

ABSTRACT
We have examined the interactions of wild-type (WT) and matrix protein-deleted (ΔMA) HIV-1 precursor Gag (PrGag) proteins in virus-producing cells using a biotin ligase-tagging approach. To do so, WT and ΔMA PrGag proteins were tagged with the Escherichia coli promiscuous biotin ligase (BirA*), expressed in cells, and examined. Localization patterns of PrGag proteins and biotinylated proteins overlapped, consistent with observations that BirA*-tagged proteins biotinylate neighbor proteins that are in close proximity. Results indicate that BirA*-tagged PrGag proteins biotinylated themselves as well as WT PrGag proteins in trans. Previous data have shown that the HIV-1 Envelope (Env) protein requires an interaction with MA for assembly into virions. Unexpectedly, ΔMA proteins biotinylated Env, whereas WT BirA*-tagged proteins did not, suggesting that the presence of MA made Env inaccessible to biotinylation. We also identified over 50 cellular proteins that were biotinylated by BirA*-tagged PrGag proteins. These included membrane proteins, cytoskeleton-associated proteins, nuclear transport factors, lipid metabolism regulators, translation factors, and RNA-processing proteins. The identification of these biotinylated proteins offers new insights into HIV-1 Gag protein trafficking and activities and provides new potential targets for antiviral interference.
IMPORTANCE We have employed a novel strategy to analyze the interactions of the HIV-1 structural Gag proteins, which involved tagging wild-type and mutant Gag proteins with a biotin ligase. Expression of the tagged proteins in cells allowed us to analyze proteins that came in close proximity to the Gag proteins as they were synthesized, transported, assembled, and released from cells. The tagged proteins biotinylated proteins encoded by the HIV-1 pol gene and neighbor Gag proteins, but, surprisingly, only the mutant Gag protein biotinylated the HIV-1 Envelope protein. We also identified over 50 cellular proteins that were biotinylated, including membrane and cytoskeletal proteins and proteins involved in lipid metabolism, nuclear import, translation, and RNA processing. Our results offer new insights into HIV-1 Gag protein trafficking and activities and provide new potential targets for antiviral interference.
INTRODUCTION
The HIV-1 precursor Gag (PrGag) protein and its mature products, matrix (MA), capsid (CA), nucleocapsid (NC), and p6, play a central role in the assembly of HIV-1 virus particles and serve other functions at subsequent stages of virus replication. PrGag itself is cotranslationally myristoylated at its N-terminal MA residue and travels via a still incompletely elucidated pathway to plasma membrane (PM) assembly sites that are enriched in cholesterol and phosphatidylinositol-(4,5)-bisphosphate (PI[4,5]P2) (1–8). The PrGag MA domain binds PI(4,5)P2, which, in part, explains how PM targeting occurs (4, 9–12). MA also binds to RNA, and models suggest that MA-RNA binding serves a chaperone function, preventing PrGag from binding to intracellular membranes prior to arrival at the PI(4,5)P2-rich PM (3, 4, 9–11, 13, 14). During transit to the PM, PrGag associates with full-length HIV-1 viral RNAs (vRNAs), which is controlled, in part, by virtue of a preferential binding of NC domains to the vRNA encapsidation (E) or Psi (Ψ) element (14–20). Delivery of PrGag proteins and vRNAs to the PM would appear to require interaction with and reconfiguration of the cellular cytoskeleton, including the cortical actin network, but details regarding these interactions are scant (21–26). However, once at the PM, major PrGag-PrGag contacts appear to be mediated by CA domains, while budding of membrane-coated virus particles is facilitated by p6 recruitment of cellular endosomal sorting complexes required for transport (ESCRT) and ESCRT-associated factors (20, 27–31). At that stage, incorporation of HIV-1 envelope (Env) proteins into virions requires an interaction between Env and the PrGag MA domains, while accessory and pol gene products use a variety of strategies for virus incorporation (20, 27–32). Analysis of HIV-1 particles indicates that a number of cellular proteins are assembled into virions, either through specific associations or as innocent bystanders (20, 32–36).
During the entry phase of HIV-1 replication, mature HIV Gag proteins have demonstrated additional functions. Multiple studies have demonstrated that NC is essential for efficient reverse transcription in newly infected cells (17, 18, 20, 37). Early studies additionally implicated MA in the nuclear localization of preintegration complexes (PICs) in nondividing cells (38). More recently, PIC-associated CA has been shown to bind to karyopherin substrate CPSF6 and nucleoporin proteins Nup153 and Nup358 to facilitate the nuclear localization of PICs (39–42). CA proteins on incoming PICs are also the targets of innate immune system antiviral proteins Trim5a and MX2, but it is noteworthy that cellular antiviral proteins also have been shown to inhibit viral functions at the assembly stage of the HIV-1 life cycle (43–47).
While major mutations of HIV-1 Gag proteins have proven to be predominantly lethal to the virus, one exception relates to MA. In particular, viruses with MA deletions that retain only the N-terminal myristylation signal and MA-CA juncture sequences were shown to be conditionally infectious, as long the viruses employed either pseudotyped envelope proteins or HIV-1 Env proteins bearing cytoplasmic tail (CT) truncations (30, 32, 48). These observations support a model in which an Env CT-MA interaction is necessary for proper Env assembly into virions but do not explain how deletion MA (ΔMA) PrGag proteins are transported to the PM and or how they manage to perform the essential functions of wild-type (WT) PrGag proteins (27, 28, 30, 32, 48, 49). Taking advantage of the relative insensitivity of HIV-1 MA to certain genetic perturbations, researchers also have generated PrGag proteins that carry protein insertions near the C terminus of MA (14, 50–52). Such PrGag proteins have required coexpression of WT PrGag proteins to produce fully infectious viruses, but the tagged proteins have proven useful in localization and binding studies (14, 50, 51).
In an effort to extend previous observations on the roles and interactions of WT and ΔMA PrGag proteins, we have employed an alternative strategy, employing the promiscuous Escherichia coli biotin ligase BirA* (53–55). With BirA*, the activated biotinoyl-5′-AMP (bioAMP) species is prematurely released by the enzyme and reacts with exposed lysines on neighbor proteins in a proximity-dependent fashion, estimated to be in the range of 10 to 30 nm (53, 55). Because there are only a few endogenously biotinylated proteins in mammalian cells and because BirA* expression does not appear to be toxic (53, 55), BirA* tagging permits the analysis of protein interactions, taking advantage of the high affinity (dissociation constant [Kd] = ∼10−15 M) of streptavidin reagents for biotin. We have tagged WT and ΔMA PrGag proteins with BirA* and have examined the activities and interactions of these proteins in an otherwise WT HIV-1 genetic background. Our data show that BirA* PrGag proteins biotinylated WT PrGag proteins in trans and, remarkably, that ΔMA-BirA* proteins preferentially labeled coexpressed vesicular stomatitis virus (VSV) glycoproteins (G) and HIV-1 Env proteins. We also have identified over 50 cellular proteins that were preferentially biotinylated by BirA*-tagged PrGag proteins, approximately half of which have been noted previously as HIV-1-interacting factors. These results provide new insights into the activities and associations of PrGag and MA.
MATERIALS AND METHODS
Recombinant DNA constructs.
The NL4-3 HIV-1 proviral construct has been described previously (56), as has the pVSV-G expression vector (57). For expression of WT PrGag in the absence of protease (PR−), the 2498T mutant construct, which also has been described previously (58), was employed. MA-BirA* is an NL4-3-based plasmid in which the coding region (residues 2 to 321) for the humanized (54) promiscuous biotin ligase (BirA*) from pLEW100-Myc-BirA* (55) was inserted between MA codons 119 and 120. The N-terminal juncture residues (after MA codon 119) include a Myc tag (underlined) and are GAKLH MEQKL ISEED DLD, and the C-terminal juncture residues (before MA codon 121) are LEGS. ΔMA-BirA* is identical to MA-BirA* except that MA codons 19 to 120 were deleted and the juncture residues between MA codon 18 and the Myc tag are GAGAK LHM.
Cell culture, transfections, infections and immunoblotting.
HEK 293T (59) and TZM-bl (10) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES (pH 7.4), penicillin, and streptomycin at 37°C and 5% CO2. MT-4 T cells (48, 57, 60) were maintained in RPMI medium with the same supplements and under the same conditions. Analysis of cellular protein expression and virus particle assembly and release was performed at 72 h after calcium phosphate transfections of HEK 293T cells (5 million cells per 10-cm-diameter plate) by using a total of 24 μg plasmid DNA constructs as described previously (49, 52, 57, 60, 61). In cotransfection experiments, 16 μg MA-BirA* or ΔMA-BirA* was transfected with 8 μg WT NL4-3, 2498T, or pVSV-G. To generate virus for infections, 18 μg NL4-3-based plasmids was cotransfected with 6 μg pVSV-G into HEK293T cells, and at 3 days posttransfection, viral supernatants were filtered through 0.45-μm-pore-size filters. Concentrations of virus stocks were normalized for Gag protein levels as described previously (57) and used to infect either MT-4 or TZM-bl cells. For MT-4 infections, aliquots (20%) of infected cell cultures were collected at intervals, centrifuged for 3 min at 16,000 × g, lysed in cold IPB (20 mM Tris-hydrochloride [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulfate [SDS], 0.5% sodium deoxycholate [DOC], 1.0% Triton X-100, 0.02% sodium azide), and stored at −80°C prior to analysis. Infections were tracked by immunoblot detection of infected-cell CA levels as described below. For TZM-bl infections, cells were infected with virus dilutions for 48 h and collected, lysed, and monitored for β-galactosidase reporter activity as described previously (10).
For virus protein analysis, virus samples were pelleted through 20% sucrose cushions in phosphate-buffered saline (PBS; 9.5 mM sodium potassium phosphate [pH 7.4], 137 mM NaCl, 2.7 mM KCl) for 45 min at 273,000 × g at 4°C (49, 52, 57, 60, 61). Pelleted viruses were carefully resuspended in cold PBS and stored at −80°C. For cellular protein analysis, cells were washed in cold PBS and then lysed in cold IPB. To remove insoluble debris, lysates were centrifuged at 4°C for 15 min at 16,000 × g. Supernatants were collected and stored at −80°C prior to use. Virus samples and cell samples (20% aliquots of total samples) were mixed with equal volumes of 2× sample loading buffer (12.5 mM Tris-HCl [pH 6.8], 2% SDS, 20% glycerol, 0.25% bromphenol blue, 10% β-mercaptoethanol), fractionated via 10% to 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) in parallel with prestained molecular size standards (Bio-Rad catalog no. 161-0318), and electroblotted onto nitrocellulose (Bio-Rad Laboratories). For HIV-1 Gag protein immunoblotting, a mouse anti-CA monoclonal antibody, Hy183 (kindly provided by Bruce Chesebro), was used at a 1:15 dilution (from culture media) or an anti-MA antibody (Capricorn Products; HIV-018-48170) was used at a 1:1,000 dilution. For HIV-1 Env transmembrane (TM) protein detection, human anti-TM antibody 2F5 (NIH AIDS Reagent Program catalog no. 1474) was used at a 1:1,000 dilution. For detection of the VSV G protein, a rabbit anti-G polyclonal antibody (Sigma; kindly provided by David Johnson, Oregon Health Science University [OHSU]) was used at a 1:5,000 dilution. Detection of proteins was achieved using secondary alkaline phosphatase-conjugated anti-mouse or anti-human antibodies (Promega) at 1:15,000 dilutions and subsequent color reactions for visualization of antibody-bound bands with nitroblue tetrazolium–5-bromo-4-chloro-3-indolyl phosphate–100 mM Tris-hydrochloride (pH 9.5)–100 mM NaCl–5 mM MgCl2. For detection of biotinylated proteins, a streptavidin-alkaline phosphatase conjugate (Invitrogen catalog no. 434322) was used at a 1:3,000 dilution and was followed via color reactions as described above. Protein levels were quantified densitometrically using NIH ImageJ software (62), and molecular masses were estimated based on migrations compared to log (molecular mass)-versus-mobility plots of size standards run in parallel. Virus particle release levels were calculated as done previously (49, 57, 60, 61) from at least two independent transfections by quantification of viral versus cellular Gag levels, and normalization of these levels relative to WT HIV-1 transfection data was performed in parallel. In experiments that involved ΔMA-BirA* or MA-BirA* cotransfection with WT or PR− HIV-1 vectors, to distinguish between the HIV-1 variants, only the Gag precursor proteins for the two BirA* constructs were included in calculations.
Fluorescence localization of proteins.
Transfected HEK 293T cells were split 24 h posttransfection onto 22-mm-by-22-mm glass coverslips in six-well plates, previously treated with 0.3 ml polylysine (Sigma) (0.1 mg/ml) for 10 min at room temperature. After an additional 40 to 48 h of culturing, the medium was aspirated and cells were washed with PBS prior to fixation with 4% paraformaldehyde–PBS for 30 min at 4°C. Cells then were washed with PBS and permeabilized using 0.2% Triton X-100–PBS for 10 min at room temperature. After permeabilization, the cells were washed with PBS for 5 min and incubated with complete media for 30 min at 37°C. Cells were then incubated with the undiluted anti-CA cell culture media for 1 h at 37°C and washed twice in complete media. Following the primary anti-CA antibody incubation and washes, cells were incubated for 1 h at 37°C with 1:1,000 dilutions (in complete media) of Alexa Fluor 488-conjugated anti-mouse IgG (H+L) (Life Technologies) plus Alexa Fluor 594-conjugated streptavidin (Life Technologies). After this step, cells were washed once in complete media followed by two washes in PBS. Coverslips were then mounted using Fluoromount-G antifade reagent (Southern Biotech) and imaged with a Zeiss Axio Observer Z1 inverted microscope equipped with filter set 10 (excitation band-pass range [BP], 450 to 490 nm; emission BP, 515 to 565) and filter set 20 (excitation BP, 546 to 612 nm; emission BP, 575 to 640). For detection of signals in transfected cells, 15-ms exposure times were used. For detection of biotinylated proteins in untransfected cells, 350-ms exposure times were used. For colocalization quantitation, Pearson's correlation coefficients, which range from −1 (uncorrelated) to +1 (completely correlated), were calculated using NIH Image J software (62, 63).
Isolation of biotinylated proteins.
Biotinylated proteins from transfected and mock-transfected cells were isolated at 4°C using a modification of previous procedures (55). Briefly, cell pellets of about 0.2 ml from three confluent 10-cm-diameter plates were resuspended in 0.5 ml of lysis buffer (10 mM Tris [pH 7.4], 500 mM NaCl, 0.4% SDS, 1 mM dithiothreitol [DTT]) containing a cocktail of protease inhibitors (3 mM phenylmethylsulfonyl fluoride [PMSF], 30 μg/ml aprotinin, 72 μg/ml chicken egg white trypsin inhibitor, 72 μg/ml soybean trypsin inhibitor, 60 μg/ml benzamidine, 150 μg/ml leupeptin, 75 μg/ml pepstatin A) and subjected to vortex mixing for 1 min. Mixtures were supplemented with 125 μl 10% Triton X-100 and subjected to vortex mixing for 1 min and then supplemented with 700 μl 50 mM Tris (pH 7.4) and again subjected to vortex mixing for 1 min. Lysates were clarified by centrifugation at 16,000 × g for 15 min at 4°C, and supernatants were collected and mixed with 100 μl (packed volume) of streptavidin-agarose beads (Novagen catalog no. 69203) that had been preequilibrated twice with a mixture of 500 μl lysis buffer, 125 μl Triton X-100, and 700 μl 50 mM Tris (pH 7.4). Bead-lysate mixtures were incubated with rocking at 4°C overnight, after which beads were pelleted (16,000 × g, 1 min) and successively washed (1 ml, 8 min per wash) with wash buffer 1 (2% SDS), wash buffer 2 (50 mM HEPES [pH 7.5], 0.1% DOC, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA), wash buffer 3 (10 mM Tris [pH 8.0], 0.5% Nonidet P-40 [NP-40], 0.5% DOC, 1 mM EDTA, 250 mM LiCl), and wash buffer 4 (50 mM Tris [pH 7.4], 50 mM NaCl). After a second wash buffer 4 wash, bound proteins were eluted five times for 10 min each time at 98°C with 100 μl of Laemmli buffer (25 mM Tris, 192 mM glycine, 0.1% SDS) containing saturating levels of biotin (∼1 mM).
MS.
Protein fractions isolated from streptavidin bead purifications were processed for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis by the Oregon Health & Science University Proteomics Shared Resource. Purified fractions were applied to NuPAGE 10% Bis-Tris SDS-PAGE gels (NP0301BOX), electrophoresed for 6 min at 200 V to remove impurities, and stained for 30 min with Imperial Blue protein stain (catalog no. 24615; Thermo Scientific) to assess sample concentration and quality. Protein-containing bands were destained, excised from gels, and processed for reduction, alkylation, and trypsin digestion as described previously (64). For the reduction and alkylation steps, gel pieces were washed with acetonitrile for 10 min, reduced for 30 min at 56°C with 10 mM DTT–100 mM ammonium bicarbonate, chilled to 22°C, washed again with acetonitrile, incubated for 20 min at room temperature in the dark with 55 mM iodoacetamide–100 mM ammonium bicarbonate, and washed again with acetonitrile. Trypsin digestion was performed at 37°C overnight with 13 μg/ml trypsin (Promega V5111)–10 mM ammonium bicarbonate–10% acetonitrile. Peptides were extracted from gels by incubation with 100 μl of 1.67% formic acid–acetonitrile for 15 min in a 37°C shaker and dried by vacuum centrifugation.
Tryptic peptide pellets were suspended in 5% formic acid, filtered (Millipore Ultrafree-MC) (0.45-μm-pore-size filter), separated using liquid chromatography with an NanoAcquity ultraperformance LC (UPLC) system (Waters), and then delivered to an LTQ Velos dual-pressure linear ion trap mass spectrometer (Thermo Fisher) using electrospray ionization with a captive spray source (Microm Biosciences) fitted with a 20-μm-inner-diameter tapered spray tip and a 1.0-kV source voltage. Xcalibur version 2.1 was used to control the system. Samples were applied at 15 μl/min to a Symmetry C18 trap cartridge (Waters) for 10 min and then switched to a 75-μm-by-250-mm NanoAcquity BEH 130 C18 column with 1.7-μm-diameter particles (Waters) using mobile phases of water (phase A) and acetonitrile (phase B) containing 0.1% formic acid, a 7.5% to 30% acetonitrile gradient over 60 min, and a 300 nl/min flow rate. A normalized collision energy level of 30 was used. Data-dependent collection of MS/MS spectra used the dynamic exclusion feature of the instrument's control software (repeat count equal to 1, exclusion list size of 500, exclusion duration of 24 s, and exclusion mass width of −1 to +4) to obtain MS/MS spectra of the 10 most abundant parent ions (minimum signal of 5,000) following each survey scan from m/z 400 to 1,400. The tune file was configured with no averaging of microscans, a maximum injection time of 200 ms, and automatic gain control targets of 3 × 104 in MS1 mode and 1 × 104 in MS2 mode.
Sequest (version 28, revision 12; Thermo Scientific) was used to search MS2 spectra against a May 2014 version of the Sprot human FASTA protein database, with added sequences for E. coli BirA* and HIV-1 strain NL4-3, concatenated sequence-reversed entries to estimate error thresholds, and 179 common contaminant sequences and their reversed forms. The database processing was performed with Python scripts that have been described previously (65). Searches for all samples were performed with trypsin enzyme specificity. The average parent ion mass tolerance was 2.5 Da. The monoisotopic fragment ion mass tolerance was 1.0 Da. A static modification of +57.02 Da was added to all cysteine residues. A variable modification of +16 Da on methionine residues was also allowed, with a maximum of 3 modifications per peptide. A linear discriminant transformation was used to improve the identification sensitivity from the SEQUEST analysis (65, 66). SEQUEST scores were combined into linear discriminant function scores, and discriminant score histograms were created separately for each peptide charge state (1+, 2+, and 3+). Separate histograms were created for matches to forward sequences and for matches to reversed sequences for all peptides of 7 amino acids or longer. Scores of histograms for reversed matches were used to estimate peptide false-discovery rates (FDR) and set score thresholds for each peptide class that achieved the desired protein FDR (approximately 1.5% to 5.3%) for each sample. Relative numbers of proteins in transfected versus mock cell samples were normalized by comparison of spectral counts corresponding to peptide sequences of the proteins, with endogenously biotinylated proteins (acetyl-coenzyme A [CoA] carboxylase plus methylcrotonoyl-CoA carboxylase plus proprionyl-CoA carboxylase plus pyruvate carboxylase [A + M + P1 + P2]). After normalization, “hits” were removed if their normalized percentages (100 × [A + M + P1 + P2]) were less than 0.5% in all MS runs. Background proteins present in mock samples were subsequently removed if normalized experimental levels were less than four times the untransfected cell levels in any MS run; these background proteins included endogenously biotinylated proteins and standard artifacts and are listed in Table 1. Following the exclusion of background proteins, signal levels in transfected cell samples were renormalized to BirA* signals to account for transfection efficiencies. At this point, protein “hits” that gave signals of <7% of the BirA* signal levels in both MA-BirA* and ΔMA-BirA* samples were discarded to remove candidates that scored too closely to the normalized false-discovery rate (FDR) signal range of 1.5% to 5.3%. The final list of specifically biotinylated proteins is provided in Table 2, ordered by the ratio of BirA*-normalized MA-BirA* sample levels to the ΔMA-BirA* sample levels.
TABLE 1.
Background proteinsa
| Protein ID | Protein description | Normalized level (%) |
|---|---|---|
| AATM_HUMAN | Aspartate aminotransferase | 0.9 |
| ACACA_HUMAN | Acetyl-CoA carboxylase | 28.9 |
| ACTIN* | Actins | 1.7 |
| ADT2_HUMAN | ADP/ATP translocase | 0.9 |
| ALDOA_HUMAN | Fructose-bisphosphate aldolase | 1.2 |
| ANXA2_HUMAN | Annexin A2 | 1.0 |
| APEX1_HUMAN | DNA lyase | 0.8 |
| C1TC_HUMAN | C-1-tetrahydrofolate synthase | 1.1 |
| CAH2_HUMAN | Carbonic anhydrase | 1.0 |
| HS PROTEINS* | Heat shock proteins | 2.0 |
| TRYPSIN | Trypsin precursor | 4.6 |
| KERATIN* | Keratins | 26.8 |
| DCD_HUMAN | Dermcidin | 3.9 |
| DESP_HUMAN | Desmoplakin | 5.4 |
| DSC1_HUMAN | Desmocollin-1 | 1.4 |
| DSG1_HUMAN | Desmoglein-1 | 2.3 |
| EF* | Elongation factors | 4.9 |
| ENOA_HUMAN | Alpha-enolase | 4.7 |
| FUBP2_HUMAN | Far upstream element BP | 1.7 |
| G3P_HUMAN | G-3-phosphate dehydrogenase | 2.5 |
| GBLP_HUMAN | Guanine nucleotide BP | 2.4 |
| GRP75_HUMAN | Stress-70 protein | 1.8 |
| GRP78_HUMAN | 78-kDa glucose-regulated protein | 2.8 |
| HNRPK_HUMAN | Het nuclear ribonucleoprotein K | 2.2 |
| HUWE1_HUMAN | E3 ubiquitin-protein ligase | 1.1 |
| IGHA1_HUMAN | Ig alpha-1 chain C region | 1.5 |
| KPYM_HUMAN | Pyruvate kinase | 2.9 |
| LDH* | l-Lactate dehydrogenases | 1.0 |
| MCCA_HUMAN | Methylcrotonoyl-CoA carboxylase | 9.6 |
| MDHM_HUMAN | Malate dehydrogenase | 1.3 |
| MOES_HUMAN | Moesin | 1.0 |
| NUCL_HUMAN | Nucleolin | 1.9 |
| PARP1_HUMAN | Poly[ADP-ribose] polymerase | 2.0 |
| PCCA_HUMAN | Propionyl-CoA carboxylase | 29.6 |
| PGK1_HUMAN | Phosphoglycerate kinase | 2.2 |
| PLAK_HUMAN | Junction plakoglobin | 3.4 |
| PLST_HUMAN | Plastin-3 | 0.9 |
| PPIA_HUMAN | Peptidyl-prolyl isomerase A | 1.8 |
| PERIREDOXIN* | Peroxiredoxins | 1.1 |
| PYC_HUMAN | Pyruvate carboxylase | 31.9 |
| PYR1_HUMAN | CAD protein | 0.8 |
| RAN_HUMAN | GTP-binding protein Ran | 1.3 |
| RCC2_HUMAN | Protein RCC2 | 1.1 |
| 60S PROTEIN* | 60S ribosomal proteins | 0.9 |
| SAHH_HUMAN | Adenosylhomocysteinase | 0.9 |
| TUBULIN | Tubulins | 1.8 |
| TGM3_HUMAN | Protein glutamyltransferase | 1.2 |
| TKT_HUMAN | Transketolase | 1.2 |
| TLN1_HUMAN | Talin-1 | 0.9 |
| TPIS_HUMAN | Triosephosphate isomerase | 1.4 |
Proteins from MS analyses were designated background proteins as described in Materials and Methods and include endogenously biotinylated proteins and standard artifacts. The left column gives the UniProt ID name; the center column gives the protein description; the right column gives the MS signals, normalized as percentages relative to the summed signals of acetyl-CoA carboxylase plus methylcrotonyl-CoA carboxylase plus proprionyl-CoA carboxylase plus pyruvate carboxylase and averaged over all MS runs. The asterisks indicate that the values given represent normalized averaged signals for the following: actins (ACTA and ACTB), heat shock proteins (CH60, HSL71L, HS90A, HS90B, HSP71, HSP7C, and TRAP1), keratins (K1C2, K1C9, K1C10, K1C13, K1C14, K1C17, K22E, K2C1, K2C1B, K2C5, K2C8, K2C78, KRT5, KRT8, KRT10, and Q13092), elongation factors (EF1A1, EF2, and EFTU), lactate dehydrogenases (LDHA and LDHB), peridoxins (PRDX1 and PRDX2), 60S ribosomal proteins (RL5 and RL6), and tubulins (TBA1A, TBA1B, TBA1C, TBB1, TBB4B, and TBB5).
TABLE 2.
Biotinylated proteinsa
| Protein ID | Protein description | WT MA-BirA* (%) | ΔMA-BirA* (%) | Ratio | Reference(s) |
|---|---|---|---|---|---|
| ATX2L_HUMAN | Ataxin-2-like protein | 21.2 | 0 | UD | 50 |
| CD2AP_HUMAN | CD2-associated protein | 12.6 | 0 | UD | None |
| CDV3_HUMAN | Protein CDV3 homolog | 10.6 | 0 | UD | None |
| DDX3X_HUMAN | ATP-dependent RNA helicase DDX3X | 8.1 | 0 | UD | 22, 36, 50, 67, 68 |
| E41L2_HUMAN | Band 4.1-like protein 2 | 12.6 | 0 | UD | None |
| GEMI5_HUMAN | Gem-associated protein 5 | 14.4 | 0 | UD | None |
| H12_HUMAN | Histone H1.2 | 9.1 | 0 | UD | 36, 50, 69 |
| HAP28_HUMAN | 28-kDa heat- and acid-stable phosphoprotein | 7.6 | 0 | UD | None |
| IF4G1_HUMAN | Eukaryotic translation initiation factor 4 gamma 1 | 12.1 | 0 | UD | 50, 69 |
| IF5_HUMAN | Eukaryotic translation initiation factor 5 | 9.6 | 0 | UD | 70 |
| KI67_HUMAN | Antigen KI-67 | 9.3 | 0 | UD | 67, 69 |
| LARP1_HUMAN | La-related protein 1 | 15.2 | 0 | UD | 50 |
| NUFP2_HUMAN | Nuclear fragile X mental retardation-interacting protein 2 | 12.6 | 0 | UD | 50 |
| PKP1_HUMAN | Plakophilin-1 | 7.6 | 0 | UD | None |
| PRC2A_HUMAN | Protein PRRC2A | 21.0 | 0 | UD | None |
| PRC2C_HUMAN | Protein PRRC2C | 27.5 | 0 | UD | None |
| RBM2 6_HUMAN | RNA-binding protein 26 | 7.4 | 0 | UD | None |
| RL40_HUMAN | Ubiquitin-60S ribosomal protein L40 | 11.7 | 0 | UD | 35, 69, 71 |
| SEMG1_HUMAN | Semenogelin-1 | 10.3 | 0 | UD | 72 |
| SEMG2_HUMAN | Semenogelin-2 | 12.4 | 0 | UD | 72 |
| SYEP_HUMAN | Bifunctional glutamate/proline-tRNA ligase | 9.9 | 0 | UD | 50, 69 |
| TOX4_HUMAN | TOX high-mobility-group box family member 4 | 16.5 | 0 | UD | None |
| UBAP2_HUMAN | Ubiquitin-associated protein 2 | 16.3 | 0 | UD | None |
| VIGLN_HUMAN | Vigilin | 12.2 | 0 | UD | None |
| XRN1_HUMAN | 5′–3′ exoribonuclease 1 | 12.6 | 0 | UD | 73 |
| UBP2L_HUMAN | Ubiquitin-associated protein 2-like | 28.9 | 1.1 | 27.4 | None |
| MAP4_HUMAN | Microtubule-associated protein 4 | 38.7 | 1.6 | 24.5 | 50, 67, 74–76 |
| IRS4_HUMAN | Insulin receptor substrate 4 | 16.7 | 1.1 | 15.8 | None |
| PERQ2_HUMAN | PERQ amino acid-rich with GYF domain-containing protein 2 | 22.7 | 1.6 | 14.4 | 50 |
| AFAD_HUMAN | Afadin | 13.6 | 1.1 | 13.0 | 69 |
| IF4B_HUMAN | Eukaryotic translation initiation factor 4B | 19.5 | 1.6 | 12.4 | None |
| FLNA_HUMAN | Filamin-A | 426.5 | 44.2 | 9.6 | 35, 67, 69, 77 |
| PAIRB_HUMAN | Plasminogen activator inhibitor 1 RNA-binding protein | 19.4 | 2.1 | 9.2 | None |
| 41_HUMAN | Protein 4.1 | 24.1 | 2.6 | 9.2 | 72 |
| DSG2_HUMAN | Desmoglein-2 | 18.7 | 2.6 | 7.1 | None |
| SRC8_HUMAN | Src substrate cortactin | 14.8 | 2.1 | 7.0 | 72 |
| HORN_HUMAN | Hornerin | 18.2 | 2.6 | 6.9 | None |
| E41L3_HUMAN | Band 4.1-like protein 3 | 17.4 | 2.6 | 6.6 | None |
| TCPQ_HUMAN | T-complex protein 1 subunit theta | 52.1 | 7.9 | 6.6 | None |
| AHNK_HUMAN | Neuroblast differentiation-associated protein AHNAK | 70.1 | 11.1 | 6.3 | 35 |
| TPR_HUMAN | Nucleoprotein TPR | 84.1 | 13.7 | 6.1 | 42, 69, 72 |
| FAS_HUMAN | Fatty acid synthase | 75.8 | 13.2 | 5.8 | 22, 36, 50, 69 |
| HNRPM_HUMAN | Heterogeneous nuclear ribonucleoprotein M | 8.9 | 1.6 | 5.7 | 22, 36, 50, 69 |
| AAAT_HUMAN | Neutral amino acid transporter B(0) | 8.8 | 1.6 | 5.6 | None |
| CSDE1_HUMAN | Cold shock domain-containing protein E1 | 19.8 | 3.7 | 5.4 | 50, 69, 71 |
| AT1A1_HUMAN | Sodium/potassium-transporting ATPase subunit alpha-1 | 12.5 | 2.6 | 4.8 | 22, 35, 67, 69 |
| FLNB_HUMAN | Filamin-B | 7.0 | 1.6 | 4.4 | 68, 69 |
| EF1A1_HUMAN | Elongation factor 1-alpha 1 | 46.9 | 15.8 | 3.0 | 50, 69, 71, 78 |
| LYRIC_HUMAN | Protein Lyric | 7.8 | 2.6 | 3.0 | 50, 69, 79 |
| POL | HIV-1 Pol | 7.8 | 3.2 | 2.5 | NA |
| GAG | HIV-1 Gag | 136.9 | 56.8 | 2.4 | NA |
| ZCCHV_HUMAN | Zinc finger CCCH-type antiviral protein 1 | 57.7 | 35.3 | 1.6 | 50, 69, 80 |
| PRKDC_HUMAN | DNA-dependent protein kinase catalytic subunit | 8.3 | 5.3 | 1.6 | 22, 50, 67, 69, 71 |
| BIRA* | E. coli bifunctional ligase/repressor BirA* | 100 | 100 | 1 | NA |
| 4F2_HUMAN | 4F2 cell surface antigen heavy chain | 8.3 | 8.4 | 1.0 | 35, 67, 69 |
| LAP2B_HUMAN | Lamina-associated polypeptide 2, isoforms beta/gamma | 15.5 | 16.3 | 0.9 | 50, 69 |
| ENV | HIV-1 Env | 0 | 10.5 | 0 | NA |
Proteins specifically biotinylated in transfected cell samples were determined as described in Materials and Methods. The left two columns give protein UniProt ID names and descriptions. The third and fourth columns give MS signals in WT MA-BirA*-transfected and ΔMA-BirA*-transfected cells, respectively: these values are percentages of the BirA* protein signals in the respective transfected cell samples. The list is ordered based on the values in the ratio column, which lists the percent WT BirA*/percent ΔMA BirA* ratios, where UD means “undefined.” The rightmost column indicates references to publications of research exploring whether a protein has or has not been previously implicated as an effector of HIV-1 replication, as an HIV-1 binding protein, or as present in HIV-1 virus particles. Numbers in this column refer to cited reference numbers. The HIV-1 Env peptides identified in the ΔMA-BirA* sample all mapped to the TM CT and represented the following env codons: 724 to 738, 746 to 759, 760 to 768, 769 to 778, 779 to 785, 817 to 826, and 827 to 836. Note that these represent sequenced peptides from the biotinylated TM protein but do not necessarily represent the specifically biotinylated peptides. None, no previous references; NA, not applicable.
RESULTS
Infectivities and cellular expression of tagged viruses and proteins.
To foster the analysis of HIV-1 PrGag protein activities and interactions, we generated proviral constructs based on the WT NL4-3 HIV-1 proviral construct (56). To do so, the open reading frame (ORF) for the promiscuous E. coli biotin ligase, BirA*, was inserted near the MA C terminus, 12 codons upstream of the MA-CA cleavage junction (Fig. 1). The WT version of the tagged construct is referred to as MA-BirA*. We also generated a BirA*-tagged ΔMA proviral construct (ΔMA-BirA*) in which NL4-3 MA residues 19 to 120 were deleted, retaining the MA myristoylation signal (2, 30) and MA-CA juncture sequences. The primary PrGag translation products for MA-BirA* and ΔMA-BirA* are predicted to be 93 kDa and 82 kDa, respectively.
FIG 1.

Recombinant DNA constructs. Depicted at the top of the figure are the wild-type (WT) NL4-3 strain of HIV-1 and its WT PrGag translation product. MA-BirA* and ΔMA-BirA* are variants of NL4-3 in which the open reading frame (ORF) for the promiscuous biotin ligase (BirA*) has been inserted into WT (MA-BirA*) or MA-deleted (ΔMA-BirA*) version of NL4-3. In MA-BirA*, the BirA* ORF has been inserted 12 codons upstream from the NL4-3 MA-CA cleavage junction, and the primary translation product is predicted to be a myristoylated PrGag protein of approximately 93 kDa. In ΔMA-BirA*, the BirA* ORF is also 12 codons upstream from the MA-CA cleavage junction and begins after codon 18 of MA, deleting MA residues 19 to 120; the primary translation product is predicted to be a myristoylated PrGag protein of approximately 82 kDa. LTR, long terminal repeat.
Previous studies have shown that HIV-1 PrGag proteins tagged near their MA C termini appeared to be trafficked normally but yielded viruses with compromised infectivities (51, 52). We thus initially examined the abilities of MA-BirA* and ΔMA-BirA* constructs to assemble and release virus particles. As shown in Fig. 2A, virus particle release was detected for MA-BirA* and, to a lesser extent, for ΔMA-BirA*, as indicated by the observation of particle-associated CA and precursor Gag (PrMABirA* and PrΔMABirA*) proteins (Fig. 2A, left side). Consistent with previous results (27–32), we also detected the HIV-1 Env transmembrane (TMl; gp41) protein in viruses encoded by MA-BirA* but not in those encoded by ΔMA-BirA* (Fig. 2A, right side).
FIG 2.
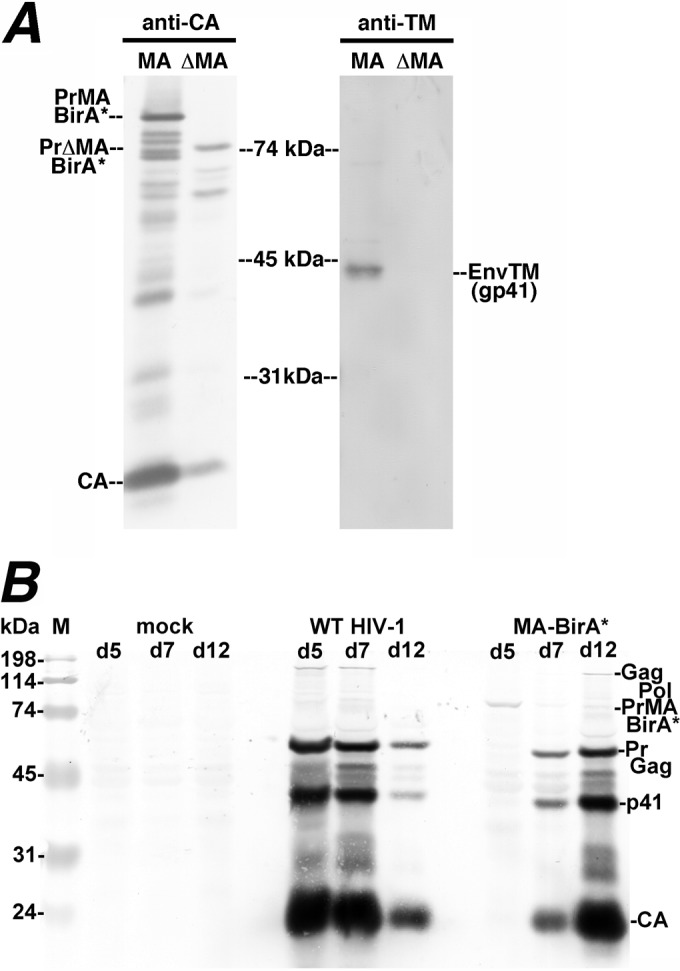
Analysis of MA-BirA* virus infection. (A) Proteins from virus particles released from HEK 293T cells transfected with MA-BirA* (MA) or ΔMA-BirA* (ΔMA) constructs were electrophoresed and detected by immunoblotting with antibodies to either CA or TM (gp41). PrMABirA*, PrΔMABirA*, CA, and TM proteins are as indicated. (B) MT4 T cells were either mock infected or infected with capsid-normalized amounts of virus from transfections of 293T cells with WT or MA-BirA* NL4-3 proviral HIV-1 constructs. In both cases, the individual proviral constructs were cotransfected with an expression construct for the VSV G protein. Infections were monitored at days 5, 7, and 12 (d5, d7, and d12) posttransfection via electrophoretic separation of proteins from infected cell aliquots and immunoblot (Western) detection of Gag proteins using an anti-CA primary antibody. As shown, peak levels of WT PrGag, p41, and CA proteins were detected by 5 days postinfection with the WT virus, after which levels diminished, due to cell death. For the MA-BirA* virus, the precursor MA-BirA* protein (PrMABirA*) was detected at day 5, while bands corresponding to WT PrGag, p41, and CA proteins were detected at day 7 and day 12, indicating a deletion of the BirA* element and reversion of the MA-BirA* virus. Note that the mobilities of the PrGag and p41 proteins in the day 7 and day 12 MA-BirA* revertant samples were slightly faster than those of their WT virus counterparts, suggesting a deletion of some MA residues during reversion. Note also that infectivity of VSV G-pseudotyped ΔMA-BirA* viruses was undetectable in MT4 cells.
We next tested the infectivities of VSV-G-pseudotyped MA-BirA* and ΔMA-BirA* viruses produced in the absence of WT HIV-1 virus by transfection of HEK 293T cells in single-round infection assays as described in Materials and Methods. Not surprisingly, infectivity of the G protein-pseudotyped MA-BirA* virus was less than 1% of that of similarly pseudotyped WT HIV-1, while the infectivity of the ΔMA-BirA* virus was undetectable (data not shown). Infection assays with G-pseudotyped viruses were also carried out in MT-4 T cells. Here again, ΔMA-BirA* viruses were noninfectious. In contrast, MA-BirA* variants again showed low but detectable levels of infection. As illustrated in Fig. 2B, within 5 days of infection with WT HIV-1, MT-4 T cells showed high PrGag, p41, and CA levels which diminished by day 12 postinfection as a consequence of cell death. Interestingly, at day 5 postinfection with the MA-BirA* virus, the 92-kDa MA-BirA* PrGag protein (PrMABirA*) was observed in infected cells, but by day 7 of infection, the 92-kDa band was replaced by apparently WT PrGag, p41, and CA bands (Fig. 2B). These results are consistent with a model in which the BirA* coding region of poorly infectious MA-BirA* viruses has been selected for deletion in the infected MT-4 cells.
To examine whether BirA*-tagged proteins retained their biotinylation activities (Fig. 3), the ΔMA-BirA* or MA-BirA* proviral constructs were transfected in the absence (lanes B, E, I, and L) or presence of WT (C, F, J, and M) or protease-negative (PR−; D, G, K, and N) HIV-1 into HEK 293T cells, after which cell samples were collected and subjected to detection of Gag proteins with an anti-CA antibody and biotinylated proteins with an alkaline phosphatase-streptavidin conjugate. Note that the WT and PR− cotransfections were performed to assess whether BirA* proteins could biotinylate Gag proteins in trans (see below). Mock-transfected cell samples (M) showed no Gag proteins (lane A) but showed endogenously biotinylated bands that migrated near the 114-kDa and 74-kDa markers (lane H, bands 4 and 5). These are approximately the expected sizes of the mammalian biotinylated polypeptide pyruvate carboxylase (130 kDa) and of the 80-kDa alpha chains of proprionyl-CoA and methylcrotonyl-CoA carboxylase (81). Importantly, all samples transfected with either ΔMA-BirA* or MA-BirA* yielded a multiplicity of biotinylated protein bands (lanes I to N) at much higher levels than the mock samples. Comparison of lanes E to G and L to N indicates that the MA-BirA* PrGag protein (PrMABirA*) was biotinylated. Because the 82-kDa ΔMA-BirA* PrGag protein (PrΔMABirA*; lanes B to D and lanes I to K) migrated so closely to the 80-kDa proprionyl-CoA and methylcrotonyl-CoA carboxylase bands (lane H, band 5), it was not possible to verify that it also was biotinylated in this gel, but evidence presented below indicates that it was. Other noteworthy bands appear to be processed forms of PrΔMABirA* and PrMABirA*. In particular, PrΔMABirA*-derived bands 1 and 2 (lane B) were biotinylated (lane I) and may correspond to BirA*-CA and CA-NC-p6 processing intermediates. Another PrΔMABirA* processing product migrated between the 31-kDa and 45-kDa markers (lanes I to K), was not detected with the anti-CA antibody, and appears to be the processed ΔMA-BirA* protein. For MA-BirA*, a MA-BirA*-CA-sized intermediate was observed (lane E, band 3), but it was not possible to assess its biotinylation status due to comigration with the endogenously biotinylated proteins (lane L). Finally, in cotransfections with WT and PR− HIV-1 (lanes C and D and lanes F and G), the WT PrGag proteins were readily detected with the anti-CA probe. However, because the ΔMA-BirA* and MA-BirA* proteins biotinylated cellular proteins in the 50-kDa to 60-kDa size range (lanes I and L), it was not possible to assess the biotinylation of WT PrGag proteins in this experiment; evidence with regard to PrGag biotinylation in trans is provided below.
FIG 3.

Detection of viral proteins and biotinylation in cells. ΔMA-BirA* or MA-BirA* proviral constructs were transfected into 293T cells alone or along with WT (+WT) or PR− (+PR−) HIV-1 proviral constructs. At 3 days posttransfection, protein samples from transfected cells and mock-transfected cells (M; lanes A and H) were electrophoretically separated, and subjected to detection of viral proteins (anti-CA; lanes A to G) or biotinylated proteins (streptavidin; lanes H to N). Molecular size markers, as indicated, were run in parallel lanes. (Left panel) In lanes B to D and E to G, the anti-CA antibody detected the precursor PrΔMA-BirA* and PrMA-BirA* proteins and capsid proteins, respectively. Also detected were PrGag proteins in samples cotransfected with WT and PR− HIV-1 constructs (lanes C, D, F, and G). In addition to the PrΔMA-BirA* band, the ΔMA-BirA* construct yielded CA-reactive bands 1 and 2: band 1 migrates at approximately 60-kDa and potentially corresponds to a BirA*-CA processing intermediate, while band 2, at about 41 kDa, potentially corresponds to a CA-NC-p6 intermediate. Similarly, band 3, detected in transfections containing the MA-BirA* construct, migrates at about 74 kDa, which corresponds to the predicted size of a MA-BirA*-CA processing intermediate. (Right panel) Bands in the mock lane (H) near the 114-kDa (band 4) and 74-kDa (band 5) markers correspond to the endogenously biotinylated pyruvate carboxylase (130-kDa) protein and to the alpha chains of proprionyl-CoA carboxylase (80 kDa) and methylcrotonyl-CoA carboxylase (80 kDa). All the transfected cell sample lanes show numerous biotinylated protein bands, including the PrMABirA* band (lanes L to M). Note that the 82-kDa biotinylated PrΔMA-BirA* band (lanes I to K) comigrates with the endogenously biotinylated carboxylase alpha chains. Note that these results are representative of nine separate experiments.
Given that cells transfected with either ΔMA-BirA* or MA-BirA* gave significantly higher levels of biotinylated proteins than untransfected cells (Fig. 3), it was of interest to examine the localization patterns of Gag proteins and biotinylated proteins in cells expressing these constructs. To do so, HEK 293T cells were transfected with MA-BirA* (MA) or ΔMA-BirA* (ΔMA) and subjected to simultaneous localization of Gag proteins, using a primary anti-CA antibody, and of biotinylated proteins, using a fluorescently tagged streptavidin conjugate (Fig. 4). As expected, even with long exposure times, Gag signals were not detectable in untransfected cells (data not shown). However, long exposure times (350 ms) permitted the visualization of endogenously biotinylated proteins in untransfected cells (Fig. 4A, right panel). Because the majority of these proteins (proprionyl-CoA carboxylase, pyruvate carboxylase, methylcrotonyl-CoA carboxylase, and acetyl-CoA carboxylase 2) are mitochondrial (81, 82), localization patterns of endogenously biotinylated proteins were assumed to be predominantly mitochondrial (Fig. 4A), although we did not costain with alternative mitochondrial markers to test this assumption.
FIG 4.

Fluorescent localization of biotinylated proteins. (A) Biotinylated proteins in untransfected 293T cells were detected with an Alexa Fluor 594-tagged streptavidin conjugate as described in Materials and Methods at an exposure time of 15 ms (left panel) or 350 ms (right panel). (B) In cells transfected with MA-BirA* (MA) or ΔMA-BirA* (ΔMA) constructs, Gag proteins (green) were detected via indirect immunofluorescence using a primary anti-CA antibody and an Alexa Fluor-488-tagged secondary antibody, while biotinylated (red) proteins were detected with an Alexa Fluor 594-tagged streptavidin conjugate. The right (overlay) column indicates an overlay of the Gag and biotin signals (yellow). Note that the low levels of endogenously biotinylated proteins were barely detectable due to use of a short (15-ms) exposure time and that Gag and biotin signals colocalized with Pearson's overlap coefficient values of 0.96 ± 0.02 for the MA-BirA* samples (n = 25) and 0.97 ± 0.01 for the ΔMA-BirA* samples (n = 25). Note also the occurrence of cells with enhanced perinuclear or vesicular staining patterns (top cell, ΔMA sample) was slightly higher for the ΔMA sample (17.1%; 25/146 cells) than for the MA sample (6.2%; 6/97 cells). Despite the high degree of Gag and biotin signal colocalization for both samples, 40.2% of MA-BirA* cells and 51.4% of ΔMA-BirA* cells showed cytoplasmic dots (white arrows) that stained for biotin but not for Gag. (C) Dots that stained for biotin but not Gag are enlarged from the overlay images shown at the bottom rows for the MA and ΔMA samples. Of MA-BirA* cells showing such dots, 33% had one dot, 28% had two dots, and 39% had three or more dots. For ΔMA-BirA* cells, the numbers were 40% with one dot, 28% with two dots, and 32% with three or more dots.
With short exposure times (15 ms), the endogenously biotinylated proteins were barely detectable (Fig. 4A, left panel) and it was possible to visualize Gag proteins as well as proteins biotinylated by the MA-BirA* and ΔMA-BirA* vectors (Fig. 4B). Consistent with previous observations (30, 51, 52, 83), Gag proteins expressed from MA-BirA* and ΔMA-BirA* gave predominantly heterogenously cytoplasmic staining patterns, extending to cell edges (Fig. 4B, left column). With ΔMA-BirA* versus MA-BirA* cells, we observed slightly enhanced perinuclear or vesicular staining patterns (see Fig. 4B, ΔMA, top panel, and the Fig. 4 legend), but the significance of this observation is undetermined. However, with both ΔMA-BirA* and MA-BirA* cells, the localization patterns of biotinylated proteins (Fig. 4B, center column) closely matched that of the Gag proteins and overlapped to a high degree (Fig. 4B, right column). Indeed, Pearson's overlap coefficients (63) for Gag and biotin signals were 0.96 for the MA-BirA* cells and 0.97 for the ΔMA-BirA* cells. These data demonstrate that at the resolution of conventional fluorescence microscopy, Gag proteins and biotinylated proteins colocalize, which is consistent with previous studies on the promiscuous biotin ligase (53, 55). The one exception to this rule was the observance of an occasional bright cytoplasmic dot that was detectable with the streptavidin reagent but not with anti-CA (Fig. 4B, right column; Fig. 4C, white arrows). The dots were observed in 40% to 50% of MA-BirA*-transfected or ΔMA-BirA*-transfected cells, and while most cells with dots had either one or two dots, about a third of the cells showed three or more (see the Fig. 4 legend). Our assumption is that the cytoplasmic dots contain biotinylated proteins that migrated after their encounters with BirA* proteins, and the nature of these dots is discussed in more detail below.
Biotinylated proteins in virus particles.
To analyze biotinylated proteins in virus particles, viruses released from cells transfected with MA-BirA* or ΔMA-BirA* in the absence or presence of WT or PR− HIV-1 were collected. Proteins in these samples were subjected to gel electrophoresis, immunoblot detection of CA-reactive and MA-reactive proteins, and identification of biotinylated proteins with alkaline phosphatase-conjugated streptavidin (Fig. 5). One immediate observation was that MA-BirA* virus particle release was efficient whereas ΔMA-BirA* particle release in the absence of WT or PR− HIV-1 was poor. This perhaps is most readily seen in the anti-CA blots by comparison of lane D to lanes E and F (for ΔMA-BirA*) and of lane M to lanes N and O (for MA-BirA*). Indeed, calculations of virus release levels (see Materials and Methods) indicated that relative, to WT HIV-1 release, particle release levels for MA-BirA* and ΔMA-BirA* were 70% ± 19% and 15% ± 11%, respectively. Notably, cotransfection of the BirA* constructs with WT or PR− HIV-1 vectors increased virus release levels for MA-BirA* and ΔMA-BirA* to 100% ± 11% and 92% ± 20% relative to WT HIV-1, respectively. These results imply that WT PrGag proteins associate with the ΔMA-BirA* and MA-BirA* Gag proteins during transit and/or release from transfected cells.
FIG 5.

Biotinylated proteins in virus particles. ΔMA-BirA* (lanes A to I) or MA-BirA* (lanes J to R) proviral constructs were transfected into 293T cells alone or along with WT (+WT) or PR− (+PR−) HIV-1 proviral constructs. At 3 days posttransfection, proteins from collected virus particles were electrophoretically separated and subjected to detection of biotinylated proteins with streptavidin (lanes A to C and J to L), viral proteins with an anti-CA antibody (lanes D to F and M to O), or viral proteins with an anti-MA antibody (lanes G to I and P to R). Molecular size markers, as indicated, were run in parallel lanes. PrΔMABirA*, PrMABirA*, PrGag, CA, and MA bands are as indicated. To match figure sizes of streptavidin and CA blots, portions of MA blots that showed no immunoreactive bands between CA and MA were truncated. As shown, MA-BirA* proteins in viruses were readily detectable, while ΔMA-BirA* virus particle release was inefficient (lanes A and D) in the absence of cotransfected HIV-1 WT or PR− constructs. Relative to WT HIV-1, virus particle release levels for MA-BirA* and ΔMA-BirA* were 70% ± 19% and 15% ± 11%, respectively, while cotransfection with WT HIV-1 increased MA-BirA* normalized release levels to 100% ± 11% and ΔMA-BirA* levels to 92% ± 20%. Note that the PrGag-sized band (band 1) in lane A may correspond to a processed BirA*-CA protein, although it stained marginally with anti-CA, while band 2 (lane A) migrated at the size of the BirA* protein (38 kDa). In the ΔMA-BirA* anti-MA blot, band 3 appears to be an anomaly, because PrΔMABirA* bands were not detected in lanes H and I. For the MA-BirA* virus, bands 4 and 5 had mobilities of 50 kDa and 46 kDa, strongly reacted with the anti-MA antibody (lane P), and appear to correspond to MA-BirA* (band 4) and a processed variant of MA-BirA* (band 5). Note also that band 6 comigrated with CA (lane M) and an anti-MA-reactive band (band 7). Data are representative of the results of nine separate experiments.
With regard to biotinylated viral proteins, for ΔMA-BirA*, the precursor Gag protein (PrΔMABirA*) was detected with streptavidin (lanes A to C), as were proteins that may correspond to the sizes of BirA*-CA (lane A, band 1) and BirA*(lane A, band 2). Additionally, the WT PrGag protein in the cotransfection with PR− HIV-1 appeared to be biotinylated in trans (lane C), but because PrGag migrated so closely to band 1 in lane A, it was not possible to determine this definitively. For MA-BirA*, a number of biotinylated viral proteins were observed. One of these was the precursor Gag protein, PrMABirA* (lanes J to L), and one comigrated with the processed CA band (lane J, band 6). Two other biotinylated processed MA-BirA* proteins were observed. These proteins (lane J, bands 4 and 5) were detected by our MA antibody (lanes P to R) and appear to correspond to MA-BirA* (band 4) and a processed variant of MA-BirA* (band 5). Interestingly, in MA-BirA* cotransfections with WT and PR− HIV-1 (lanes K and L, N and O, and Q and R), our data strongly indicate that the WT PrGag proteins were biotinylated in trans (lanes K and L), demonstrating that MA-BirA* and WT HIV-1 Gag proteins were in close proximity (48, 75) during trafficking and/or release.
Because of the complexity of cellular (Fig. 3) and viral (Fig. 5) streptavidin blots, it was not possible to assess possible ΔMA-BirA* or MA-BirA* biotinylation of the HIV-1 Env proteins: this issue is addressed below. However, it was possible to examine biotinylation of the VSV G protein in pseudotyped particles. To do so, ΔMA-BirA* or MA-BirA* constructs were cotransfected with a VSV G protein expression construct, and virus particles from these transfections were monitored for protein biotinylation and G protein content (Fig. 6). Because the G protein is passively assembled into HIV-1 particles (20, 32–34), it was not surprising to see that it was incorporated into ΔMA-BirA* (lane D) or MA-BirA* (lane H) virions. Streptavidin blots also demonstrated that the G protein was biotinylated in both types of virus (lanes B and F), although biotinylation levels appeared higher with ΔMA-BirA* than with MA-BirA*. As normalized to PrGag biotinylation levels, G protein biotinylation levels in ΔMA-BirA* viruses were measured to be 3.2 times greater than those in MA-BirA* viruses: this could have been due to higher BirA* ligase activities in ΔMA-BirA* cells or viruses and/or to greater G protein accessibilities in ΔMA-BirA* cells or viruses.
FIG 6.
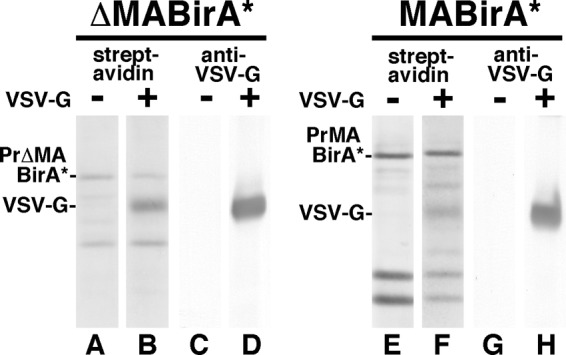
Detection of biotinylated VSV G protein in virus particles. ΔMA-BirA* (lanes A to D) and MA-BirA* (lanes E to H) constructs were transfected alone (lanes A, C, E, and G) or cotransfected with a VSV G protein expression construct (lanes B, D, F, and H). Three days posttransfection, proteins from virus particles were separated electrophoretically and blotted to detect biotinylated proteins (lanes A and B and lanes E and F) or the VSV G protein (lanes C and D and lanes G and H). PrΔMABirA*, PrMABirA*, and VSV G bands are as indicated.
Identification of biotinylated cellular proteins.
Use of BirA* tags permits the convenient identification of potential interacting proteins via a mass spectrometry (MS) approach, with an estimated biotinylation labeling range of 10 to 30 nm (53, 55). Thus, to identify putative cellular associates of HIV-1 Gag proteins, biotinylated proteins in mock-transfected cells and in cells transfected with either ΔMA-BirA* or MA-BirA* were purified on streptavidin columns and identified after trypsin digestion, liquid chromatography (LC), and MS/MS as described in Materials and Methods. Note that this protocol identifies proteins that have been biotinylated but does not identify specific biotinylated residues (53, 55). One advantage of the BirA*-tagging approach is that endogenously biotinylated proteins can serve as internal controls for protein recovery levels in mock treatment versus transfected-cell LC MS/MS samples. Accordingly, normalized spectral counts for each protein in each MS run were calculated as percentages of the combined number of spectral counts for the four major endogenously biotinylated species (acetyl-CoA carboxylase, methylcrotonyl-CoA carboxylase, proprionyl-CoA carboxylase, and pyruvate carboxylase) (81, 82). Protein “hits” were removed if their normalized signals were <0.5% of the combined endogenous signal. After this step, background proteins were removed if normalized signals for transfected cell samples were less than four times the level of the untransfected cell sample signals in any MS run. A list of these background proteins is provided in Table 1. In addition to endogenously biotinylated proteins, these include standard MS contaminants, such as keratins and heat shock proteins, as well as ribosomal proteins and some elongation factors. Importantly, the list of background proteins also includes proteins that have been implicated as Gag-interacting proteins such as actins and tubulins (35, 36, 84). In this regard, it should be emphasized that the appearance of a protein on Table 1 does not indicate that a protein is not a Gag-interacting protein but only that our methodology was unable to confirm such a possibility.
After subtraction of background hits, a secondary normalization step was applied in an effort to account for transfection efficiencies. Here, spectral counts were normalized to the spectral counts for the BirA* ligase itself in a given transfection, and proteins with signals of less than 7% of the level of the the BirA* signals in both MA-BirA* and ΔMA-BirA* samples were discarded as being potentially too close to false-discovery signals (see Materials and Methods). After this step, we obtained a list of over 50 proteins that were preferentially biotinylated in MA-BirA*-transfected cells and/or ΔMA-BirA*-transfected cells (Table 2). Proteins were ranked based on their ratios of representation in MA-BirA* samples divided by their representation in ΔMA-BirA* samples, but it is notable that because BirA* ligase spectral counts were lower in the MA versus the ΔMA samples, MA/ΔMA ratios are skewed to the high side, and the list includes a number of proteins that were identified in MA-BirA* but not ΔMA-BirA* transfections.
Interestingly, over half of the biotinylated proteins have been observed in HIV-1 particles, observed as binding partners with HIV-1 proteins, or implicated as HIV-1 interacting factors in genetic screens (Table 2, far right column) (22, 26, 35, 36, 42, 50, 67–80, 85). As can be seen, the list includes a variety of translation factors, cytoskeleton-associated proteins, membrane proteins, and RNA-processing proteins: these are discussed in more detail below. Consistent with the results presented in Fig. 3 and 5, Gag proteins were also biotinylated in both sets of samples, as were pol gene products. However, remarkably, the HIV-1 envelope protein was found to be biotinylated in ΔMA-BirA*-transfected cells but not in MA-BirA*-transfected cells (Table 2). Interestingly, all seven Env peptides biotinylated by the ΔMA-BirA* ligase mapped to the TM (gp41) CT (Table 2 legend). This observation was wholly unexpected, since multiple studies have demonstrated an interaction between MA and Env (20, 27–30, 48, 61). The failure of the biotin ligase encoded by MA-BirA* to biotinylate Env was not due to a defect in Env protein expression, because the Env transmembrane (TM; gp41) protein was readily detected in MA-BirA* virions, whereas it was not detected in ΔMA-BirA* proteins (Fig. 2A). Alternative models for this unexpected observation are discussed below.
DISCUSSION
The HIV-1 PrGag proteins traffic to the PMs of infected cells, where they organize the assembly and budding of virus particles. The PrGag MA domains facilitate this process by binding to PI(4,5)P2-enriched regions of the PM and by fostering Env protein assembly into virus particles (3, 4, 6–11, 27–30, 32, 48, 61). Nevertheless, PrGag proteins with large MA deletions are still able to direct the assembly of infectious viruses, as long as they are pseudotyped with alternative envelope proteins or matched with CT-truncated HIV-1 Env proteins (30, 48). Previous studies also have shown that PrGag proteins tolerate protein insertions near the C terminus of MA, at least for the purposes of virus particle assembly (14, 51, 52).
We have taken advantage of relative resilience of the matrix coding region with respect to genetic perturbation by tagging WT and ΔMA PrGag proteins with the promiscuous BirA* biotin ligase; this has afforded us the opportunity to biotinylate proteins that are in close proximity (53, 55) of the PrGag proteins during the translation, transport, and assembly stages of the viral life cycle, with the caveat that our tagged viruses were poorly infectious. Our results clearly show that MA-BirA* and ΔMA-BirA* PrGag proteins have the capacity of biotinylating the PrGag proteins and pol gene-derived proteins, presumably in cis (Fig. 3 and 5 and Table 2). Evidence also demonstrated that at least the MA-BirA* PrGag protein could biotinylate WT PrGag proteins in trans (Fig. 5). Because WT PrGag proteins facilitated the release of ΔMA-BirA* PrGag proteins from cotransfected cells (Fig. 5), we assume that these proteins associate during transport and/or assembly as well. Perhaps our most surprising observation regarding the biotinylation of viral proteins is that the HIV-1 Env protein was biotinylated with ΔMA-BirA* but not with MA-BirA* (Table 2). Given that previous work has demonstrated an interaction between MA and the Env CT (27–30, 32, 48, 61), this is exactly the opposite of what we expected. We do not know which Env residues are biotinylated by the ΔMA-BirA* ligase, but the sequenced peptides represented TM (gp41) and not the surface envelope glycoprotein (SU) (gp120) (see footnote a of Table 2). To explain how ΔMA-BirA* proteins might biotinylate Env proteins, we conjecture that the two proteins are targeted independently to the same intracellular sites but that the two are unable to interact productively. Relevant to this hypothesis is that VSV G proteins in pseudotyped virions appeared to be biotinylated more efficiently with ΔMA-BirA* than with MA-BirA* proteins (Fig. 6); this may indicate that biotinylation of G proteins, which are passively assembled (20, 32–34) into virus particles, may be influenced by potential differences in intrinsic biotin ligase activities and/or accessibilities to the activated bioAMP species. Why do WT MA-BirA* proteins fail to biotinylate Env proteins to detectable levels? One possibility is that the PrMA-BirA* ligase is less active than the PrΔMA-BirA* ligase when coassembled with other PrMA-BirA* proteins. Alternatively, MA domains and Env CTs may form a lattice (29, 32) in which Env CT lysines are not readily accessible to the activated biotin species. If this is the case, it may be fruitful to examine Env TM biotinylation in the context of MA and Env CT mutations with known effects on MA-Env interactions (27–32).
In addition to analysis of viral protein biotinylation, the use of BirA*-tagged proteins has permitted the identification of cellular proteins that potentially interact with HIV-1 Gag proteins. We have identified over 50 such candidates, over half of which have been implicated as effectors of HIV-1 replication, as binding partners, or as present in virions (Table 2). We have categorized these roughly into seven classes (Table 3). By far, the most specifically biotinylated cellular protein was the cytoskeleton-associated protein, Filamin A. Previous research has shown that the PrGag CA domain is necessary for efficient binding to Filamin A and supports a model in which binding of PrGag to Filamin A induces a reorganization of the cortical actin network to permit PrGag delivery to the PM via actin filaments (77). Our MS results support this model, although we did observe a difference between the MA-BirA* and ΔMA-BirA* samples in the levels of Filamin A biotinylation; this may simply denote a relative preference of the ΔMA-BirA* PrGag protein for self-biotinylation versus biotinylation in trans or it may indicate that, although MA does not bind to Filamin A, it is partially responsible for trafficking PrGag proteins to Filamin A binding sites. Besides Filamin A, nine other cytoskeleton-associated proteins were also biotinylated in our study. Of these, desmoglein-2 interacts with intermediate filaments, while protein 4.1, cortactin, afadin, Filamin B, the CD2-associated protein (CD2AP), and band 4.1-like protein 2 all have been implicated as involved in actin cytoskeleton arrangements (26, 68, 72, 84); this presumably underscores the importance of actin filament and membrane reorganization during virus assembly and potentially identifies targets of virus assembly interference. It also may pertain to the regulation of budding, since cortactin and CD2AP bind ESCRT-associated proteins Alix and TSG101 (26, 86). One of the remaining two proteins in the cytoskeletal group, AHNAK, is recruited to cholesterol-rich domains at the PM by annexin 2, which plays a cell type-dependent role in the production of infectious HIV-1 (87, 88). The final member of this group, microtubule-associated protein 4 (MAP4), promotes microtubule assembly, and knockdown of this protein was shown to inhibit HIV-1 replication at a postentry, preintegration stage of replication (75). Because our BirA*-expressing viruses were poorly infectious (Fig. 2B), we assume that MAP4-interaction sites on incoming PICs are also present on newly translated PrGag proteins or that MAP4 may play an additional role in the assembly stage of HIV-1 replication. Similar considerations pertain to the nuclear transport-related TPR proteins and the lamina-associated protein (LAP2B; TMPO). Whereas recent studies have shown that multimeric CA binding sites on incoming PICs interact with nuclear import/pore proteins TNPO3, NUP153, and NUP358 (39–42), it is possible that LAP2B and TPR biotinylation reflects a Gag protein interaction at either the assembly stage or the entry stage of the HIV-1 life cycle.
TABLE 3.
Cellular biotinylated proteinsa
| Protein |
|---|
| Cytoskeletal |
| Filamin-A |
| Neuroblast differentiation-associated protein AHNAK |
| Microtubule-associated protein 4 |
| Protein 4.1 |
| Desmoglein-2 |
| Src substrate cortactin |
| Afadin |
| CD2-associated protein |
| Band 4.1-like protein 2 |
| Filamin-B |
| Membrane |
| Hornerin |
| 4F2 cell-surface antigen heavy chain |
| Sodium/potassium-transporting ATPase subunit alpha-1 |
| Neutral amino acid transporter B(0) |
| Hornerin |
| Lipid regulation |
| Fatty acid synthase |
| Vigilin |
| Nuclear transport |
| Nucleoprotein TPR |
| Lamina-associated polypeptide 2, isoforms beta/gamma |
| Translation |
| Elongation factor 1-alpha 1 |
| PERQ amino acid-rich with GYF domain-containing protein 2 |
| Eukaryotic translation initiation factor 4B |
| La-related protein 1 |
| Eukaryotic translation initiation factor 4 gamma 1 |
| Ubiquitin-60S ribosomal protein L40 |
| Bifunctional glutamate/proline-tRNA ligase |
| Eukaryotic translation initiation factor 5 |
| RNA processing |
| Zinc finger CCCH-type antiviral protein 1 |
| Ubiquitin-associated protein 2-like |
| Protein PRRC2C |
| Cold shock domain-containing protein E1 |
| Plasminogen activator inhibitor 1 RNA-binding protein |
| Ataxin-2-like protein |
| Protein PRRC2A |
| Ubiquitin-associated protein 2 |
| Gem-associated protein 5 |
| 5′–3′ exoribonuclease 1 |
| Nuclear fragile X mental retardation-interacting protein 2 |
| Heterogenous nuclear ribonucleoprotein M |
| ATP-dependent RNA helicase DDX3X |
| RNA-binding protein 26 |
| Orphans |
| T-complex protein 1 subunit theta |
| Band 4.1-like protein 3 |
| Insulin receptor substrate 4 |
| TOX high-mobility-group box family member 4 |
| DNA-dependent protein kinase catalytic subunit |
| Semenogelin-2 |
| Protein CDV3 homolog |
| Protein Lyric |
| Semenogelin-1 |
| Antigen KI-67 |
| Histone H1.2 |
| 28-kDa heat- and acid-stable phosphoprotein |
Observed cellular biotinylated proteins were roughly categorized into groups as cytoskeletal, membrane, lipid regulation, nuclear transport, translation, RNA processing, and orphan proteins. Within each group, lists are ordered based on summed observed detection signals (percent WT BirA* plus percent ΔMA BirA* from Table 2).
Table 3 also lists five membrane-associated proteins that were biotinylated. However, at this point, although the Na/K-transporting ATPase and the 4F2 protein have been found in virions (35), it is not yet clear whether either plays an active role in HIV-1 replication. Two additional proteins play roles in lipid regulation. Fatty acid synthase catalyzes the formation of long-chain fatty acids and has been found in HIV-1 particles (36). Vigilin, which was biotinylated by MA-BirA*, but not by ΔMA-BirA* (Table 2), regulates sterol metabolism and may play a role in protecting cells from cholesterol overaccumulation. This raises the intriguing possibility that MA may contribute to the observed HIV-1 upregulation of cholesterol biosynthesis (89) to supply enough cholesterol for the generation of infectious HIV-1 particles (1, 5, 8).
Two additional large groups of observed biotinylated proteins were translation factors and RNA-processing proteins (Table 3). In this regard, it is worthwhile to note the interconnections between translation and RNA-processing proteins. In particular, the destinations of viral and cellular RNAs include actively translating ribosomes; stress granules (SGs), where mRNAs on stalled preinitiation complexes are stored or sorted; and processing bodies (PBs), involved in RNA silencing and decay (22, 90–95). Previously, HIV-1 Gag proteins were found to associate with elongation factor 1 alpha, via RNA bridges to MA and NC, and this interaction was proposed to induce a shift from Gag protein translation to vRNA encapsidation (26). PrGag also was shown to associate with the Staufen-1 SG protein (22, 90, 91), which may foster both virus assembly and vRNA encapsidation. Our data show that several PB or SG proteins (22, 69, 90–92, 94), including translation initiation factors 4B and 4 gamma, the 5′-3′ XRN exonuclease, and the P2 nuclear fragmentation protein, were biotinylated. Interestingly, the sizes, locations, and numbers of streptavidin-stained but anti-CA-negative dots observed in Fig. 4 are consistent with their assignment as PBs or SGs, suggesting that components of these ribonucleoproteins (RNPs) migrated to RNPs after biotinylation. In principle, it seems likely that PrGag proteins bind to ribosomal, PB, or SG proteins to exploit or subvert their functions, as for encapsidation (16, 19, 91). However, associations may also represent host defense mechanisms against viral infection, as in the case of zinc finger CCCH antiviral protein (69). We believe that investigation of the potential roles of the previously and newly identified biotinylated translation and RNA-processing factors will be fruitful.
As shown in Table 3, a number of proteins have been classified as orphans, for convenience. Several of these already have been implicated as HIV-1-interacting proteins. For instance, histone H1.2 has been found in HIV-1 particles (36), and the DNA-dependent PK catalytic subunit has been found in Gag-positive, Staufen-1-positive RNPs (22). Recently, the protein Lyric was shown to bind to PrGag proteins and to be incorporated into virions, but the mechanism by which it might regulate virus infectivity will require further investigation (79). Further investigation also appears warranted for other orphan or otherwise-classified biotinylated proteins that we have identified. We suggest that further use of BirA*-tagged WT and mutant HIV-1 proteins will be an interesting approach to examine potential virus-host interactions. Moreover, it may be possible to adopt this method for the identification of the exposed lysines that are biotinylated on target proteins, which would afford the opportunity to probe protein structures in vivo.
ACKNOWLEDGMENTS
We are grateful to Ayna Alfadhli, Scott Landfear, and Claudia Lopez for assistance and advice and to Rachel Sloan for help in training and assistance. Mass spectrometry was performed through the OHSU Proteomics Shared Resource, and we are particularly grateful to Larry David, John Klimek, and Ashok Reddy for their invaluable help.
Our research was supported by NIH grants R01 GM060170 and R01 GM101983 and by OHSU School of Medicine Research Core Initiative funding.
REFERENCES
- 1.Brügger B, Glass B, Haberkant P, Leibrecht I, Wieland F, Krausslich H-G. 2006. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci U S A 103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant M, Ratner L. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A 87:523–527. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. 2008. Interaction between HIV-1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag-membrane binding. J Virol 82:2405–2417. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A 107:1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham D, Chertova E, Hilburn J, Arthur L, Hildreth J. 2003. Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with beta-cyclodextrin inactivates and permeabilizes the virions: evidence for virion-associated lipid rafts. J Virol 77:8237–8248. doi: 10.1128/JVI.77.15.8237-8248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol(4,5)bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ono A, Freed EO. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A 98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ono A, Waheed A, Freed EO. 2007. Depletion of cellular cholesterol inhibits membrane binding and higher-order multimerization of human immunodeficiency virus type 1 Gag. Virology 360:27–35. doi: 10.1016/j.virol.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alfadhli A, Still A, Barklis E. 2009. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol 83:12196–12203. doi: 10.1128/JVI.01197-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alfadhli A, McNett H, Eccles J, Tsagli S, Noviello C, Sloan R, Lopez C, Peyton D, Barklis E. 2013. Analysis of small molecule ligands targeting the HIV-1 matrix protein-RNA binding site. J Biol Chem 288:666–676. doi: 10.1074/jbc.M112.399865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alfadhli A, McNett H, Tsagli S, Bachinger HP, Peyton DH, Barklis E. 2011. HIV-1 matrix protein binding to RNA. J Mol Biol 410:653–666. doi: 10.1016/j.jmb.2011.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A 103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones CP, Datta SA, Rein A, Rouzina I, Musier-Forsyth K. 2011. Matrix domain modulates HIV-1 Gag's nucleic acid chaperone activity via inositol phosphate binding. J Virol 85:1594–1603. doi: 10.1128/JVI.01809-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kutluay S, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, Bieniasz P. 2014. Global changes in the RNA binding specificity of HIV-1 Gag regulate virion genesis. Cell 159:1096–1109. doi: 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aldovini A, Young RA. 1990. Mutations of RNA and protein sequences involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J Virol 64:1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berkowitz R, Fisher J, Goff SP. 1996. RNA packaging. Curr Top Microbiol Immunol 214:177–218. [DOI] [PubMed] [Google Scholar]
- 17.Buckman J, Bosche W, Gorelick R. 2003. Human immunodeficiency virus type 1 nucleocapsid Zn2+ fingers are required for efficient reverse transcription, initial integration processes, and protection of newly synthesized viral DNA. J Virol 77:1469–1480. doi: 10.1128/JVI.77.2.1469-1480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorelick R, Chabot D, Rein A, Henderson L, Arthur LO. 1993. The two zinc fingers in the human immunodeficiency virus type 1 nucleocapsid protein are not functionally equivalent. J Virol 67:4027–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaye J, Lever A. 1996. Trans-acting proteins involved in RNA encapsidation and viral assembly in human immunodeficiency virus type 1. J Virol 70:880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swanstrom R, Wills JW. 1997. Synthesis, assembly, and processing of viral proteins, p 263–334 InCoffin J, Hughes S, Varmus H (ed), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- 21.Garrus J, von Schwedler U, Pornillos O, Morham S, Zavitz K, Wang H, Wettstein D, Stray K, Côté M, Rich R, Myszka D, Sundquist W. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65. doi: 10.1016/S0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 22.Milev M, Ravichandran M, Khan M, Schriemer D, Mouland A. 2012. Characterization of staufen1 ribonucleoproteins by mass spectrometry and biochemical analyses reveal the presence of diverse host proteins associated with human immunodeficiency virus type 1. Front Microbiol 3:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strack B, Calistri A, Craig S, Popova E, Gottlinger H. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699. doi: 10.1016/S0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 24.VerPlank L, Bouamr F, LaGrassa T, Agresta B, Kikonyogo A, Leis J, Carter C. 2001. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc Natl Acad Sci U S A 98:7724–7729. doi: 10.1073/pnas.131059198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Schwedler U, Stuchell M, Müller B, Ward D, Chung H, Morita E, Wang H, Davis T, He G, Cimbora D, Scott A, Kräusslich H-G, Kaplan J, Morham S, Sundquist W. 2003. The protein network of HIV budding. Cell 114:701–713. doi: 10.1016/S0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe S, Chen M-H, Khan M, Ehrlich L, Kemal KS, Weiser B, Shi B, Chen C, Powell M, Anastos K, Burger H, Carter CA. 2013. The S40 residue in HIV-1 Gag p6 impacts local and distal budding determinants, revealing additional late domain activities. Retrovirology 10:143. doi: 10.1186/1742-4690-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freed EO, Martin MA. 1996. Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J Virol 70:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murakami T, Freed EO. 2000. Genetic evidence for an interaction between human immunodeficiency virus type 1 matrix and α-helix 2 of the gp41 cytoplasmic tail. J Virol 74:3548–3554. doi: 10.1128/JVI.74.8.3548-3554.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tedbury PR, Ablan SD, Freed EO. 2013. Global rescue of defects in HIV-1 envelope glycoprotein incorporation: implications for matrix structure. PLoS Pathog 9:e1003739. doi: 10.1371/journal.ppat.1003739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, Zhang Y, McDermott J, Barklis E. 1993. Conditional infectivity of a human immunodeficiency virus matrix domain deletion mutant. J Virol 67:7067–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, Yuan X, Matsuda Z, Lee TH, Essex M. 1992. The matrix protein of human immunodeficiency virus type 1 is required for incorporation of viral envelope protein into mature virions. J Virol 66:4966–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Checkley MA, Luttge B, Freed EO. 2011. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 410:582–608. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burns JC, Friedmann T, Driever W, Burrascano M, Yee J-K. 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci U S A 90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgenson RL, Vogt VM, Johnson MC. 2009. Foreign glycoproteins can be actively recruited to virus assembly sites during pseudotyping. J Virol 83:4060–4067. doi: 10.1128/JVI.02425-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ott D. 2008. Cellular proteins detected in HIV-1. Rev Med Virol 18:159–175 http://ncifrederick.cancer.gov/research/avp/protein_db.asp. doi: 10.1002/rmv.570. [DOI] [PubMed] [Google Scholar]
- 36.Santos S, Obukhov Y, Nekhai S, Bukrinsky M, Iordanskiy S. 2012. Virus-producing cells determine the host protein profiles of HIV-1 virion cores. Retrovirology 9:65. doi: 10.1186/1742-4690-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berthoux L, Pechoux C, Ottmann M, Morel G, Darlix JL. 1997. Mutations in the N-terminal domain of human immunodeficiency virus type 1 nucleocapsid protein affect virion core structure and proviral DNA synthesis. J Virol 71:6973–6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bukrinsky MI, Haggerty S, Dempsey MP, Sharova N, Adzhubel A, Spitz L, Lewis P, Goldfarb D, Emerman M, Stevenson M. 1993. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature 365:666–669. doi: 10.1038/365666a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamorte L, Titolo S, Lemke C, Goudreau N, Mercier J, Wardrop E, Shah V, von Schwedler U, Langelier C, Banik S, Aiken C, Sundquist W, Mason S. 2013. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob Agents Chemother 57:4622–4631. doi: 10.1128/AAC.00985-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee K, Ambrose Z, Martin T, Oztop I, Mulky A, Julias J, Vandegraaff N, Baumann J, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman D, Coffin J, Hughes S, Unutmaz D, Engelman A, KewalRamani V. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monette A, Panté N, Mouland A. 2011. HIV-1 remodels the nuclear pore complex. J Cell Biol 193:619–631. doi: 10.1083/jcb.201008064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price A, Jacques D, McEwan W, Fletcher A, Essig S, Chin J, Halambage U, Aiken C, James L. 2014. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog 10:e1004459. doi: 10.1371/journal.ppat.1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goujon C, Moncorgé O, Bauby H, Doyle T, Ward C, Schaller T, Hué S, Barclay W, Schulz R, Malim M. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kane M, Yadav S, Bitzegeio J, Kutluay S, Zang T, Wilson S, Schoggins J, Rice C, Yamashita M, Hatziioannou T, Bieniasz P. 2013. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, Liang C. 2013. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 46.Malim M, Bieniasz P. 2012. HIV restriction factors and mechanisms of evasion. Cold Spring Harb Perspect Med 2:a006940. doi: 10.1101/cshperspect.a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohmine S, Sakuma R, Sakuma T, Thatava T, Takeuchi H, Ikeda Y. 2011. The antiviral spectra of TRIM5α orthologues and human TRIM family proteins against lentiviral production. PLoS One 6:e16121. doi: 10.1371/journal.pone.0016121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reil H, Bukovsky AA, Gelderblom HR, Gottlinger HG. 1998. Efficient HIV-1 replication can occur in the absence of the viral matrix protein. EMBO J 17:2699–2708. doi: 10.1093/emboj/17.9.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scholz I, Arvidson B, Huseby D, Barklis E. 2005. Virus particle core defects caused by mutations in the human immunodeficiency virus capsid N-terminal domain. J Virol 79:1470–1479. doi: 10.1128/JVI.79.3.1470-1479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engeland C, Brown N, Börner K, Schümann M, Krause E, Kaderali L, Müller G, Kräusslich HG. 2014. Proteome analysis of the HIV-1 Gag interactome. Virology 460–461:194–206. doi: 10.1016/j.virol.2014.04.038. [DOI] [PubMed] [Google Scholar]
- 51.Müller B, Daecke J, Fackler O, Dittmar M, Zentgraf H, Kräusslich H-G. 2004. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J Virol 78:10803–10813. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scholz I, Still A, Dhenub TC, Coday K, Webb M, Barklis E. 2008. Analysis of human immunodeficiency virus matrix domain replacements. Virology 371:322–335. doi: 10.1016/j.virol.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim D, Birendra K, Zhu W, Motamedchaboki K, Doye V, Roux K. 2014. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci U S A 111:E2453-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mechold U, Gilbert C, Ogryzko V. 2005. Codon optimization of the BirA enzyme gene leads to higher expression and an improved efficiency of biotinylation of target proteins in mammalian cells. J Biotechnol 116:245–249. doi: 10.1016/j.jbiotec.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 55.Roux K, Kim D, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adachi A, Gendelman H, Koenig S, Folks T, Willey R, Rabson A, Martin M. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noviello C, López C, Kukull B, McNett H, Still A, Eccles J, Sloan R, Barklis E. 2011. Second-site compensatory mutations of HIV-1 capsid mutations. J Virol 85:4730–4738. doi: 10.1128/JVI.00099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Barklis E. 1997. Effects of nucleocapsid mutations on human immunodeficiency virus assembly and RNA encapsidation. J Virol 71:6765–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DuBridge R, Tang P, Hsia H, Leong P, Miller J, Calos M. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol 7:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.López C, Tsagli S, Sloan R, Eccles J, Barklis E. 2013. Second site reversion of a mutation near the amino terminus of the HIV-1 capsid protein. Virology 447:95–103. doi: 10.1016/j.virol.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.López C, Sloan R, Cylinder I, Kozak S, Kabat D, Barklis E. 2014. RRE-dependent HIV-1 Env RNA effects on Gag protein expression, assembly and release. Virology 462–463:126–134. doi: 10.1016/j.virol.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adler J, Parmryd I. 2010. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A 77:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 64.Shevchenko A, Tomas H, Havli J, Olsen J, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 65.Wilmarth P, Riviere M, David LL. 2009. Techniques for accurate protein identification in shotgun proteomic studies of human, mouse, bovine, and chicken lenses. J Ocul Biol Dis Infor 2:223–234. doi: 10.1007/s12177-009-9042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keller A, Nesvizhskii A, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 67.Bushman F, Malani N, Fernandes J, D'Orso I, Cagney G, Diamond T, Zhou H, Hazuda D, Espeseth A, König R, Bandyopadhyay S, Ideker T, Goff S, Krogan N, Frankel A, Young J, Chanda S. 2009. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog May 5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shoeman RL, Hartig R, Hauses C, Traub P. 2002. Organization of focal adhesion plaques is disrupted by action of die HIV-1 protease. Cell Biol Int 26:529–539. doi: 10.1006/cbir.2002.0895. [DOI] [PubMed] [Google Scholar]
- 69.Ptak R, Fu W, Sanders-Beer B, Dickerson J, Pinney J, Robertson D, Rozanov M, Katz K, Maglott D, Pruitt K, Dieffenbach C. 2008. Cataloguing the HIV-1 human protein interaction network. AIDS Res Hum Retroviruses 24:1497–1502 http://www.ncbi.nlm.nih.gov/genome/viruses/retroviruses/hiv-1/interactions/. doi: 10.1089/aid.2008.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoque M, Hanauske-Abel H, Palumbo P, Saxena D, Gandolfi DD, Park MH, Pe'ery T, Mathews MB. 2009. Inhibition of HIV-1 gene expression by Ciclopirox and Deferiprone, drugs that prevent hypusination of eukaryotic initiation factor 5A. Retrovirology 6:90. doi: 10.1186/1742-4690-6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jäger S, Cimermancic P, Gulbahce N, Johnson J, McGovern K, Clarke S, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang G, Roth S, Akiva E, Marlett J, Stephens M, D'Orso I, Fernandes J, Fahey M, Mahon C, O'Donoghue A, Todorovic A, Morris J, Maltby D, Alber T, Cagney G, Bushman F, Young J, Chanda S, Sundquist W, Kortemme T, Hernandez R, Craik C, Burlingame A, Sali A, Frankel A, Krogan N. 2012. Global landscape of HIV-human protein complexes. Nature 481:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yeung ML, Houzet L, Venkat S, Yedavalli RK, Jeang K-T. 2009. A genome-wide short hairpin RNA screening of Jurkat T-cells for human proteins contributing to productive HIV-1 replication. J Biol Chem 284:19463–19473. doi: 10.1074/jbc.M109.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chable-Bessia C, Meziane O, Latreille D, Triboulet R, Zamborlini A, Wagschal A, Jacquet J-M, Reynes J, Levy Y, Saib A, Bennasser Y, Benkirane M. 2009. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 6:26. doi: 10.1186/1742-4690-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brass A, Dykxhoorn D, Benita Y, Yan N, Engelman A, Xavier R, Lieberman J, Elledge S. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 75.Gallo D, Hope T. 2012. Knockdown of MAP4 and DNAL1 produces a post-fusion and pre-nuclear translocation impairment in HIV-1 replication. Virology 422:13–21. doi: 10.1016/j.virol.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.König R, Zhou Y, Elleder D, Diamond T, Bonamy G, Irelan J, Chiang C, Tu B, De Jesus P, Lilley C, Seidel S, Opaluch A, Caldwell J, Weitzman M, Kuhen K, Bandyopadhyay S, Ideker T, Orth A, Miraglia L, Bushman F, Young J, Chanda S. 2008. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cooper J, Liu L, Woodruff EA, Taylor HE, Goodwin JS, D'Aquila RT, Spearman P, Hildreth JE, Dong X. 2011. Filamin A protein interacts with human immunodeficiency virus type 1 Gag protein and contributes to productive particle assembly. J Biol Chem 286:28498–28510. doi: 10.1074/jbc.M111.239053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cimarelli A, Luban J. 1999. Translation elongation factor 1-alpha interacts specifically with the human immunodeficiency virus type 1 Gag polyprotein. J Virol 73:5388–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Engeland C, Oberwinkler H, Schümann M, Krause E, Müller G, Kräusslich H-G. 2011. The cellular protein lyric interacts with HIV-1 Gag. J Virol 85:13322–13332. doi: 10.1128/JVI.00174-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Impens F, Timmerman E, Staes A, Moens K, Ariën K, Verhasselt B, Vandekerckhove J, Gevaert K. 2012. A catalogue of putative HIV-1 protease host cell substrates. Biol Chem 393:915–931. doi: 10.1515/hsz-2012-0168. [DOI] [PubMed] [Google Scholar]
- 81.Ingaramo M, Beckett D. 2012. Selectivity in post-translational biotin addition to five human carboxylases. J Biol Chem 287:1813–1822. doi: 10.1074/jbc.M111.275982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abu-Elheiga L, Brinkley W, Zhong L, Chirala S, Woldegiorgis G, Wakil S. 2000. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci U S A 97:1444–1449. doi: 10.1073/pnas.97.4.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fäcke M, Janetzko A, Shoeman R, Krausslich H-G. 1993. A large deletion in the matrix domain of the human immunodeficiency virus gag gene redirects virus particle assembly from the plasma membrane to the endoplasmic reticulum. J Virol 67:4972–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jolly C, Mitar I, Sattentau Q. 2007. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J Virol 81:5547–5560. doi: 10.1128/JVI.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou H, Xu M, Huang Q, Gates A, Zhang X, Castle J, Stec E, Ferrer M, Strulovici B, Hazuda D, Espeseth A. 2008. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4:495. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 86.Morita E, Sandrin V, Chung H, Morham S, Gygi S, Rodesch C, Sundquist W. 2007. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J 26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benaud C, Gentil BJ, Assard N, Court M, Garin J, Delphin C, Baudier J. 2004. AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J Cell Biol 164:133–144. doi: 10.1083/jcb.200307098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rai T, Mosoian A, Resh M. 2010. Annexin 2 is not required for human immunodeficiency virus type 1 particle production but plays a cell type-dependent role in regulating infectivity. J Virol 84:9783–9792. doi: 10.1128/JVI.01584-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van't Wout A, Swain JV, Schindler M, Rao U, Pathmejeyan M, Mullins J, Kirchhoff F. 2005. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1-infected T cells. J Virol 79:10053–10058. doi: 10.1128/JVI.79.15.10053-10058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abrahamyan L, Chatel-Chaix L, Ajamian L, Milev M, Monette A, Clément J, Song R, Lehmann M, DesGroseillers L, Laughrea M, Boccaccio G, Mouland A. 2010. Novel Staufen1 ribonucleoproteins prevent formation of stress granules but favour encapsidation of HIV-1 genomic RNA. J Cell Sci 123:369–383. doi: 10.1242/jcs.055897. [DOI] [PubMed] [Google Scholar]
- 91.Chatel-Chaix L, Clément J, Martel C, Bériault V, Gatignol A, DesGroseillers L, Mouland A. 2004. Identification of Staufen in the human immunodeficiency virus type 1 Gag ribonucleoprotein complex and a role in generating infectious viral particles. Mol Cell Biol 24:2637–2648. doi: 10.1128/MCB.24.7.2637-2648.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chatel-Chaix L, Germain M-A, Motorina A, Bonneil E, Thibault P, Baril M, Lamarre D. 2013. A host YB-1 ribonucleoprotein complex is hijacked by hepatitis C virus for the control of NS3-dependent particle production. J Virol 87:11704–11720. doi: 10.1128/JVI.01474-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen A, Sengupta P, Waki K, Van Engelenburg S, Ochiya T, Ablan S, Freed EO, Lippincott-Schwartz J. 2014. MicroRNA binding to the HIV-1 Gag protein inhibits Gag assembly and virus production. Proc Natl Acad Sci U S A 111:E2676-83. doi: 10.1073/pnas.1408037111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lorgeoux RP, Guo F, Liang C. 2012. From promoting to inhibiting: diverse roles of helicases in HIV-1 replication. Retrovirology 9:79. doi: 10.1186/1742-4690-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reed J, Molter B, Geary C, McNevin J, McElrath J, Giri S, Klein K, Lingappa J. 2012. HIV-1 Gag co-opts a cellular complex containing DDX6, a helicase that facilitates capsid assembly. J Cell Biol 198:439–456. doi: 10.1083/jcb.201111012. [DOI] [PMC free article] [PubMed] [Google Scholar]