

ABSTRACT
Previously, we reported that a mutant of Tat referred to as Nullbasic inhibits HIV-1 reverse transcription although the mechanism of action is unknown. Here we show that Nullbasic is a reverse transcriptase (RT) binding protein that targets the reverse transcription complex rather than directly inhibiting RT activity. An interaction between Nullbasic and RT was observed by using coimmunoprecipitation and pulldown assays, and a direct interaction was measured by using a biolayer interferometry assay. Mixtures of recombinant 6×His-RT and Nullbasic-FLAG-V5-6×His at molar ratios of up to 1:20,000 did not inhibit RT activity in standard homopolymer primer template assays. An analysis of virus made by cells that coexpressed Nullbasic showed that Nullbasic copurified with virus particles, indicating that it was a virion protein. In addition, analysis of reverse transcription complexes (RTCs) isolated from cells infected with wild type or Nullbasic-treated HIV-1 showed that Nullbasic reduced the levels of viral DNA in RTC fractions. In addition, a shift in the distribution of viral DNA and CAp24 to less-dense non-RTC fractions was observed, indicating that RTC activity from Nullbasic-treated virus was impaired. Further analysis showed that viral cores isolated from Nullbasic-treated HIV undergo increased disassembly in vitro compared to untreated HIV-1. To our knowledge, this is the first description of an antiviral protein that inhibits reverse transcription by targeting the RTC and affecting core stability.
IMPORTANCE HIV-1 infection is treated by using combinations of antiretroviral drugs that target independent steps of virus replication. A newly described antiviral protein called Nullbasic can also inhibit a combination of different steps in virus replication (transcription, reverse transcription, and Rev-mediated viral mRNA transport), although the precise mechanism of action is unknown. This study shows that Nullbasic can inhibit reverse transcription by binding to the viral enzyme called reverse transcriptase, which results in accelerated uncoating of the viral core and instability of the viral apparatus called the reverse transcription complex (RTC). This unique antiviral activity may inform development of other RTC inhibitors, as well as providing a unique investigative tool for dissecting the RTC cellular composition.
INTRODUCTION
Like all retroviruses, HIV-1 has a single positive-sense strand of RNA genome that is converted into double-strand proviral DNA by a hallmark process called reverse transcription. Proviral DNA is subsequently integrated into the host chromosomes and is transcribed by RNA polymerase II producing viral mRNA. The mechanisms regulating reverse HIV-1 transcription have been described in detail elsewhere (1). Briefly, the viral mRNA genome annealed to host cell tRNALys3 form a ribonucleoprotein complex with viral proteins, including reverse transcriptase (RT), integrase (IN), and nucleocapsid to form a prototypical reverse transcription complex (RTC) (2). The initiation of reverse transcription by the RTC begins shortly after cell infection after cytoplasmic nucleotides become available. Using tRNALys3 as a primer, DNA synthesis by RT produces a short strand of DNA called negative-strand strong stop DNA (−sssDNA). Degradation of the viral RNA strand by RT RNase H activity liberates −sssDNA that is transferred to the 3′ end of the viral RNA by annealing of complementary nucleotide sequences, a step called first-strand transfer. The synthesis of the remaining negative-strand DNA can then be completed by RT. The complete synthesis of double-strand proviral DNA follows additional DNA synthesis following additional priming reactions and strand displacement DNA synthesis by RT. Cellular factors, including eEF1A, associate with the RTC and play an important role in the reverse transcription process (3, 4).
Many virion proteins, including Tat, affect the efficiency of reverse transcription. Tat is an HIV-1 regulatory protein with pleiotropic effects on various cellular and viral functions. As examples, Tat stimulates HIV-1 gene expression through interaction with a cellular transcription factor called pTEFb, composed of cyclin T1 and CDK9, and histone deacetylases (5). Tat regulates at least two steps of HIV-1 mRNA processing, including cotranscriptional capping by Mce1 (6) and mRNA splicing through interactions with p32, an ASF/SF-2 splicing cofactor (5). Harrich et al. demonstrated that native Tat stimulated HIV-1 reverse transcription (7), and Apolloni et al. showed that Tat improved the binding of reverse transcriptase to the RNA template, which required intact Tat activation and basic domains (8), although a precise role for Tat in reverse transcription has been controversial (9).
Previously, we described a mutant of the two-exon HIV-1 Tat protein, termed Nullbasic, which can potently inhibit multiple steps of the HIV replication cycle (10). Nullbasic was created by replacing the entire arginine-rich basic domain of wild-type Tat with glycine/alanine residues. Given that Tat has reported ability to enhance HIV-1 reverse transcription (7, 8, 11, 12), is present in HIV-1 virions (13), and can interact with reverse transcriptase (8), we performed experiments to determine whether mutant forms of Tat could antagonize this Tat function. We showed that Nullbasic did strongly inhibited HIV-1 reverse transcription (10, 14), but, unexpectedly, Nullbasic effectively decreased the steady-state levels of unspliced and singly spliced viral mRNA, an activity caused by inhibition of HIV Rev (10, 15). Both human T cell lines and primary human CD4+ cells constitutively expressing Nullbasic were protected from a spreading infection by HIV-1 (14), indicating that Nullbasic is a potent HIV-1 inhibitor. Very few single antiviral agents that block HIV replication at multiple steps of the life cycle have been described (16).
Like some related one-exon Tat mutants (17, 18), Nullbasic exhibited trans-dominant negative (TDN) effects on Tat-dependent HIV gene expression (10). TDN inhibitors (TDNi) are defined as a mutant proteins that lack an intrinsic activity that can inhibit function of the wild-type protein in trans (19). Early studies showed that Tat TDNi inhibited HIV-1 trans-activation of transcription if expressed in excess compared to wild-type Tat (17, 18). The proposed mechanism of Tat TDNi inhibition of HIV-1 transcription involves sequestration of the cellular protein complex PTEF-b (18, 20). Tat TDNi can interact with PTEF-b but due to mutation of the basic domain cannot bind HIV-1 TAR RNA (21), which is necessary for hyperphosphorylation of the RNA polymerase II C-terminal domain by PTEF-b, leading to efficient transcriptional elongation. Subsequent studies showed that first-generation Tat TDNi modestly protected human T cells lines from HIV-1 replication (22), and coexpression of a Tat and Rev TDN (RevM10) inhibited HIV-1 infection and replication better than individual expression (22), presumably by additive or multiplicative effect due to inhibiting two steps of virus replication. RevM10 is a competitive inhibitor wild-type Rev (23) that binds CRM1 and inhibits export of Rev-mRNA complexes from the nucleus (24). We showed that coexpression of Nullbasic and Rev caused a CRM1-dependent change of Rev localization in cells and defective Rev function (10, 15).
Nullbasic can also inhibit HIV-1 reverse transcription that significantly diminishes infectivity of HIV-1, as well virus-like particles (VLPs) of lentiviral vector systems typically based on HIV-1 made by cells expressing Nullbasic (10, 14). Nullbasic inhibits HIV-1 and VLP infectivity 4- to >10-fold in a dose-dependent manner. Defective HIV-1 and VLP infection caused by Nullbasic has been observed in different target cells, including primary human CD4 lymphocytes, Jurkat, HeLa, TZM-bl, and HEK293T cells (10, 14). With respect to early replication, the affected HIV-1 or VLPs are defective for synthesis of the first product of reverse transcription called negative-strand strong stop DNA (−sssDNA) as determined by monitoring endogenous reverse transcription reactions and viral DNA synthesis following cell infection (10, 14). While Nullbasic affects HIV-1 and HIV-1-based VLPs, it does not affect murine leukemia virus based retroviral vectors indicating that Nullbasic is a specific HIV-1 inhibitor (14). Whether this is due to direct or indirect effects of Nullbasic on RT, the RTC or another mechanism has not been determined.
In this study, we further investigated how Nullbasic inhibits reverse transcription. We show here that Nullbasic can directly bind HIV-1 RT. Although this does not appear to downregulate RT activity in a standard in vitro RT assay, our experiments indicate that Nullbasic affects viral core disassembly, leading to defective viral DNA synthesis by the reverse transcription complex. To our knowledge, this makes Nullbasic a protein inhibitor of the reverse transcription with a novel mechanism of action.
MATERIALS AND METHODS
Cell lines and HIV-1NL4.3.
HEK293T cell lines were grown in Dulbecco modified Eagle medium supplemented with 10% heat-inactivated newborn bovine serum, penicillin-streptomycin 50 to 100 IU/ml. All cells were incubated at 37°C in 5% CO2 incubator. A stock of HIV-1NL4.3was generated as previously described (3) by transfection of the corresponding proviral DNA using X-tremeGENE HP DNA transfection reagent (Roche Applied Science) into HEK293T cells according to the manufacturer's recommendations. Cell culture supernatants were removed at 48 h posttransfection and centrifuged (200 × g, 10 min), and the supernatant was filtered (0.45-μm pore size) and stored in 1-ml aliquots at −80°C.
Plasmids.
For expression of HIV RT subunits p51and p66 in cells, the coding DNA sequences were amplified from pHGPsyn (25), which contains a Gag/Pol codon-optimized sequence, and the PCR fragment was inserted into pDONR vector using Gateway system (Invitrogen) by BP recombination reaction. The inserts were then transferred into destination mammalian expression vectors that contain a V5 tag by LR recombination reactions. The pCDNA3.1-Nullbasic-FLAG and pCDNA3.1-Nullbasic-FLAG-mCherry expression plasmids were previously described (10, 15). A Nullbasic-FLAG DNA fragment lacking a stop codon was produced by PCR using the Gateway system and inserted into pDEST42. The plasmid pDEST42-Nullbasic-FLAG was used to make recombinant Nullbasic-FLAG-V5-6×His protein in Escherichia coli. To make the pGCHΔENV plasmid, the pGCH plasmid (10, 15) was digested with BamHI and SalI to make a vector. A corresponding BamHI and SalI DNA fragment was obtained from the plasmid pNL4-3.Luc.R−.E− (NIH AIDS Reagent Program catalog number 3418) and ligated into the pGCH vector.
Recombinant proteins.
The recombinant RT protein was kindly provided by Stuart Le Grice (HIV Drug Resistance Program, Center for Cancer Research, National Cancer Institute). The recombinant Nullbasic-FLAG-V5-6×His was produced in E. coli strain BL21-AI (Invitrogen) transformed by pDEST42-Nullbasic-FLAG plasmid. A 200-ml culture was grown in Luria broth to log phase and induced with 0.2% arabinose and 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After 4 h, the E. coli was collected by centrifugation and immediately processed. First, 1× FastBreak cell lysis reagent (Promega) was added to the cell pellet for 15 min, and the lysate was mixed with 2 ml of HisLink protein purification resin (Promega) for 1 min. The beads were placed into a column and 6 ml of wash buffer (100 mM HEPES [pH 7.5], 10 mM imidazole) was applied six times. Then, 2 ml of elution buffer (100 mM HEPES [pH 7.5], 500 mM imidazole) was added and collected after 1 min. The eluted protein was dialyzed twice for 90 min each in 1 liter of storage buffer (20% glycerol [vol/vol], 50 mM Tris-HCl [pH 8.0], and 1 mM ZnSO4, adding fresh 2 mM 1,4-dithioerythritol every 30 min). The protein was stored in aliquots in liquid nitrogen.
Coimmunoprecipitation (co-IP), pull-down, and Western blot analyses.
HEK293T cells were transfected with each plasmid alone or in combination as shown. The cells were lysed at 48 h posttransfection in S100 buffer (10 mM Tris [pH 7.4], 1.5 mM MgCl2, 10 mM KCl, 1× complete protease inhibitor cocktail [Roche Applied Science], 0.5 mM β-mercaptoethanol) using a Dounce homogenizer. The lysate was cleared by centrifugation in a refrigerated microcentrifuge at 4°C and 13,500 × g for 30 min. The supernatant (500 μl) was incubated with 10 μl of anti-FLAG antibody coated beads (Sigma, IL) for 2 h at 4°C. The immunoprecipitate was washed twice with S100 buffer plus 0.01 to 0.02% Triton X-100, followed by Western blot analysis with anti-Tat or anti-RT antibodies. For the pulldown experiment, FLAG-beads were saturated with recombinant Nullbasic-FLAG-V5-6×His protein and washed with 1× phosphate-buffered saline (PBS) to remove unbound protein. The loaded beads were incubated with 500 μl of RTp51 and RTp66 lysates. As a control, a lysate containing RTp51 and RTp66 were incubated with unbound FLAG-beads. Otherwise, pulldown assays were performed as described for the co-IP assays.
Biolayer interferometry (BLI) assay.
RT (50 μg) was biotinylated using EZ-Link Sulfo-NHS-Biotin according to the manufacturer's instructions (Pierce Biotechnology, IL). To immobilize the biotinylated protein onto biosensors, biosensors coupling with streptavidin (Pall ForteBio, CA) were incubated in the biotinylated protein at a final concentration of 1 μM for 15 min in 1× kinetic buffer using the OctetRed system (Pall ForteBio). The association of the two proteins was measured by incubating ligand biosensors (biotinylated protein) into another protein solution (analyte, nonbiotinylated) with shaking at 1,000 rpm. The concentration of analytes Nullbasic-FLAG-V5-6×His and bovine serum albumin (BSA) ranged from 270 nM to 10 nM in 3-fold serial dilutions. The dissociation was determined by moving the ligand biosensor from analyte to the kinetic buffer only. The kinetic buffer (1 mM phosphate, 15 mM NaCl, 0.002% Tween 20, 1.5 μM gelatin) was used in all experiments. The data were analyzed using the ForteBio data analysis program (Pall ForteBio), and a mean kd and ka for each protein-protein interaction was calculated from three independent experiments, where Kd = kd/ka. A chi-squared test, least-squares fit analysis was determined by the using ForteBio data analysis program.
RT assay.
The RT assay (Roche Applied Science) was performed according to the manufacturer's instructions with exceptions. Protein mixtures containing recombinant HIV-1 RT heterodimer at 1.5 nM and recombinant Nullbasic-FLAG-V5-6×His at 3 nM, 30 nM, 300 nM, and 3 μM in 1× PBS (pH 7.5) were incubated at room temperature for 30 min. A separate sample included the RT inhibitor nevirapine used at a final concentration of 200 nM. The remaining reaction components provided by the manufacturer were added, and the samples were incubated at 37°C for 10 min. The DNA products were processed according to the protocol provided.
Immunofluorescence microscopy.
HEK293T cells (0.5 × 106) were grown on glass coverslips in six-well plates and transfected with 1 μg of Nullbasic-FLAG-mCherry plasmid using X-tremeGENE HP DNA transfection reagent according to the manufacturer's instructions. After 24 h, the cells were fixed in 3% (wt/vol) paraformaldehyde at room temperature for 10 min and quenched with 50 mM NH4Cl for 5 min. The cells were then permeabilized with 0.1% (vol/vol) Triton X-100 for 15 min. Nuclei were stained with 1 μM DAPI (4′,6′-diamidino-2-phenylindole; Life Technologies). Finally, the coverslips were mounted onto slides with ProLong Gold antifade reagent (Life Technologies). Fluorescent images were captured using a Leica TCS SP2 confocal scanning microscope (Leica Microsystems) with ×63 objective lenses and standard lasers and filters for mCherry and DAPI fluorescence.
Purification of HIV-1 particles by velocity gradients.
HEK293T cells were cotransfected with 5 μg of pGCH proviral plasmid and 3.3 μg of pcDNA3.1/Nullbasic-FLAG or pcDNA3.1 plasmid. Cell culture supernatants were harvested 48 h after transfection and filtered through 0.45-μm-pore-size filters. Viruses in supernatants were concentrated by ultracentrifugation through a 20% (vol/vol) sucrose cushion at 125,000 × g for 1 h. The pellet was washed with 1× PBS and resuspended in 500 μl of PBS overnight at 4°C. The concentrated viral particles were further purified by velocity gradient ultracentrifugation using Optiprep (Axis-Shield) diluted from 6 to 18% as described previously (26). Gradient fractions (1 ml) were collected from the top of the tube and assayed for p24 antigen using the RETROtek HIV-1 p24 antigen enzyme-linked immunosorbent assay (ELISA) (Zeptometrix Corp.). The purified virions in each gradient fraction were concentrated again by ultracentrifugation and subjected to Western blot analysis. Nullbasic-FLAG protein was detected using a rabbit anti-DYKDDDDK Tag polyclonal antibody (Cell Signaling Technology).
Subtilisin analysis.
The subtilisin assay used was adapted from a published method (27). Briefly HIV-1 was concentrated by ultracentrifugation through 20% sucrose at 125,000 × g for 1 h at 4°C. The pellet was resuspended in PBS at a ratio of 1:100. Concentrated virus preparation was added to an equal volume of subtilisin digestion buffer at a ratio of 1:1, followed by incubation at 37°C for 2 h. The enzymatic reaction was stopped by the addition of 5 μg of phenylmethylsulfonyl fluoride/ml. The digested virions were pelleted through another 20% sucrose cushion by ultracentrifugation as previously described. The pelleted samples were solubilized in 2× sodium dodecyl sulfate (SDS) loading dye and used in Western blot assays with an anti-HIVIG (NIH AIDS Reagent Program), anti-CA (Santa Cruz), anti-MA (Santa Cruz), and M2 anti-FLAG (Sigma) antibodies and the appropriate secondary antibody conjugated to horseradish peroxidase (HRP).
RTC purification.
Purification of HIV RTC has been described previously (28). Briefly, VSV-G envelope-pseudotyped HIV-1 was produced by cotransfecting HEK293T cells in a 10-cm dish that were 70% confluent with 5 μg of pGCHΔENV proviral plasmid, 2 μg of pCMV-VSV-G plasmid, and 3.3 μg of pcDNA3.1/Nullbasic-FLAG or pcDNA3.1 using X-tremeGENE HP DNA transfection reagent according to the manufacturer's instructions. At 48 h posttransfection, supernatants containing viruses were harvested, filtered through 0.45-μm-pore-size filters, and treated with DNase I for 30 min at 37°C. Viral CAp24 concentrations were determined by ELISA. HEK293T cells were grown in a T-175 flask until they were 80% confluent and then incubated with HIV-1 supernatant having 5 μg of CAp24 or the same amount of heat-inactivated HIV-1 using Polybrene (8 μg/ml) at 4°C for 2 h and then at 37°C for 4 h. Cell lysates prepared as previously described (3) were placed on top of a linear sucrose gradient (20 to 70% [wt/wt]) and subjected to centrifugation at 145,000 × g for 18 h at 4°C. Then, 1-ml fractions were collected from the bottom of tube and assayed by qPCR for HIV-1 −sssDNA as previously described (29) using 0.2-μl portions of each fraction. The CAp24 concentrations in each fraction were assayed by ELISA.
HIV-1 entry assay.
HEK293 cells grown in 10-cm dishes until 80% confluent were treated with 1 μM nevirapine (NVP) for 2 h at 37°C and then incubated with VSV-G-pseudotyped HIV-1 or HIV-1-Nullbasic viral supernatant containing 500 ng of CA for 2 h at 4°C to facilitate virus attachment. The cells were incubated at 37°C to initiate envelope and cell membrane fusion and then incubated for a further 2 h. Each sample was collected using TRIzol reagent (Life Technologies), and the RNA fraction was column purified using a Direct-Zol miniprep kit (Zymo Research) and treated on the column with DNase I prior to elution. The purified RNAs were used in RT-PCRs in the presence or absence of SuperScript III reverse transcriptase (Life Technologies) using random hexamer oligonucleotides for the first-strand DNA synthesis. Second-strand DNA and PCR was performed using oligonucleotides specific for full-length viral mRNA as previously described (10). All samples were used in qPCRs to measure eEF1A mRNA using the oligonucleotides 5′-CTGGAGCCAAGTGTCTAATATGCC (forward) and 5′-GCCAGGCTTGAGAACACCAGTC (reverse). A plasmid containing EEF1A1 was used to generate a standard curve. A Rotor-Gene 600 was used for DNA amplification at 95°C for 15 s and annealing and elongation at 60°C for 1 min. A negative control without cDNA template was included in every assay.
In vitro core stability assay.
HIV-1 cores were purified as previously described with minor modification (30). Briefly, high titer HIV-1 described above was pelleted by ultracentrifugation at 125,000 × g for 1 h through 20% sucrose and resuspended in 1× PBS–1 mM EDTA overnight at 4°C. The virus particles underwent a “spin-thru” detergent treatment as previously described (30) in a 30 to 70% sucrose in 1× PBS–1 mM EDTA buffer. Gradients were formed using a Biocomp gradient master (BioComp Instruments, Inc.). The gradients were fractionated from the bottom of the gradient collecting 1-ml fractions. Fractions containing CA with a density of 1.24 to 1.27 were used in in vitro uncoating assays as previously described (30) using a Beckman TLA100.3 rotor.
Electron microscopy analysis.
HEK293T cells were cotransfected with 2 μg of pGCH proviral plasmid and 1.7 μg of pCDNA3.1/Nullbasic-FLAG or pcDNA3.1 plasmid. At 48 h posttransfection, the cells were fixed with 5% (vol/vol) glutaraldehyde (Electron Microscopy Sciences) at room temperature for 2 h. Fixed cells were contrasted with 1% osmium tetroxide and 4% uranyl acetate prior to dehydration and embedding in LX-112 resin (31). Sections (60 nm) were cut using an ultramicrotome (UC64; Leica), and viral particles were visualized at ×60,000 using a transmission electron microscope (model 1011; JEOL) equipped with a Morada cooled charge-coupled device camera and iTEM AnalySIS software. Three samples of Nullbasic-treated and untreated cells were inspected collecting sets of 60 to 160 images of virus particle from each independent sample. An unpaired Student t test was used to calculate P values.
RESULTS
Nullbasic directly interacts with RT.
We previously reported that the HIV-1 Tat protein could associate with RT (8), suggesting the possibility that Nullbasic (10), a mutant of Tat, may also interact with RT. To test this possibility, co-IP experiments were undertaken using plasmids expressing carboxyl-FLAG epitope tagged Nullbasic and plasmids expressing codon-optimized reverse transcriptase p66 and p51 genes each having carboxyl-6×His and V5 epitope tags (25). HEK293T cells were transfected with each plasmid alone or in combination as shown (Fig. 1A). After 48 h, lysates were prepared, and Nullbasic was immunoprecipitated with anti-FLAG antibody-coated beads. We observed that RT subunits, RT p66 and p51, expressed separately were coimmunoprecipitated, indicating that Nullbasic could associate with either RT subunit. Pulldown experiments performed in parallel using recombinant Nullbasic-FLAG-V5-6×His protein purified from E. coli (shown in Fig. 1C) also showed that Nullbasic could associate with each RT subunit (Fig. 1B, lanes 6 and 7). Neither RT subunit bound to the anti-FLAG beads in the absence of Nullbasic (Fig. 1 lane 4). These experiments indicate that Nullbasic can associate with RT p66 and p51 subunits in vitro.
FIG 1.
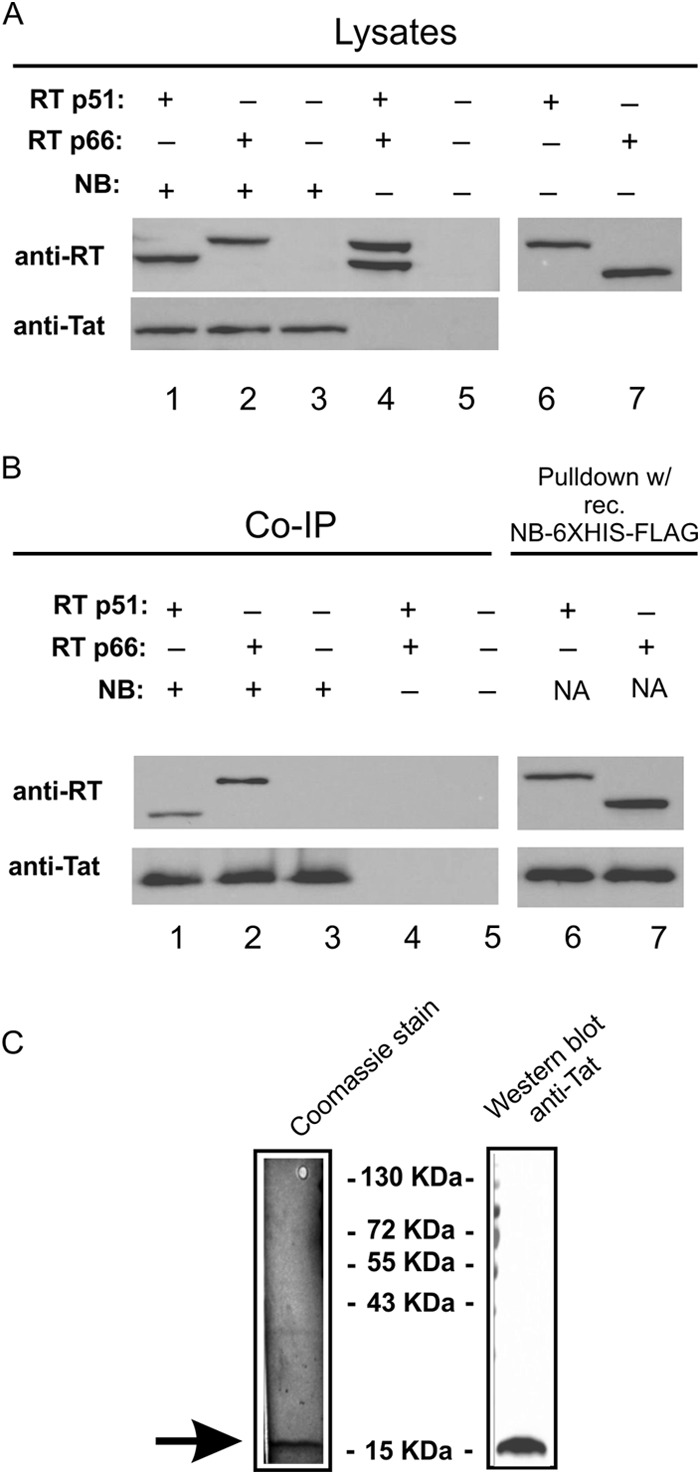
Co-IP of RT subunits with Nullbasic. HEK293T cells were transfected with plasmids that can express RT subunits p51 (RT p51) and p66 (RT p66). The RT subunits have carboxy-terminal 6×His and V5 epitope tags. (A) As indicated for lanes 1 to 7, a plasmid expressing Nullbasic-FLAG (NB) was transfected alone or in combination with an RT expression plasmid(s). Cell lysates were made from transfected cells, and 20 μg of total protein was analyzed by SDS-PAGE and Western blotting with rabbit anti-RT or anti-Tat antibody. (B) In lanes 1 to 5, immunoprecipitation reactions were performed with 100 μg of total protein, and beads coated with a mouse anti-FLAG monoclonal antibody were incubated at 4°C for 2 h. In lanes 6 and 7, pull-down reactions were performed with 100 μg of total protein, and beads coated with recombinant Nullbasic-FLAG-6×His protein were incubated at 4°C for 2 h. The captured protein underwent SDS-PAGE and Western blot analysis with the same anti-RT and anti-Tat antibodies. The results of a representative experiment of four experiments that all had similar results are shown. (C) Nullbasic-FLAG-V5-6×His protein was purified from E. coli. Approximately 50 ng of Nullbasic was applied to a SDS–10% PAGE gel and stained with Coomassie blue (left panel). A parallel lane on the same gel that underwent Western blot analysis with a rabbit anti-Tat antibody is shown (right panel).
Next, using the Octet Red96 system, BLI was used to determine whether protein association between recombinant Nullbasic (Fig. 1C) and RT was direct. A streptavidin-coated biosensor was saturated with biotinylated RT, incubated with Nullbasic-FLAG-V5-6×His or BSA protein in a range of 10 to 270 nM, and subsequently immersed in disassociation buffer. The biosensor sensograms were recorded using Octet Red96 system. The sensograms clearly showed that Nullbasic bound to immobilized recombinant RT under these experimental conditions (Fig. 2A). The kinetic interactions between Nullbasic-FLAG-V5-6×His and an RT heterodimer probe showed that interaction was satisfactorily fitted by a 1:1 binding model. The goodness of the fitted curves was indicated by a low apparent χ2 of 1.87, where values of <10 are considered acceptable (32). The calculated Kd of the interaction was ∼40 nM under the conditions tested. No binding between the RT probe and BSA was detected, indicating that the interaction between RT and Nullbasic was specific (Fig. 2B). The BLI data supported the results obtained by co-IP and pulldown experiments and further indicated that the interaction between RT and Nullbasic is direct.
FIG 2.
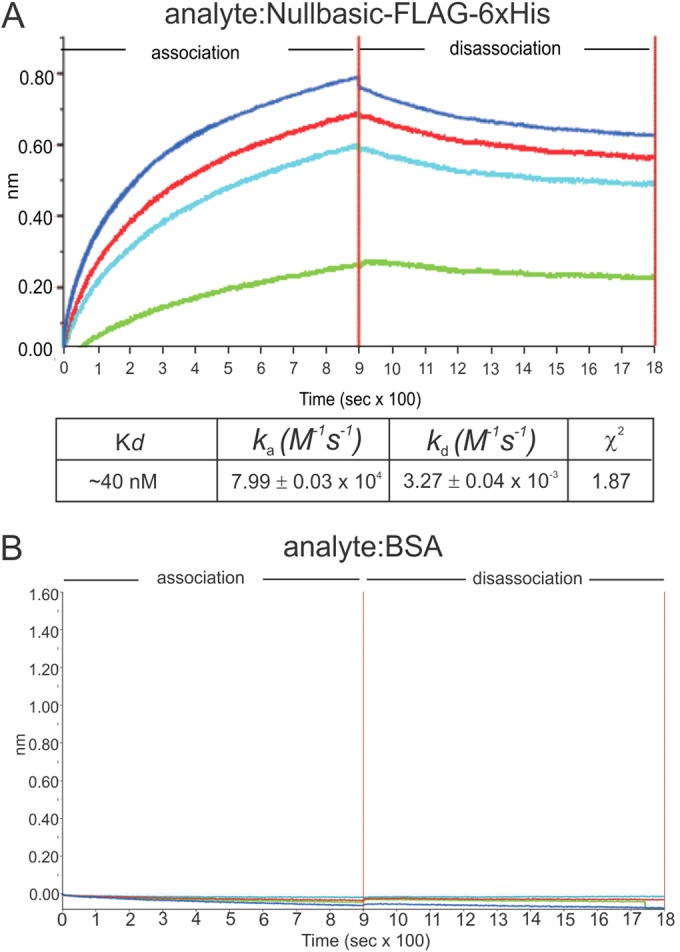
BLI sensograms of binding events between Nullbasic and RT. The Octet Red96 system was used to measure binding events between Nullbasic and RT. (A and B) BLI sensograms. Biotinylated RT p51/66 was bound to a streptavidin biosensor and applied to solutions containing 10 nM (green), 30 nM (light blue), 90 nM (red), or 270 nM (dark blue) of Nullbasic-FLAG-V5-6×His (A) or (B) BSA. The BLI experiment was repeated at three times with similar results. The results of a representative sensogram are shown.
Recombinant Nullbasic does not affect RT activity in vitro.
We undertook in vitro experiments to address whether Nullbasic inhibited RT activity in a standard in vitro HIV-1 RT assay. The assay uses a homopolymer RNA template and oligonucleotide primer as a substrate for DNA synthesis by RT. RT (1.5 nM) was mixed with same Nullbasic-FLAG-V5-6×His used in previous experiments (Fig. 1C) or BSA at molar ratios of 1:2, 1:20, 1:200, 1:2,000, and 1:20,000 for 30 min at room temperature prior to addition to the RT assay. The assays were incubated at 37°C for 10 min, and the amount of DNA made was compared to DNA levels made by standard concentrations of RT run in parallel. The results showed no significant inhibition of RT activity in the presence of Nullbasic or BSA at the concentrations tested (Fig. 3). As a control, side by side assays were incubated with nevirapine at 200 nM that produced no detectable DNA (data not shown). The results indicate that recombinant Nullbasic does not directly inhibit RT activity in vitro in a standard assay.
FIG 3.
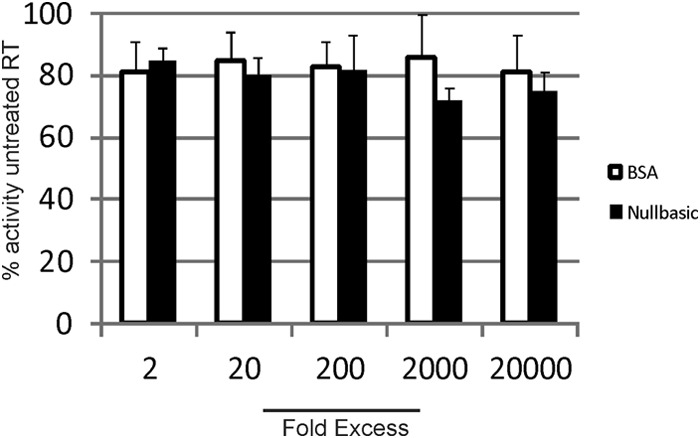
Nullbasic does not inhibit RT enzymatic activity in vitro. HIV-1 RT was preincubated with Nullbasic or BSA for 30 min at room temperature, as indicated. The remaining reaction components were added to the protein mixture, followed by incubation for 10 min at 37°C. The reverse transcription DNA products were measured according to the manufacturer's instructions. The experiment was performed in triplicate and repeated three times. The results of a representative experiment are shown. Mean values and standards error of the mean are shown.
Nullbasic is present in HIV-1 particles.
Confocal imaging of Nullbasic-FLAG-mCherry fusion proteins show that they are diffusely distributed in the cytoplasm and nucleus when expressed in human HeLa cells (10, 15) and HEK293T cells (Fig. 4A). It seemed possible that Nullbasic could be packaged in virions during budding given its wide distribution in the cytoplasm, its proximity to the cell plasma membrane, and its interaction with RT. Also, HIV-1 made by cells expressing Nullbasic are defective in endogenous reverse transcription reactions and in reverse transcription after cell infection compared to HIV-1 made by untreated cells (10, 14). To test this possibility, we made HIV-1 in cells by cotransfection of pGCH, a proviral HIV-1 expression plasmid, and a Nullbasic-FLAG expression plasmid. The virus particles were purified using iodixanol velocity gradients that separate HIV-1 from contaminating cellular microvesicles (26). Virus in supernatants were pelleted through 20% sucrose, resuspended in 1× PBS and placed onto a 6 to 18% iodixanol velocity gradient. Fractions collected from the top of the gradient were assayed for CA by ELISA (Fig. 4) and for the presence of Nullbasic-FLAG by Western blotting. The elution profile for capsid was similar for HIV-1 irrespective of whether the virus producing cell expressed Nullbasic (Fig. 4). It was observed that Nullbasic was detected in the same fractions that contained peak CA in fractions 4 and 5 consistent with its incorporation in virus particles.
FIG 4.

Nullbasic is present in HIV-1 virions. (A) HEK293T cells were transfected with a Nullbasic-FLAG-mCherry expression plasmid. After 24 h, the cells were stained with DAPI. The cells were visualized by fluorescent confocal microscopy, and the white bar in the left panel indicates 10 μm. (B) Isolation of HIV-1 by velocity gradient centrifugation and detection of Nullbasic. HIV-1 was produced by cotransfection of a proviral plasmid with a Nullbasic-FLAG expression plasmid (right panel) or an empty expression plasmid (left panel). The virus was pelleted through 20% sucrose cushion, collected in 1× PBS, placed on the top of a linear 6 to 18% iodixanol gradient, and centrifuged for 1.5 h at 250,000 × g. Fractions were collected from the top and were assayed for CAp24 by ELISA and Nullbasic-FLAG by Western blot analysis with an anti-FLAG antibody. The experiment was repeated three times with similar results. The results of a representative experiment are shown.
We performed subtilisin treatment to confirm that Nullbasic was present in virus particles. As shown in Fig. 5, coexpression of Nullbasic and HIV-1 by cells produced particles that copelleted with Nullbasic after passing through a 20% sucrose cushion (Fig. 5A, lane 2). Nullbasic was not detected in control HIV-1 (Fig. 5A, lane 1). Western blot analysis of HIV-1 treated with subtilisin showed significant reduction in levels of gp120 compared to untreated HIV-1 (Fig. 5B and C, top panels), but the levels of CA or MA were similar, confirming that subtilisin did not degrade proteins within virus particles. Nullbasic was also detected in the same Western blot, indicating that Nullbasic was inside virus particles and thus protected from subtilisin digestion. The combined experiments indicate that expression of Nullbasic in HIV-1-infected cells results in the incorporation of Nullbasic inside virus particles.
FIG 5.
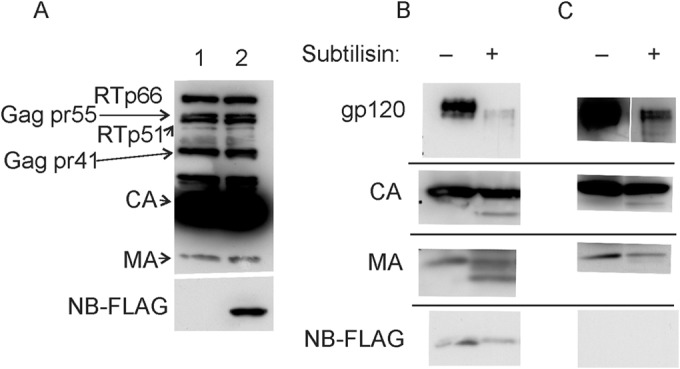
Subtilisin digestion of HIV-1 indicates Nullbasic is packaged in virions. HIV-1 virus stocks described used for velocity gradient analysis were pelleted through a 20% sucrose cushion. Samples of each concentrated virus were incubated with or without 1× subtilisin digestion buffer and analyzed by Western blot analysis. (A) Concentrated HIV-1 produced in the absence (lane 1) or presence (lane 2) of Nullbasic underwent Western blot analysis with an anti-HIV antibody (top panel) or the M2 anti-FLAG antibody (bottom panel). (B) Western blot analysis of Nullbasic-treated HIV-1 with antibodies specific for gp120 (top panel), CA (second panel), MA (third panel), and FLAG (that detects Nullbasic) in the presence or absence of subtilisin as indicated. (C) A Western blot as described for panel B but using HIV-1 made by cells not coexpressing Nullbasic. The experiment was repeated twice with similar results, and the results of a representative experiment are shown.
Viral DNA association with the RTC is reduced by Nullbasic.
Since recombinant Nullbasic had no effect on RT activity in vitro (Fig. 3) but can downregulate −sssDNA synthesis during reverse transcription in ERT assays and in cells (10, 14), we examined whether Nullbasic affected DNA synthesis by changing of the composition or the activity of RTCs. The RTCs were isolated from HIV-1-infected cells by using equilibrium density centrifugation, as previously described (3, 28, 33). VSV-G envelope-pseudotyped HIV-1 made in cells in the presence or absence of Nullbasic was used to synchronously infect HEK293T cells. Cells incubated with equivalent amounts of heat-inactivated HIV-1 were used as negative controls for the experiment. The cells were incubated for 4 h, and cytoplasmic lysates prepared from the infected cells were fractionated using a 20 to 70% linear sucrose gradient and equilibrium density centrifugation. Fractions collected from the bottom of the gradient were assayed for HIV-1 −sssDNA by qPCR and for CA by ELISA (Fig. 6). The viral RTC, which has a density of 1.32 to 1.34 g/ml, contains viral and cellular proteins, as well as viral RNA and DNA (33). Fraction 5, which had a density of 1.32 g/ml, corresponded to the fraction containing RTCs and also had peak viral −sssDNA, as determined by qPCR (3, 28, 33, 34). No viral DNA was detected in gradient fractions using heat-inactivated HIV-1 (data not shown), as reported previously (3). Nullbasic-treated HIV-1 produced ∼3-fold less viral DNA than untreated HIV-1, which is consistent with but slightly lower than defects we reported previously (10, 14). This is most likely due to levels Nullbasic expression plasmid used here at minimal levels sufficient to observe defects in infectivity. Although the distribution of −sssDNA was observed in fractions 1 to 8, Nullbasic-treated HIV-1 had 36% of the total viral −sssDNA in fraction 5 compared to 56% for untreated HIV-1. Analysis of CA in each fraction showed that untreated virus had a clear peak amount in fraction 5 (Fig. 6B). However, HIV-1 containing Nullbasic had a majority of CA in fractions with lower densities (fractions 6 to 8). The data suggest that the redistribution of viral DNA and CA to non-RTC fractions is due to a Nullbasic-induced change in the composition or organization of RTCs or increased uncoating.
FIG 6.
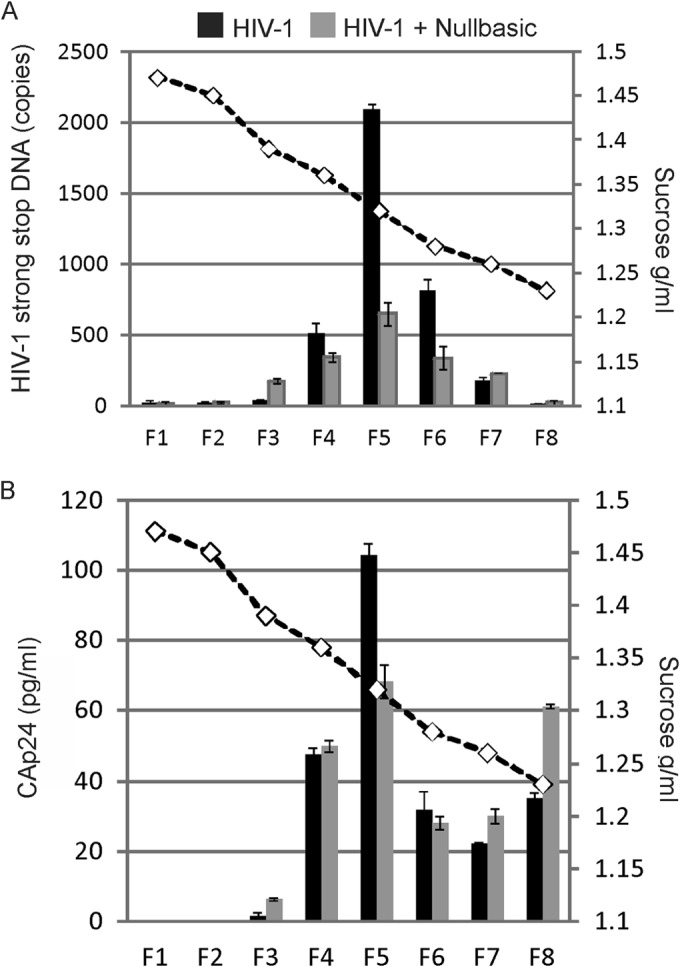
Inhibition of RTC DNA synthesis by Nullbasic. HIV-1 was produced by cotransfection of a proviral plasmid with a Nullbasic-FLAG expression plasmid (■) or an empty expression plasmid (■) as described in Materials and Methods. HEK293T cells were infected with VSV-G-pseudotyped HIV-1. Cell lysates were prepared 4 h postinfection and layered on top of a 20 to 70% linear sucrose gradient. After centrifugation, fractions were collected from the bottom and the density of each fraction was measured (dashed line). A sample from each fraction was assayed for HIV-1 −sssDNA by qPCR (top graph) and for CAp24 by ELISA (bottom graph). The experiment was repeated twice with similar results, and the results of a representative experiment are shown.
HIV-1 in vitro core disassembly is increased by Nullbasic.
The efficiency and timing of HIV-1 reverse transcription can be affected by stability of the viral core (35, 36). For example, HIV-1 having point mutations in CA can have increased or decreased core stability and which have reduced reverse transcription efficiency in cells. The possibility that Nullbasic affected core architecture was assessed by thin-section electron microscopy (Fig. 7). The results show that most virus particles produced without Nullbasic had a mature morphology with an intact viral core (60%). Virus particles produced in the presence of Nullbasic showed a slight but not statistically significant reduction in levels virions with mature morphology (54%) and a commensurate increase in virions with an immature morphology. A Western blot of the virus producing cells confirmed the expression of Nullbasic-FLAG, as expected (Fig. 7C). The disassembly of virion cores was measured using a in vitro uncoating assay (30), with cores purified from HIV-1 and Nullbasic-containing HIV-1. Samples containing equivalent amounts cores from sucrose gradient fractions were incubated at either 4 or 37°C for 0, 60, or 120 min. The samples underwent ultracentrifugation to sediment intact cores, leaving CA from disassembled cores in the supernatant. As shown in Fig. 8, Western blotting of pelleted cores with an anti-CA antibody consistently showed decreased amounts of CA in pelleted cores isolated from Nullbasic-treated HIV-1 at 120 min compared to untreated HIV-1 cores. The results obtained here, combined with analysis of the CA content in RTC fractions, indicate that Nullbasic incorporation can lead to accelerated core uncoating in vitro.
FIG 7.

Effect of Nullbasic on virion morphology. (A) Representative electron micrograph images illustrating mature, eccentric, and immature particle morphologies. (B) Frequencies of the different core morphologies for HIV-1 produced by HEK293T cells transfected with pGCH proviral plasmid and pcDNA3.1-FLAG (black bars) or pcDNA3.1-Nullbasic-FLAG (gray bars). Mean values and standard errors of the mean were determined by comparing three independent sets of 60 to 180 virus particles each. Statistical analysis was performed using a Student t test from at least three independent measurements. The statistical significance was set at P < 0.05. (C) Western blot of lysates from the transfected cells using anti-capsid, anti-FLAG, or anti-tubulin antibodies.
FIG 8.
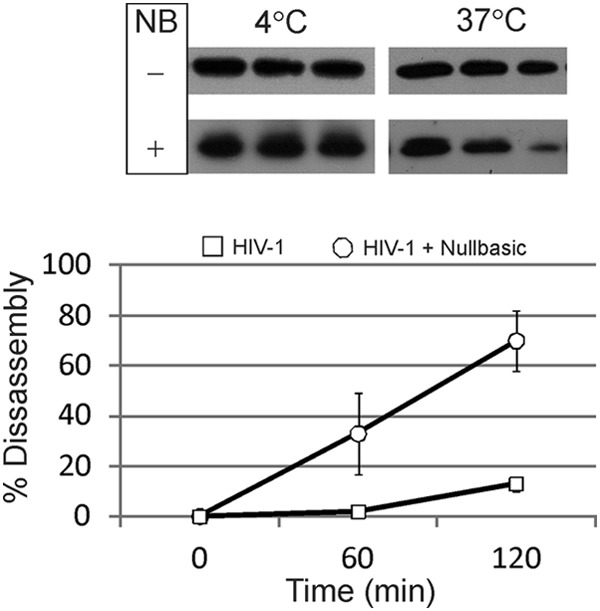
Nullbasic incorporation in HIV-1 leads to increased core disassembly kinetics in vitro. Purified cores from HIV-1 or HIV-1 containing Nullbasic were incubated at 4 or 37°C for 0, 60, or 120 min. (A) Western blot analysis of treated cores. The treated samples underwent ultracentrifugation, and the protein sediment was solubilized in an SDS–2% PAGE loading dye and analyzed by Western blotting with a monoclonal anti-CA antibody and a secondary antibody conjugated to HRP. The blots were developed using chemiluminescence. (B) Disassembly kinetics of viral cores in vitro at 37°C. Quantification of the protein bands from all experiments was calculated using digital images and ImageJ software. The percent disassembly was calculated by comparing values at 60 and 120 min to those at time zero. The results show mean values and standard errors of the mean. The experiment was repeated three times, and the results of a representative result are shown.
Nullbasic does not affect viral entry of VSV-G-pseudotyped HIV-1.
We previously showed that Nullbasic has no effect on entry of lentiviral particles pseudotyped with VSV-G envelope (14). The disruption of entry of pseudotyped HIV-1 in HEK293T cells was tested (Fig. 9). The experiment can detect viral genomic RNA postinfection as an indicator of viral entry. HEK293T cells were treated with 1 μM NVP for 2 h and then incubated with each HIV-1 sample at 4°C for 2 h. A heat-inactivated sample of each HIV-1 was used as a negative control. NVP was used to block reverse transcription and degradation of viral genomic RNA by RT RNase H activity. The cells were incubated at 37°C for 2 h to permit viral entry, and the total cellular RNA was collected. RT-PCR was used to measure the levels of viral genomic RNA that were normalized to levels of cellular eEF1A mRNA in each sample. The results show that similar levels of viral genomic RNA were detected irrespective of the presence of absence of Nullbasic in virus particles.
FIG 9.
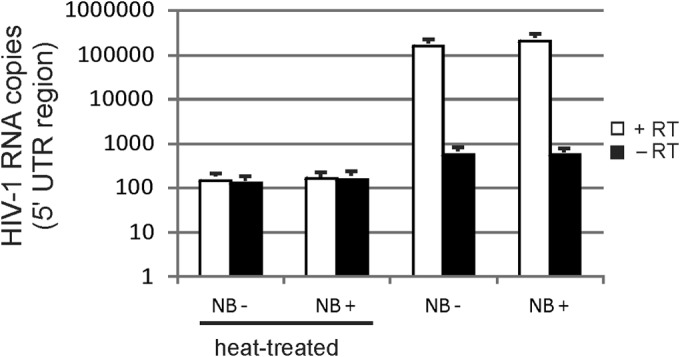
Entry of VSV-G-pseudotyped HIV-1 is not affected by Nullbasic. HEK293 cells were treated with 1 μM nevirapine (to prevent reverse transcription and degradation of viral mRNA by RNase H activity) for 2 h at 37°C and then incubated with VSV-G-pseudotyped HIV-1 or HIV-1-Nullbasic viral supernatant containing 500 ng of CA for 2 h at 4°C to facilitate virus attachment. Heat-treated virus was used as a control for attachment and entry. The cells were incubated at 37°C to initiate envelope and cell membrane fusion, followed by incubation for a further 2 h. Total cellular RNA was collected and analyzed by RT-PCR for HIV-1 full-length mRNA and cellular eEF1A mRNA in the presence or absence of RT. The relative copy number of HIV-1 RNA was normalized to levels of eEF1A mRNA in each sample. A negative control without cDNA template was included in every assay. All PCRs were performed in triplicate. The experiment was performed twice with similar results. The results of a representative experiment are shown. NB− and NB+, the absence and presence of Nullbasic, respectively.
DISCUSSION
Our previous studies showed that HIV-1 or HIV-1-based lentiviral virus-like particles produced by cells expressing Nullbasic are defective for synthesis of −sssDNA by 4- to 10-fold in a dose-dependent manner (10, 14). The experiments described here support a model in which a direct interaction between Nullbasic and RT leads to defective core disassembly and reverse transcription. First, coexpression of Nullbasic and HIV-1 in cells results in virion containing Nullbasic. Second, co-IP and pulldown assays indicate interaction between Nullbasic and RT, and BLI assays indicated a direct interaction. Incorporation of Nullbasic by HIV-1 leads to accelerated core disassembly and defective reverse transcription postinfection. This is supported by in vitro uncoating assays, by reduced amounts of viral cDNA in RTC fractions, and by increased redistribution of CA into non-RTC fractions. In sharp contrast, we observed no inhibition of RT activity by recombinant Nullbasic in a standard homopolymer RT assay even when added at a 20,000-fold molar excess compared to RT. Given the outcomes reported here and the observation that Nullbasic can inhibit −sssDNA synthesis in endogenous and cell-based reverse transcription assays (10, 14), we propose that Nullbasic inhibits reverse transcription by disrupting assembly of the prototypical RTC in virions, rather than by directly inhibiting RT activity, and this results in accelerated uncoating and fewer functional RTC following cell infection. To our knowledge, only one other antiviral molecule, a methylated oligoribonucleotide, targeting the RTC has been reported (37).
We showed that Nullbasic can bind RT and affects core stability in vitro and activity of RTCs in cells. It is possible that interaction between RT and Nullbasic affects other RTC components. Matrix, CA, nucleocapsid, Vpr, Vif, and integrase have been detected in the partially purified RTC (33, 34, 38, 39), and roles of each RTC component in early virus replication have been discussed in detail elsewhere (1). The present study shows that Nullbasic may can be a useful tool for investigation on the biology of uncoating, RTC activity that may reveal novel antiviral strategies to target the RTC.
Tat is a virion protein (13) that has reported paradoxical abilities to positively (7, 8, 11, 12, 40–43) or negatively (9, 44, 45) affect DNA synthesis by, or molecular components important for, reverse transcription. The opposing effects of Tat on reverse transcription leave open possibilities that Nullbasic may be either a TDNi form of Tat or a better Tat-based inhibitor of reverse transcription. Some observations and further experiments may help to resolve the issue. First, our experience is that inclusion of the basic domain greatly increases steady-state levels of Nullbasic compared to Tat (unpublished observation). Since Nullbasic downregulates reverse transcription in a dose-dependent manner (10), increased steady-state levels of Nullbasic in the cells may enhance the concentrations of Nullbasic and hence its antiviral effects. Second, the basic domain of Tat is capable of direct interaction with the TAR RNA stem-loop structure (46–48), which is formed by viral genomic RNA (49) and has been implicated as important for the nucleic acid annealing activity of Tat (9, 41–43, 50, 51). One possibility is that Tat binding to RT and its RNA annealing activities help to coordinate RTC assembly, which is disrupted by the basic domain mutation in Nullbasic. This scenario shares elements of the mechanism attributed to Tat TDN mutants, which can bind pTEFb but are unable to direct pTEFb to TAR RNA for efficient transcription (20). Structural studies of the viral RNA/tRNAlys3 RNA complex in the presence of RT or the RT-Nullbasic complex (49, 52) should help to further clarify a role for Tat in −sssDNA synthesis. In addition, analysis of point mutants in Nullbasic should identify a domain(s) required for interaction with RT and may determine the relative importance of RT binding and RNA annealing functions of Tat in reverse transcription.
The complexity of abundant interactions between Tat and other viral and cellular proteins are well known (53). Although Nullbasic has potential as an antiviral agent by inhibiting three steps of HIV replication, possibly using a gene therapy approach, it also has utility as a probe to investigate fundamental mechanisms that underlie interaction between HIV proteins.
ACKNOWLEDGMENTS
We thank Stuart LeGrice for kindly providing the recombinant RT protein. The plasmid pNL4-3.Luc.R−.E− was provided by Nathaniel Landau through the NIH/NIAID AIDS Reagent Program, Division of AIDS.
The research was supported by an Australian Research Council Future Fellowship award to D.H.
REFERENCES
- 1.Hu WS, Hughes SH. 2012. HIV-1 reverse transcription. Cold Spring Harb Perspect Med 2:a006882. doi: 10.1101/cshperspect.a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warrilow D, Tachedjian G, Harrich D. 2009. Maturation of the HIV reverse transcription complex: putting the jigsaw together. Rev Med Virol 19:324–337. doi: 10.1002/rmv.627. [DOI] [PubMed] [Google Scholar]
- 3.Warren K, Wei T, Li D, Qin F, Warrilow D, Lin MH, Sivakumaran H, Apolloni A, Abbott CM, Jones A, Anderson JL, Harrich D. 2012. Eukaryotic elongation factor 1 complex subunits are critical HIV-1 reverse transcription cofactors. Proc Natl Acad Sci U S A 109:9587–9592. doi: 10.1073/pnas.1204673109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warren K, Warrilow D, Meredith L, Harrich D. 2009. Reverse transcriptase and cellular factors: regulators of HIV-1 reverse transcription. Viruses 1:873–894. doi: 10.3390/v1030873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karn J, Stoltzfus CM. 2012. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med 2:a006916. doi: 10.1101/cshperspect.a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu YL, Coronel E, Ho CK, Shuman S, Rana TM. 2001. HIV-1 Tat protein interacts with mammalian capping enzyme and stimulates capping of TAR RNA. J Biol Chem 276:12959–12966. doi: 10.1074/jbc.M007901200. [DOI] [PubMed] [Google Scholar]
- 7.Harrich D, Ulich C, Garcia-Martinez LF, Gaynor RB. 1997. Tat is required for efficient HIV-1 reverse transcription. EMBO J 16:1224–1235. doi: 10.1093/emboj/16.6.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Apolloni A, Meredith LW, Suhrbier A, Kiernan R, Harrich D. 2007. The HIV-1 Tat protein stimulates reverse transcription in vitro. Curr HIV Res 5:473–483. doi: 10.2174/157016207781662443. [DOI] [PubMed] [Google Scholar]
- 9.Kameoka M, Morgan M, Binette M, Russell RS, Rong L, Guo X, Mouland A, Kleiman L, Liang C, Wainberg MA. 2002. The Tat protein of human immunodeficiency virus type 1 (HIV-1) can promote placement of tRNA primer onto viral RNA and suppress later DNA polymerization in HIV-1 reverse transcription. J Virol 76:3637–3645. doi: 10.1128/JVI.76.8.3637-3645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meredith LW, Sivakumaran H, Major L, Suhrbier A, Harrich D. 2009. Potent inhibition of HIV-1 replication by a Tat mutant. PLoS One 4:e7769. doi: 10.1371/journal.pone.0007769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ulich C, Dunne A, Parry E, Hooker CW, Gaynor RB, Harrich D. 1999. Functional domains of Tat required for efficient human immunodeficiency virus type 1 reverse transcription. J Virol 73:2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hooker CW, Scott J, Apolloni A, Parry E, Harrich D. 2002. Human immunodeficiency virus type 1 reverse transcription is stimulated by tat from other lentiviruses. Virology 300:226–235. doi: 10.1006/viro.2002.1554. [DOI] [PubMed] [Google Scholar]
- 13.Chertova E, Chertov O, Coren LV, Roser JD, Trubey CM, Bess JW Jr, Sowder RC II, Barsov E, Hood BL, Fisher RJ, Nagashima K, Conrads TP, Veenstra TD, Lifson JD, Ott DE. 2006. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol 80:9039–9052. doi: 10.1128/JVI.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Apolloni A, Lin MH, Sivakumaran H, Li D, Kershaw MH, Harrich D. 2013. A mutant Tat protein provides strong protection from HIV-1 infection in human CD4+ T cells. Hum Gene Ther 24:270–282. doi: 10.1089/hgtb.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin MH, Sivakumaran H, Apolloni A, Wei T, Jans DA, Harrich D. 2012. Nullbasic, a potent anti-HIV Tat mutant, induces CRM1-dependent disruption of HIV Rev trafficking. PLoS One 7:e51466. doi: 10.1371/journal.pone.0051466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sivakumaran H, Cutillas V, Harrich D. 2013. Revisiting trans-dominant-negative proteins in HIV gene therapy. Future Virol 8:757–768. doi: 10.2217/fvl.13.65. [DOI] [Google Scholar]
- 17.Pearson L, Garcia J, Wu F, Modesti N, Nelson J, Gaynor R. 1990. A transdominant Tat mutant that inhibits tat-induced gene expression from the human immunodeficiency virus long terminal repeat. Proc Natl Acad Sci U S A 87:5079–5083. doi: 10.1073/pnas.87.13.5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modesti N, Garcia J, Debouck C, Peterlin M, Gaynor R. 1991. Trans-dominant Tat mutants with alterations in the basic domain inhibit HIV-1 gene expression. New Biol 3:759–768. [PubMed] [Google Scholar]
- 19.Herskowitz I. 1987. Functional inactivation of genes by dominant negative mutations. Nature 329:219–222. doi: 10.1038/329219a0. [DOI] [PubMed] [Google Scholar]
- 20.Orsini MJ, Debouck CM. 1996. Inhibition of human immunodeficiency virus type 1 and type 2 Tat function by transdominant Tat protein localized to both the nucleus and cytoplasm. J Virol 70:8055–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones KA. 1997. Taking a new TAK on tat transactivation. Genes Dev 11:2593–2599. doi: 10.1101/gad.11.20.2593. [DOI] [PubMed] [Google Scholar]
- 22.Ulich C, Harrich D, Estes P, Gaynor RB. 1996. Inhibition of human immunodeficiency virus type 1 replication is enhanced by a combination of transdominant Tat and Rev proteins. J Virol 70:4871–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malim MH, McCarn DF, Tiley LS, Cullen BR. 1991. Mutational definition of the human immunodeficiency virus type 1 Rev activation domain. J Virol 65:4248–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollard VW, Malim MH. 1998. The HIV-1 Rev protein. Annu Rev Microbiol 52:491–532. doi: 10.1146/annurev.micro.52.1.491. [DOI] [PubMed] [Google Scholar]
- 25.Wagner R, Graf M, Bieler K, Wolf H, Grunwald T, Foley P, Uberla K. 2000. Rev-independent expression of synthetic gag-pol genes of human immunodeficiency virus type 1 and simian immunodeficiency virus: implications for the safety of lentiviral vectors. Hum Gene Ther 11:2403–2413. doi: 10.1089/104303400750038507. [DOI] [PubMed] [Google Scholar]
- 26.Dettenhofer M, Yu XF. 1999. Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J Virol 73:1460–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ott DE. 2009. Purification of HIV-1 virions by subtilisin digestion or CD45 immunoaffinity depletion for biochemical studies. Methods Mol Biol 485:15–25. [DOI] [PubMed] [Google Scholar]
- 28.Fassati A. 2009. Methods of preparation and analysis of intracellular reverse transcription complexes. Methods Mol Biol 485:107–119. doi: 10.1007/978-1-59745-170-3_2. [DOI] [PubMed] [Google Scholar]
- 29.Warrilow D, Warren K, Harrich D. 2010. Strand transfer and elongation of HIV-1 reverse transcription is facilitated by cell factors in vitro. PLoS One 5:e13229. doi: 10.1371/journal.pone.0013229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aiken C. 2009. Cell-free assays for HIV-1 uncoating. Methods Mol Biol 485:41–53. doi: 10.1007/978-1-59745-170-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin S, Harper CB, May LM, Coulson EJ, Meunier FA, Osborne SL. 2013. Inhibition of PIKfyve by YM-201636 dysregulates autophagy and leads to apoptosis-independent neuronal cell death. PLoS One 8:e60152. doi: 10.1371/journal.pone.0060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herschhorn A, Oz-Gleenberg I, Hizi A. 2008. Quantitative analysis of the interactions between HIV-1 integrase and retroviral reverse transcriptases. Biochem J 412:163–170. doi: 10.1042/BJ20071279. [DOI] [PubMed] [Google Scholar]
- 33.Fassati A, Goff SP. 2001. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J Virol 75:3626–3635. doi: 10.1128/JVI.75.8.3626-3635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nermut MV, Fassati A. 2003. Structural analyses of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J Virol 77:8196–8206. doi: 10.1128/JVI.77.15.8196-8206.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci U S A 108:9975–9980. doi: 10.1073/pnas.1014522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grigorov B, Bocquin A, Gabus C, Avilov S, Mely Y, Agopian A, Divita G, Gottikh M, Witvrouw M, Darlix JL. 2011. Identification of a methylated oligoribonucleotide as a potent inhibitor of HIV-1 reverse transcription complex. Nucleic Acids Res 39:5586–5596. doi: 10.1093/nar/gkr117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iordanskiy S, Berro R, Altieri M, Kashanchi F, Bukrinsky M. 2006. Intracytoplasmic maturation of the human immunodeficiency virus type 1 reverse transcription complexes determines their capacity to integrate into chromatin. Retrovirology 3:4. doi: 10.1186/1742-4690-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carr JM, Coolen C, Davis AJ, Burrell CJ, Li P. 2008. Human immunodeficiency virus 1 (HIV-1) virion infectivity factor (Vif) is part of reverse transcription complexes and acts as an accessory factor for reverse transcription. Virology 372:147–156. doi: 10.1016/j.virol.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 40.Apolloni A, Hooker CW, Mak J, Harrich D. 2003. Human immunodeficiency virus type 1 protease regulation of tat activity is essential for efficient reverse transcription and replication. J Virol 77:9912–9921. doi: 10.1128/JVI.77.18.9912-9921.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boudier C, Humbert N, Chaminade F, Chen Y, de Rocquigny H, Godet J, Mauffret O, Fosse P, Mely Y. 2014. Dynamic interactions of the HIV-1 Tat with nucleic acids are critical for Tat activity in reverse transcription. Nucleic Acids Res 42:1065–1078. doi: 10.1093/nar/gkt934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Godet J, Boudier C, Humbert N, Ivanyi-Nagy R, Darlix JL, Mely Y. 2012. Comparative nucleic acid chaperone properties of the nucleocapsid protein NCp7 and Tat protein of HIV-1. Virus Res 169:349–360. doi: 10.1016/j.virusres.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boudier C, Storchak R, Sharma KK, Didier P, Follenius-Wund A, Muller S, Darlix JL, Mely Y. 2010. The mechanism of HIV-1 Tat-directed nucleic acid annealing supports its role in reverse transcription. J Mol Biol 400:487–501. doi: 10.1016/j.jmb.2010.05.033. [DOI] [PubMed] [Google Scholar]
- 44.Kameoka M, Rong L, Gotte M, Liang C, Russell RS, Wainberg MA. 2001. Role for human immunodeficiency virus type 1 Tat protein in suppression of viral reverse transcriptase activity during late stages of viral replication. J Virol 75:2675–2683. doi: 10.1128/JVI.75.6.2675-2683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo X, Kameoka M, Wei X, Roques B, Gotte M, Liang C, Wainberg MA. 2003. Suppression of an intrinsic strand transfer activity of HIV-1 Tat protein by its second-exon sequences. Virology 307:154–163. doi: 10.1016/S0042-6822(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 46.Feng S, Holland EC. 1988. HIV-1 tat trans-activation requires the loop sequence within tar. Nature 334:165–167. doi: 10.1038/334165a0. [DOI] [PubMed] [Google Scholar]
- 47.Berkhout B, Jeang KT. 1989. trans activation of human immunodeficiency virus type 1 is sequence specific for both the single-stranded bulge and loop of the trans-acting-responsive hairpin: a quantitative analysis. J Virol 63:5501–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia JA, Harrich D, Soultanakis E, Wu F, Mitsuyasu R, Gaynor RB. 1989. Human immunodeficiency virus type 1 LTR TATA and TAR region sequences required for transcriptional regulation. EMBO J 8:765–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6:e96. doi: 10.1371/journal.pbio.0060096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuciak M, Gabus C, Ivanyi-Nagy R, Semrad K, Storchak R, Chaloin O, Muller S, Mely Y, Darlix JL. 2008. The HIV-1 transcriptional activator Tat has potent nucleic acid chaperoning activities in vitro. Nucleic Acids Res 36:3389–3400. doi: 10.1093/nar/gkn177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doetsch M, Furtig B, Gstrein T, Stampfl S, Schroeder R. 2011. The RNA annealing mechanism of the HIV-1 Tat peptide: conversion of the RNA into an annealing-competent conformation. Nucleic Acids Res 39:4405–4418. doi: 10.1093/nar/gkq1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lanchy JM, Ehresmann C, Le Grice SF, Ehresmann B, Marquet R. 1996. Binding and kinetic properties of HIV-1 reverse transcriptase markedly differ during initiation and elongation of reverse transcription. EMBO J 15:7178–7187. [PMC free article] [PubMed] [Google Scholar]
- 53.Van Duyne R, Kehn-Hall K, Carpio L, Kashanchi F. 2009. Cell-type-specific proteome and interactome: using HIV-1 Tat as a test case. Expert Rev Proteomics 6:515–526. doi: 10.1586/epr.09.73. [DOI] [PubMed] [Google Scholar]