

ABSTRACT
The genotype at polymorphic codon 129 of the PRNP gene has a profound influence on both phenotypic expression and prion strain susceptibility in humans. For example, while the most common sporadic Creutzfeldt-Jakob disease (CJD) subtype, sporadic CJD-MM1 (M1 strain), induces a single phenotype after experimental transmission regardless of the codon 129 genotype of the recipient animal, the phenotype elicited by sporadic CJD-VV2 (V2 strain), the second most common subtype, varies according to the host codon 129 genotype. In particular, the propagation of the V2 strain in codon 129 methionine homozygotes has been linked only to acquired forms of CJD such as plaque-type dura mater graft-associated CJD (dCJD), a subgroup of iatrogenic CJD with distinctive phenotypic features, but has never been observed in sporadic CJD cases. In the present report, we describe atypical CJD cases carrying codon 129 methionine homozygosity, in a neurosurgeon and in a patient with a medical history of neurosurgery without dural grafting, showing the distinctive phenotypic features and transmission properties of plaque-type dCJD. These findings raise the possibility that the two cases, previously thought to represent sporadic CJD, might actually represent acquired CJD caused by infection with the V2 strain. Thus, careful analyses of phenotypic features and transmission properties in atypical cases may be useful to distinguish acquired from sporadic cases of CJD.
IMPORTANCE Susceptibility to and phenotypic expression of Creutzfeldt-Jakob disease (CJD) depend on both the prion strain and genotype at polymorphic codon 129 of the PRNP gene. For example, propagation of the second most common sporadic CJD strain (V2 strain) into codon 129 methionine homozygotes has been linked to plaque-type dura mater graft-associated CJD (dCJD), a subgroup of iatrogenic CJD with distinctive phenotypic features, but has never been observed in sporadic CJD. In the present report, we describe atypical CJD cases in a neurosurgeon and in a patient with a medical history of neurosurgery without dural grafting, showing the distinctive phenotypic features and transmission properties of plaque-type dCJD. These findings raise the possibility that the two cases, previously considered to represent sporadic CJD, might actually represent acquired CJD caused by infection with the V2 strain.
INTRODUCTION
Creutzfeldt-Jakob disease (CJD) is a fatal transmissible neurodegenerative disease that may originate either from an apparently spontaneous generation of an abnormal isoform of prion protein (PrPSc) as in sporadic CJD (sCJD) and genetic CJD or from a PrPSc infection as in acquired CJD. Patients with sCJD show wide phenotypic heterogeneity, and their clinicopathological phenotype is determined by both the genotype (methionine [M] or valine [V]) at polymorphic codon 129 of the PRNP gene and the type (1 or 2) of PrPSc in the brain (1). PrPSc types 1 and 2 are distinguishable according to the size of the proteinase K-resistant core of the protein (21 and 19 kDa, respectively). Accordingly, sCJD is currently classified into six subtypes, i.e., MM1, MV1, VV1, MM2, MV2, and VV2 (1).
Phenotypic heterogeneity can also be recognized in transmission experiments using PrP-humanized knock-in mice, depending on both the inoculated CJD strain and the codon 129 genotype of the recipient animals (2–4). For example, sCJD-MM1 or sCJD-MV1 prions, representing the most common CJD strain (denoted strain M1 [4]), induce the same phenotype, i.e., diffuse synaptic-type PrP deposition and type 1 PrPSc accumulation resembling those observed in sCJD-MM1 or sCJD-MV1 patients, regardless of the mouse codon 129 genotype. In contrast, sCJD-VV2 or sCJD-MV2 prions, representing the second most common CJD strain (denoted strain V2 [4]), induce heterogeneous phenotypes with distinctive features depending on the codon 129 genotypes of the recipient animals. While animals carrying codon 129 valine homozygosity show plaque-type PrP deposition and type 2 PrPSc accumulation resembling those of human sCJD-VV2 cases, the animals carrying codon 129 methionine homozygosity develop numerous amyloid plaques (kuru plaques) and a unique PrPSc with electrophoretic mobility of about 20 kDa, which is intermediate in size between types 1 and 2 PrPSc and thus designated an intermediate type (type i) (5). It is noteworthy that the equivalent to the mouse phenotype with kuru plaques and type i PrPSc, reflecting transmission of the V2 sCJD strain to codon 129 methionine homozygotes, has been observed only in acquired prion diseases, especially in a subgroup of dura mater graft-associated iatrogenic CJD (plaque-type dCJD) cases in Japan, but not in sCJD cases (5–9). Thus, in addition to phenotypic expression, the codon 129 genotype may also influence prion strain susceptibility depending on the disease etiology, e.g., acquired versus sporadic. The finding that the plaque-type phenotype associated with codon 129 methionine homozygosity can be observed in iatrogenic dCJD but not in sCJD suggests that 129M PrPC may be converted into type i PrPSc by the V2 sCJD strain in acquired CJD, while, in contrast, it would not be susceptible to a spontaneous conversion into the conformation of type i PrPSc.
Assuming that the combination of kuru plaques and type i PrPSc in CJD patients with codon 129 methionine homozygosity can be a reliable indication of transmission of the V2 sCJD strain, we have searched the literature for CJD cases reported as sCJD despite showing distinctive features resembling those of plaque-type dCJD. Furthermore, we have performed a transmission study to verify that the identified CJD cases were identical to plaque-type dCJD cases in transmission properties also.
MATERIALS AND METHODS
Ethics statement.
Brain tissues were obtained at autopsy after receiving written informed consent for research use. Animal experiments were performed in strict accordance with the Regulations for Animal Experiments and Related Activities at Tohoku University and in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions of the Ministry of Education, Culture, Sports, Science and Technology in Japan, notice no. 71. The protocol was approved by the Institutional Animal Care and Use Committee of Tohoku University.
Patients.
We found two atypical CJD cases that showed widespread kuru plaques despite their 129M/M genotype. Details of these atypical CJD cases have been reported elsewhere (10, 11). In addition, clinically, genetically, and histopathologically proven sCJD and dCJD cases with the 129M/M genotype were also included in this study. The diagnosis of CJD, the PRNP genotype, and PrPSc type were confirmed by immunohistochemistry, Western blot analysis, and PRNP sequence analysis as described previously (12–14).
Transmission experiments.
The production of knock-in mice expressing human PrP with the 129M/M or 129M/V or 129V/V genotype has been reported previously (15). Intracerebral inoculation was performed as described previously (5). The inoculated mice were sacrificed at a predefined clinical endpoint or died as a consequence of accident. One hemisphere of the brain was fixed in 10% buffered formalin for immunohistochemistry, and the other hemisphere was immediately frozen for Western blotting or serial passage. Incubation times are expressed as means ± standard errors of the means (SEM).
Immunohistochemistry.
Formalin-fixed mouse brain tissues were treated with 60% formic acid for 1 h to inactivate the infectivity and were embedded in paraffin. Tissue sections were pretreated by hydrolytic autoclaving before PrP immunohistochemistry testing (12). PrP-N anti-PrP antiserum (12) was used as the primary antibody, and goat anti-rabbit immunoglobulin polyclonal antibody labeled with a peroxidase-conjugated dextran polymer, EnVision+ (Dako), was used as the secondary antibody. For morphometric analysis, at least four representative digital microscopy images of the cerebral cortices, white matter, basal ganglia, hippocampus, thalamus, and cerebellar cortices were obtained from each mouse. Data were collected from all diseased mice. These images were analyzed using ImageJ software (http://rsb.info.nih.gov/ij/). For quantification of PrP plaques, the number of plaques was manually counted on PrP-immunostained sections. Data are presented as means ± SEM. Differences between groups were analyzed by Student's t test using JMP Pro software version 10.0 (SAS Institute Inc., USA). P values of <0.05 were considered significant.
Western blotting.
PrPSc was obtained from cerebral cortex after tissue homogenization, collagenase digestion, and Sarkosyl-NaCl extraction (16). For deglycosylation of PrPSc, samples were digested with PNGaseF (New England BioLabs) (5). Protein samples were subjected to SDS-PAGE and Western blotting (14, 15). PrPSc type-specific polyclonal antibodies (designated Tohoku 1 [T1] or Tohoku 2 [T2]) (2) or anti-PrP monoclonal antibodies 3F4 (Signet) and #71 (17) were used as the primary antibodies. Anti-rabbit EnVision+ and anti-mouse EnVision+ were used as the secondary antibodies.
RESULTS
The clinical and neuropathological features of the two atypical cases of CJD are summarized in Table 1 (10, 11, 18). One patient (case 1) was a neurosurgeon, whereas the other (case 2) had a medical history of neurosurgery without dura mater grafting. Both patients were homozygous for methionine at polymorphic codon 129 (129M/M) and homozygous for glutamate at polymorphic codon 219 of the PRNP gene, the latter being a rare glutamate/lysine polymorphism in Asian and Pacific populations which modulates the susceptibility to the CJD, similar to the codon 129 genotype (19). In addition, case 2 carried a 24-bp deletion in the octapeptide repeat region of the PRNP gene, which is a nonpathogenic polymorphism (20). It is noteworthy that both patients showed widespread PrP amyloid plaques despite their 129M/M genotype. Therefore, we examined the biochemical properties of PrPSc in their brain to test the possibility that these cases might have the intermediate type (type i) PrPSc similar to that seen with plaque-type dCJD. Western blot analysis of PrPSc revealed that the PrPSc in the two atypical CJD cases was smaller than the type 1 PrPSc (21 kDa) in sCJD-MM1 and larger than the type 2 PrPSc (19 kDa) in sCJD-MM2, resembling the intermediate-size type i PrPSc (20 kDa) found in plaque-type dCJD (Fig. 1A and B) (18). In Western blot analysis using the conventional 3F4 anti-PrP monoclonal antibody, discrimination between type 1 and type i PrPSc is rather difficult due to subtle differences in the electrophoretic mobility. Therefore, we also performed PrPSc fragment analysis using a monoclonal antibody, #71, directed against the carboxyl terminus of PrP (17). In this analysis, the PrPSc fragment patterns of the atypical CJD cases were quite different from those of sCJD-MM1 (∼12 kDa) and identical to those of plaque-type dCJD (∼10 kDa) (Fig. 1C and D). Thus, the two atypical CJD cases showed neuropathological and biochemical features identical to those of the plaque-type dCJD, which is caused by the transmission of sCJD-VV2 or sCJD-MV2 to individuals with the 129M/M genotype.
TABLE 1.
Patients' histories and clinicopathological features of the atypical CJD casesa
| Feature | Case 1 | Case 2 |
|---|---|---|
| Gender | Male | Female |
| Age at onset (yrs) | 54 | 75 |
| Occupational history | Neurosurgeon consulted on a case of CJD in which a myelogram was performed but not known to have participated in the procedure (7 yrs before onset) | |
| Medical historyb | Surgical operations for perinephric abscess, inguinal herniorrhaphy, and removal of ureteric calculus (28 yrs, 8 yrs, and 32 mos before onset, respectively) | Neurosurgery for cerebellar hemangioblastoma (14 yrs before onset) |
| Initial symptomsc | Abnormal sleep patterns, hallucinations, myoclonic twitching of the extremities, paresthesia of the toes, loss of vibration sense, gait disturbance | Drowsiness, gait disturbance |
| Duration of illness (mos) | 17 | 11 |
| PSWCs on EEGd | Negative | Negative |
| PRNP genee | 129M/M, 219E/E; no pathogenic mutations | 129M/M, 219E/E; no pathogenic mutations (24 bp deletion in octapeptide repeat region) |
| PrP depositionf | Kuru plaques | Kuru plaques |
Details of the clinicopathological features of these patients have been reported elsewhere (10, 11). Underlined data were not described in the original reports and were obtained in the present study.
There was no medical history of the use of dura mater grafts, pituitary hormone, corneal transplants, or electroencephalogram (EEG) needles in either patient.
Case 1 had necrotizing cutaneous lesions with vasculitis in the lower extremities. A part of his initial symptoms might have been related to these lesions.
PSWCs on EEG, the presence of periodic sharp-wave complexes (PSWCs) in electroencephalogram (EEG) results, which is a characteristic of sCJD-MM1/MV1.
PRNP gene, the genotype at polymorphic codon 129 (methionine [M] or valine [V]) or codon 219 (glutamate [E] or lysine [K]) of the PRNP gene.
PrP deposition, the patterns of PrP deposition in the brain.
FIG 1.

Biochemical features of the two atypical CJD cases. (A and B) Western blot analysis of PrPSc in the proteinase K-digested brain homogenates using 3F4, a conventional anti-PrP monoclonal antibody. The PrPSc in the atypical CJD cases (cases 1 and 2) was smaller than the type 1 PrPSc (21 kDa) in sCJD-MM1 but was similar in size to the type i PrPSc (20 kDa) in plaque-type dCJD. (C) Western blot analysis of deglycosylated PrPSc fragments in the proteinase K-digested and PNGase F-treated brain homogenates using a monoclonal antibody, #71, which can detect carboxyl-terminal PrPSc fragments in addition to full-length PrPSc (17). The carboxyl-terminal PrPSc fragments in the atypical CJD cases (∼10 kDa; black arrowhead) were smaller than those in sCJD-MM1 (∼12 kDa; white arrowhead). sCJD-MM2 contained only trace amounts of the carboxyl-terminal PrPSc fragments that were identical in size to those in sCJD-MM1. (D) The carboxyl-terminal PrPSc fragments in the plaque-type dCJD (∼10 kDa; black arrowhead) were also smaller than those in sCJD-MM1 or non-plaque-type dCJD (∼12 kDa; white arrowhead). Plaque-type, plaque-type dCJD; Non-plaque-type, non-plaque-type dCJD.
Next, we performed animal experiments using PrP-humanized knock-in mice carrying human PrP with the 129M/M, 129M/V, or 129V/V genotype to exclude the possibility that the atypical CJD cases might represent a very rare variation of the sCJD subgroups with the 129M/M genotype. Transmission study is still the gold standard in prion strain typing and can be a powerful tool to identify the causative origin of acquired prion diseases (15, 21). The transmissibility of the atypical CJD cases to the PrP-humanized mice was identical to that of plaque-type dCJD (Fig. 2A). In particular, although the incompatibility of the codon 129 genotypes between host and inoculum usually results in a prolonged incubation period (5), the 129V/V mice inoculated with brain homogenates from the atypical CJD cases or plaque-type dCJD showed incubation periods shorter than those of the other mouse lines. Moreover, the neuropathological and biochemical features in the inoculated mice were also identical between the atypical CJD and plaque-type dCJD. Briefly, after challenge with brain homogenates from the atypical CJD cases or plaque-type dCJD, the 129M/M mice showed widespread PrP plaques in the cerebral cortices and thalamus, whereas the 129V/V mice showed PrP plaques restricted to regions within the white matter (Fig. 2B and C). PrP-humanized mice inoculated with sCJD-MM1 material showed diffuse synaptic-type PrP deposition regardless of their codon 129 genotype. In Western blot analysis, after challenge with brain homogenates from the atypical CJD cases or plaque-type dCJD, the 129M/M mice produced type i PrPSc (20 kDa), the 129V/V mice produced type 2 PrPSc (19 kDa), and the 129M/V mice produced both types i and 2 PrPSc (Fig. 2D and E). PrP-humanized mice inoculated with sCJD-MM1 material produced type 1 PrPSc (21 kDa) regardless of their codon 129 genotype. Since the transmission properties of plaque-type dCJD are similar to those of sCJD-VV2 or sCJD-MV2 (2, 3), these findings are in full agreement with a previous study showing neuropathological and biochemical similarities among nonhuman primates inoculated with brain homogenates from case 1, sCJD-VV2, or sCJD-MV2 (18). In summary, the transmission properties of the two atypical CJD cases were quite different from those of sCJD-MM1 and identical to those of plaque-type dCJD, sCJD-VV2, or sCJD-MV2, indicating that their causative origin might be sCJD-VV2 or sCJD-MV2.
FIG 2.

Transmission properties of the two atypical CJD cases. (A) Transmissibility to the PrP-humanized knock-in mice carrying human PrP with the 129M/M or 129M/V or 129V/V genotype. The properties of transmissibility to each mouse line were similar among the atypical CJD cases and plaque-type dCJD. The mean incubation periods from intracerebral inoculation to the onset of disease and attack rates (number of mice positive for PrP accumulation in immunohistochemical analysis/number of inoculated mice) were as follows: case 1 in 129M/M, 663 ± 36 days (5/5); case 1 in 129M/V, 725 ± 39 days (4/5); case 1 in 129V/V, 313 ± 10 days (6/6); case 2 in 129M/M, 590 ± 81 days (4/5); case 2 in 129M/V, 699 ± 75 days (5/5); case 2 in 129V/V, 296 ± 9 days (6/6); plaque-type dCJD in 129M/M, 685 ± 51 days (5/5); plaque-type dCJD in 129M/V, 818 ± 20 days (4/4); plaque-type dCJD in 129V/V, 259 ± 6 days (6/6); sCJD-MM1 in 129M/M, 467 ± 24 days (8/8); sCJD-MM1 in 129M/V, 490 ± 26 days (5/5); sCJD-MM1 in 129V/V, 775 ± 32 days (6/6); sCJD-VV2 in 129M/M, 633 ± 49 days (6/6); sCJD-VV2 in 129M/V, 788 ± 30 days (4/4); and sCJD-VV2 in 129V/V, 302 ± 9 days (7/7). (B) Neuropathological features in the inoculated mice. Immunohistochemical analysis of PrP in the brains revealed that the 129M/M mice inoculated with brain homogenates from the atypical CJD cases or plaque-type dCJD showed widespread PrP plaques in the cerebral cortices, whereas the 129V/V mice inoculated with the same brain homogenates showed PrP plaques in the white matter (arrowheads). PrP-humanized mice inoculated with sCJD-MM1 material showed diffuse synaptic-type PrP deposition regardless of their codon 129 genotype. Scale bar, 100 μm. (C) Comparison of brain regional distributions of PrP plaques between the 129M/M mice and the 129V/V mice. *, P < 0.05. (D) Biochemical features in the inoculated mice. Western blot analysis of PrPSc in the brains using the conventional 3F4 anti-PrP antibody, type 1/type i PrPSc-specific T1 antibody, or type 2 PrPSc-specific T2 antibody revealed that the biochemical characteristics were identical among the mice inoculated with brain homogenates from the atypical CJD cases, plaque-type dCJD, or sCJD-VV2. After challenge with these materials, the 129M/M mice produced predominantly T1-reactive type i PrPSc with trace amounts of T2-reactive type 2 PrPSc, whereas the 129V/V mice produced predominantly T2-reactive type 2 PrPSc with small amounts of T1-reactive type i PrPSc. The 129M/V mice produced a mixture of T1-reactive type i PrPSc and T2-reactive type 2 PrPSc, visible as doublets in Western blot analysis using the 3F4 antibody that reacts with both types i and 2 PrPSc (3). In contrast, PrP-humanized mice inoculated with sCJD-MM1 material produced predominantly T1-reactive PrPSc regardless of their codon 129 genotype. (E) Western blot analysis of deglycosylated PrPSc in the mouse brain using the conventional 3F4 anti-PrP antibody. Type i PrPSc (20 kDa) was reproduced in the 129M/M mice inoculated with brain homogenates from the atypical CJD cases or plaque-type dCJD. Details of the transmission properties of plaque-type dCJD, sCJD-MM1, or sCJD-VV2 have been reported previously (2, 3, 5). Plaque-type, plaque-type dCJD.
We also examined the effects of serial passage on the transmission properties of case 2 (Fig. 3). The transmissibility, neuropathological features, and biochemical characteristics in each mouse line were identical to those in the primary passage. The codon 129 genotype of mice in the primary passage did not affect the transmission properties in the secondary passage. Thus, we confirmed that the characteristic transmission properties were maintained after serial passage through various codon 129 genotypes.
FIG 3.
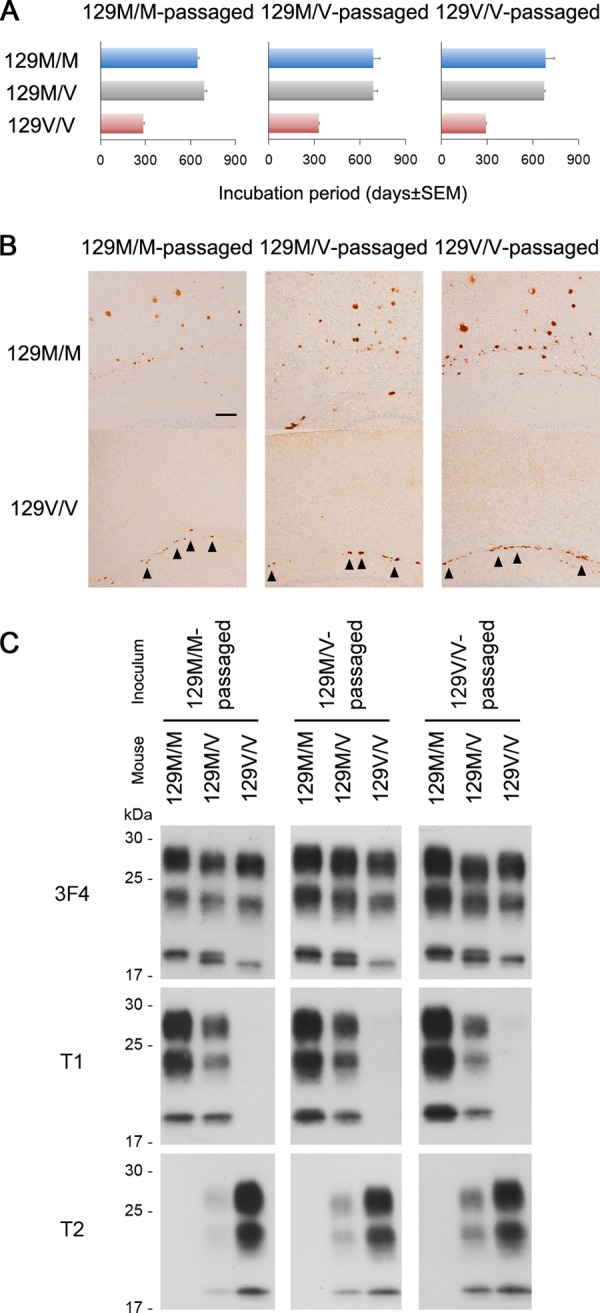
Transmission properties in secondary passage of the atypical CJD case. (A) Transmissibility to the PrP-humanized knock-in mice in the secondary passage of case 2. The brain homogenates of the affected mice in the primary passage were inoculated into additional knock-in mice of each genotype to examine the effects of serial passage on the transmission properties. Transmissibility to each mouse line was identical to that in the primary passage. The codon 129 genotype of mice in the primary passage did not affect the transmissibility in the secondary passage. The mean incubation periods from intracerebral inoculation to the onset of disease and attack rates (number of mice positive for PrP accumulation in immunohistochemical analysis/number of inoculated mice) were as follows: 129M/M-passaged case 2 in 129M/M, 646 ± 10 days (10/10); 129M/M-passaged case 2 in 129M/V, 691 ± 16 days (7/7); 129M/M-passaged case 2 in 129V/V, 285 ± 8 days (8/8); 129M/V-passaged case 2 in 129M/M, 687 ± 43 days (9/9); 129M/V-passaged case 2 in 129M/V, 686 ± 28 days (7/8); 129M/V-passaged case 2 in 129V/V, 328 ± 6 days (9/9); 129V/V-passaged case 2 in 129M/M, 683 ± 57 days (6/7); 129V/V-passaged case 2 in 129M/V, 674 ± 6 days (7/9); and 129V/V-passaged case 2 in 129V/V, 292 ± 9 days (9/9). (B) Neuropathological features in the secondary passage of case 2. Immunohistochemical analysis of PrP in the brains revealed that the 129M/M mice showed widespread PrP plaques in the cerebral cortices, whereas the 129V/V mice showed PrP plaques in the white matter (arrowheads) similar to those in the primary passage. The codon 129 genotype of mice in the primary passage did not affect the neuropathological features in the secondary passage. Scale bar, 100 μm. (C) Biochemical features in secondary passage of case 2. Western blot analysis of PrPSc in the brains using the conventional 3F4 anti-PrP antibody, type 1/type i PrPSc-specific T1 antibody, or type 2 PrPSc-specific T2 antibody revealed that the biochemical characteristics in each mouse line were identical to those in the primary passage. The codon 129 genotype of mice in the primary passage did not affect the biochemical features in the secondary passage. 129M/M-passaged, 129M/M mouse-passaged case 2; 129M/V-passaged, 129M/V mouse-passaged case 2; 129V/V-passaged, 129V/V mouse-passaged case 2.
DISCUSSION
In the present report, we have shown that two CJD cases previously reported as sCJD shared neuropathological and biochemical features with plaque-type dCJD, namely, PRNP codon 129 methionine homozygosity (129M/M), widespread kuru plaques, and the intermediate type (type i) PrPSc accumulation in the brain. Furthermore, the transmission properties of the two CJD cases propagated in PrP-humanized mice were also identical to those of transmitted plaque-type dCJD. In particular, the 129V/V mice showed the highest susceptibility to inocula from the two CJD cases despite the mismatched codon 129 genotypes and produced type 2 PrPSc, similarly to those observed in transmission studies of plaque-type dCJD (5). Moreover, these characteristic transmission properties were maintained after serial passage through various codon 129 genotypes. Taken together, the present results raise the possibility that these two CJD cases might have also been caused by transmission of sCJD-VV2 or sCJD-MV2 prions (V2 strain), as it is the case for plaque-type dCJD (9). Since transmission studies using PrP-humanized knock-in mice have been conducted so far on very limited numbers of typical iatrogenic CJD and sCJD cases, further analyses with larger series, including cases with atypical features, may be needed to generalize the implications of the present study. However, the main conclusion of our transmission experiments is also supported by a previous study showing identical neuropathological and biochemical features in nonhuman primates inoculated with brain homogenates from case 1 and sCJD-VV2 or from sCJD-MV2 which were clearly distinct from those of all animals inoculated from an unselected series of 55 cases of CJD-MM1 or sCJD-MV1 (18).
Iatrogenic transmission of CJD through neurosurgery has been reported in only 4 cases (22–25), and CJD transmission due to occupational exposure has not been recognized so far. In 2009, a Spanish pathologist developed CJD, and direct contact with infectious tissues during his professional activities was suspected to be the etiology of his disease because he had had a history of minor injuries during postmortem examinations (26). However, since his clinicopathological and biochemical features were indistinguishable from those of sCJD, the cause of his disease has remained undetermined. In contrast, our case 1, a neurosurgeon, showed distinctive phenotype and transmission properties that have been linked only to acquired CJD caused by infection with the V2 sCJD strain. The most plausible explanation for his disease etiology may be infection through occupational exposure over a period of many decades in which he had operated upon multiple patients with CJD. Furthermore, the other patient in the present study (case 2), who also showed distinctive phenotype and transmission properties similar to those of plaque-type dCJD, had a medical history of neurosurgery without dura mater grafting, suggesting that she might have also been infected through cross-contamination from operative instruments. During the period 1999 to 2008, the Japanese CJD Surveillance registry listed 6 of 760 sCJD patients who had undergone neurosurgery after the onset but before the diagnosis of sCJD (27). Although none of the individuals exposed to possibly contaminated instruments has developed CJD (as of September 2014), the ensemble of these observations suggests that the potential risk of iatrogenic transmission via neurosurgical procedures may be greater than is presently appreciated.
On the basis of the findings from the present study, together with data from previous studies (2, 3, 5), we propose the neuropathological and biochemical criteria that may help distinguish acquired CJD caused by transmission of the V2 sCJD strain to individuals with the 129M/M genotype, here denoted acquired CJD-MMiK (the 129 M/M genotype, type i PrPSc, and kuru plaques), from sporadic CJD. Since widespread kuru plaques have also been reported in pituitary hormone-associated iatrogenic CJD cases with the 129M/M genotype (28, 29), the type of PrPSc in these cases needs to be examined in the future. In addition to these neuropathological and biochemical clues, clinical features of acquired CJD-MMiK, such as slow progression of disease and absence or late occurrence of periodic sharp-wave complexes on electroencephalogram, may also be distinctive as revealed by a comprehensive analysis of plaque-type dCJD (6, 7). Moreover, the experimental transmission data of our study suggest that PrP-humanized mice are also useful to evaluate transmission properties of acquired CJD-MMiK even after serial passage through various codon 129 genotypes. The diagnostic approach outlined above would help to distinguish acquired CJD from sporadic cases in the future as well as aid retrospective studies to investigate acquired CJD cases overlooked in past examinations.
Several issues remain to be addressed, the first of which is the potential for instrument cross-contamination of patients subjected to subsequent operations. Therefore, improvement of case recognition and ascertainment of the medical records of CJD patients are our future tasks. Second, it is uncertain whether the current decontamination/disinfection procedures are effective against all CJD strains, including the V2 sCJD strain that can cause acquired CJD-MMiK. The current procedures were developed using scrapie and CJD isolates before genotyping became possible (30–32). However, different prion strains can show different susceptibilities to the decontamination procedures (33, 34). Therefore, the efficacy of the current decontamination procedures needs to be tested on the entire spectrum of CJD strains. Finally, the findings presented in this report have potential implications for the etiological classification of CJD, namely, the distinction between acquired and sporadic CJD. Indeed, while the detection of acquired CJD-MMiK cases may be facilitated by their distinctive neuropathological and biochemical features, this subgroup may account for only a minority of total acquired CJD cases. This is because the combined incidence rates of sCJD-VV2 and sCJD-MV2, the origins of acquired CJD-MMiK, are only 26% in European countries and the United States (35) and 3.2% in Japan (8) among total sCJD cases. In addition, the frequencies of the 129M/M genotype in the general population are 37% in European countries and the United States (1) and 92% in Japan (36). Therefore, the theoretical probabilities of occurrence of acquired CJD-MMiK, i.e., transmission of the V2 sCJD strain to individuals with the 129M/M genotype, are only 9.6% (= 26% × 37%) and 2.9% (= 3.2% × 92%) of the total proportions of possible iatrogenic transmission, respectively. The other acquired CJD cases, i.e., the other combinations of source of infection and host codon 129 genotype, may be indistinguishable from sCJD cases with respect to neuropathological and biochemical features, as suggested by animal experiments (2, 3). These facts raise the disturbing possibility that more acquired CJD cases may remain unrecognized.
In conclusion, we identified two CJD cases that were reported as sCJD but that showed distinctive phenotypic features and transmission properties of plaque-type dCJD, which is caused by the transmission of V2 sCJD strain to individuals with the 129M/M genotype. The available clinical records of the two cases did not show the direct evidence of exposure to prion-contaminated neurosurgical instruments, and other potential routes of infection could not be rigorously investigated because of the insufficient clinical information. Nevertheless, the results of the present study may indicate the need to reconsider the potential risk of CJD transmission via occupational exposure as well as neurosurgery. Further investigation of acquired CJD cases and their routes of infections may help to identify novel risk factors of CJD transmission in the future.
ACKNOWLEDGMENTS
This study was supported by Grants-in-Aid from the Ministry of Health, Labor and Welfare of Japan (A.K. and S.M.), Grants-in-Aid for Scientific Research from JSPS (A.K. and T.K.), a grant from MEXT for the Joint Research Program of the Research Center for Zoonosis Control, Hokkaido University (T.K.), and a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) from MEXT (T.K.). Research for development of decontamination/disinfection procedures against various sCJD subgroups has been supported by a Grant-in-Aid from the Research Committee of Surveillance and Infection Control of Prion Disease, the Ministry of Health, Labor and Welfare of Japan (T.K.).
We thank Y. Ishikawa, H. Kudo, M. Yamamoto, and A. Yamazaki for their excellent technical assistance and B. Bell for critical reviews of the manuscript.
REFERENCES
- 1.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. 1999. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. [PubMed] [Google Scholar]
- 2.Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T. 2010. Experimental verification of a traceback phenomenon in prion infection. J Virol 84:3230–3238. doi: 10.1128/JVI.02387-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi A, Iwasaki Y, Otsuka H, Yamada M, Yoshida M, Matsuura Y, Mohri S, Kitamoto T. 2013. Deciphering the pathogenesis of sporadic Creutzfeldt-Jakob disease with codon 129 M/V and type 2 abnormal prion protein. Acta Neuropathol Commun 1:74. doi: 10.1186/2051-5960-1-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bishop MT, Will RG, Manson JC. 2010. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107:12005–12010. doi: 10.1073/pnas.1004688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi A, Asano M, Mohri S, Kitamoto T. 2007. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem 282:30022–30028. doi: 10.1074/jbc.M704597200. [DOI] [PubMed] [Google Scholar]
- 6.Noguchi-Shinohara M, Hamaguchi T, Kitamoto T, Sato T, Nakamura Y, Mizusawa H, Yamada M. 2007. Clinical features and diagnosis of dura mater graft associated Creutzfeldt-Jakob disease. Neurology 69:360–367. doi: 10.1212/01.wnl.0000266624.63387.4a. [DOI] [PubMed] [Google Scholar]
- 7.Yamada M, Noguchi-Shinohara M, Hamaguchi T, Nozaki I, Kitamoto T, Sato T, Nakamura Y, Mizusawa H. 2009. Dura mater graft-associated Creutzfeldt-Jakob disease in Japan: clinicopathological and molecular characterization of the two distinct subtypes. Neuropathology 29:609–618. doi: 10.1111/j.1440-1789.2008.00987.x. [DOI] [PubMed] [Google Scholar]
- 8.Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F, Shiga Y, Kuroiwa Y, Nishizawa M, Kuzuhara S, Inuzuka T, Takeda M, Kuroda S, Abe K, Murai H, Murayama S, Tateishi J, Takumi I, Shirabe S, Harada M, Sadakane A, Yamada M. 2010. Prospective 10-year surveillance of human prion diseases in Japan. Brain 133:3043–3057. doi: 10.1093/brain/awq216. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi A, Matsuura Y, Mohri S, Kitamoto T. 2014. Distinct origins of dura mater graft-associated Creutzfeldt-Jakob disease: past and future problems. Acta Neuropathol Commun 2:32. doi: 10.1186/2051-5960-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoene WC, Masters CL, Gibbs CJ Jr, Gajdusek DC, Tyler HR, Moore FD, Dammin GJ. 1981. Transmissible spongiform encephalopathy (Creutzfeldt-Jakob disease). Atypical clinical and pathological findings. Arch Neurol 38:473–477. [DOI] [PubMed] [Google Scholar]
- 11.Ishida C, Kakishima A, Okino S, Furukawa Y, Kano M, Oda Y, Nakanishi I, Makifuchi T, Kitamoto T, Yamada M. 2003. Sporadic Creutzfeldt-Jakob disease with MM1-type prion protein and plaques. Neurology 60:514–517. doi: 10.1212/01.WNL.0000044403.41041.A4. [DOI] [PubMed] [Google Scholar]
- 12.Kitamoto T, Shin RW, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi J. 1992. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol 140:1285–1294. [PMC free article] [PubMed] [Google Scholar]
- 13.Kitamoto T, Ohta M, Doh-ura K, Hithoshi S, Terao Y, Tateishi J. 1993. Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun 191:709–714. doi: 10.1006/bbrc.1993.1275. [DOI] [PubMed] [Google Scholar]
- 14.Notari S, Capellari S, Giese A, Westner I, Baruzzi A, Ghetti B, Gambetti P, Kretzschmar HA, Parchi P. 2004. Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. J Biol Chem 279:16797–16804. doi: 10.1074/jbc.M313220200. [DOI] [PubMed] [Google Scholar]
- 15.Asano M, Mohri S, Ironside JW, Ito M, Tamaoki N, Kitamoto T. 2006. vCJD prion acquires altered virulence through trans-species infection. Biochem Biophys Res Commun 342:293–299. doi: 10.1016/j.bbrc.2006.01.149. [DOI] [PubMed] [Google Scholar]
- 16.Grathwohl KUD, Horiuchi M, Ishiguro N, Shinagawa M. 1996. Improvement of PrPSc-detection in mouse spleen early at the preclinical stage of scrapie with collagenase-completed tissue homogenization and Sarkosyl-NaCl extraction of PrPSc. Arch Virol 141:1863–1874. doi: 10.1007/BF01718200. [DOI] [PubMed] [Google Scholar]
- 17.Muramoto T, Tanaka T, Kitamoto N, Sano C, Hayashi Y, Kutomi T, Yutani C, Kitamoto T. 2000. Analyses of Gerstmann-Straussler syndrome with 102Leu219Lys using monoclonal antibodies that specifically detect human prion protein with 219Glu. Neurosci Lett 288:179–182. doi: 10.1016/S0304-3940(00)01232-5. [DOI] [PubMed] [Google Scholar]
- 18.Parchi P, Cescatti M, Notari S, Schulz-Schaeffer WJ, Capellari S, Giese A, Zou WQ, Kretzschmar H, Ghetti B, Brown P. 2010. Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 133:3030–3042. doi: 10.1093/brain/awq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. 1998. Protective prion protein polymorphisms against sporadic Creutzfeldt-Jakob disease. Lancet 351:419. [DOI] [PubMed] [Google Scholar]
- 20.Palmer MS, Mahal SP, Campbell TA, Hill AF, Sidle KC, Laplanche JL, Collinge J. 1993. Deletions in the prion protein gene are not associated with CJD. Hum Mol Genet 2:541–544. doi: 10.1093/hmg/2.5.541. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi A, Asano M, Mohri S, Kitamoto T. 2009. A traceback phenomenon can reveal the origin of prion infection. Neuropathology 29:619–624. doi: 10.1111/j.1440-1789.2008.00973.x. [DOI] [PubMed] [Google Scholar]
- 22.Nevin S, McMenemey WH, Behrman S, Jones DP. 1960. Subacute spongiform encephalopathy – a subacute form of encephalopathy attributable to vascular dysfunction (spongiform cerebral atrophy). Brain 83:519–564. doi: 10.1093/brain/83.4.519. [DOI] [PubMed] [Google Scholar]
- 23.Foncin JF, Gaches J, Cathala F, El Sherif E, Le Beau J. 1980. Transmission iatrogène interhumaine possible de maladie de Creutzfeldt-Jakob avec atteinte des grains du cervelet. Rev Neurol (Paris) 136:280. [Google Scholar]
- 24.Will RG, Matthews WB. 1982. Evidence for case-to-case transmission of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 45:235–238. doi: 10.1136/jnnp.45.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.el Hachimi KH, Chaunu MP, Cervenakova L, Brown P, Foncin JF. 1997. Putative neurosurgical transmission of Creutzfeldt-Jakob disease with analysis of donor and recipient: agent strains. C R Acad Sci III 320:319–328. doi: 10.1016/S0764-4469(97)82774-6. [DOI] [PubMed] [Google Scholar]
- 26.Alcalde-Cabero E, Almazán-Isla J, Bradel JP, Breithaupt M, Catarino J, Collins S, Hayback J, Hoftberger R, Kahana E, Kovacs GG, Ladogana A, Mitrova E, Molesworth A, Nakamura Y, Pocchiari M, Popovic M, Ruiz-Tovar M, Taratuto A, van Duijn C, Yamada M, Will RG, Zerr I, de Pedro Cuesta J. 2012. Health professionals and risk of sporadic Creutzfeldt-Jakob disease, 1965 to 2010. Euro Surveill 17:pii=20144 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20144. [PubMed] [Google Scholar]
- 27.Hamaguchi T, Noguchi-Shinohara M, Nozaki I, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Yamada M. 2009. Medical procedures and risk for sporadic Creutzfeldt-Jakob disease, Japan, 1999–2008. Emerg Infect Dis 15:265–271. doi: 10.3201/eid1502.080749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delisle MB, Fabre N, Rochiccioli P, Doerr-Schott J, Rumeau JL, Bes A. 1993. Creutzfeldt-Jakob disease after treatment with human extracted growth hormone. A clinicopathological study. Rev Neurol (Paris) 149:524–527 (In French.) [PubMed] [Google Scholar]
- 29.Billette de Villemeur T, Gelot A, Deslys JP, Dormont D, Duyckaerts C, Jardin L, Denni J, Robain O. 1994. Iatrogenic Creutzfeldt-Jakob disease in three growth hormone recipients: a neuropathological study. Neuropathol Appl Neurobiol 20:111–117. doi: 10.1111/j.1365-2990.1994.tb01169.x. [DOI] [PubMed] [Google Scholar]
- 30.Brown P, Gibbs CJ Jr, Amyx HL, Kingsbury DT, Rohwer RG, Sulima MP, Gajdusek DC. 1982. Chemical disinfection of Creutzfeldt-Jakob disease virus. N Engl J Med 306:1279–1282. doi: 10.1056/NEJM198205273062107. [DOI] [PubMed] [Google Scholar]
- 31.Taguchi F, Tamai Y, Uchida K, Kitajima R, Kojima H, Kawaguchi T, Ohtani Y, Miura S. 1991. Proposal for a procedure for complete inactivation of the Creutzfeldt-Jakob disease agent. Arch Virol 119:297–301. doi: 10.1007/BF01310679. [DOI] [PubMed] [Google Scholar]
- 32.Tateishi J, Tashima T, Kitamoto T. 1991. Practical methods for chemical inactivation of Creutzfeldt-Jakob disease pathogen. Microbiol Immunol 35:163–166. doi: 10.1111/j.1348-0421.1991.tb01544.x. [DOI] [PubMed] [Google Scholar]
- 33.Kimberlin RH, Walker CA, Millson GC, Taylor DM, Robertson PA, Tomlinson AH, Dickinson AG. 1983. Disinfection studies with two strains of mouse-passaged scrapie agent. J Neurol Sci 59:355–369. doi: 10.1016/0022-510X(83)90021-7. [DOI] [PubMed] [Google Scholar]
- 34.Taylor DM, Fraser H, McConnell I, Brown DA, Brown KL, Lamza KA, Smith GR. 1994. Decontamination studies with the agents of bovine spongiform encephalopathy and scrapie. Arch Virol 139:313–326. doi: 10.1007/BF01310794. [DOI] [PubMed] [Google Scholar]
- 35.Parchi P, Strammiello R, Giese A, Kretzschmar H. 2011. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol 121:91–112. doi: 10.1007/s00401-010-0779-6. [DOI] [PubMed] [Google Scholar]
- 36.Doh-ura K, Kitamoto T, Sakaki Y, Tateishi J. 1991. CJD discrepancy. Nature 353:801–802. [DOI] [PubMed] [Google Scholar]