

ABSTRACT
Viral semaphorins are semaphorin 7A (sema7A) mimics found in pox- and herpesviruses. Among herpesviruses, semaphorins are encoded by gammaherpesviruses of the Macavirus genus only. Alcelaphine herpesvirus 1 (AlHV-1) is a macavirus that persistently infects wildebeest asymptomatically but induces malignant catarrhal fever (MCF) when transmitted to several species of susceptible ruminants and the rabbit model. MCF is caused by the activation/proliferation of latently infected T lymphocytes. Viral semaphorins have been suggested to mediate immune evasion mechanisms and/or directly alter host T cell function. We studied AlHV-sema, the viral semaphorin encoded by the A3 gene of AlHV-1. Phylogenetic analyses revealed independent acquisition of pox- and herpesvirus semaphorins, suggesting that these proteins might have distinct functions. AlHV-sema showed a predicted three-dimensional structure very similar to sema7A and conserved key residues in sema7A-plexinC1 interaction. Expression analyses revealed that AlHV-sema is a secreted 93-kDa glycoprotein expressed during the early phase of virus replication. Purified AlHV-sema was able to bind to fibroblasts and dendritic cells and induce F-actin condensation and cell retraction through a plexinC1 and Rho/cofilin-dependent mechanism. Cytoskeleton rearrangement was further associated with inhibition of phagocytosis by dendritic cells and migration to the draining lymph node. Next, we generated recombinant viruses and demonstrated that the lack of A3 did not significantly affect virus growth in vitro and did not impair MCF induction and associated lymphoproliferative lesions. In conclusion, AlHV-sema has immune evasion functions through mechanisms similar to poxvirus semaphorin but is not directly involved in host T cell activation during MCF.
IMPORTANCE Whereas most poxviruses encode viral semaphorins, semaphorin-like genes have only been identified in few gammaherpesviruses belonging to the Macavirus genus. Alcelaphine herpesvirus 1 (AlHV-1) is a macavirus carried asymptomatically by wildebeest but induces a latency-associated lymphoproliferative disease of T lymphocytes in various ruminant species, namely, malignant catarrhal fever (MCF). Viral semaphorins have been hypothesized to have immune evasion functions and/or be involved in activating latently infected T cells. We present evidence that the viral semaphorin AlHV-sema inhibits dendritic cell phagocytosis and migration to the draining lymph node, both being indispensable mechanisms for protective antiviral responses. Next, we engineered recombinant viruses unable to express AlHV-sema and demonstrated that this protein is dispensable for the induction of MCF. In conclusion, this study suggests that herpesvirus and poxvirus semaphorins have independently evolved similar functions to thwart the immune system of the host while AlHV-sema is not directly involved in MCF-associated T-cell activation.
INTRODUCTION
Semaphorins are members of a large family of secreted, membrane-anchored and transmembrane glycoproteins that can be found in invertebrate (classes 1 and 2) and vertebrate (classes 3 to 7) species, as well as viruses (class 8), such as poxviruses and some gammaherpesviruses (1). Although originally identified as axon guidance cues, semaphorins have been implicated in a wide variety of biological processes in many different organ systems, including the brain and the cardiovascular and immune systems (2). Immune semaphorins have been involved in various phases of the immune response, from initiation to terminal inflammatory processes (3). Most of semaphorins signal through plexin receptors to mediate their activity (4). Semaphorin 7A (sema7A) is highly pleiotropic and is the only glycosylphosphatidylinositol (GPI)-anchored member of the semaphorin family. This protein has been implicated in several biological processes such as neural development, bone homeostasis, cancer, and in the immune system (2). Although sema7A has been shown to mediate crucial functions in the regulation of immune responses through different signaling pathways, the exact roles of the protein in the immune system are not completely identified. Sema7A expression is induced in activated T cells, and the protein has been shown to induce proinflammatory cytokine production in monocytes/macrophages (5–7). Though sema7A can signal through plexinC1, a growing body of evidence has shown that the protein can also bind with high affinity to β1 integrins through the RGD motives (Arg267-Gly268-Asp269) present in its SEMA domain (7, 8). Signaling of sema7A through integrins and recruitment at the immunological synapse has been shown to mediate pro- and anti-inflammatory responses in macrophages after binding to different α-integrin subunits (7, 9).
Sema7A sequence has initially been identified based on its sequence similarity with viral semaphorins (10). Viral semaphorin homologs are all predicted to be secreted proteins and are found in Poxviridae and in some Gammaherpesirinae (10). Whereas the function of herpesvirus semaphorins has never been studied, the role of A39R semaphorin homolog has been investigated. Sema7A and viral semaphorins can bind to the same receptor, namely, plexinC1 (11, 12). Because viral semaphorins do not have RGD motives, it has been suggested that while sema7A can signal through either plexinC1 or β1 integrins, its viral homologs are restricted to plexinC1 signaling. A39R binding through plexinC1 results in cytoskeleton rearrangement in dendritic cells (DCs) (11–13) and inhibition of phagocytosis and transwell migration (14). These effects were explained by deactivation of focal adhesion kinase and cofilin-dependent inhibition of F-actin turnover (13, 14). A39R has therefore been suggested to thwart the host immune response rather than directly regulate inflammation. However, A39R has also been shown to induce inrterleukin-6 (IL-6) and IL-8 production in monocytes (11) and one report using recombinant vaccinia virus strains suggested that A39R could induce a proinflammatory response in a murine intradermal infection model (15). Other than this last study, the role of viral semaphorins has never been addressed during virus infection in vivo, and the exact roles of these proteins during infection remain to be resolved.
Herpesvirus semaphorins are only encoded by particular gammaherpesviruses all belonging to the Macavirus genus. Ovine herpesvirus 2 (OvHV-2) and alcelaphine herpesvirus 1 (AlHV-1) are both macaviruses that cause no apparent disease in their natural host species, suggesting that these viruses have evolved immune evasion strategies to persist in their respective hosts. Sheep are naturally infected by OvHV-2, whereas AlHV-1 naturally infects and persists in wildebeest. Importantly, both viruses can induce a pansystemic and fatal lymphoproliferative disease upon cross-species transmission to several ruminant species, including cattle. OvHV-2 is responsible for the sheep-associated malignant catarrhal fever (MCF), whereas AlHV-1 induces wildebeest-derived MCF (16). We recently demonstrated that MCF is caused by the activation and proliferation of latently infected CD8+ T cells (17–19). Although latency has been shown to be essential for MCF induction (20), the actual mechanisms involved during AlHV-1 latent infection of CD8+ T cells that lead to their activation and proliferation are yet to be identified. MCF can be experimentally induced in rabbits, where the observed lesions are indistinguishable from those described in the MCF susceptible species, such as bovines (21). Gammaherpesviruses latency in lymphocytes has been associated with malignant lymphocyte activation and proliferation (20, 22). A recent report showed that noncoding RNAs expressed by saimirine herpesvirus 2 (SaHV-2) during latency specifically degraded host microRNA-27 (miR-27), leading to T cell activation and upregulation of gamma interferon (IFN-γ) and sema7A expression (23). A parallelism has therefore been proposed, suggesting that Macavirus semaphorins might directly be involved in the T cell activation and proliferation observed in MCF.
We sought here to determine whether AlHV-sema has immune evasion functions and/or is involved in the activation and proliferation of latently infected T lymphocytes during MCF. We first characterized the A3 gene expression during virus infection. We observed that AlHV-sema is a glycoprotein of 93 kDa that is secreted during the early phase of virus replication. We next found that purified AlHV-sema could induce cytoskeleton rearrangement through plexinC1 that interfered with DC function via Rho-dependent kinase (ROCK) signaling and inhibition of cofilin. Finally, we engineered recombinant viruses and observed that AlHV-sema expression is not essential in the induction of the lymphoproliferative lesions developing during MCF in rabbits. We conclude that despite their independent phylogenetic acquisition, pox- and herpesvirus semaphorins have likely evolved similar functions to thwart the immune system of the infected host and that AlHV-sema is not directly involved in the activation or proliferation of latently infected T lymphocytes in MCF.
MATERIALS AND METHODS
Phylogenetic analyses.
Sequences homologous to cellular sema7A were extracted from public databases (PhEVER, GenBank, and Ensembl) and analyzed using Seaview4. The sequences were aligned with MUSCLE, and a maximum-likelihood tree was computed using PhyML with a JTT model and a gamma distribution for variable substitution rates among sites. The topological space search was done combining both the nearest neighbor interchange and the subtree pruning and regrafting methods. Branch support was measured by the approximate likelihood-ratio test.
Homology modeling of AlHV-sema 3-D structure.
The three-dimensional (3-D) structure of AlHV-sema was predicted based on the crystal structure of sema7A (12) and using I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). Alignment of sema7A and AlHV-sema was performed using PyMol 1.3.
Cell lines, virus strains, and virus production.
Rabbit kidney cells (RK13, ATCC CCL-37), bovine turbinate (BT) cells (ATCC CRL-1390), bovine macrophages (BoMac) (24), bovine mammary epithelial cells (MacT) (25), and HeLa cells (ATCC CCL-2) were cultured in Dulbecco modified essential medium (DMEM; Life Technologies) supplemented with 10% fetal calf serum (FCS; Bio Whittaker) and Madin-Darby bovine kidney (MDBK) cells (ATCC CCL-22) were cultured in minimum essential medium (MEM; Life Technologies) supplemented with 10% FCS. MacT-cre cells stably expressing NLS-cre were produced as described previously (26). The pathogenic AlHV-1 C500 strain isolated from an ox with MCF (27) and the AlHV-1 C500 BAC clone (28) were used throughout the present study. The virus strains were maintained by a limited number of passages (<5). Virus was amplified in BT cells before supernatants, together with infected cells, were concentrated by ultracentrifugation (100,000 × g, 2 h, 4°C) and suspended in 1 ml of DMEM before storage at −80°C. The samples were thawed and clarified (100 × g, 20 min, 4°C), and the supernatants were titrated by plaque assay.
Plasmids.
AlHV-sema was expressed as Fc- and V5/His-tagged proteins by cloning the A3 coding sequence (RefSeq accession no. NC_002531, nucleotides [nt] 3633 to 5453) amplified from the C500 bacterial artificial chromosome (BAC) (28) into the NotI site of the pTorsten expression vector (29). The pTorsten-A3Fc was produced by amplifying the A3 coding sequence by using the primers XbaI142S and A3deltastop (see Table 1 for the primer sequence) and the AlHV-1 BAC C500 as the template DNA. The PCR product was cloned into a pGEM-T Easy vector (Promega) and then subcloned by T4 DNA ligase (Roche) into the pTorsten. Similarly, the A3 coding sequence was amplified using the primers A3142S and A3deltastop#2 to produce a pcDNA3.1-A3V5-His plasmid using the pcDNA3.1/V5-His TOPO TA expression kit (Life Technologies). This vector was then used to amplify the A3V5-His sequence by using the primers NotIA3V5S and NotIA3V5AS and subcloned into the NotI restriction site of the pTorsten plasmid using an In-Fusion HD cloning kit (Clontech) to produce the pTorsten-A3V5-His expression vector. pEX-sema7A was generously obtained from M. Miura (University of Tokyo). Plasmid DNA was further prepared for transfection (Endofree Plasmid Maxi kit; Qiagen).
TABLE 1.
Oligonucleotides used in the study
| Target sequence | Primer | Sequence (5′–3′) | Length (bp) |
|---|---|---|---|
| ORF73 | ORF73-3598F | GGACTAGACCCTCTTTATGACCC | 244 |
| ORF73-3841R | CGAGCAAAATAGGAACCCATTGC | ||
| ORF09 (DPOL) | ORF09-2170F | GTAACCCTGCAAGGCAGAACC | 469 |
| ORF09-2638R | GTGAATAGGAAAGGGTCTGG | ||
| ORF22 (gH) | ORF22-239F | GTTTACTGATCAATCGATAAGAG | 952 |
| ORF22-1190R | CATCCACAAAGTACAGTGTCTGC | ||
| A3 | A3142S | GACATGGGCACTTTGTGTGTTAG | |
| A3712AS | GGCCGGTGCATGGCGGTGTC | 570 | |
| A3-Fc | XbaIA3142S | TCTAGAATGGGCACTTTGTGTGTTAG | 1,824 |
| A3deltastop | GACATCAGTGTCACATTCACCTC | ||
| A3-V5 | A3142S | GACATGGGCACTTTGTGTGTTAG | 1,818 |
| A3deltastop#2 | CTCGAGCATCAGTGTCACATTCACCTC | ||
| NotIA3V5S | CAGGAGCTCGCGGCCATGGGCACTTTGTGTGTTAG | 1,966 | |
| NotIA3V5AS | CGATGGGATCCGCGGCCTCATCAATGGTGATGGTGATGATG | ||
| A3-galK | SEMAFgalkF | GACACACCCTGACACCCAACACTGCTCAATGTCTACTTAACATGTAGTATCATCAATCCTGTTGACAATTAATCATCGGCA | 1,345 |
| SEMAFgalkR | GGTGAGTTTTTAGATACATCATCCACTTGTAAGACAATGTTGATAACAAGGACTGTGTCAGCACTGTCCTGCTCCTT | ||
| A3-rev | SEMAF | ACACACCCTGACACCCAACACTGCTCAATGTCTACTTAACATGTAGTATCATCAAT | 2,074 |
| SEMAR | GGTGAGTTTTTAGATACATCATCCACTTGTAAGACAATGTTGATAACAAGGACTGTG | ||
| A3-ns-galK | A3-NS-galkFwd | GTTTGGGGCTGATAACAGAGTTTGTAATACATCCTGCAGCTATGGGCACTCCTGTTGACAATTAATCATCGGCA | 1,331 |
| A3-NS-galkRev | GATTTAGCACTGTGATGGC TGATAAAATCATCAGTAATCTAATACTAACTCAGCACTGTCCTGCTCCTT | ||
| A3-ns | A3-NS-oligofwd | CTGATAACAGAGTTTGTAATACATCCTGCAGCTATGGGCACTTAGTGATGAATTCGTTAGTATTAGATTACTGATGATTTTATCAGCCATCACAGCTG | 98 |
| A3-NS-oligorev | CAGCTGTGATGGCTGATAAAATCATCAGTAATCTAATACTAACGAATTCATCACTAAGTGCCCATAGCTGCAGGATGTATTACAAACTCTGTTATCAG | ||
| A3-ns-rev | preH1A3S | GCAAGAGTGTACCATGTCTCC | 597 |
| A3407AS | GGTGGGTATTATGTGTGCTAG | ||
| A3-end-galK | SemaendgalKF | GAAGATTCGGTGACAGTGAAACTGTTAGAGGTGAATGTGACACTGATGCCTGTTGACAATTAATCATCGGCA | 1,331 |
| SemaendgalKR | GTTTTTAGATACATCATCCACTTGTAAGACAATGTTGATAACAAGGACTGTGTCAGCACTGTCCTGCTCCTT | ||
| A3-Fc | SemaFcF | GAAGATTCGGTGACAGTGAAACTGTTAGAGGTGAATGTGACACTGATGTCAATCACTAGTGAATTCGCGGCCGCG | 858 |
| SemaFcR | GTTTTTAGATACATCATCCACTTGTAAGACAATGTTGATAACAAGGACTGTGTCATTTACCCGGAGACAGGGAGAGGC |
Protein production.
HeLa cell monolayers were transfected at 70% confluence with 20 μg of expression vector DNA and 120 μg of polyethyleneimine (Polysciences) in 175-cm2 flasks in DMEM supplemented with 10% FCS. After incubation overnight, the medium was replaced with serum-free Iscove modified Dulbecco medium (IMDM; Gibco). After 48 h of incubation, supernatant of transfected cells was concentrated (Amicon, Millipore) with cutoffs of 50 and 100 kDa for AlHV-sema-V5 and AlHV-sema-Fc, respectively. Next, the concentrated supernatants were purified on HiTrap Protein A HP (GE Healthcare) for AlHV-sema-Fc and on a Hispur cobalt purification kit (Thermo Scientific) for AlHV-sema-V5. The protein concentration was determined by a BCA protein assay (Thermo Scientific), and the purity was verified by SDS-PAGE and Coomassie blue staining. In further experiments, purified hIgG1 (Calbiochem) and denatured AlHV-sema-Fc (95°C, 10 min) were used as negative controls for AlHV-sema-Fc. Supernatants of transfected cells by an empty pTorsten vector were used as a negative control for AlHV-sema-V5. The purified “empty” supernatant was used at the same volume that AlHV-sema-V5.
Western blot.
Lysates were denatured by heating (95°C, 5 min) and processed by SDS-PAGE in Laemmli buffer (31.25 mM Tris-HCl [pH 6.8], 1% [wt/vol] SDS, 12.5% [wt/vol] glycerol, 0.005% [wt/vol] bromophenol blue, 10% [vol/vol] β-mercaptoethanol). Where indicated, tunicamycin (1 μg/ml; Sigma) or brefeldin A (10 μg/ml; Sigma) was added to the medium. Proteins were separated by electrophoresis on a Mini-Protean TGX precast 4 to 15% resolving gels (Bio-Rad) in SDS-PAGE running buffer (25 mM Tris-base, 192 mM glycine, 0.1% [wt/vol] SDS) and transferred onto polyvinylidene difluoride membranes (Thermo Scientific, 0.45-μm pore size). The membranes were blocked with 3% skimmed milk in phosphate-buffered saline (PBS)–0.1% Tween 20. For detection of AlHV-sema fusion proteins, membranes were blotted with anti-human IgG goat polyserum (0.8 μg/ml; Invitrogen) or anti-V5 mouse monoclonal antibody (1 μg/ml; Invitrogen). Bound antibodies were detected with horseradish peroxidase-conjugated Zymax rabbit anti-goat IgG polyserum (Invitrogen) or rabbit anti-mouse IgG polyserum (Dako Corp.). For cofilin and p-cofilin immunoblotting, rabbit polysera and HRP-conjugated anti-rabbit polyserum were used (Cell Signaling Technology). Development was performed with an enhanced chemiluminescence substrate (GE Healthcare) and exposure to X-ray film.
Glycosylation prediction and oligosaccharide digestion.
Glycosylation sites were predicted using NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) and NetOGlyc 4.0 (http://www.cbs.dtu.dk/services/NetOGlyc/) glycosylation site predictors. A protein deglycosylation mix kit (New England BioLabs) was used. Samples were denatured in glycoprotein denaturing buffer (0.5% SDS, 40 mM dithiothreitol) for 10 min at 100°C and then digested for 3 h at 37°C with 20% protein deglycosylation mix (containing PNGase F [500,000 U/ml], O-glycosidase [40,000,000 U/ml], neuraminidase [50,000 U/ml], β1-4 galactosidase [8,000 U/ml], β-N-acetylglucosaminidase [4,000 U/ml]) in G7 reaction buffer (50 mM sodium phosphate [pH 7.5]) with 1% NP-40. Reactions were stopped by the addition of Laemmli sample buffer, and proteins were analyzed by immunoblotting.
siRNA transfection and knockdown.
Small interfering RNA (siRNA) duplexes targeting two sequences of the B. taurus plexinC1 (access no. NM_001192481) (GCUCCUCUGUGAUUCUUGAdTdT and CCCACAUCUUCACUGAAGAdTdT) or irrelevant control siRNA were designed and synthesized (Eurogentec). The siRNAs were transfected into BT cells plated on six-well dishes by using HiPerFect transfection reagent (Qiagen). After 24 h of the first transfection, the cells were collected for anti-PlexinC1 staining (cross-reacting anti-human/mouse MAb, mouse IgG2b; clone 544232[R&D Systems]) by flow cytometry to monitor the knockdown efficiency of the siRNA. The cells were then plated on 24-well plates before treatment with AlHV-sema.
Mutagenesis.
The AlHV-1 BAC clone was used to produce the recombinant plasmids using the galactokinase gene (galK) as selection marker in Escherichia coli (20). An A3-Fc tagged BAC plasmid was produced (see Fig. 3D). First, we replaced the stop codon of the A3 coding sequence by the galK sequence resulting in the C500 BAC A3-galK-end plasmid using the amplicon consisting of the galK gene flanked by 48/52-bp sequences surrounding A3 open reading frame (ORF) stop codon and generated by PCR using the pgalK vector (30) as the template, and primers SemaendgalKF and SemaendgalKR (Table 1). The second recombination process consisted in replacing the galK gene with the hIgG1 Fc sequence using an amplicon generated with the primers SemaFcF and SemaFcR and the pTorsten-A3Fc as the template. To produce the A3 deleted BAC plasmid (A3−) the recombination fragment used for galK-positive selection consisted of a galK flanked by 57-bp sequences corresponding to the regions directly flanking the A3 coding sequence (accession no. NC_002531, nt 3492 to 5453) in the AlHV-1 genome. This fragment was produced by PCR using the pgalK vector as a template, the forward chimeric primer SEMAFgalkF, and the reverse chimeric primer SEMAFgalkR (see Fig. 10A). The A3 revertant BAC plasmid (A3-rev) was produced by galK-negative selection with a recombination fragment produced by PCR using the AlHV-1 C500 BAC clone as a template and the primers SEMAF and SEMAR. A similar strategy was used to introduce three multistop codons at the 5′ end of the A3 coding sequence to produce the A3ns BAC plasmid (Fig. 10B). The galK gene was introduced by using a recombination fragment generated by PCR using the pgalK plasmid as the template and chimeric primers A3-NS-galkFwd and A3-NS-galkRev to produce the A3ns galK amplicon. These primers contain the 50-bp homology sequences flanking the “ttgtgt” sequence at the 5′ end of the ORFA3 coding sequence (accession no. NC_002531, nt 3542 to 3548). The synthesized oligonucleotides A3-NS-oligofwd and A3-NS-oligorev containing three stop codons and one EcoRI restriction site were then annealed and used to produce the A3ns BAC plasmid by galK-dependent negative selection. To produce the A3ns-rev strain, the A3ns galK amplicon was used to reintroduce the galK gene before negative selection was performed in the presence of the A3-WT amplicon amplified with preH1A3S and A3407AS primers and BAC wild-type (WT) plasmid as the template. All deletions and insertions were verified by restriction profiles and Southern blotting and sequencing of the recombination sites. All strains were reconstituted in MacT-cre cells before propagation in BT cells.
FIG 3.
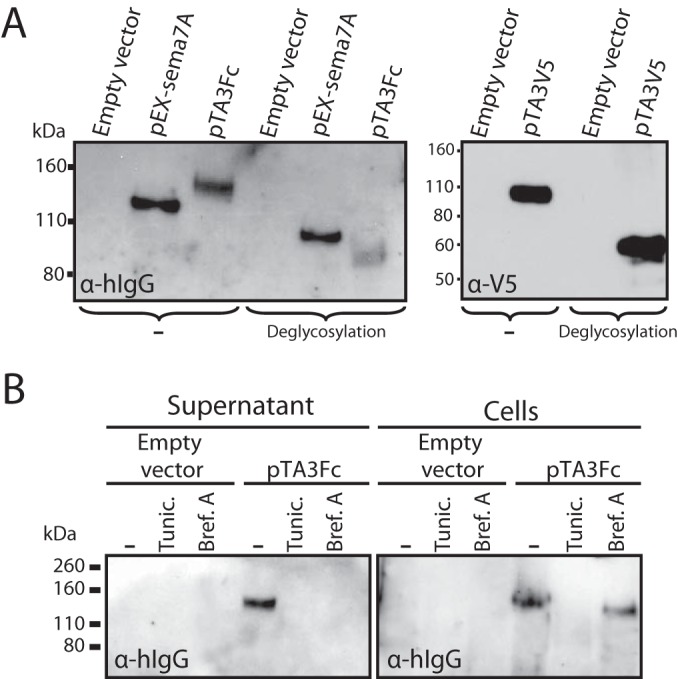
AlHV-sema is a 93-kDa secreted glycoprotein. (A) Immunoblot analysis of sema7A and AlHV-sema. HeLa cells were transfected with pEX-sema7A, pTA3Fc, pTA3V5, or empty vector. At 24 h, the proteins in the supernatant were analyzed by immunoblotting with anti-human IgG1 goat polyserum or anti-V5 MAb. Supernatants were left untreated (−) or subjected to deglycosylation treatment (PNGase F, O-glycosidase, neuraminidase, β1-4 galactosidase, β-N-acetylglucosaminidase) before SDS-PAGE and immunoblotting. The positions of molecular size markers are indicated in kilodaltons. (B) Transient expression of sema7A or AlHV-sema was performed in HeLa cells in the presence or absence of tunicamycin (Tunic.) or brefeldin A (Bref.A), and both secreted (Supernatant) or cell-associated (Cells) proteins were analyzed by immunoblotting.
FIG 10.

Impairment of A3 expression does not affect viral replication in vitro. (A and B) Recombineering methodologies used to delete the entire A3 coding sequence by galK insertion and generation of the A3− and A3-rev strains (A) or insert multistop codons at the 5′ end of the A3 coding sequence and generation of the A3ns and A3ns-rev strains (B). The produced BAC plasmids were analyzed by Southern blotting after EcoRI or HindIII restriction and ethidium bromide staining (EtBr). Probes are indicated as follows: A3, entire A3 coding sequence; galK, entire galK coding sequence. (C) Multistep growth curves of WT, A3−, A3-rev, A3ns, or A3ns-rev virus strains in BT fibroblasts. The data presented are means ± the standard deviations of results from measurements in triplicate. (D) Syncytial areas over time postinfection in MDBK cells. The data presented are means ± the standard deviations of results from syncytium size measurements (n = 25).
Southern blotting.
Southern blot analyses were performed as described previously (18).
Growth curves.
The in vitro growth kinetics of recombinant viruses were compared to those of the WT. Cells were infected (MOI = 0.01), and supernatants and infected cells were harvested at successive intervals. The total amount of infectious viral particles was determined by plaque assay on MDBK cells as described previously (18).
Syncytium size.
MDBK cells grown on glass coverslips were infected with the different strains (MOI = 3 × 10−4) and overlaid with MEM containing 10% FCS and 0.6% (wt/vol) carboxymethyl cellulose (Sigma) to obtain isolated syncytia. Syncytium areas were measured by using ImageJ software as described previously (28).
Kinetics of viral gene expression.
BT cells were incubated with cycloheximide (CHX; 100 μg/ml; Sigma-Aldrich) or phosphonoacetic acid (PAA; 300 μg/ml; Sigma-Aldrich) before infection with AlHV-1 WT strain (MOI = 0.05) in the presence of inhibitors. At 24 h postinfection, RNA was extracted (RNeasy Miniprep; Qiagen), and cDNA was synthesized using an Improm II reverse transcription system kit (Promega). AlHV-1 ORF73 (immediate-early [IE]), DNA polymerase (ORF09; early [E]), and glycoprotein H (ORF22; late [L]) were used as reference genes of the three kinetic classes (see Table 1 for primer sequences).
Animals and infection in vivo.
Six groups of specific-pathogen-free New Zealand white rabbits, each comprising six rabbits were used. Animals were inoculated intranasally with 3 × 105 PFU of the different AlHV-1 recombinant viruses in DMEM or with DMEM only for the mock-infected group. The rabbits were examined daily for clinical signs. According to bioethical rules, rabbits were euthanized when rectal temperature remained higher than 40°C for two consecutive days. The animal study performed had been accredited by the local ethics committee of the University of Liège (ethics protocols 1127 and 1571).
Quantitative PCR (qPCR).
Total DNA was extracted from peripheral blood mononuclear cells (PBMCs), popliteal lymph node (pLN) cells, or splenocytes using a QiaAmp DNA minikit (Qiagen). AlHV-1 genomic DNA copies were quantified using an AlHV-1 ORF3 real-time PCR with normalization on the cellular beta-globin genomic sequence, as described previously (17).
Leukocytic cell suspension preparation.
PBMCs were isolated from 5 ml of blood collected from the ear central artery before and at different time points after infection. Immediately after euthanasia, single-cell suspensions were prepared from the pLN and spleen as follows. Tissue biopsy specimens were delicately chopped in sterile RPMI media and passed through a 70-μm-pore-size cell strainer. Mononuclear leukocyte suspensions from peripheral blood and tissue samples were prepared with Ficoll-Paque Premium density gradient media (GE Healthcare). Then, a 5-ml single-cell suspension was diluted 1:1 in sterile PBS, overlaid onto a 5-ml Ficoll-Paque density cushion, and centrifuged (1,825 × g) for 20 min at room temperature. Mononuclear leukocytes at the interface were collected and washed in ice-cold PBS before further analysis.
Antibodies and flow cytometry.
Multicolor flow cytometry analysis of rabbit PBMCs was performed as described previously (19). Briefly, cells were incubated with a monoclonal antibody (MAb) anti-rabbit CD4 (IgG2a, KEN-4), CD8 (IgG1, 12C.7), and IgM (IgG1, NRBM) antibody cocktail and left on ice for 10 min. The cells were washed and further incubated for 10 min on ice with isotype-specific phycoerythrin (PE)-conjugated rat anti-mouse IgG1 (A85-1; BD Biosciences) and biotinylated rat anti-mouse IgG2a (R19-15; BD Biosciences) antibodies. After a third wash, the cells were incubated with fluorescein isothiocyanate-conjugated anti-rabbit T cells (KEN-5), Pacific blue anti-human CD14 (TÜK4) and allophycocyanin (APC)-conjugated streptavidin (BD Biosciences) before analysis. Antibodies were obtained from AbD-Serotec. For the binding assay of AlHV-sema, staining was revealed using Alexa Fluor 488-nm goat anti-human IgG polyserum (AlHV-sema-Fc) or MAb anti-V5 primary antibody, followed by secondary staining with Alexa Fluor 488-conjugated goat anti-mouse IgG polyserum (AlHV-sema-V5). Dendritic and macrophage cell lines were blocked with purified goat IgG (0.5 μg/ml). Anti-bovine major histocompatibility complex II (MHC-II; MAb 7C7) (31) and cross-reacting anti-human CD40 rabbit polysera (Enzo Life Sciences, a gift from F. Bureau, GIGA-ULg) were used. Acquisition was performed using a FACSAria or a FACSCanto (BD Biosciences), and data were analyzed by using FlowJo v10.0.7 software (Tree Star).
ELISA.
AlHV-1-coated plates (generous gift from Hong Li, Washington State University) were incubated with rabbit sera (diluted 1:100 in PBS [pH 7.4] containing 0.1% Tween 20 and 3% bovine serum albumin [BSA]). Bound antibodies were detected with alkaline phosphatase-conjugated goat anti-rabbit IgG polyclonal antibody (Sigma-Aldrich). Washes were performed with PBS (pH 7.4) containing 0.1% Tween 20. FAST p-nitrophenyl phosphate (Sigma-Aldrich) was used as the substrate, and the absorbance was read at 405 nm using a Bio-Rad iMark enzyme-linked immunosorbent assay (ELISA) plate reader.
IFN-γ RNA expression.
Quantification of IFN-γ expression and normalization on HPRT (hypoxanthine phosphoribosyltransferase) cellular genes were performed as described previously (32).
Generation of mouse bone marrow-derived macrophages.
Bone marrow cells were collected from BALB/c femurs and tibias. Cells were seeded into bacteriological petri dishes at 6 × 105 cells/ml in 20 ml of DMEM supplemented with 10% FCS and 30% L929 conditioned medium. At day 3, the medium was renewed, and the cells were collected at day 6 with cold PBS containing 5 mM EDTA and 4 mg of lidocaine (Sigma-Aldrich)/ml. The animal study performed had been accredited by the local ethics committee of the University of Liège (ethics protocol 1337).
Generation of bovine monocyte-derived DCs (moDCs).
Bovine PBMC (Clinique Vétérinaire Universitaire, ULg, Liège, Belgium) were isolated on Ficoll-Paque premium density gradient media (GE Healthcare) and incubated with anti-human CD14 microbeads (Miltenyi-Biotech). Labeled cells were enriched using LS columns according to the manufacturer's instructions (Miltenyi-Biotech). Cells were seeded at 106 cells/ml in six-well culture plates in IMDM (Gibco) supplemented with 10% FCS and a 5% bovine dendritic cell growth kit (AbD Serotec) containing mixed bovine recombinant interleukin-4 (rIL-4) and recombinant granulocyte-macrophage colony-stimulating factor (rGM-CSF). On day 3 of culture, nonadherent cells were harvested with cold PBS and matured for 2 h with lipopolysaccharide (LPS; 2 μg/ml; E. coli O111:B4 [Sigma-Aldrich]) before further experiments.
Adoptive transfer of rabbit BMDCs.
IIIEP/J syngeneic rabbits were used for BMDC generation and adoptive-transfer experiments (the breeders were generously provided by Neil Christensen, Pennsylvania State University College of Medicine) (33). Bone marrow-derived dendritic cells (BMDCs) were generated as described previously (34). Briefly, femurs were dissected, and the marrow was flushed out of the bone using 40 ml of IMDM. The marrow was then gently homogenized by using a 5-ml syringe plunger and passed through a 100-μm-pore-size cell strainer, and red cells were lysed with ammonium chloride lysis buffer. The cells were cultured in six-well tissue culture plates at 4 × 106 cells/ml at 37°C in IMDM containing 10% FCS. After 3 h of incubation, nonadherent cells were removed, and medium was replaced with fresh IMDM containing 20 ng of human recombinant GM-CSF/ml and 100 ng of IL-4 (R&D Systems)/ml. The cells were cultured for 7 days, and medium was replaced after 3 days with fresh medium supplemented with the appropriate cytokines. LPS (E. coli O111:B4; Sigma-Aldrich) was added the day before harvesting the cells. The loosely adherent matured DCs were harvested and the upregulation of MHC-II DQ (clone 2C4, 5 μg/ml [AbD Serotec]) controlled using flow cytometry (data not shown). Cells were counted and cultured in bacterial petri dishes at 5 × 105 cells/ml and treated with AlHV-sema-V5 (50 nM) or an adequate volume of empty vector control during 3 h at 37°C. Cells were then harvested and labeled with 1 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; CellTrace CFSE kit [Life Technologies]) according to a published protocol (35). Labeled cells were then injected subcutaneously into the footpads of anesthetized 4-month-old male IIIEP/J rabbits (35 mg of xylazine and 5 mg of ketamine/kg, given intramuscularly). Draining popliteal lymph nodes were harvested 48 h later for analysis. The animal studies performed have been accredited by the local ethics committee of the University of Liège (ethics protocols 1127 and 1571).
Cytoskeleton.
BT and bovine moDCs were grown on uncoated or poly-d-lysine (0.1 mg/ml, Sigma-Aldrich)-coated glass coverslips, respectively. Cells were treated with 50 nM the different protein productions at 37° in 5% CO2. Where indicated, cells were treated with Y-27632 ROCK inhibitor (Millipore). After treatment, F-actin was stained with Alexa Fluor 568-nm-conjugated phalloidin (Life Technologies) on paraformaldehyde-fixed cells that had been permeabilized in 0.1% NP-40 in PBS. Pictures were captured with a charge-coupled device (CCD) camera system (Leica, DM2000LED).
Phagocytosis assay.
Bovine moDCs (105 cells/well) were cultured in round-bottom 96-well plates in IMDM (Gibco) containing 10% FCS. Following 3 h of treatment, Alexa Fluor 488-conjugated E. coli (K-12 strain) BioParticles (Life Technologies) were added to reach a ratio of five particles per cell. After different incubation periods, the plates were washed three times with cold PBS to remove excess bacteria before fluorescence measurement by flow cytometry.
Transwell migration assay.
Low density cells from sheep skin lymph were obtained by pseudoafferent lymph duct cannulation as described previously (36). This protocol was approved by the Committee on the Ethics of Animal Experiments of the INRA research center in Jouy-en-Josas and AgroParisTech (0 11-019). A transwell migration assay was performed as described elsewhere (37). Sheep lymph that had been enriched in DCs by density gradient were suspended in migration medium (0.5% BSA in RPMI) and distributed in the upper chamber of a 24 Transwell system insert (5-μm pore size; Corning). The lower chamber was filled with migration medium alone or containing 100 ng of recombinant mouse XCL-1 (R&D Systems)/ml. Cells were incubated for 3 h at 37°C in 5% CO2. A constant number of polybead polystyrene microspheres (Polyscience) was added in each lower chamber before harvesting migrated cells. Migrated cells were then stained and analyzed by flow cytometry. A constant event of microspheres for each well was acquired to obtain the absolute cell numbers recovered after migration. The percentage of migrated cells was calculated by the following formula for each subset: [(number of migrated cells)/(total number of input cells)] × 100. The CD1b+ and CD26+ cells subsets were stained with mouse MAbs against CD1b (mouse IgG2a, clone TH97A) and CD26 (mouse IgG1, clone CC69), followed by biotin-labeled rat anti-IgG2a and PE-labeled rat anti-mouse IgG1 and a final incubation with streptavidin-APC (BD Biosciences) (38, 39).
Histological analysis.
Organ explants from mock-infected or infected animals were fixed in 10% buffered formalin and embedded in paraffin blocks. Sections (5 μm) were then stained with hematoxylin and eosin prior to microscopic analysis performed with a CCD camera system (Leica, DM2000LED).
Statistical analysis.
Statistical analyses were conducted using GraphPad Prism v6 software.
RESULTS
Herpesvirus semaphorins are phylogenetically close to mammalian sema7A and evolved independently of poxvirus semaphorin-like genes.
Sema7A has been identified by its sequence homology to viral semaphorins (10). To further analyze viral semaphorin peptidic similarities to sema7A, we performed phylogenetic analyses and compared known viral semaphorin peptidic sequences to sema7A orthologs of mammals, birds, reptiles, and fishes (Fig. 1). We observed that the semaphorins from herpesviruses clustered with the mammalian sema7A sequences, whereas semaphorin homologs of poxviruses showed a much higher divergence. In particular the cluster of semaphorin sequences from orthopoxviruses formed an outgroup to the sema7A cluster and avipoxvirus semaphorins branched with reptile and bird semaphorins. These analyses suggested independent acquisitions for the orthopox-, avipox-, and herpesvirus semaphorins.
FIG 1.

AlHV-sema is phylogenetically close to mammalian sema7A. Phylogenetic analysis of mammalian sema7A and viral homologs. The sequences were extracted from public databases GenBank and Ensembl and were aligned with MUSCLE, and a maximum-likelihood tree was computed as described in Materials and Methods. The accession numbers are as follows: AlHV-1, NP_065506; AlHV-2, YP_009044389; OvHV-2, YP_438129; vaccinia virus, AAA48169; variola virus, NP_042192; fowlpoxvirus, NP_039010; camelpoxvirus, NP_570549; cowpoxvirus, NP_619959; ectromelia virus, NP_671658; canarypoxvirus, NP_955088; taterapoxvirus, YP_717471; Anas platyrhynchos, ENSAPLP00000010932; Anolis carolinensis, ENSACAP00000016773; Takifugu rubripes, ENSTRUP00000034641; Oryzias latipes, ENSORLP00000009077; Gasterosteus aculeatus, ENSGACP00000006179; Tetraodon nigroviridis, ENSTNIP00000011250; Rattus norvegicus, NP_001101623; Equus caballus, XP_001917710; Bos taurus, NP_001193114; Gallus gallus, NP_001186678; Homo sapiens, NP_003603; Mus musculus, NP_035482; and Danio rerio, XP_002663156.
AlHV-sema is very similar to sema7A in its structure and has conserved domains interacting with plexinC1.
Phylogenetic divergence between pox- and herpesvirus semaphorins could presage distinct functions of the proteins. Thus, alignment of viral semaphorins to human sema3A, sema4D, and sema7A and to bovine sema7A was performed and analyzed based on recent published data (10, 40, 41) (Fig. 2A). Key residues involved in the interactions with the plexinC1 receptor were highly conserved between the herpesvirus semaphorins and human and bovine sema7A (blade 3, 4c-4d loop, and extrusion helix 2), suggesting that AlHV-sema can bind plexinC1 similarly to its cellular homolog as previously suggested (11). These residues were however more divergent in A39R, suggesting that the poxvirus semaphorin has evolved independent mechanisms to bind the same receptor (42). We next sought to predict the 3-D structure of AlHV-sema. Modeling of the 3-D structure of AlHV-sema was performed based on the recently published crystal structure of sema7A (Fig. 2B) (42). We observed a strong similarity between AlHV-sema and the seven-blade β propeller of sema7A. Together, our results demonstrate that the predicted structure of AlHV-sema is highly similar to sema7A and the predicted binding of AlHV-sema to plexinC1 is nearly identical to sema7A.
FIG 2.

Sequence alignment of human sema3A, sema4D, and sema7A, bovine sema7A and A39R, OvHV-sema, and AlHV-sema. (A) CLUSTALW was used to align peptidic sequences. Sequence analyses of sema7A and viral homologs were extrapolated from data available for other semaphorins (41). The SEMA domain is boxed in black. Extrusion and PSI domains are highlighted in light blue and boxed in blue, respectively. The 70-residue section of sema3A implicated in receptor specificity is underlined in black (53). The regions involved in the binding of sema7A and A39R to plexinC1 (blade 3, 4c-4d loop, and extrusion helix 2) are boxed in pink. Key residues involved in bonding interaction with plexinC1 are highlighted in blue for sema7A and macavirus semaphorins and in green for A39R (12). Conserved structural determinant in the 4c-4d loop are highlighted in yellow. RGD motives at the base of the sema7A 4c-4d loop are underlined in red. Cysteines conserved in the semaphorins are highlighted in red. The figure was produced with CLC Sequence Viewer v.7.0.2 and modified using Adobe Illustrator CS5. (B) 3-D prediction of the structure of AlHV-sema based on the crystal structure of human sema7A (12) (http://zhanglab.ccmb.med.umich.edu/I-TASSER/).
AlHV-sema is a 93-kDa glycoprotein expressed in the early phase of viral infection.
Whereas sema7A is expressed at the cell surface through a GPI anchor motif, AlHV-sema is predicted as a secreted protein. We transiently expressed AlHV-sema as Fc- or V5-tagged (Fig. 3) proteins in HeLa cells. Immunoblotting analysis using anti-human IgG1 or anti-V5 antibodies (Fig. 3A) detected 130- and 100-kDa glycosylated proteins, respectively. An Fc-tagged truncated human sema7A was used as positive control (pEX-sema7A) (43). matrix-assisted laser desorption ionization–mass spectrometry (MALDI-MS) analyses revealed that AlHV-sema-Fc and AlHV-sema-V5 monomers are 122- and 97-kDa proteins, respectively (data not shown). After subtraction of the human IgG1 Fc fragment and V5-His epitope molecular masses, we could infer that AlHV-sema is a 93-kDa glycoprotein. Inhibition of glycosylation with tunicamycin further confirmed the glycosylation of AlHV-sema, and brefeldin A treatment impaired the secretion of the protein (Fig. 3B). We next sought to determine the A3 gene expression during virus infection. Expression kinetics of the A3 gene was first investigated after AlHV-1 infection in presence or absence of CHX or PAA (Fig. 4A). Whereas CHX inhibited A3 gene expression, RNA could be detected in the presence of PAA, demonstrating that A3 is an early gene. ORF73, DNA polymerase, and gH were used as immediate-early, early, and late control viral genes, respectively. To determine whether A3 gene expression actually lead to AlHV-sema secretion in infected cells, we inserted the human IgG1 Fc region in frame to the A3 coding sequence to produce the BAC A3Fc recombinant virus (Fig. 4B and C). This virus replicated similarly to the WT virus in BT cells (not shown). Although gp115 complex expression in infected cells could only be detected from 18 h postinfection (p.i.), AlHV-sema expression could be observed as early as 8 h p.i. and from 22 h p.i. (weak signal, MOI = 0.01) in the supernatant (Fig. 4D and E). Together, these results indicated that AlHV-sema is a glycoprotein of 93 kDa that is secreted during the early phase of AlHV-1 infection.
FIG 4.

A3 is an early gene encoding a 93-kDa glycoprotein secreted during virus infection. (A) Determination of the A3 kinetic class of transcription. BT cells were infected with AlHV-1 and treated with CHX or PAA or left untreated (−). At 24 h p.i., the expression of ORF73 (IE gene), ORF09 (DPOL, E gene), ORF22 (gH, L gene), or A3 was determined by using a reverse transcription-PCR approach, as described in Materials and Methods. (B) Recombineering methodology used to insert a carboxy-terminal hIgG1 Fc fragment tag to the A3 gene and produce the BAC A3Fc strain. (C) The BAC constructs were analyzed by Southern blotting after EcoRI restriction. Probes are indicated: galK, entire galK coding sequence; A3, entire A3 coding sequence. (D) AlHV-sema kinetics of expression after infection with the BAC A3Fc virus (MOI = 0.01). Infected BT cells were fixed and permeabilized at the given time points postinfection before staining with Alexa Fluor 488-nm anti-human IgG1 goat polyserum for AlHV-sema-Fc detection or MAb 15-A primary antibody (specific to gp115 viral complex), followed by Alexa Fluor 488-nm goat anti-mouse IgG secondary antibody. (E) Supernatants were collected and analyzed by immunoblotting with anti-human IgG1 polyserum.
AlHV-sema binds to rabbit, bovine, ovine, and wildebeest cell surface.
In order to further investigate AlHV-sema biological functions, we sought to determine its capacity to bind to the cell surface of different cell types. Vaccinia virus A39R has been shown to bind to plexinC1-expressing myeloid cells (13). Thus, we purified AlHV-sema to test its binding to the surface of mouse bone marrow-derived macrophages, rabbit kidney cells and CD14+ blood monocytes, bovine macrophages (BoMac), bovine and wildebeest CD14+ monocytes, fibroblasts isolated from bovine nasal turbinates (BT), monocyte-derived bovine DCs, and sheep lymph CD1b+ DCs (Fig. 5A). Whereas AlHV-sema binding to murine cell surface could not be detected, AlHV-sema binding could be detected on all tested cell types of rabbit, bovine, ovine, and wildebeest origin. Further confocal analysis showed that both Fc- and V5-tagged AlHV-sema could be detected at the surface of BT cells (Fig. 5B).
FIG 5.

AlHV-sema cell surface binding. (A) Flow cytometry analysis of AlHV-sema-Fc (500 nM) binding to the cell surface of mouse bone marrow-derived macrophages (BMDM), bovine macrophage (BoMac) and bovine turbinate (BT) cell lines, rabbit kidney cells (RK13), wildebeest, bovine, and rabbit CD14+ blood monocytes, bovine monocyte-derived DCs (moDCs), and sheep CD1b+ lymph DCs. Purified human IgG1 (500 nM; Calbiochem) and PBS only were used as negative controls. Dendritic and macrophage cell lines were blocked with purified goat IgG (0.5 μg/ml). Staining was revealed using Alexa Fluor 488-nm goat anti-human IgG polyserum. The median fluorescence intensities are indicated for each condition. (B) Confocal analysis of AlHV-sema-Fc and -V5 staining (50 nM) at the surfaces of BT cells. PKH26 counterstaining is shown.
AlHV-sema induces actin filament condensation in bovine fibroblasts and monocyte-derived DCs.
Semaphorin binding to their plexin receptors induces intracellular signaling events generally resulting in the suppression of actin dynamics and inhibition of integrin-mediated adhesion to the extracellular matrix that leads to repulsion, a phenomenon that can be visualized in vitro through cell retraction and/or detachment (44). Treatment of BT cells with both AlHV-sema-Fc- and AlHV-sema-V5-tagged proteins resulted in cell retraction, loss of lamellipodia, and condensation of F-actin filaments specifically stained with phalloidin (Fig. 6A). This phenotype was not observed in cells treated with respective controls such as PBS, hIgG1, heat-denatured AlHV-sema-Fc, or empty vector control. Cell retraction due to AlHV-sema was not associated with increased cell death (data not shown). We next sought to determine whether AlHV-sema could induce repulsion in DCs. We differentiated DCs from bovine CD14+ monocytes and incubated for different duration the obtained moDCs with both forms of AlHV-sema (Fig. 6B). Likewise, bovine moDCs showed a retracted phenotype and condensation of actin filaments, suggesting that AlHV-sema induces cytoskeleton rearrangements.
FIG 6.

AlHV-sema induces actin filament condensation. (A) BT cells were seeded onto glass coverslips in 24-well plates at a density of 30,000 cells per well and left 2 h at 37°C. Purified AlHV-sema proteins and controls were then added to the wells during the indicated time points before Alexa Fluor 568-nm phalloidin staining. The percentage of condensed cells was then calculated for each condition and plotted on a graph. The results are representative of three independent experiments. (B) moDCs were differentiated from bovine CD14+ monocytes during 3 days in a GM-CSF–IL-4 cocktail. DCs were then detached by pipetting, seeded in 24-well plates at a density of 105 cells per well, and left for 2 h at 37°C in the presence of LPS (2 μg/ml) for DC maturation. Purified AlHV-sema proteins and controls were added to the wells during the indicated time points before Alexa Fluor 568-nm phalloidin staining. The percentage of condensed cells was then calculated for each condition and plotted on the graph. The results are representative of two independent experiments.
PlexinC1 knockdown impairs AlHV-sema-mediated actin filament condensation.
To determine whether AlHV-sema signals through plexinC1 to induce cytoskeleton rearrangement, we used an siRNA strategy to knockdown plexinC1 expression in BT cells. We observed plexinC1 expression in BT cells (Fig. 7A) that was significantly reduced 24 h after siRNA transfection (Fig. 7B). BT cells were then harvested, plated, and incubated with AlHV-sema-Fc. AlHV-sema treatment induced actin filament condensation in untransfected BT cells and BT cells transfected with an irrelevant control siRNA (Fig. 7C). However, BT cells transfected with siRNAs targeting plexinC1 did not display cytoskeleton rearrangement upon AlHV-sema treatment. These results suggest that AlHV-sema signals through plexinC1 to induce actin filament condensation.
FIG 7.

AlHV-sema causes actin filament condensation through plexinC1 and induces cofilin phosphorylation through a ROCK-dependent signaling pathway. (A) BT cells express plexinC1. Cells were passaged and harvested 24 h later for flow cytometry analysis using an anti-human/mouse plexinC1 cross-reacting MAb. (B) PlexinC1 knockdown in BT cells. Cells were transfected with siRNA duplexes targeting bovine plexinC1 or irrelevant control siRNA. They were harvested 24 h later, followed by analysis of the plexinC1 expression levels using flow cytometry. Expression levels are given in median fluorescence intensities. The results are representative of three independent experiments. (C) PlexinC1 knockdown impairs actin filament condensation after AlHV-sema treatment. Cells were transfected with siRNAs and treated 24 h later with AlHV-sema-Fc or control hIgG1 (50 nM, 3 h) before phalloidin staining. (D) Immunoblot of phospho-cofilin after different time points of treatment with AlHV-sema or hIgG1 (50 nM) of BT cells. (E) Immunoblot of phospho-cofilin after 30 min of Y-27632 (1 μM) treatment, followed by a 10-min treatment with AlHV-sema or hIgG1 (50 nM) of BT cells. A total cofilin immunoblot was performed to control equivalent loading. (F) Phalloidin staining of BT cells after Y-27632 treatment, followed by treatment with AlHV-sema (50 nM, 1 h) or control.
AlHV-sema inhibits cofilin through a ROCK-dependent pathway.
Cofilin phosphorylation at Ser3 inhibits its binding to actin and the promotion of polymerization/depolymerization of actin filaments (44). AlHV-sema treatment of BT cells induced a transient phosphorylation of cofilin as soon as 5 min after incubation (Fig. 7D). Cofilin phosphorylation induction by AlHV-sema was, however, inhibited after treatment with the specific inhibitor of the Rho-associated protein kinase (ROCK), Y-27632 (Fig. 7E). Inhibition of ROCK signaling pathway with Y-27632 further impaired cell condensation (Fig. 7F). These results suggested that AlHV-sema induces cytoskeleton rearrangement through ROCK-dependent signaling and cofilin phosphorylation.
Cytoskeleton rearrangement induced by AlHV-sema inhibits DC phagocytosis without affecting DC activation.
Condensation of actin filaments through plexinC1 might result in a lesser ability of DCs to mediate phagocytosis and cell migration (13, 14). We differentiated DCs from bovine CD14+ monocytes using GM-CSF and IL-4 and assayed the uptake of fluorescent bioparticles of E. coli. Uptake at 37°C significantly increased over time, whereas it remained lower when kept at 4°C (Fig. 8A). Treatment with AlHV-sema-Fc or AlHV-sema-V5 significantly reduced DC-mediated uptake of E. coli particles, as shown by the reduced fluorescence intensities of DCs at 10 and 30 min of incubation compared to control treatments (Fig. 8B). To further determine whether AlHV-sema treatment affects the activation state of DCs, we used flow cytometry to measure the expression levels of MHC-II and CD40 costimulation receptors. We observed that both receptors were similarly expressed on moDCs treated or not with AlHV-sema, suggesting the inhibition of phagocytosis did not affect DC activation (Fig. 8C). Inhibition of ROCK using Y-27632, however, restored phagocytosis to a similar level as control treatments, suggesting that the ROCK-dependent pathway is implicated in AlHV-sema-mediated inhibition of phagocytosis (Fig. 8D).
FIG 8.

AlHV-sema inhibits DC phagocytosis. (A and B) Bovine moDCs were grown to maturity in the presence of LPS (2 μg/ml) before treatment with AlHV-sema (50 nM, 3 h). Human IgG1, heat-denatured AlHV-sema-Fc, and empty vector supernatant were used as controls. Alexa Fluor 488-nm-conjugated E. coli bioparticles were then added to the culture before cell harvesting at the indicated time points. Representative histograms of flow cytometry analysis are shown with the percentages of Alexa Fluor 488-nm-positive cells (A). Mean fluorescence intensities were measured by flow cytometry and plotted on graph (B). The data show the means ± the standard errors of the mean (SEM; n = 3) and are representative of two independent experiments. A two-way analysis of variance (ANOVA) with a Bonferroni post test was performed (*, P < 0.05). (C) Bovine moDCs matured with LPS (2 μg/ml) and treated with AlHV-sema-V5 (50 nM) or empty control during 3 h were stained with anti-bovine MHC-II (MAb 7C7) and cross-reacting anti-human CD40 rabbit polysera. (D) Bovine moDCs were used in a phagocytosis assay, as in panel A, and Y-27632 ROCK inhibitor was used (1 μM) before AlHV-sema (50 nM) or control treatments where indicated. The data show means ± the standard deviations (n = 3). One-way ANOVA with a Bonferroni post test was performed (*, P < 0.05).
Cytoskeleton rearrangement induced by AlHV-sema inhibits DC migration to the draining lymph node.
In addition to the inhibition of phagocytosis, AlHV-sema might impair DC migration to the draining lymph node (LN). To test this hypothesis, BMDCs were generated from the femur bone marrow cells of IIIEP/J inbred rabbits before maturation and treatment with AlHV-sema. BMDCs were then labeled with CFSE before injected subcutaneously into the footpad of syngeneic IIIEP/J rabbits according to a published protocol (45). Draining popliteal LN were harvested 2 days later, and the absolute numbers of migrated CFSE+ DCs were analyzed by flow cytometry (Fig. 9A). BMDC treatment with AlHV-sema significantly reduced the percentages (Fig. 9B) and numbers (Fig. 9C) of CFSE+ cells in the draining LN 2 days after injection, demonstrating that the viral semaphorin action on cytoskeleton impacts DC function. A39R has been shown to inhibit mouse BMDC transwell migration (13) and phagocytosis of apoptotic bodies, indirectly inhibiting effective cross-presentation to CD8+ T cells (14). Effective activation of antiviral CD8+ T cells is dependent on DC-mediated cross-presentation, and this cross-priming mechanism relies on XCL-1 interaction with XCR-1-expressing cross-presenting DCs (46, 47). Thus, we used an established transwell migration assay of lymph sheep CD1b+ CD26+ DCs, a cell subset homologous to specialized cross-presenting mouse CD8α+ DCs (37). We observed that treatment with AlHV-sema inhibited transwell migration of CD1b+ CD26+ DCs (Fig. 9D), suggesting that cross-presenting DC migration is affected. Together, these results indicated that AlHV-sema interferes with DC antigen processing and presentation to T cells in the draining LN.
FIG 9.

AlHV-sema inhibits DC migration. (A to C) Bone marrow-derived DCs were differentiated from the femurs of a IIIEP/J male rabbit as described in Materials and Methods. BMDCs were matured overnight using E. coli LPS (2 μg/ml) before harvest and treatment with AlHV-sema-V5 (50 nM, 5 × 105 cells/ml) or empty vector control at 37°C during 3 h in bacterial petri dishes. Cells were then collected, counted, CFSE labeled, and injected in the footpads of IIIEP/J syngeneic male rabbits (2 × 106 cells in 0.5 ml per footpad), together with 5 μg of AlHV-sema-V5. Popliteal draining LNs and axillary nondraining LNs were harvested 2 days later, the cells were put into a single-cell suspension and counted, and the percentages of the migrated CFSE+ DCs were measured using flow cytometry (A). Dot plots show representative results for one rabbit. The percentages (B) and absolute numbers (C) of CFSE+ cells that migrated to the pLNs are plotted. The data are shown as means ± the SEM (n = 4). A Mann-Whitney unpaired t test was performed (*, P < 0.05). (D) Transwell migration assay on enriched sheep lymph subjected to recombinant mouse XCL-1 (100 ng/ml) and AlHV-sema (50 nM) treatment. Human IgG1, heat-denatured AlHV-sema-Fc, and empty vector supernatant were used as controls. Migrated cells were subjected to CD1b and CD26 double staining before flow cytometry analysis and the acquisition of 2,500 polystyrene microspheres. Migration is expressed as the percentage of input cells in the CD1b+ CD26+ gate. Bars show means ± the SEM of independent analyses in duplicates of three different sheep lymph samples. One-way ANOVA with a Bonferroni post test was performed (*, P < 0.05; **, P < 0.01).
Absence of A3 expression does not affect viral replication in vitro and does not impair the development of MCF in rabbits.
AlHV-sema could affect MCF lymphoproliferative lesions through immune evasion mechanisms and/or direct effect on latently infected lymphocyte activation (23). Thus, the role of AlHV-sema in the pathogenesis of MCF was addressed by producing A3 deletion (A3−) and A3 nonsense (A3ns) recombinant viruses, together with their respective revertant strains (Fig. 10A and B). The lack of A3 expression did not affect viral growth in vitro (Fig. 10C) and did not affect plaque sizes over time (Fig. 10D). We then used the rabbit model to determine the hypothetical role of AlHV-sema during MCF. After intranasal infection with 3 × 105 PFU, the development of MCF was monitored. Rabbits infected with the A3− or A3ns recombinant viruses developed MCF typical hyperthermia, LN and spleen hypertrophy, and histopathological lesions and did not survive the infection (Fig. 11A to C). We further observed that rabbits infected with the A3− or A3ns recombinant strains showed CD8+ T cell expansion similar to control WT and revertant viruses (Fig. 11D). We next determined the viral load in vivo following rabbit infection in lymphoid tissues by quantitative PCR (qPCR). We observed similar viral loads at the time of euthanasia in PBMCs, pLNs, and spleens from rabbits infected with each virus strains (Fig. 11E). Guo et al. (23) suggested that upregulation of sema7A in SaHV-2-infected lymphocytes was associated with increased IFN-γ production. As expected, we observed increased IFN-γ gene expression levels in MCF-developing rabbits (19); however, we did not observe any significant reduction of IFN-γ expression in the absence of A3 (Fig. 11F). Finally, we tested the ability of A3− or A3ns recombinant strains to induce antiviral specific antibodies (Fig. 12). Similar levels of anti-AlHV-1 antibodies could be detected in the serum of all rabbit infected groups. Together, these results demonstrated that AlHV-sema is not essential for the induction of MCF and that the lack of A3 does not affect expansion or activation of latently infected CD8+ T cells and does not alter the humoral immune response against the virus infection in rabbits.
FIG 11.

Malignant catarrhal fever induction in the absence of AlHV-sema. (A) Cumulative incidence of survival of the rabbits of each group. (B) Popliteal LNs and spleen masses at the time of euthanasia. Bars represent means ± the SEM. A one-way ANOVA with Bonferroni's post test was performed (*, P < 0.05; **, P < 0.01). (C) Histopathological characterization of MCF lesions. Kidney, liver, and lung sections of one rabbit representative of each group are shown. White arrowheads indicate typical infiltrations of lymphoblastoid cells. Abbreviations: A, arterioles; Av, alveoli; B, small bile ducts; Hp, hepatocytes; RC, renal corpuscles; T, uriniferous tubules; V, veins, Br, bronchi. Equivalent results were obtained in two independent experiments. Original magnification, ×200. (D) Percentages of CD8+ cells in the gated T cell population analyzed by flow cytometry at regular intervals throughout the experiment. Rabbits were identified according to the day of euthanasia postinfection (see numbers, n = 5 to 6). (E) qPCR of viral genome copies in PBMCs, pLNs, and spleens at the time of euthanasia. Real-time PCR quantification was normalized on β-globin cellular genomic sequence. The data are plotted as individual measurements. Bars show means ± the SEM (n = 5 to 6). (F) Relative IFN-γ RNA expression at time of euthanasia in the spleen. The relative expression levels were normalized to the HPRT copy numbers. Bars show means ± the SEM (n = 5 to 6).
FIG 12.
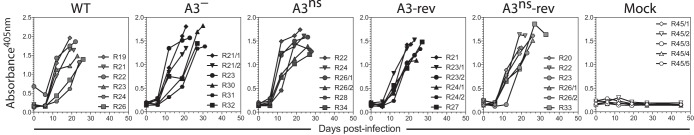
Humoral immune response in the absence of AlHV-sema. Serum samples were collected at different time points postinfection for each rabbit, and the relative quantities of anti-AlHV-1 antibodies were measured by indirect ELISA. The data are plotted as individual measurements. Rabbits were identified according to the day of euthanasia postinfection (see numbers, n = 5 to 6).
DISCUSSION
We have addressed the role of AlHV-sema, a gammaherpesvirus semaphorin encoded by the A3 gene of AlHV-1. We have shown that AlHV-sema is a 93-kDa glycoprotein that is secreted during the early phase of virus infection. Phylogenetic and bioinformatics analyses further demonstrated that AlHV-sema shares high sequence and structural similarities with cellular sema7A, in particular in the sema7A-plexinC1 interaction domains. Phylogenetic analyses revealed that poxvirus and macavirus semaphorins have been acquired independently during evolution, suggesting that they might have distinct activities.
A39R has been suggested to inhibit the activation of antiviral CD8+ cytotoxic T cells through impairment of DC phagocytosis of apoptotic bodies, resulting in the inhibition of antigen cross-presentation (14). Thus, the capability of AlHV-sema to also mediate immune evasion properties targeting DC functions was addressed. AlHV-sema binding to mouse macrophages could not be detected (Fig. 5A), and we did not observe any significant effect of the protein on mouse BMDC phagocytosis (data not shown). However, AlHV-sema was able to specifically bind to the surface of rabbit, bovine, ovine, and wildebeest cells such as fibroblasts, macrophages, monocytes, and DCs. AlHV-sema has been shown to display weaker binding to human plexinC1 compared to A39R (11). Therefore, we chose to use ruminant or rabbit cells to investigate AlHV-sema biological activities. In fibroblasts and DCs, AlHV-sema treatment induced repulsion with cell contraction and F-actin condensation, and this phenotype was impaired after siRNA-mediated knockdown of plexinC1 expression in BT cells, further suggesting that AlHV-sema signals through plexinC1. Cytoskeleton rearrangement was explained by actin condensation through inhibition of cofilin that involved ROCK. Cofilin-dependent cytoskeleton rearrangement was observed after A39R treatment of mouse DCs (13) and cofilin phosphorylation has also been reported in melanocytes after sema7A signaling through plexinC1 (48, 49) or sema3A signaling in neurons (50). Cofilin phosphorylation results in the inactivation of actin dynamics by inhibition of F-actin severing and nucleation (44). ROCK mediates Lin-11/Isl-1/Mec-3 kinase (LIMK) phosphorylation that in turn mediates the phosphorylation of cofilin (51, 52). Inhibition of ROCK impaired cofilin phosphorylation by AlHV-sema signaling and also impaired AlHV-sema-mediated cell condensation in BT cells. ROCK has also been shown to phosphorylate myosin light chain leading to the contraction of actin fibers (51), a phenomenon that together with inhibition of actin dynamics could explain the observed cell contraction. ROCK activation triggered by AlHV-sema likely results from signaling through plexinC1 cytoplasmic segmented GTPase-activating protein domains and GTPase-binding domain resulting in activation of a Rho-dependent signaling cascade, ultimately leading to cofilin phosphorylation and cell contraction.
Using bovine moDCs and rabbit BMDCs, we further demonstrated that AlHV-sema-mediated cytoskeleton rearrangement in DCs resulted in the inhibition of their capability to mediate phagocytosis and migrate to the draining lymph node. The expression levels of surface MHC-II or CD40 costimulatory receptor were not affected in bovine moDCs, suggesting that AlHV-sema does not affect DC activation. ROCK inhibition impaired AlHV-sema-mediated inhibition of phagocytosis by bovine moDCs, further suggesting that the ROCK/LIMK/cofilin regulation pathway is triggered by AlHV-sema in DCs. Based on these results, we further addressed the effect of AlHV-sema on the migratory capability of a DC subset specialized in antigen cross-presentation. XCL-1 is a potent chemoattractant for XCR-1-expressing DC subset in several species (37), and effective antigen cross-presentation to CD8+ T cells is dependent on XCL-1–XCR-1 interactions (46). Crozat et al. demonstrated that sheep CD1b+ CD26+ skin DCs are homologous to cross-presenting mouse CD8α+ DCs through the expression of high levels of XCR-1 and selective attraction by XCL-1 (37). Targeting transwell migration of this specific cell subset, we demonstrated that AlHV-sema inhibits XCL-1-dependent migration of cross-presenting DCs. Thus, AlHV-sema affects DC function and might therefore thwart the immune system of the host during AlHV-1 primary infection and/or reactivation by affecting DC antigen processing and activation of antigen-specific T cells. Interestingly, these results would suggest that poxvirus and herpesvirus semaphorins have independently evolved similar mechanisms to affect DC function.
Guo et al. hypothesized that AlHV-sema could directly alter activation of latently infected T cells (23). We generated A3− and A3ns recombinant viruses, both impaired for the expression of AlHV-sema. The lack of AlHV-sema expression did not affect viral growth in bovine nasal fibroblasts, suggesting that A3 is not necessary for virus replication. Moreover, A3− and A3ns recombinant viruses induced typical MCF clinical signs, lesions, and expansion of CD8+ T cells after nasal infection of rabbits. In addition, virus persistence was not affected by the lack of AlHV-sema expression, suggesting that the expansion and infiltration of lymphoblastoid cells in the tissue was the consequence of the proliferation of infected T cells as observed with the WT virus (17–20). These results demonstrated that AlHV-sema is not a virulent factor in the pathogenesis of MCF and challenged the hypothesis, suggesting that macaviruses might have acquired semaphorin-encoding genes to alter host T cell function upon latent infection (23). Although the lack of A3 did not modify CD8+ T cell proliferation, T cell activation could have been affected. However, RNA expression levels of IFN-γ were similarly increased in all infected groups, suggesting that AlHV-sema does not interfere with T cell activation or proliferation. Sema7A has been shown to be upregulated in activated T cells (7), but to our knowledge how this protein is involved in T cell activation remains unknown. Although Guo et al. showed that activation of latently infected T cells is a direct consequence of miR-27 degradation by HSUR-1 (23), sema7A expression might be an indirect consequence rather than actually represent a driving force of lymphocyte activation.
AlHV-1 genome tiling array analysis during productive infection in vitro revealed a strong expression of the A3 transcript region, whereas the expression of this region was not detected in MCF-developing calves (20). AlHV-sema expression during lytic infection was further confirmed in the present study by the detection of protein in the supernatant of cells infected with the BAC A3Fc recombinant virus (Fig. 3E). AlHV-sema could therefore mediate immune evasion functions during the productive phase of replication in vivo. This hypothesis, however, postulates that AlHV-1 effectively replicates at the first stages of infection in MCF-susceptible hosts such as cattle. We did not observe any delay in the development of MCF in the absence of A3, and the lack of A3 did not affect the humoral antiviral response (Fig. 12). Although this observation could suggest that AlHV-sema might not affect B cell antiviral responses during MCF, it is also possible that AlHV-sema is not sufficiently produced in MCF-susceptible species to effectively exert its biological activities. Preliminary results using AlHV-1-luc+ virus seem to support the latter hypothesis (unpublished results).
Gammaherpesviruses have coevolved with their natural host species and acquired specific genes for their adaptation to their host. AlHV-1 has coevolved with wildebeest and has become so adapted to its natural host that it is able to persist without causing any clinical sign or lesion, resulting in the infection of virtually the entire wildebeest population. MCF could be seen as an accidental occurrence in AlHV-1 evolution due to cross-species transmission. Thus, AlHV-1 would have primarily developed mechanisms to persist in its natural host and the acquisition of a gene encoding a semaphorin-like protein could be an asset for achieving such an adaptation. Based on this hypothesis, immune evasion properties mediated by AlHV-sema could be restricted to the natural host but not in MCF-susceptible species.
The results presented in the present study demonstrated that AlHV-sema is a 93-kDa glycoprotein that is very similar in its structure to the host sema7A and that is secreted during the early phase of AlHV-1 infection. We then brought evidence that AlHV-sema affects DC function through cytoskeleton rearrangement, leading to the inhibition of phagocytosis and cell migration to the lymph node, a mechanism that involves plexinC1 signaling and the ROCK/LIMK/cofilin pathway. Next, two recombinant viruses impaired for AlHV-sema expression were used to demonstrate that this protein is dispensable for the activation and proliferation of latently infected T cells in MCF. Based on these results, we can hypothesize that AlHV-sema secretion during virus replication in primary infection and/or reactivation in wildebeest might reduce the capability of DCs to process viral antigens and migrate to exert their activity. Thus, AlHV-sema could generate an ideal environment for the virus to mediate a long-lasting persistence in its natural host.
ACKNOWLEDGMENTS
B.G.D. and F.M. are Research Associate and Research Fellow of the Fonds de la Recherche Scientifique (FNRS), respectively. O.S. is Research Fellow of the Fonds pour la Formation à la Recherche dans l'Industrie et dans l'Agriculture (FRIA). A.V. is a member of the BELVIR consortium (IAP, phase VII) granted by the Belgian Science Policy Office (BELSPO, Belgium).
We thank the Clinique Vétérinaire Universitaire and, in particular, Arnaud Sartelet of the Faculty of Veterinary Medicine, University of Liège, for providing bovine blood, Jean-Christophe Bertho of the Monde Sauvage in Aywaille for providing wildebeest blood, Nicolas Smargiasso (GIGA-ULg) for MALDI-MS analyses, Neil Christensen for providing IIIEP/J inbred rabbits, and Céline Urien and Michel Bonneau for their contributions from a sheep lymph collection (INRA Jouy-en-Josas), as well as Justine Javaux, Emeline Deglaire, Jérémy Dumoulin, and Cédric Delforge for technical assistance (FARAH ULg). We also thank L. Gillet for helpful discussions.
REFERENCES
- 1.Committee SN. 1999. Unified nomenclature for the semaphorins/collapsins. Cell 97:551–552. doi: 10.1016/S0092-8674(00)80766-7. [DOI] [PubMed] [Google Scholar]
- 2.Jongbloets BC, Ramakers GM, Pasterkamp RJ. 2013. Semaphorin7A and its receptors: pleiotropic regulators of immune cell function, bone homeostasis, and neural development. Semin Cell Dev Biol 24:129–138. doi: 10.1016/j.semcdb.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki K, Kumanogoh A, Kikutani H. 2008. Semaphorins and their receptors in immune cell interactions. Nat Immunol 9:17–23. doi: 10.1038/ni1553. [DOI] [PubMed] [Google Scholar]
- 4.Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, Tessier-Lavigne M, Comoglio PM. 1999. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99:71–80. doi: 10.1016/S0092-8674(00)80063-X. [DOI] [PubMed] [Google Scholar]
- 5.Holmes S, Downs AM, Fosberry A, Hayes PD, Michalovich D, Murdoch P, Moores K, Fox J, Deen K, Pettman G, Wattam T, Lewis C. 2002. Sema7A is a potent monocyte stimulator. Scand J Immunol 56:270–275. doi: 10.1046/j.1365-3083.2002.01129.x. [DOI] [PubMed] [Google Scholar]
- 6.Mine T, Harada K, Matsumoto T, Yamana H, Shirouzu K, Itoh K, Yamada A. 2000. CDw108 expression during T-cell development. Tissue Antigens 55:429–436. doi: 10.1034/j.1399-0039.2000.550505.x. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki K, Okuno T, Yamamoto M, Pasterkamp RJ, Takegahara N, Takamatsu H, Kitao T, Takagi J, Rennert PD, Kolodkin AL, Kumanogoh A, Kikutani H. 2007. Semaphorin 7A initiates T-cell-mediated inflammatory responses through α1β1 integrin. Nature 446:680–684. doi: 10.1038/nature05652. [DOI] [PubMed] [Google Scholar]
- 8.Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL. 2003. Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature 424:398–405. doi: 10.1038/nature01790. [DOI] [PubMed] [Google Scholar]
- 9.Kang S, Okuno T, Takegahara N, Takamatsu H, Nojima S, Kimura T, Yoshida Y, Ito D, Ohmae S, You DJ, Toyofuku T, Jang MH, Kumanogoh A. 2012. Intestinal epithelial cell-derived semaphorin 7A negatively regulates development of colitis via αvβ1 integrin. J Immunol 188:1108–1116. doi: 10.4049/jimmunol.1102084. [DOI] [PubMed] [Google Scholar]
- 10.Xu X, Ng S, Wu ZL, Nguyen D, Homburger S, Seidel-Dugan C, Ebens A, Luo Y. 1998. Human semaphorin K1 is glycosylphosphatidylinositol-linked and defines a new subfamily of viral-related semaphorins. J Biol Chem 273:22428–22434. doi: 10.1074/jbc.273.35.22428. [DOI] [PubMed] [Google Scholar]
- 11.Comeau MR, Johnson R, DuBose RF, Petersen M, Gearing P, VandenBos T, Park L, Farrah T, Buller RM, Cohen JI, Strockbine LD, Rauch C, Spriggs MK. 1998. A poxvirus-encoded semaphorin induces cytokine production from monocytes and binds to a novel cellular semaphorin receptor, VESPR. Immunity 8:473–482. doi: 10.1016/S1074-7613(00)80552-X. [DOI] [PubMed] [Google Scholar]
- 12.Liu H, Juo ZS, Shim AH, Focia PJ, Chen X, Garcia KC, He X. 2010. Structural basis of semaphorin-plexin recognition and viral mimicry from Sema7A and A39R complexes with PlexinC1. Cell 142:749–761. doi: 10.1016/j.cell.2010.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walzer T, Galibert L, Comeau MR, De Smedt T. 2005. Plexin C1 engagement on mouse dendritic cells by viral semaphorin A39R induces actin cytoskeleton rearrangement and inhibits integrin-mediated adhesion and chemokine-induced migration. J Immunol 174:51–59. doi: 10.4049/jimmunol.174.1.51. [DOI] [PubMed] [Google Scholar]
- 14.Walzer T, Galibert L, De Smedt T. 2005. Poxvirus semaphorin A39R inhibits phagocytosis by dendritic cells and neutrophils. Eur J Immunol 35:391–398. doi: 10.1002/eji.200425669. [DOI] [PubMed] [Google Scholar]
- 15.Gardner JD, Tscharke DC, Reading PC, Smith GL. 2001. Vaccinia virus semaphorin A39R is a 50-55 kDa secreted glycoprotein that affects the outcome of infection in a murine intradermal model. J Gen Virol 82:2083–2093. [DOI] [PubMed] [Google Scholar]
- 16.Plowright W. 1990. Malignant catarrhal fever virus, p 123–150 InDinter ZMB. (ed), Virus infections of ruminants. Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 17.Dewals B, Boudry C, Farnir F, Drion PV, Vanderplasschen A. 2008. Malignant catarrhal fever induced by alcelaphine herpesvirus 1 is associated with proliferation of CD8+ T cells supporting a latent infection. PLoS One 3:e1627. doi: 10.1371/journal.pone.0001627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dewals B, Myster F, Palmeira L, Gillet L, Ackermann M, Vanderplasschen A. 2011. Ex vivo bioluminescence detection of alcelaphine herpesvirus 1 infection during malignant catarrhal fever. J Virol 85:6941–6954. doi: 10.1128/JVI.00286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dewals BG, Vanderplasschen A. 2011. Malignant catarrhal fever induced by Alcelaphine herpesvirus 1 is characterized by an expansion of activated CD3+ CD8+ CD4− T cells expressing a cytotoxic phenotype in both lymphoid and non-lymphoid tissues. Vet Res 42:95. doi: 10.1186/1297-9716-42-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmeira L, Sorel O, Van Campe W, Boudry C, Roels S, Myster F, Reschner A, Coulie PG, Kerkhofs P, Vanderplasschen A, Dewals BG. 2013. An essential role for gammaherpesvirus latency-associated nuclear antigen homolog in an acute lymphoproliferative disease of cattle. Proc Natl Acad Sci U S A 110:E1933–E1942. doi: 10.1073/pnas.1216531110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buxton D, Reid HW. 1980. Transmission of malignant catarrhal fever to rabbits. Vet Rec 106:243–245. doi: 10.1136/vr.106.11.243. [DOI] [PubMed] [Google Scholar]
- 22.Ensser A, Fleckenstein B. 2005. T-cell transformation and oncogenesis by gamma2-herpesviruses. Adv Cancer Res 93:91–128. doi: 10.1016/S0065-230X(05)93003-0. [DOI] [PubMed] [Google Scholar]
- 23.Guo YE, Riley KJ, Iwasaki A, Steitz JA. 2014. Alternative capture of noncoding RNAs or protein-coding genes by herpesviruses to alter host T cell function. Mol Cell 54:67–79. doi: 10.1016/j.molcel.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stabel JR, Stabel TJ. 1995. Immortalization and characterization of bovine peritoneal macrophages transfected with SV40 plasmid DNA. Vet Immunol Immunopathol 45:211–220. doi: 10.1016/0165-2427(94)05348-V. [DOI] [PubMed] [Google Scholar]
- 25.Huynh HT, Robitaille G, Turner JD. 1991. Establishment of bovine mammary epithelial cells (MAC-T): an in vitro model for bovine lactation. Exp Cell Res 197:191–199. doi: 10.1016/0014-4827(91)90422-Q. [DOI] [PubMed] [Google Scholar]
- 26.Gillet L, Daix V, Donofrio G, Wagner M, Koszinowski UH, China B, Ackermann M, Markine-Goriaynoff N, Vanderplasschen A. 2005. Development of bovine herpesvirus 4 as an expression vector using bacterial artificial chromosome cloning. J Gen Virol 86:907–917. doi: 10.1099/vir.0.80718-0. [DOI] [PubMed] [Google Scholar]
- 27.Plowright W, Herniman KA, Jessett DM, Kalunda M, Rampton CS. 1975. Immunization of cattle against the herpesvirus of malignant catarrhal fever: failure of inactivated culture vaccines with adjuvant. Res Vet Sci 19:159–166. [PubMed] [Google Scholar]
- 28.Dewals B, Boudry C, Gillet L, Markine-Goriaynoff N, de Leval L, Haig DM, Vanderplasschen A. 2006. Cloning of the genome of Alcelaphine herpesvirus 1 as an infectious and pathogenic bacterial artificial chromosome. J Gen Virol 87:509–517. doi: 10.1099/vir.0.81465-0. [DOI] [PubMed] [Google Scholar]
- 29.Spiller OB, Mark L, Blue CE, Proctor DG, Aitken JA, Blom AM, Blackbourn DJ. 2006. Dissecting the regions of virion-associated Kaposi's sarcoma-associated herpesvirus complement control protein required for complement regulation and cell binding. J Virol 80:4068–4078. doi: 10.1128/JVI.80.8.4068-4078.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Letesson JJ, Coppe P, Lostrie-Trussart N, Depelchin A. 1983. A bovine ‘Ia-like' antigen detected by a xenogeneic monoclonal antibody. Anim Blood Groups Biochem Genet 14:239–250. [PubMed] [Google Scholar]
- 32.Godornes C, Leader BT, Molini BJ, Centurion-Lara A, Lukehart SA. 2007. Quantitation of rabbit cytokine mRNA by real-time RT-PCR. Cytokine 38:1–7. doi: 10.1016/j.cyto.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hein DW, Smolen TN, Fox RR, Weber WW. 1982. Identification of genetically homozygous rapid and slow acetylators of drugs and environmental carcinogens among established inbred rabbit strains. J Pharmacol Exp Ther 223:40–44. [PubMed] [Google Scholar]
- 34.Cody V, Shen H, Shlyankevich M, Tigelaar RE, Brandsma JL, Hanlon DJ. 2005. Generation of dendritic cells from rabbit bone marrow mononuclear cell cultures supplemented with hGM-CSF and hIL-4. Vet Immunol Immunopathol 103:163–172. doi: 10.1016/j.vetimm.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 35.Quah BJ, Parish CR. 2010. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J Vis Exp 88:e51627. doi: 10.3791/51627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Contreras V, Urien C, Guiton R, Alexandre Y, Vu Manh TP, Andrieu T, Crozat K, Jouneau L, Bertho N, Epardaud M, Hope J, Savina A, Amigorena S, Bonneau M, Dalod M, Schwartz-Cornil I. 2010. Existence of CD8alpha-like dendritic cells with a conserved functional specialization and a common molecular signature in distant mammalian species. J Immunol 185:3313–3325. doi: 10.4049/jimmunol.1000824. [DOI] [PubMed] [Google Scholar]
- 37.Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J, Boudinot P, Hosmalin A, Schwartz-Cornil I, Dalod M. 2010. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8α+ dendritic cells. J Exp Med 207:1283–1292. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gliddon DR, Howard CJ. 2002. CD26 is expressed on a restricted subpopulation of dendritic cells in vivo. Eur J Immunol 32:1472–1481. doi:. [DOI] [PubMed] [Google Scholar]
- 39.Parsons KR, MacHugh ND. 1991. Individual antigens of cattle: bovine CD1 (BoCD1). Vet Immunol Immunopathol 27:37–41. doi: 10.1016/0165-2427(91)90076-O. [DOI] [PubMed] [Google Scholar]
- 40.Antipenko A, Himanen JP, van Leyen K, Nardi-Dei V, Lesniak J, Barton WA, Rajashankar KR, Lu M, Hoemme C, Puschel AW, Nikolov DB. 2003. Structure of the semaphorin-3A receptor binding module. Neuron 39:589–598. doi: 10.1016/S0896-6273(03)00502-6. [DOI] [PubMed] [Google Scholar]
- 41.Love CA, Harlos K, Mavaddat N, Davis SJ, Stuart DI, Jones EY, Esnouf RM. 2003. The ligand-binding face of the semaphorins revealed by the high-resolution crystal structure of SEMA4D. Nat Struct Biol 10:843–848. doi: 10.1038/nsb977. [DOI] [PubMed] [Google Scholar]
- 42.Liu BP, Strittmatter SM. 2001. Semaphorin-mediated axonal guidance via Rho-related G proteins. Curr Opin Cell Biol 13:619–626. doi: 10.1016/S0955-0674(00)00260-X. [DOI] [PubMed] [Google Scholar]
- 43.Ohsawa S, Hamada S, Kuida K, Yoshida H, Igaki T, Miura M. 2010. Maturation of the olfactory sensory neurons by Apaf-1/caspase-9-mediated caspase activity. Proc Natl Acad Sci U S A 107:13366–13371. doi: 10.1073/pnas.0910488107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J. 2013. Functions of cofilin in cell locomotion and invasion. Nature reviews. Mol Cell Biol 14:405–415. doi: 10.1038/nrm3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MartIn-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. 2003. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med 198:615–621. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Guttler S, Hutloff A, Mages HW, Ranke K, Schaefer M, Jack RS, Henn V, Kroczek RA. 2009. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 31:823–833. doi: 10.1016/j.immuni.2009.08.027. [DOI] [PubMed] [Google Scholar]
- 47.Lei Y, Takahama Y. 2012. XCL1 and XCR1 in the immune system. Microbes Infect 14:262–267. doi: 10.1016/j.micinf.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Scott GA, McClelland LA, Fricke AF, Fender A. 2009. Plexin C1, a receptor for semaphorin 7a, inactivates cofilin and is a potential tumor suppressor for melanoma progression. J Investig Dermatol 129:954–963. doi: 10.1038/jid.2008.329. [DOI] [PubMed] [Google Scholar]
- 49.Ma B, Herzog EL, Lee CG, Peng X, Lee CM, Chen X, Rockwell S, Koo JS, Kluger H, Herbst RS, Sznol M, Elias J. 2014. Role of chitinase 3-like-1 and semaphorin 7A in pulmonary melanoma metastasis. Cancer Res 75:1–10. doi: 10.1158/0008-5472.CAN-13-3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I. 2001. Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci 4:367–373. doi: 10.1038/86011. [DOI] [PubMed] [Google Scholar]
- 51.Jaffe AB, Hall A. 2005. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 52.Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. 2000. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem 275:3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 53.Koppel AM, Feiner L, Kobayashi H, Raper JA. 1997. A 70 amino acid region within the semaphorin domain activates specific cellular response of semaphorin family members. Neuron 19:531–537. doi: 10.1016/S0896-6273(00)80369-4. [DOI] [PubMed] [Google Scholar]