

ABSTRACT
The RNA genome of respiratory syncytial virus (RSV) is constitutively encapsidated by the viral nucleoprotein N, thus forming a helical nucleocapsid. Polymerization of N along the genomic and antigenomic RNAs is concomitant to replication and requires the preservation of an unassembled monomeric nucleoprotein pool. To this end, and by analogy with Paramyxoviridae and Rhabdoviridae, it is expected that the viral phosphoprotein P acts as a chaperone protein, forming a soluble complex with the RNA-free form of N (N0-P complex). Here, we have engineered a mutant form of N that is monomeric, is unable to bind RNA, still interacts with P, and could thus mimic the N0 monomer. We used this N mutant, designated Nmono, as a substitute for N0 in order to characterize the P regions involved in the N0-P complex formation. Using a series of P fragments, we determined by glutathione S-transferase (GST) pulldown assays that the N and C termini of P are able to interact with Nmono. We analyzed the functional role of amino-terminal residues of P by site-directed mutagenesis, using an RSV polymerase activity assay based on a human RSV minireplicon, and found that several residues were critical for viral RNA synthesis. Using GST pulldown and surface plasmon resonance assays, we showed that these critical residues are involved in the interaction between P[1-40] peptide and Nmono in vitro. Finally, we showed that overexpression of the peptide P[1-29] can inhibit the polymerase activity in the context of the RSV minireplicon, thus demonstrating that targeting the N0-P interaction could constitute a potential antiviral strategy.
IMPORTANCE Respiratory syncytial virus (RSV) is the leading cause of lower respiratory tract illness in infants. Since no vaccine or efficient antiviral treatment is available against RSV, it is essential to better understand how the viral machinery functions in order to develop new antiviral strategies. RSV phosphoprotein P, the main RNA polymerase cofactor, is believed to function as a chaperon protein, maintaining N as a nonassembled, RNA-free protein (N0) competent for RNA encapsidation. In this paper, we provide the first evidence, to our knowledge, that the N terminus of P contains a domain that binds specifically to this RNA-free form of N. We further show that overexpression of a small peptide spanning this region of P can inhibit viral RNA synthesis. These findings extend our understanding of the function of RSV RNA polymerase and point to a new target for the development of drugs against this virus.
INTRODUCTION
The human respiratory syncytial virus (HRSV) is the leading cause of severe respiratory tract infections in newborn children worldwide (1). HRSV infects close to 100% of infants within the first 2 years of life and is the main cause of bronchiolitis. It is also recognized as a significant cause of severe respiratory infections in the elderly. The virus belongs to the Mononegavirales order and constitutes the prototype virus of the Pneumovirus genus of the Paramyxoviridae family. As for all the viruses belonging to the Mononegavirales order, its genome consists of a nonsegmented negative-strand RNA that is bundled by the nucleoprotein (N) within a helical nucleocapsid (NC) (2) and is thus protected from RNases or cellular sensors involved in antiviral responses during infection. This ribonucleoprotein complex constitutes the template for viral transcription and replication by the viral RNA-dependent RNA polymerase (RdRp) (reviewed in reference 3). Two main consequences ensue from this specific protection of the viral genome. First, the NC structure impairs the direct accessibility of the RNA to the viral polymerase (L). Although the viral phosphoprotein (P) acts as the main cofactor of the L polymerase, allowing the L/P complex to bind to NC through specific P-N interactions, the strategy developed to gain access to the viral genome still remains unclear. However, a recent study revealed that the N-terminal domain of P of mumps virus (PNTD) binds the NC and should induce timely uncoiling of the helical NC (4). Second, protection of the viral genome requires a continuous supply of nucleoprotein during replication to encapsidate neosynthesized single-stranded genomic RNA (−RNA) and antigenomic RNA (+RNA). This suggests that N has to be prevented from binding to cellular RNAs but also from self-oligomerizing. To this avail, and by analogy with other paramyxoviruses and rhabdoviruses, the RSV P protein is thought to play the role of a chaperone protein to maintain N in an unassembled and RNA-free form named N0, the capsid protein protomer (5). It is also expected that N0-P interaction involves domain(s) of P different from those involved in the NC-P interaction and that a specific N0 binding domain exists in the N-terminal region of RSV P. Recently, bioinformatics analysis of Mononegavirales P proteins revealed the presence of a motif within the first 40 amino acids (aa) of RSV P that could be involved in the specific interaction with the N0 protein (6).
For RSV, an N0-P complex had not yet been isolated and characterized. The RSV P protein is composed of 241 amino acids and constitutes the shortest P protein among paramyxoviruses. Human RSV P (strain Long) is phosphorylated mainly at S232 (7, 8), although other minor phosphorylation sites have been identified (S30, S39, S45, T46, S54, T108, S116, S117, S119, S237) (9, 10). The precise role of phosphorylation for P activity remains unclear, since (i) unphosphorylated P is competent for oligomerization and binding to N-RNA (11, 12) and (ii) substitution of all the phosphorylated residues has little effect on viral transcription and replication (13, 14). However, P phosphorylation could play a role in NC encapsidation in viral particles (10, 13). Although there is no sequence similarity between the P proteins of Pneumovirinae and other related families and subfamilies in the Mononegavirales order, all share similar organization, structure, and functions. RSV P forms homotetramers of elongated shape and has a modular organization with a protease-resistant central domain containing a predicted coiled coil (aa 120 to 160) involved in oligomerization, flanked by two intrinsically disordered regions (aa 1 to 120 and 161 to 241) (11, 15–17). The C-terminal region of P (aa 231 to 241) is involved in the interaction with the NC (12, 18). Some experiments performed with the close homolog bovine RSV (BRSV) P protein have suggested that the aa 161 to 180 region could also participate in N-P interactions (19) and that the N terminus of P (residues 1 to 40) interacts with N (20).
Here, our aim was to determine and characterize the region(s) of P capable of binding to N0 and involved in the formation of a N0-P complex. We used the crystallographic structure of the decameric N-RNA rings (21) to rationally design N mutations to disrupt the interaction with RNA and thus destabilize the oligomeric organization of N-RNA rings. This work resulted in the purification of a recombinant double mutant NK170A/R185A protein that (i) does not interact with nucleic acids, (ii) is monomeric, and (iii) still interacts with the P protein. We showed that the N-terminal residues (aa 1 to 29) of P are sufficient to bind to the monomeric N, and we identified residues of P critical for this interaction. Our results strongly suggest that the P binding domain on N0 is a molecular recognition element (MoRE). Furthermore, using an RSV minireplicon, we found that overexpression of the peptide P[1-29] inhibits viral RNA synthesis.
MATERIALS AND METHODS
Plasmid constructions.
All the viral sequences were derived from the human RSV strain Long, ATCC VR-26 (GenBank accession number AY911262.1). The full-length P gene, the sequences of P with N-terminal deletions, or internal domains of P were PCR amplified using Pfu DNA polymerase (Stratagene, Les Ulis, France) (primer sequences available on request) and cloned into pGEX-4T-3 at BamHI-XhoI sites to engineer the pGEX-P plasmids. The C-terminal deletion mutants of P were obtained by introducing stop codons at the appropriate site in the coding sequence of pGEX-P, and point mutations K170A and R185A were introduced into pET-N-His by site-directed mutagenesis, using the Quikchange site-directed mutagenesis kit (Stratagene). pET-N and pET-NK170A/R185A were used to produce N-derived proteins with a C-terminal poly-His tag. Sequence analysis was carried out to check the integrity of all the constructions.
Plasmids for eukaryotic expression of the HRSV proteins N, P, M2-1, and L, designated pN, pP, pM2-1, and pL, have been described previously (12, 22). The pM/Luc subgenomic replicon, which contains the firefly luciferase (Luc) gene under the control of the M/SH gene start sequence was derived from the pM/SH subgenomic replicon (23) and has been described previously (24). The replication-defective minigenome pMΔ/Luc was constructed by deleting 24 nucleotides from the trailer region of the plasmid pM/Luc (25, 26). Point mutations were introduced in pP by site-directed mutagenesis as described above. The plasmid pP[1-29] was generated by replacing residue 30 of P by a stop codon. The sequence coding for mCherry was fused in frame upstream of P[1-29] in order to generate the plasmid pmCherry-P[1-29].
Antibodies.
The following primary antibodies were used for immunoblotting: a mouse monoclonal anti-N protein (Serotec, Oxford, United Kingdom), rabbit anti-P and anti-N antisera, previously described (11), and a mouse monoclonal anti-β-tubulin (Sigma). Secondary antibodies directed against mouse and rabbit IgG coupled to horseradish peroxidase (HRP; P.A.R.I.S., Compiègne, France) were used for immunoblotting experiments.
Expression and purification of recombinant proteins.
Escherichia coli BL21(DE3) bacteria (Novagen, Madison, WI) transformed with pGEX-P fragment plasmids were grown at 37°C for 8 h in 100 ml of Luria-Bertani (LB) medium containing 100 μg/ml ampicillin. Bacteria transformed with pET-N-derived plasmids alone or together with pGEX-P-derived plasmids were grown in LB medium containing kanamycin (50 μg/ml) or ampicillin and kanamycin, respectively. The same volume of LB was then added, and protein expression was induced by adding 80 μg/ml isopropyl-β-d-thio-galactoside (IPTG) to the medium. The bacteria were incubated for 15 h at 28°C and then harvested by centrifugation. For GST fusion protein purification, bacterial pellets were resuspended in lysis buffer (50 mM Tris-HCl [pH 7.8], 60 mM NaCl, 1 mM EDTA, 2 mM dithiothreitol [DTT], 0.2% Triton X-100, 1 mg/ml lysozyme) supplemented with complete protease inhibitor cocktail (Roche, Mannheim, Germany), incubated for 1 h on ice, sonicated, and centrifuged at 4°C for 30 min at 10,000 × g. Glutathione-Sepharose 4B beads (GE Healthcare, Uppsala, Sweden) were added to clarified supernatants and incubated at 4°C for 15 h. Beads were then washed two times in lysis buffer and three times in 1× phosphate-buffered saline (PBS) and stored at 4°C in an equal volume of PBS. To isolate GST-free P[1-40]-NK170AR185 complex, beads containing bound complex were incubated with biotinylated thrombin (Novagen) for 16 h at 20°C. Thrombin was then removed using the cleavage capture kit according to the manufacturer's instructions (Novagen). For purification of 6×His fusion proteins, bacterial pellets were resuspended in lysis buffer (20 mM Tris-HCl [pH 8], 500 mM NaCl, 0.1% Triton X-100, 10 mM imidazole, 1 mg/ml lysozyme) supplemented with complete protease inhibitor cocktail (Roche). After sonication and centrifugation, lysates were incubated for 1 h with chelating Sepharose Fast Flow beads charged with Ni2+ (GE Healthcare). Finally, beads were successively washed in the washing buffer (20 mM Tris-HCl, pH 8, 500 mM NaCl) containing increasing concentrations of imidazole (25, 50, and 100 mM), and proteins were eluted in the same buffer with 500 mM imidazole. Purified recombinant N proteins were loaded onto a Sephacryl S-200 HR 16/30 column (GE Healthcare) and eluted in 20 mM Tris-HCl (pH 8.5)–150 mM NaCl. The presence of RNA was determined by measuring the optical density at 200 nm (OD260)/OD280 nm absorption ratio, which is 2 for pure RNA and 0.57 for nucleic acid-free proteins.
Cross-linking analysis.
Purified recombinant N proteins were dialyzed against 20 mM HEPES (pH 8.5)–150 mM NaCl and centrifuged for 10 min at 100,000 × g. Samples containing 5 μg of protein were incubated with increasing amounts of ethylene glycol disuccinate (EGS) for 30 min at 20°C. Reactions were stopped by the addition of glycine to a final concentration of 50 mM. The cross-linked products were analyzed by SDS-PAGE and silver staining.
Pulldown assays.
GST or GST-P fragment fusion proteins fixed on beads were incubated in the presence of 30 μg of purified recombinant N proteins, in a final volume of 200 μl in 20 mM Tris (pH 8.5)–50 mM NaCl. After overnight incubation under agitation at 4°C, beads were extensively washed with 20 mM Tris (pH 8.5)–50 mM NaCl, boiled in 30 μl Laemmli buffer, and analyzed by SDS-PAGE and Coomassie blue staining.
Cell culture and transfections.
BHK-21 cells (clone BSRT7/5) constitutively expressing the T7 RNA polymerase (27) were grown in Dulbecco modified essential medium (Lonza, Cologne, Germany) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, and antibiotics. Cells were transfected using Lipofectamine 2000 (Invitrogen, Cergy-Pontoise, France) as described by the manufacturer.
Minigenome replication assay (minireplicon).
Cells at 90% confluence in 24-well dishes were transfected with Lipofectamine 2000 (Invitrogen) with a plasmid mixture containing 0.25 μg of pM/Luc, 0.25 μg of pN, 0.25 μg of pP, 0.125 μg of pL, and 62.5 ng of pM2-1 (24) as well as 62.5 ng of pRSV-β-Gal (where β-Gal is β-galactosidase; Promega) to normalize transfection efficiencies. Transfections were done in duplicate, and each independent transfection was performed three times. Cells were harvested 24 h posttransfection and then lysed in luciferase lysis buffer (30 mM Tris [pH 7.9], 10 mM MgCl2, 1 mM DTT, 1% Triton X-100, and 15% glycerol). Luciferase activities were determined for each cell lysate with an Anthos Lucy 3 luminometer (Bio Advance, Bussy Saint Martin, France) and normalized based on β-Gal expression.
Isothermal titration calorimetry assays.
Thermodynamic analysis of the P-N (wild type and NK170A/R185A) interactions was performed with a MicroCal ITC200 microcalorimeter (Microcal, Northampton, MA) at 20°C. For the interaction between wild-type N and P, samples were dialyzed against 20 mM Tris-HCl (pH 8.5)–150 mM NaCl. The P concentration in the microcalorimeter cell (200 μl) was 60 μM, and a total of 20 injections of 2 μl of N solution (concentration, 300 μM) were carried out at 180-s intervals, with stirring at 1,000 rpm. For the interaction between NK170A/R185A and P, samples were dialyzed against 20 mM Tris (pH 8.5)–150 mM NaCl–5% glycerol. The NK170A/R185A concentration in the microcalorimeter cell was 38 μM, and a total of 20 injections of 2 μl of P solution (concentration, 255 μM) were carried out under the same conditions as above.
The raw data were integrated to generate curves in which the areas under the injection peaks were plotted against the ratio of injected sample to cell content. The data were analyzed according to the one-binding-site model using the MicroCal Origin software provided by the manufacturer. Changes in the free energy and entropy upon binding were calculated from determined equilibrium parameters using the following equation: −RT ln (Ka) = ΔG = ΔH − TΔS, where R is the universal gas constant, T is the temperature in kelvins, Ka is the association constant, ΔG is the change in Gibbs free energy, ΔH is the change in enthalpy, and ΔS is the change in entropy. The binding constant is expressed as 1/Ka = Kd for more clarity.
CD spectropolarimetry.
Circular dichroism (CD) experiments were performed on a J-810 spectropolarimeter (Jasco, Tokyo, Japan) in a thermostated cell holder at 20°C. Purified recombinant N proteins were dialyzed against 5 mM phosphate–100 mM NaF (pH 8.5). Far-UV spectra (180 to 260 nm) were recorded using a bandwidth of 1 nm and an integration time of 1s, with proteins at a concentration of 20 μM. Each spectrum was the average of 6 scans, with a scan rate of 100 nm/min. The spectra were corrected by subtracting the signal from the buffer, smoothed using the fast Fourier transform (FFT) filter (Jasco Software, Tokyo, Japan), and treated as previously described (28).
SPR assays.
Real-time surface plasmon resonance (SPR) assays were carried out using a ProteOn XPR36 (Bio-Rad) instrument equilibrated at 25°C in 20 mM Tris-HCl (pH 8.5)–150 mM NaCl–5% glycerol–0.01% Tween 20. A goat anti-GST antibody (Biacore GST Capture kit) was covalently coupled to a gas-liquid chromatography (GLC) sensor chip, using the Amine Coupling kit (GE Healthcare), reaching an immobilization density of around 4,500 resonance units (RU; 1 RU ≈ 1 pg · mm−2). The antibody-functionalized surface was used to capture tightly GST-fused P[1-40] mutants (or GST as a control) to a density of 100 to 160 RU or, as a control, GST (800 RU). Nmono (19 μM, 6.33 μM, 2.1 μM, 703 nM, and 234 nM) was then injected in duplicate over the GST-P[1-40] (wild type or mutants) and GST surfaces for 1 min at a flow rate of 50 μl · min−1. The surfaces were then regenerated by washing with a 10 mM glycine–HCl (pH 1.5) for 2 min and 0.05% SDS for 1 min. The real-time interaction profiles were double referenced using the ProteOn manager software (Bio-Rad); that is, both the signals from the reference surface (with GST captured on the anti-GST antibody) and those from buffer blank experiments were subtracted. The SPR steady-state responses were plotted against the Nmono concentration and fitted using the ProteOn Manager 3.1 software (Bio-Rad).
RESULTS
Generation of a monomeric, RNA-free mutant of N.
In order to characterize the region of P that specifically interacts with N to maintain it in an RNA-free form, named N0, one must first be able to obtain such an RNA-free N0 monomer. This is not a straightforward task given that, when expressed in bacteria alone or together with P, the RSV N protein can be purified only in the form of soluble 10-mer and 11-mer rings containing RNA (12). In order to isolate the N0 monomer, we designed a strategy to prevent N from interacting with RNA. This strategy was based on the hypothesis that N oligomerization is coupled or directly linked to RNA binding. Based on the crystal structure of N (21), residues K170, D175, R184, R185, Y337, and R338 of N, which are known to be involved in the interaction with RNA, were replaced (alone or combined) by alanine (Fig. 1A). Mutant N proteins with a C-terminal hexa-His tag were expressed in E. coli, purified, and screened for their capacity to bind RNA by measuring the OD260/OD280 nm absorption ratio. We thus selected the double mutant NK170A/R185A, for which the observed OD260/OD280 was 0.6 (compared to 1.44 for N-RNA rings), confirming the absence of RNA. Purified recombinant wild-type N and NK170A/R185A were then loaded onto an S200 column. The Nwt was found to elute from the column in one single peak with an apparent molecular mass of ∼500 kDa, as expected for N-RNA rings (Fig. 1B). The NK170A/R185A eluted mainly in one peak with an apparent molecular mass of ∼ 50 kDa, as expected for the monomeric form of N (44 kDa). The gel filtration profile of NK170A/R185A also revealed the presence of minor peaks corresponding to higher molecular masses likely due to partial aggregation (Fig. 1B). To confirm the monomeric state of purified NK170A/R185A recombinant protein, we performed cross-linking assays. Purified N-RNA rings and NK170A/R185A proteins were cross-linked by increasing concentrations of EGS. As revealed in Fig. 1C, both forms of N were purified to homogeneity (>95%) and migrated in SDS-polyacrylamide gel with an expected apparent molecular mass of 44 kDa. For N-RNA rings, bands migrating with apparent masses of ∼80 kDa, ∼120 kDa, and higher molecular mass (>300 kDa) appeared upon increasing EGS concentration. These masses are compatible with N dimers, trimers, and bigger oligomers, respectively. Under the same conditions, no cross-linked products were observed for the NK170A/R185A protein, and we thus concluded that this mutant was present as a monomer. Finally, the folding of purified NK170A/R185A recombinant protein was analyzed by circular dichroism (CD). As shown in Fig. 1D, far-UV CD spectra of N-RNA rings and NK170A/R185A were similar and presented one positive peak at 190 nm and two negative peaks at 208 and 222 nm, typical of secondary structures with a mainly α-helical content. These results thus show that no major secondary structure change occurs in the mutated NK170A/R185A variant with respect to the N-RNA state.
FIG 1.

Characterization of the NK170A/R185A recombinant protein. (A) Closeup view of the N-RNA interactions based on the three-dimensional (3D) crystal structure (Protein data bank file 2WJ8). The NNTD and NCTD domains of the N protein are colored in yellow and red, respectively, and residues implicated in the interaction with RNA are colored in blue. RNA is displayed in gray. Mutated residues K170 and R185 are in bold letters. (B) Gel filtration elution profiles of Nwt (blue) and NK170A/R185A (red). (C) Chemical cross-linking of N proteins. Samples of 5 μg of purified N proteins were subjected to cross-linking with increasing concentrations of EGS as indicated above each lane. The reaction products were resolved by SDS-PAGE and silver stained. Molecular mass markers are indicated (kDa). (D) Far-UV CD spectra showing the absence of significant conformational changes between N-RNA rings (blue) and monomeric NK170A/R185A (red).
Altogether, our data show that the NK170A/R185A recombinant protein (i) does not interact with nucleic acids, (ii) is monomeric, and (iii) presents a secondary structure similar to that of the wild-type N. We thus used this mutant, which was designated Nmono, as an N0 substitute for further experiments.
Stoichiometry and thermodynamics of the P-N interactions followed by ITC.
We investigated the interaction between P and N-RNA rings or Nmono, using isothermal titration calorimetry (ITC). This approach can give access to the stoichiometry of complexes, their equilibrium constants (KD), and the variation of enthalpy (ΔH) and entropy (ΔS) associated with complex formation (29). For the couple N-RNA/P, the P protein was loaded into the calorimeter sample cell and titrated with N-RNA rings. Due to the propensity of Nmono to aggregate at high concentration, the corresponding titration for the Nmono/P couple was performed by loading Nmono into the calorimeter sample cell and P in the titration syringe. Binding isotherm titrations of both couples are exothermic, as shown by the negative heat flow patterns (Fig. 2, upper panels). Integration of the heat flow revealed that N-RNA and P form a 0.5:1 stoichiometric complex with a KD of ∼2.5 μM, whereas P binds to Nmono with a stoichiometry of 1:1 and a KD of ∼0.8 μM (Fig. 2). Taking into account the tetrameric state of P, the stoichiometries determined for both P-N couples suggest that one tetramer of P could interact with two N protomers in the context of N-RNA, whereas the N0-P complex would consist of one P tetramer bound to four N0. These data also revealed that the N-RNA/P interaction is driven by both enthalpic and entropic components (ΔH = −3.25 kcal/mol, and −TΔS = −4.25 kcal/mol). As for the Nmono-P interaction, it is driven by a strong favorable enthalpy (ΔH = −14.43 kcal/mol), partially compensated by an unfavorable entropic contribution (−TΔS = 6.47 kcal/mol). The last observation suggests that the Nmono-P interaction could involve some degree of structural reorganization.
FIG 2.

Isothermal titration calorimetry characterization of complex formation between RSV P protein and Nwt (A) and Nmono (B). (A) Data obtained with the following initial concentrations: 60 μM P (cell) and 300 μM N-RNA (syringe). (B) Data obtained with the following initial concentrations: 38 μM Nmono (cell) and 255 μM P (syringe). Upper panels correspond to heat flow traces produced by injections of N (A) and P (B), respectively. Each peak corresponds to the injection of 2 μl of proteins into the ITC reaction cell at 20°C. Graphs shown in the bottom part of each panel correspond to integrated and corrected ITC data fit to a single site model. The filled squares represent the experimental data, and the solid lines correspond to the fitted model. The derived equilibrium constant (KD) and the stoichiometric ratio (N) are indicated.
N- and C-terminal extremities of P interact with Nmono in solution.
Next, to identify the domain(s) of P capable of binding to Nmono, we performed GST pulldown assays of purified Nmono protein using serial deletions of P fused with GST. Based on previous mapping of the oligomerization domain of P between residues ∼120 and ∼160 (11, 16, 17, 30), different fragments of P corresponding to the N- and C-terminal domains of P including or not the oligomerization domain were fused to GST (Fig. 3A). GST-P fusion proteins bound to glutathione-Sepharose beads were then incubated with Nmono purified separately (Fig. 3B, left panel). Analysis of the interactions between GST-P proteins and Nmono revealed the presence of two binding sites on P (Fig. 3B, right panel). First, the C-terminal domain of P alone or associated with the oligomerization domain (GST-P[127-241] and GST-P[161-241] constructs) interacted with Nmono, and deletion of the last 12 C-terminal residues of P (GST-P[127-229]) was sufficient to abrogate this interaction. Second, Nmono was also capable of interacting with GST-P[1-163], including the N-terminal domain of P and the oligomerization domain. Of note, the interaction between Nmono and this construct could not be detected at first, because GST-P[1-126] and N have a similar apparent molecular weight when analyzed by SDS-PAGE. However, this interaction was validated after separating P[1-126] and the GST tag by thrombin cleavage (Fig. 3C). Finally, no N-P interaction was observed with GST-P[60-126], suggesting that the second Nmono binding domain is located upstream of residue 60 of P (Fig. 3B). To confirm these results, and since the last 10 C-terminal amino acids of P (aa 231 to 241) were previously shown to be implicated in the interaction with N-RNA (12, 21), we compared GST pulldown of either N-RNA rings or Nmono with GST-P[1-230] and GST-P[231-241]. As shown in Fig. 3D, GST-P[231-241] interacted with both N-RNA and Nmono, confirming that the last 10 C-terminal residues of P are sufficient to interact with both forms of N. On the other hand, GST-P[1-230] interacted only with Nmono.
FIG 3.

Deletion mapping of Nmono binding domain(s) on P. (A) Schematic illustration of the wild-type and the truncated RSV P proteins used in this study. The oligomerization domain of P is represented as a gray box, and numbers indicate amino acid positions. Deletion mutants of P harboring a GST tag at the N terminus were purified on glutathione-Sepharose beads and incubated in the presence of Nmono. For each deletion mutant, the ability to interact with Nmono is summarized on the right. (B, C, D) GST-P-derived proteins were analyzed by SDS-PAGE before or after incubation with Nmono (B, C) or N-RNA rings (D), as indicated. (C) For GST-P[1-126], the presence of N0 after pulldown was verified after thrombin cleavage of GST tag (P, purification products; S and B, supernatant and beads after thrombin cleavage, respectively) (E, F) Identification of the minimal domain of P involved in the interaction with Nmono. (E) Analysis of copurification products of Nmono with P[231-241] and N-terminal fragments of P (GST-PΔ). (F) Study of the fixation of both N and C termini of P on Nmono. Recombinant N-RNA rings, Nmono, or P[1-40]+Nmono complex were incubated with GST or GST-P[161-241] bound to beads, and interactions were analyzed by SDS-PAGE and Coomassie blue staining.
Altogether, these results suggest the presence of two N binding sites on P, the first one located in the C-terminal domain of P (PCTD) and capable of binding to both N-RNA and Nmono and the second one located in the N-terminal domain of P (PNTD) and specific for Nmono.
Determination of the minimal N-terminal domain of P required for interaction with Nmono.
We then focused on the characterization of the minimal N-terminal domain of P required for Nmono binding. To this end, Nmono was coexpressed in bacteria with N-terminal fragments of P (PΔ) fused with GST, and the ability of Nmono to copurify with GST–PΔ proteins was evaluated by SDS-PAGE and Coomassie blue staining (Fig. 3E). Our data showed that Nmono copurified with GST-P[1-29] and GST-P[1-40] but not with GST-P[1-10], GST-P[1-20], GST-P[10-30], or GST-P[10-40]. These results revealed that an Nmono-specific binding site is located between amino acids 1 and 29 of P. It is noteworthy that residues 1 to 10, if not sufficient to interact with Nmono, are required for the interaction. Finally, we observed that Nmono also copurified with GST-P[231-241] used as a control but less efficiently than with GST-P[1-29] and GST-P[1-40], in a reproducible way.
The last observation led us to determine whether the N- and C-terminal domains of P can bind concomitantly to Nmono. We performed GST pulldown assays with GST-P[161-241] using either separately purified N-RNA rings, Nmono, or the P[1-40]+Nmono complex. Analysis of the interactions by SDS-PAGE revealed that GST-P[161-241] can interact with both N-RNA rings and Nmono, although the last interaction seemed weak (Fig. 3F). On the contrary, no interaction was observed between GST-P[161-241] and the preformed P[1-40]+Nmono complex. These data revealed that binding of P[1-40] to the monomeric N inhibited the binding of the C terminus of P. However, the N[31-252] domain, which was previously shown to contain the binding site for PCTD (18), did not copurify with GST-P[1-40] (not shown). It is thus unlikely that PNTD and PCTD bind to the same pocket on N.
In conclusion, these results show that the first 29 N-terminal and the last 10 C-terminal residues of P can independently bind to the RNA-free N monomer. However, although two distinct P binding sites exist for Nmono in solution, the interaction with the PCTD appears to be weaker, and binding of the PNTD inhibits the interaction with the PCTD.
Effect of N-terminal mutations of P on RNA polymerase activity.
Based on these results, we investigated the potential role of N-terminal residues (aa 1 to 29) of P on the polymerase complex activity by site-directed mutagenesis using an RSV plasmid-based minireplicon system (24). Preventing the formation of an N0-P complex competent for genomic or antigenomic RNA encapsidation would result in a decrease of N-RNA template formation and amplification and therefore also of mRNA transcription and expression of the luciferase (Luc) reporter. We replaced one by one residues E2 to F28 of P by alanines (except residues Ala5, Ala13, and Ala17), and we tested the functionality of these P mutants within the polymerase complex. As shown in Fig. 4A, among these 24 mutants, 11 displayed a reduction of Luc activity of about or less than 50% compared to the wild-type P protein. The E2A, F4A, F8A, G10A, and F20A substitutions had the strongest effect, with a reduction of 80 to 90% of polymerase activity. As assessed by Western blotting, all P mutants displaying a reduction of Luc activity were expressed in amounts similar to those of wild-type P protein in BSRT7/5 cells (Fig. 4B).
FIG 4.

Identification of N-terminal residues of P critical for RSV polymerase activity. (A) Polymerase activity assays in the presence of P mutants. BSRT7/5 cells were transfected with plasmids encoding the wild-type (WT) N, M2-1, and L proteins, the pMT/Luc minigenome, and WT or mutant P proteins, together with pCMV-βGal for transfection standardization. Viral RNA synthesis was quantified by measuring the Luc activity after cell lysis 24 h after transfection. Each luciferase minigenome activity value was normalized based on β-galactosidase expression and is the average of results from three independent experiments performed in triplicate. Error bars represent standard deviations calculated based on three independent experiments made in triplicate. (B) Western blot analysis showing efficient expression of P mutant (E2A to G10A and F20A to G26A) proteins in BSRT7/5 cells.
These data revealed that the 11 residues E2, F4, E7, F8, G10, F20, L21, E22, I24, K25, and G26 are critical for polymerase activity, and could be directly involved in the interaction between P and the monomeric RNA-free N protein. These results also suggest the presence of two potential critical stretches of residues, one located between residues E2 and G10 and the second between residues F20 and G26.
Identification of N-terminal residues of P involved in the interaction with the Nmono.
We therefore attempted to determine whether the drop in polymerase activity induced by substitutions in the E2-F28 region of P was correlated to a defect of interaction between P and monomeric N. Mutations of all the residues E2 to F28 were introduced into the plasmid expressing GST-P[1-40] to generate 24 mutant proteins. The Nmono mutant protein was coexpressed with the wild-type or mutant forms of GST-P[1-40] in E. coli and purified by using the GST tag. Purified complexes were analyzed by SDS-PAGE. Mutations F4A, F8A, G10A, F20A, L21A, and I24A totally or nearly abrogated the interaction of GST-P[1-40] with Nmono (Fig. 5). The substitutions E7A and K25A attenuated partially the interaction compared to the wild-type P[1-40]. The other mutations and more specifically E2A, E22A, and G26A, which were shown to induce a decrease of polymerase activity, did not significantly modify the interaction between P[1-40] and Nmono.
FIG 5.

Identification of N-terminal residues of P involved in the interaction with Nmono. Copurification of Nmono with GST-P[1-40] mutants from E. coli. GST-P[1-40] constructs (wild type or mutants E2A to F28A) were coexpressed with Nmono protein in bacteria, and complexes were pulled down using the GST tag. Interaction of Nmono with P[1-40] mutants was analyzed by SDS-PAGE and Coomassie blue staining.
In order to confirm these results and to quantify the impact of P mutations E2A-G10A and F20A-G26A on the interaction with Nmono, we then characterized the specific interaction between GST-P[1-40] (wild type or mutants) and Nmono by surface plasmon resonance (SPR). GST-P[1-40] proteins were captured on an anti-GST antibody surface, and serial dilutions of Nmono were injected. We first characterized the specific interaction between wild-type GST-P[1-40] and Nmono. The interaction was transient, with a very high dissociation rate and a Kd of 4 to 5 μM (Fig. 6; see also Fig. 8). Similar affinities were obtained for mutants K3A, P6A, H9A, and S23A. Mutations F8A and F20A had the strongest effect on the interaction with Nmono, with calculated KDs of 41 μM and >100 μM, respectively. The affinity for Nmono was also affected by mutations of residues F4, E7, L21, and I24 (10 μM < KD < 20 μM) and to a lesser extent by mutations of residues G10, E22, and K25 (5 μM < KD < 10 μM). Finally, the affinity of GST-P[1-40] for Nmono was increased nearly 2-fold by mutations E2A and G26A, with KDs of 1.9 μM and 2.6 μM, respectively.
FIG 6.
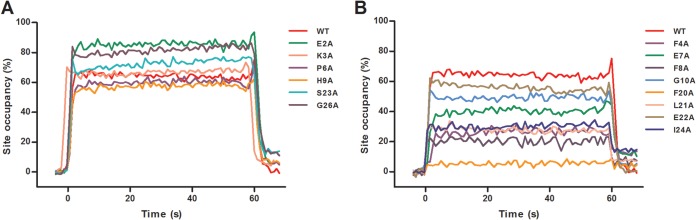
Characterization of Nmono binding to GST-P[1-40] mutants by SPR. Real-time association and dissociation profiles corresponding to the injection over immobilized GST-P[1-40] (wild type or mutants) of Nmono at 6.3 μM. (A) Comparison of profiles for wild-type P[1-40] and mutants of similar (K3A, P6A, H9A, and S23A) or higher (E2A and G26) affinity for Nmono. (B) Comparison of profiles for wild-type P[1-40] and mutants of lower affinity for Nmono (F4A, E7A, F8A, G10A, F20A, L21A, E22A, I24A, and K25A).
FIG 8.

Sequence of the N terminus of P and summary of effects of point mutations on RdRp activity and P-N0 interactions. (A) The sequence of the first 40 N-terminal residues of P is indicated at the top. Residues of the predicted mir motif are boxed. The location of the predicted α-helix is indicated by a blue rectangle below the sequence. The left-hand column indicates the point mutations. Right-hand columns summarize the impact of mutations on (i) the polymerase activity, (ii) in vitro copurification of GST-P[1-40] mutants with Nmono, and (iii) affinity of Nmono for GST-P[1-40] mutants; nd, not determined. Residues identified as critical for both polymerase and interaction with Nmono based on pulldown and SPR data are in red in the sequence. (B) Helical wheel representation (HeliQuest online program) of the putative α-helix located between residues 11 and 28 of P. Residues critical for N0-binding are indicated by a star, and the putative site of interaction with the Nmono is indicated by a black half-circle under the wheel. Positively charged residues are in blue, negatively charged residues in red, and hydrophobic residues in yellow.
Altogether, these results reveal that the 11 residues of P previously identified as critical for polymerase activity are directly involved in the interaction with the soluble monomeric RNA-free N protein.
Overexpression of peptide P[1-29] inhibits viral RNA synthesis.
In our minireplicon system, replication and transcription of RNA by RSV RdRp rely on the correct encapsidation by N0 of both the minigenome and the antiminigenome and the formation of an RNA-N template. It is thus expected that inhibition of the formation of an N0-P complex, which is competent for RNA encapsidation, would result in a decrease of RNA synthesis as Luc expression. Likewise, it is expected that, by binding to N0, a short peptide corresponding to the N-terminal domain of P could compete with full-length P and inhibit the formation of an N0-P complex and so RNA replication and transcription. To evaluate whether P[1-29] could interfere with RSV RNA synthesis, a plasmid encoding P[1-29] was transfected in BSRT7/5 cells in the context of the minireplicon. RdRp activity assayed by measuring Luc activity was reduced in a dose-dependent manner by the P[1-29]-expressing plasmid (Fig. 7A). Similar results were obtained when using a plasmid encoding mCherry-P[1-29] (Fig. 7A), for which expression can be easily visualized by epifluorescence.
FIG 7.
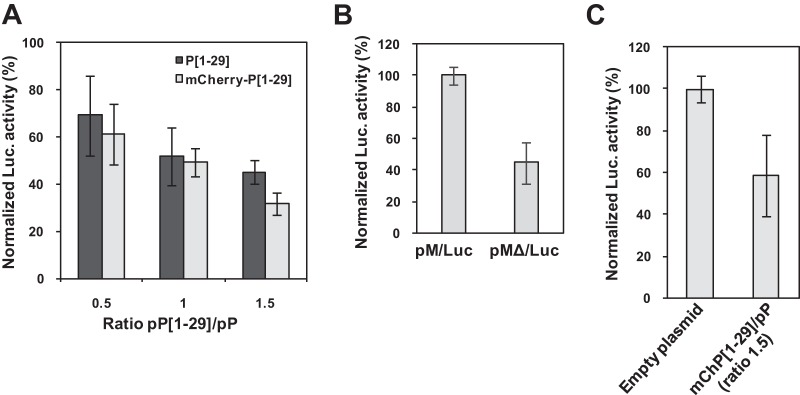
Inhibition of RSV replication by P[1-29]. (A) BSRT7/5 cells were transfected with pP, pN, pM2-1, pL plasmids, and pM/Luc, together with pCMV-βGal for transfection standardization, and various ratios of pP[1-29]/pP or pmCherry-P[1-29]/pP. (B) Comparison of Luc activity using pM/Luc and pMΔ/Luc vectors coding for replication-competent and replication-defective minigenomes, respectively. (C) Replication-defective minigenome assay with a pmCherry-P[1-29]/pP ratio of 1.5. Viral RNA synthesis was quantified by measuring the Luc activity after cell lysis 24 h after transfection. Each Luc activity value was normalized based on β-galactosidase expression and is the average of results from three independent experiments performed in duplicate. Error bars represent standard deviations calculated based on three independent experiments made in duplicate.
To test whether P[1-29] could interfere with RSV transcription, we used an altered minireplicon (pMΔ/luc) with a deleted trailer sequence, still efficient for transcription but defective for replication. Hence, using this system, Luc activity should reflect only the transcription activity. As previously observed (25, 26), Luc activity was reduced to less than one-half when using the pMΔ/Luc vector compared to pM/Luc (Fig. 7B). We then performed a minigenome replication-defective assay in the context of the plasmid expressing mCherry-P[1-29], using a pmCherry-P[1-29]/pP ratio of 1.5. Expression of the mCherry-P[1-29] peptide reduced the transcriptional activity to 50% (Fig. 7C). Thus, the short peptide P[1-29] is capable of inhibiting both RNA replication and transcription by RdRp in mammalian cells.
DISCUSSION
Strategy to obtain an RSV N0-like protein.
By analogy with Paramyxovirinae and Rhabdoviridae, it is assumed that pneumovirus replication requires a constant supply of monomeric, unassembled N (N0) to encapsidate neosynthesized single-stranded genomic RNA (−RNA) and antigenomic RNA (+RNA) (31). However, when expressed alone in mammalian cells, insect cells, or bacteria, nucleoproteins of Mononegavirales bind to cellular RNA and form only stable RNA-N complexes (for a review, see reference 5). The phosphoprotein P plays a critical role in maintaining N0 unassembled during infection. However, purification of N0-P complexes is tricky and the existence of this theoretical complex has never been shown for RSV or other pneumoviruses. Structural characterization of the RSV N0-P complex constitutes a major challenge. It could be useful to gain insight into the mechanisms of regulation that sustain the switch between N0 and N-RNA but also to develop a rational search for inhibitors targeting this complex.
Among Mononegavirales, the crystal structures of N0-P complexes have been resolved for one rhabdovirus (vesicular stomatitis virus [VSV]) and one paramyxovirus (Nipah virus), allowing to clarify the role of P as a chaperone protein (32, 33). For VSV, the complex was reconstituted in vitro after purification of a recombinant N protein deleted of its 21 N-terminal residues, which was monomeric although still capable of interacting with RNA (32). A second step consisted of displacing RNA by a peptide, P[1-60], corresponding to residues 1 to 60 of the P protein. The crystal structure of this complex revealed that the binding domain of P[1-60] on N partially overlaps the RNA binding site and that the N terminus of P blocks the backside groove of N, preventing N oligomerization. For Nipah virus, the structure of a complex between a monomeric N mutant with its N and C termini deleted and a peptide corresponding to the sequence of the 50 N-terminal residues of P was also obtained (33). In this complex, the N terminus of P was shown to interact only with the C-terminal domain of N. In this case, the binding of P should prevent N oligomerization and keep N0 in an open conformation ready to interact with RNA. Finally, although the N0-P complex of RSV has not yet been characterized, an RNA-free N protein has been isolated by deletion of the first 12 N-terminal residues (34).
In the present study, based on the crystallographic structure of RSV RNA-N rings (21) and on the hypothesis that preventing RNA binding would also prevent N oligomerization, we generated the RNA-free N mutant protein K170A/R185A. This recombinant N mutant protein was shown to be monomeric at concentrations of up to 40 μM (Fig. 1B and C). This result shows that disrupting N-RNA interactions can partially prevent RSV N self-oligomerization. This first observation supports the debated theory according to which N RNA binding and oligomerization are coupled (5, 35, 36). We have also shown that this RNA-free N protein presents a secondary structure similar to that of N assembled with RNA (Fig. 1D). Therefore, it is expected that the switch between N0 and N-RNA does not require major secondary structure changes. This result is in agreement with those obtained for VSV and Nipah virus (32, 33), for which it was shown that compared to the oligomerized RNA-bound form of N, the N- and C-terminal globular domains of N0 present similar structures. Finally, the RSV recombinant NK170A/R185A mutant can still interact with the P protein. The last observation strongly suggests that residues K170 and R185, which are located in the RNA groove of N and are critical for RNA binding, are not critical for N0-P interaction. Thus, a different set of residues should be involved in RNA binding and in P binding. Since bacterially expressed recombinant RSV P protein is not phosphorylated, these results also show that P phosphorylation is not required for N0-P complex formation.
P-N stoichiometries.
The interactions between P and N-RNA rings or Nmono protein were compared by isothermal titration calorimetry (Fig. 2). The calculated stoichiometry for both binding pairs suggests that one tetramer of P could interact with two N protomers in the context of NC, whereas it could interact with four N0. Recently, an analytical ultracentrifugation experiment and a small-angle X-ray scattering (SAXS) study of the entire VSV N0-P complex suggested that a dimer of P associates with only one N0 (36). Therefore, our results suggest that N0-P complexes of RSV and rhabdoviruses do not have the same stoichiometry. Isothermal titration calorimetry experiments also revealed that P presents similar affinities for both N-RNA and Nmono proteins. The calculated KD for these two N-P interactions are in the micromolar range. It is postulated that, as a general feature for Mononegavirales, RSV replication requires the switch from N0-P to N-RNA-P, P freeing N0 for RNA encapsidation just after its synthesis next to the replicase L-P. It is thus expected that such a mechanism requires low affinities between P and N. It is worth noting that, based on our results, we cannot exclude that a tetramer of P could concomitantly interact with N0 and NC.
An N0-specific binding domain is located at the N terminus of P.
In parallel, identification of the Nmono binding domain on P was investigated by either coincubating or coexpressing GST-fused P fragments with N. Altogether, the data demonstrated that the RNA-free N monomer presents binding sites for both PCTD and PNTD and that the oligomerization domain of P is not required for these interactions (Fig. 3). Furthermore, the binding of PNTD to Nmono is specific to the N monomer, as no binding was observed with N-RNA rings. These results also revealed that the 10 C-terminal and the 29 N-terminal residues of P are sufficient to interact with Nmono. The interaction between Nmono and the C terminus of P correlates with recent data from Shapiro et al., who showed that the C terminus of P binds to a monomeric RNA-free N protein, deleted from the 12 first N-terminal residues (37). However, we have further shown that PCTD binds very poorly to Nmono and that binding of the PNTD to Nmono impairs the binding of PCTD (Fig. 3F). The last observation correlates with ITC data, since the experimental titration curve of Nmono by P can be fitted well, assuming a single binding site (Fig. 2B). Two hypotheses could explain these observations. First, PCTD and PNTD could bind to the same site of N. However, the NNTD truncated variant, which was previously shown to contain the PCTD binding site (18), did not copurify with PNTD (not shown). This result strongly suggests the existence of two distinct P binding sites on Nmono. This observation is supported by the fact that PNTD and PCTD do not present sequence homologies. It is thus unlikely that these domains interact with the same N binding site. However, we cannot exclude the possibility that the PNTD binding site partially overlaps the PCTD binding site on N. Furthermore, although the NNTD is a globular, alpha-helical domain that should not undergo major conformational changes between N0 and N-RNA forms, PNTD binding on Nmono could induce a local conformational change that renders the PCTD binding site inaccessible. In any case, our results concerning the RSV N0-PNTD interaction are in agreement with data obtained previously with BRSV showing a direct interaction between PNTD (residues 1 to 40) and N using the yeast two-hybrid system (20). Since P and N are highly conserved between BRSV and HRSV, it is likely that PNTD is the main N0 binding site for both viruses. Our results are also supported by data from the literature. Indeed, using sensitive sequence similarity search programs, Karlin and Belshaw (6) found that all Paramyxovirinae share a short conserved motif in the first 40 amino acids of P (aa 11 to 26), which was called the soyuz motif and should be involved in N0 binding. Conserved motifs were also found within Pneumovirinae (mir motif), Filoviridae (sputnik motif), and Rhabdoviridae. The presence of N0 binding domains within the N terminus of P was confirmed by experimental data for SeV (residues 33 to 41) (38), HPIV3 (first 40 N-terminal residues) (39), RABV (residues 4 to 40) (40), VSV (residues 11 to 30) (41), and Nipah virus (residues 1 to 35) (33).
The N0-binding domain of P could fold upon interaction.
Based on our results, we then focused on the characterization of the Nmono binding domain located at the N terminus of P and more specifically on the potential involvement of residues 1 to 29 of P in the Nmono-P interaction by Ala scanning (Fig. 8A). Since this interaction is required for maintaining N in an RNA-free form (N0) competent for RNA encapsidation and formation of an RNA-N template used for replication and transcription, we first used the functional HRSV minireplicon system to screen for residues critical for polymerase activity. This approach highlighted the critical role of 11 residues of P, located between residues E2-G10 and F20-G26 (Fig. 4A). None of these mutations fully abolished RNA synthesis. Pulldown and SPR experiments were then combined to determine whether the effect of mutations on viral RNA synthesis correlated with a defect in Nmono-P interaction. Using these approaches, we confirmed that the Ala substitution of the 9 residues F4, E7, F8, G10, F20, L21, E22, I24, and K25 totally or partially altered the Nmono-P interaction, whereas mutations of residues E2 and G26 increased the affinity of Nmono for the N terminus of P (Fig. 8A). These data highlight a direct role of these 11 residues in the interaction with the putative N0. The localization of these critical residues suggests the presence of two main domains of interaction of P on N0, located between residues E2-G10 and F20-G26 and separated by a stretch of 9 residues. The structural study of P predicted the presence of an α-helix in the region D12-F20 (16), and as previously evoked, the N0 binding site was predicted to be located between residues 11 and 26 (6).
Taking into account these data and the periodicity (i, i + 3; i, i + 4) of the critical residues F20-G26 of P, we thus modeled a putative short α-helix between residues E11 and F28 (Fig. 8B). This representation shows that residues F20, L21, I24, and K25, for which mutation strongly affected the interaction with Nmono, should be located on the same side of the putative helix. It is worth noting that the unfavorable entropic contribution determined by ITC for the Nmono-P interaction also argues for the involvement of structural constraints during the establishment of the interaction, which could be explained by the folding of the N terminus of P. Such a mechanism of folding of intrinsically disordered regions upon interaction has already been described for other paramyxoviruses such as measles virus (reviewed in reference 42) and for the rhabdovirus VSV N0-P interaction. More precisely, the nuclear magnetic resonance (NMR) study of the N-terminal domain of VSV P protein revealed that it contains two transient α helices (43), which were subsequently shown to constitute molecular recognition elements (MoRE) for the interaction with N0 (32). A MoRE is a small segment of a protein that is intrinsically disordered but folds upon binding to a molecular partner (44). Thus, the N-terminal domain of the RSV P protein, which is located in a predicted disordered region extending from residues 1 to ∼120, could contain such a MoRE.
The N0-binding site is a potential target for antiviral compounds.
Because P plays a central role in the functioning of the RNA polymerase, this protein could be a target of choice for antiviral strategies aiming to destabilize the complex and inhibit viral replication. We have demonstrated that coexpression of the peptide P[1-29] encompassing the N0 binding site together with the minireplicon is sufficient to inhibit the replication activity of the polymerase complex, in a dose-dependent manner. This small peptide thus constitutes an interesting basis upon which to develop antiviral approaches. A proof of concept has already been developed for rabies virus (RABV) (45) and more recently for Nipah virus (33), for which peptides that mimic the N terminus of the P protein were shown to present an antiviral effect. In addition, a study describing the prevention of nasopulmonary infection by mucosal delivery of a peptide that interferes with the fusion step of RSV (46) demonstrates the power of similar antiviral strategies.
Conserved mechanisms among Mononegavirales for RNA encapsidation.
Altogether, the data presented here demonstrate for the first time that RSV P can bind to an RNA-free, monomeric form of N, thus forming an N0-P like complex, and reinforce previous observations that the P proteins of Rhabdoviridae, Paramyxovirinae, and Pneumovirinae share similar functions and modular organization, although they have no detectable sequence similarity. All the P proteins are oligomeric and contain intrinsically disordered regions upstream and downstream of the oligomerization domain. From a functional point of view, they interact with many partners, including the L polymerase, N-RNA, and N0, the main nucleocapsid (RNA-N) and N0 binding domains being located at the C and N termini of P, respectively. However, a study of the P-N interaction of MuV recently reopened the debate concerning the role of P as a chaperone (4). Using electron microscopy, the authors have indeed shown that incubation of PNTD with nucleocapsid (NC) induces uncoiling of MuV NC. They also showed that both PNTD and PCTD bind to the NC and that overexpression of PNTD in cells increases the polymerase activity. These two results are different from our data since RSV PNTD did not bind to N-RNA rings whereas PNTD and PCTD can both bind to Nmono and overexpression of PNTD led to a decrease of the RSV polymerase activity.
Finally there seems to be a fundamental difference between Paramyxoviridae and Rhabdoviridae concerning the organization of the functional domains of the P protein. Indeed, the L binding site has also been localized in the N-terminal part of rhabdovirus P protein (47, 48). For rabies virus, it was proposed that these two functional domains do not overlap but are juxtaposed, composed of residues 4 to 40 for N0 and 40 to 70 for L. In contrast, for Paramyxoviridae, the L main binding site has been localized at the C terminus of P (49, 50), but the N terminus of P could also bind to L (38). Although the L binding domain of RSV does not appear to be located in the N-terminal region of P but rather in either the oligomerization domain or the C-terminal region (19, 30), it would be worthwhile to revisit this issue, since it could be of importance to fully understand the mechanism of N0 release by P in the context of L and RNA synthesis.
ACKNOWLEDGMENTS
We thank John Barr and David Karlin for critical readings of the manuscript, Origène Nyanguile and Christina Sizun for helpful discussions, and Damien Vitour for providing polyclonal rabbit anti-N antibody. Molecular graphics images were produced using Pymol software.
This work was carried out with the financial support of the French Agence Nationale de la Recherche, specific program ANR Blanc 2011 “Bronchioliteaser” (ANR-11-BSV8-0024).
REFERENCES
- 1.Collins PL, Crowe JE. 2007. Respiratory syncytial virus and metapneumovirus, p 1601–1646 InKnipe DM, Howley PM (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Bakker SE, Duquerroy S, Galloux M, Loney C, Conner E, Eleouet JF, Rey FA, Bhella D. 2013. The respiratory syncytial virus nucleoprotein-RNA complex forms a left-handed helical nucleocapsid. J Gen Virol 94:1734–1738. doi: 10.1099/vir.0.053025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cowton VM, McGivern DR, Fearns R. 2006. Unravelling the complexities of respiratory syncytial virus RNA synthesis. J Gen Virol 87:1805–1821. doi: 10.1099/vir.0.81786-0. [DOI] [PubMed] [Google Scholar]
- 4.Cox R, Pickar A, Qiu S, Tsao J, Rodenburg C, Dokland T, Elson A, He B, Luo M. 2014. Structural studies on the authentic mumps virus nucleocapsid showing uncoiling by the phosphoprotein. Proc Natl Acad Sci U S A 111:15208–15213. doi: 10.1073/pnas.1413268111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruigrok RW, Crepin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol 14:504–510. doi: 10.1016/j.mib.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 6.Karlin D, Belshaw R. 2012. Detecting remote sequence homology in disordered proteins: discovery of conserved motifs in the N-termini of Mononegavirales phosphoproteins. PLoS One 7:e31719. doi: 10.1371/journal.pone.0031719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barik S, McLean T, Dupuy LC. 1995. Phosphorylation of Ser232 directly regulates the transcriptional activity of the P protein of human respiratory syncytial virus: phosphorylation of Ser237 may play an accessory role. Virology 213:405–412. doi: 10.1006/viro.1995.0013. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez-Seco MP, Navarro J, Martinez R, Villanueva N. 1995. C-terminal phosphorylation of human respiratory syncytial virus P protein occurs mainly at serine residue 232. J Gen Virol 76(Part 2):425–430. doi: 10.1099/0022-1317-76-2-425. [DOI] [PubMed] [Google Scholar]
- 9.Navarro J, Lopez-Otin C, Villanueva N. 1991. Location of phosphorylated residues in human respiratory syncytial virus phosphoprotein. J Gen Virol 72(Part 6):1455–1459. doi: 10.1099/0022-1317-72-6-1455. [DOI] [PubMed] [Google Scholar]
- 10.Asenjo A, Rodriguez L, Villanueva N. 2005. Determination of phosphorylated residues from human respiratory syncytial virus P protein that are dynamically dephosphorylated by cellular phosphatases: a possible role for serine 54. J Gen Virol 86:1109–1120. doi: 10.1099/vir.0.80692-0. [DOI] [PubMed] [Google Scholar]
- 11.Castagne N, Barbier A, Bernard J, Rezaei H, Huet JC, Henry C, Da Costa B, Eleouet JF. 2004. Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J Gen Virol 85:1643–1653. doi: 10.1099/vir.0.79830-0. [DOI] [PubMed] [Google Scholar]
- 12.Tran TL, Castagne N, Bhella D, Varela PF, Bernard J, Chilmonczyk S, Berkenkamp S, Benhamo V, Grznarova K, Grosclaude J, Nespoulos C, Rey FA, Eleouet JF. 2007. The nine C-terminal amino acids of the respiratory syncytial virus protein P are necessary and sufficient for binding to ribonucleoprotein complexes in which six ribonucleotides are contacted per N protein protomer. J Gen Virol 88:196–206. doi: 10.1099/vir.0.82282-0. [DOI] [PubMed] [Google Scholar]
- 13.Lu B, Ma CH, Brazas R, Jin H. 2002. The major phosphorylation sites of the respiratory syncytial virus phosphoprotein are dispensable for virus replication in vitro. J Virol 76:10776–10784. doi: 10.1128/JVI.76.21.10776-10784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villanueva N, Hardy R, Asenjo A, Yu Q, Wertz G. 2000. The bulk of the phosphorylation of human respiratory syncytial virus phosphoprotein is not essential but modulates viral RNA transcription and replication. J Gen Virol 81:129–133 http://vir.sgmjournals.org/content/81/1/129.long. [DOI] [PubMed] [Google Scholar]
- 15.Asenjo A, Villanueva N. 2000. Regulated but not constitutive human respiratory syncytial virus (HRSV) P protein phosphorylation is essential for oligomerization. FEBS Lett 467:279–284. doi: 10.1016/S0014-5793(00)01171-6. [DOI] [PubMed] [Google Scholar]
- 16.Llorente MT, Garcia-Barreno B, Calero M, Camafeita E, Lopez JA, Longhi S, Ferron F, Varela PF, Melero JA. 2006. Structural analysis of the human respiratory syncytial virus phosphoprotein: characterization of an alpha-helical domain involved in oligomerization. J Gen Virol 87:159–169. doi: 10.1099/vir.0.81430-0. [DOI] [PubMed] [Google Scholar]
- 17.Llorente MT, Taylor IA, Lopez-Vinas E, Gomez-Puertas P, Calder LJ, Garcia-Barreno B, Melero JA. 2008. Structural properties of the human respiratory syncytial virus P protein: evidence for an elongated homotetrameric molecule that is the smallest orthologue within the family of paramyxovirus polymerase cofactors. Proteins 72:946–958. doi: 10.1002/prot.21988. [DOI] [PubMed] [Google Scholar]
- 18.Galloux M, Tarus B, Blazevic I, Fix J, Duquerroy S, Eleouet JF. 2012. Characterization of a viral phosphoprotein binding site on the surface of the respiratory syncytial nucleoprotein. J Virol 86:8375–8387. doi: 10.1128/JVI.00058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khattar SK, Yunus AS, Samal SK. 2001. Mapping the domains on the phosphoprotein of bovine respiratory syncytial virus required for N-P and P-L interactions using a minigenome system. J Gen Virol 82:775–779 http://vir.sgmjournals.org/content/82/4/775.long. [DOI] [PubMed] [Google Scholar]
- 20.Mallipeddi SK, Lupiani B, Samal SK. 1996. Mapping the domains on the phosphoprotein of bovine respiratory syncytial virus required for N-P interaction using a two-hybrid system. J Gen Virol 77(Part 5):1019–1023. doi: 10.1099/0022-1317-77-5-1019. [DOI] [PubMed] [Google Scholar]
- 21.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eleouet JF, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. doi: 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- 22.Fix J, Galloux M, Blondot ML, Eleouet JF. 2011. The insertion of fluorescent proteins in a variable region of respiratory syncytial virus L polymerase results in fluorescent and functional enzymes but with reduced activities. Open Virol J 5:103–108. doi: 10.2174/1874357901105010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardy RW, Wertz GW. 1998. The product of the respiratory syncytial virus M2 gene ORF1 enhances readthrough of intergenic junctions during viral transcription. J Virol 72:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran TL, Castagne N, Dubosclard V, Noinville S, Koch E, Moudjou M, Henry C, Bernard J, Yeo RP, Eleouet JF. 2009. The respiratory syncytial virus M2-1 protein forms tetramers and interacts with RNA and P in a competitive manner. J Virol 83:6363–6374. doi: 10.1128/JVI.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peeples ME, Collins PL. 2000. Mutations in the 5′ trailer region of a respiratory syncytial virus minigenome which limit RNA replication to one step. J Virol 74:146–155. doi: 10.1128/JVI.74.1.146-155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fearns R, Collins PL, Peeples ME. 2000. Functional analysis of the genomic and antigenomic promoters of human respiratory syncytial virus. J Virol 74:6006–6014. doi: 10.1128/JVI.74.13.6006-6014.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chenal A, Nizard P, Forge V, Pugniere M, Roy MO, Mani JC, Guillain F, Gillet D. 2002. Does fusion of domains from unrelated proteins affect their folding pathways and the structural changes involved in their function? A case study with the diphtheria toxin T domain. Protein Eng 15:383–391. doi: 10.1093/protein/15.5.383. [DOI] [PubMed] [Google Scholar]
- 29.Freyer MW, Lewis EA. 2008. Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol 84:79–113. doi: 10.1016/S0091-679X(07)84004-0. [DOI] [PubMed] [Google Scholar]
- 30.Asenjo A, Mendieta J, Gomez-Puertas P, Villanueva N. 2008. Residues in human respiratory syncytial virus P protein that are essential for its activity on RNA viral synthesis. Virus Res 132:160–173. doi: 10.1016/j.virusres.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 31.Kolakofsky D, Le Mercier P, Iseni F, Garcin D. 2004. Viral DNA polymerase scanning and the gymnastics of Sendai virus RNA synthesis. Virology 318:463–473. doi: 10.1016/j.virol.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 32.Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA Jr, Jensen MR, Blackledge M, Ruigrok RW, Jamin M. 2011. Structure of the vesicular stomatitis virus N(0)-P complex. PLoS Pathog 7:e1002248. doi: 10.1371/journal.ppat.1002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yabukarski F, Lawrence P, Tarbouriech N, Bourhis JM, Delaforge E, Jensen MR, Ruigrok RW, Blackledge M, Volchkov V, Jamin M. 2014. Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat Struct Mol Biol 21:754–759 http://www.nature.com/nsmb/journal/v21/n9/full/nsmb.2868.html. [DOI] [PubMed] [Google Scholar]
- 34.El Omari K, Scott K, Dhaliwal B, Ren J, Abrescia NG, Budworth J, Lockyer M, Powell KL, Hawkins AR, Stammers DK. 2008. Crystallization and preliminary X-ray analysis of the human respiratory syncytial virus nucleocapsid protein. Acta Crystallogr F Struct Biol Crystall Commun 64:1019–1023. doi: 10.1107/S1744309108031059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Green TJ, Tsao J, Qiu S, Luo M. 2008. Role of intermolecular interactions of vesicular stomatitis virus nucleoprotein in RNA encapsidation. J Virol 82:674–682. doi: 10.1128/JVI.00935-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green TJ, Cox R, Tsao J, Rowse M, Qiu S, Luo M. 2014. Common mechanism for RNA encapsidation by negative-strand RNA viruses. J Virol 88:3766–3775. doi: 10.1128/JVI.03483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shapiro AB, Gao N, O'Connell N, Hu J, Thresher J, Gu RF, Overman R, Hardern IM, Sproat GG. 2014. Quantitative investigation of the affinity of human respiratory syncytial virus phosphoprotein C-terminus binding to nucleocapsid protein. Virol J 11:191. doi: 10.1186/s12985-014-0191-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curran J, Marq JB, Kolakofsky D. 1995. An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J Virol 69:849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De BP, Hoffman MA, Choudhary S, Huntley CC, Banerjee AK. 2000. Role of NH(2)- and COOH-terminal domains of the P protein of human parainfluenza virus type 3 in transcription and replication. J Virol 74:5886–5895. doi: 10.1128/JVI.74.13.5886-5895.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mavrakis M, Iseni F, Mazza C, Schoehn G, Ebel C, Gentzel M, Franz T, Ruigrok RW. 2003. Isolation and characterisation of the rabies virus N degrees-P complex produced in insect cells. Virology 305:406–414. doi: 10.1006/viro.2002.1748. [DOI] [PubMed] [Google Scholar]
- 41.Chen M, Ogino T, Banerjee AK. 2007. Interaction of vesicular stomatitis virus P and N proteins: identification of two overlapping domains at the N terminus of P that are involved in N0-P complex formation and encapsidation of viral genome RNA. J Virol 81:13478–13485. doi: 10.1128/JVI.01244-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Longhi S. 2012. The measles virus N(TAIL)-XD complex: an illustrative example of fuzziness. Adv Exp Med Biol 725:126–141. doi: 10.1007/978-1-4614-0659-4_8. [DOI] [PubMed] [Google Scholar]
- 43.Leyrat C, Jensen MR, Ribeiro EA Jr, Gerard FC, Ruigrok RW, Blackledge M, Jamin M. 2011. The N(0)-binding region of the vesicular stomatitis virus phosphoprotein is globally disordered but contains transient alpha-helices. Protein Sci 20:542–556. doi: 10.1002/pro.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuxreiter M, Simon I, Friedrich P, Tompa P. 2004. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol 338:1015–1026. doi: 10.1016/j.jmb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 45.Castel G, Chteoui M, Caignard G, Prehaud C, Mehouas S, Real E, Jallet C, Jacob Y, Ruigrok RW, Tordo N. 2009. Peptides that mimic the amino-terminal end of the rabies virus phosphoprotein have antiviral activity. J Virol 83:10808–10820. doi: 10.1128/JVI.00977-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bird GH, Boyapalle S, Wong T, Opoku-Nsiah K, Bedi R, Crannell WC, Perry AF, Nguyen H, Sampayo V, Devareddy A, Mohapatra S, Mohapatra SS, Walensky LD. 2014. Mucosal delivery of a double-stapled RSV peptide prevents nasopulmonary infection. J Clin Invest 124:2113–2124. doi: 10.1172/JCI71856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emerson SU, Schubert M. 1987. Location of the binding domains for the RNA polymerase L and the ribonucleocapsid template within different halves of the NS phosphoprotein of vesicular stomatitis virus. Proc Natl Acad Sci U S A 84:5655–5659. doi: 10.1073/pnas.84.16.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chenik M, Schnell M, Conzelmann KK, Blondel D. 1998. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J Virol 72:1925–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liston P, DiFlumeri C, Briedis DJ. 1995. Protein interactions entered into by the measles virus P, V, and C proteins. Virus Res 38:241–259. doi: 10.1016/0168-1702(95)00067-Z. [DOI] [PubMed] [Google Scholar]
- 50.Smallwood S, Ryan KW, Moyer SA. 1994. Deletion analysis defines a carboxyl-proximal region of Sendai virus P protein that binds to the polymerase L protein. Virology 202:154–163. doi: 10.1006/viro.1994.1331. [DOI] [PubMed] [Google Scholar]