association with the gp41 transmembrane glycoprotein. J Virol 65:2119–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell 37:656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci U S A 107:1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung AW, Isitman G, Navis M, Kramski M, Center RJ, Kent SJ, Stratov I. 2011. Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc Natl Acad Sci U S A 108:7505–7510. doi: 10.1073/pnas.1016048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrari G, Pollara J, Kozink D, Harms T, Drinker M, Freel S, Moody MA, Alam SM, Tomaras GD, Ochsenbauer C, Kappes JC, Shaw GM, Hoxie JA, Robinson JE, Haynes BF. 2011. An HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody-dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV-1 serum. J Virol 85:7029–7036. doi: 10.1128/JVI.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, Fouts TR, Gallo RC, DeVico AL, Lewis GK. 2013. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc Natl Acad Sci U S A 110:E69–E78. doi: 10.1073/pnas.1217609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol 88:2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomaras GD, Ferrari G, Shen X, Alam SM, Liao HX, Pollara J, Bonsignori M, Moody MA, Fong Y, Chen X, Poling B, Nicholson CO, Zhang R, Lu X, Parks R, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Rerks-Ngarm S, Gilbert PB, Kim JH, Michael NL, Montefiori DC, Haynes BF. 2013. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc Natl Acad Sci U S A 110:9019–9024. doi: 10.1073/pnas.1301456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med 366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acharya P, Tolbert WD, Gohain N, Wu X, Yu L, Liu T, Huang W, Huang CC, Kwon YD, Louder RK, Luongo TS, McLellan JS, Pancera M, Yang Y, Zhang B, Flinko R, Foulke JS Jr, Sajadi MM, Kamin-Lewis R, Robinson JE, Martin L, Kwong PD, Guan Y, DeVico AL, Lewis GK, Pazgier M. 2014. Structural definition of an antibody-dependent cellular cytotoxicity response implicated in reduced risk for HIV-1 infection. J Virol 88:12895–12906. doi: 10.1128/JVI.02194-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pham TN, Lukhele S, Hajjar F, Routy JP, Cohen EA. 2014. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 11:15. doi: 10.1186/1742-4690-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, Evans DT. 2014. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A 111:6425–6430. doi: 10.1073/pnas.1321507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. 2013. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, Wilson IA, Ward AB. 2013. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342:1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GB, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. 2014. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP Jr, Bjorkman PJ, Wilen CB, Doms RW, O'Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 2013. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci U S A 110:6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis GK, Guan Y, Kamin-Lewis R, Sajadi M, Pazgier M, Devico AL. 2014. Epitope target structures of Fc-mediated effector function during HIV-1 acquisition. Curr Opin HIV AIDS 9:263–270. doi: 10.1097/COH.0000000000000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore JP, McCutchan FE, Poon SW, Mascola J, Liu J, Cao Y, Ho DD. 1994. Exploration of antigenic variation in gp120 from clades A through F of human immunodeficiency virus type 1 by using monoclonal antibodies. J Virol 68:8350–8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.