

ABSTRACT
RNA viruses exist within a host as a population of mutant sequences, often referred to as quasispecies. Within a host, sequences of RNA viruses constitute several distinct but interconnected pools, such as RNA packed in viral particles, double-stranded RNA, and virus-derived small interfering RNAs. We aimed to test if the same representation of within-host viral population structure could be obtained by sequencing different viral sequence pools. Using ultradeep Illumina sequencing, the diversity of two coexisting Potato virus Y sequence pools present within a plant was investigated: RNA isolated from viral particles and virus-derived small interfering RNAs (the derivatives of a plant RNA silencing mechanism). The mutational landscape of the within-host virus population was highly similar between both pools, with no notable hotspots across the viral genome. Notably, all of the single-nucleotide polymorphisms with a frequency of higher than 1.6% were found in both pools. Some unique single-nucleotide polymorphisms (SNPs) with very low frequencies were found in each of the pools, with more of them occurring in the small RNA (sRNA) pool, possibly arising through genetic drift in localized virus populations within a plant and the errors introduced during the amplification of silencing signal. Sequencing of the viral particle pool enhanced the efficiency of consensus viral genome sequence reconstruction. Nonhomologous recombinations were commonly detected in the viral particle pool, with a hot spot in the 3′ untranslated and coat protein regions of the genome. We stress that they present an important but often overlooked aspect of virus population diversity.
IMPORTANCE This study is the most comprehensive whole-genome characterization of a within-plant virus population to date and the first study comparing diversity of different pools of viral sequences within a host. We show that both virus-derived small RNAs and RNA from viral particles could be used for diversity assessment of within-plant virus population, since they show a highly congruent portrayal of the virus mutational landscape within a plant. The study is an important baseline for future studies of virus population dynamics, for example, during the adaptation to a new host. The comparison of the two virus sequence enrichment techniques, sequencing of virus-derived small interfering RNAs and RNA from purified viral particles, shows the strength of the latter for the detection of recombinant viral genomes and reconstruction of complete consensus viral genome sequence.
INTRODUCTION
RNA viruses are one of the fastest-evolving biological entities known. Due to their high mutation and recombination rates, viral populations exist within hosts as a cloud of nonidentical but similar sequences, often referred to as viral quasispecies (1). The generated variability, coupled with natural selection, population bottlenecks, and stochasticity, shape the structure of virus populations, which was shown to have important implications in virus fitness and pathogenicity (1). With the advent of next-generation sequencing (NGS), in-depth studies of viral populations within a host became possible. In the past few years, several in-depth population studies have been conducted on human (2) or animal pathogenic viruses using NGS (3–5). Less attention has been given to plant-infecting viruses; several studies have been carried out using Sanger amplicon sequencing of a limited number of molecular clones (6–10). All NGS in-depth within-plant virus population studies reported to date employed amplicon sequencing, focusing on only a particular part of the viral genome (11–14). However, different parts of the viral genome can be subjected to different selection pressures (15, 16); thus, whole-genome characterization of virus populations would give a more complete picture.
High background levels of host nucleic acids and the high diversity of viral populations complicate the reconstruction of a complete consensus viral genome sequence from NGS data. Moreover, within-host viral population studies demand high sequencing coverage (10,000× and more). Most of such studies include a preamplification step for the amplification of complete or partial viral genomes (2). Such an approach allows us to achieve great sequencing depth easily; however, it can distort the variation occurring in primer-annealing regions (17) and affect the detection of other variants, which are epistatically connected with those regions. Moreover, this approach requires specific knowledge about viral genome sequence, since the sequences of PCR primers used for the amplification of the viral genome have to be known in advance.
A more generic solution is the enrichment for viral nucleic acids before sequencing library preparation, employing the characteristics of different viral sequence pools within the host. In plants, the sequences of RNA virus origin constitute several different pools: RNA molecules packed within the virions, double-stranded RNA (dsRNA) molecules, and virus-derived small interfering RNAs (vsiRNAs). dsRNA molecules can arise through replication or the action of plant RNA-dependent RNA polymerases. Shorter stretches of dsRNA-like structures also can be produced by intramolecular base pairing between different parts of the viral genome. In plants and invertebrates, viral dsRNA molecules represent a substrate for the generation of 21- to 24-nucleotides (nt)-long virus-derived small interfering RNAs (vsiRNAs), which result from the activity of dicer-like proteins (18).
Deep sequencing of vsiRNAs has been used efficiently for the reconstruction of consensus viral genome sequences from plants (19) and invertebrates (20). It is a relatively time-efficient, affordable, and generic method, and as such, it is widely applicable. It was observed that different variants could be detected in the pool of vsiRNAs (21). However, no comparisons have been made yet to test if the variation observed in vsiRNAs reflects the full diversity of viral populations. To fill this gap, we have used a sequence-independent, preamplification-free deep sequencing approach to investigate and compare two different pools of RNA virus sequences from the infected plants: single-stranded RNA (ssRNA) isolated from purified viral particles (VP pool) and vsiRNAs (sRNA pool). Deep sequencing of viral RNA directly from purified viral particles was performed only in a few cases and was shown to be highly efficient for enrichment for viral nucleic acids (4, 22, 23). Here, we have used the recently developed CIM monolithic chromatography-based approach for fast purification of viral particles (24).
Potato virus Y (PVY)-potato was used as a model system. PVY, a single-stranded positive-sense RNA virus and a member of the genus Potyvirus (family Potyviridae), is one of the most important potato pathogens and is distributed in potato-growing regions worldwide (25). In this study, the highly pathogenic recombinant strain NTN was investigated.
Our two main aims were to (i) compare the efficiency of the approaches for the consensus virus genome sequence generation and (ii) compare the within-host population structure inferred from the two viral sequence pools. Single-nucleotide polymorphisms (SNPs) were compared between the samples using a stringent filtering strategy; a low-frequency SNP detection algorithm and technical replicates were used to discriminate real SNPs from reverse transcription (RT), PCR, and sequencing artifacts. The VP pool was investigated additionally for recombination events. We hypothesized that the diversity of vsiRNAs and RNA isolated from viral particles show a highly similar mutational landscape.
This study presents the most comprehensive whole-genome characterization of within-plant virus population to date and allows independent validation and comparison of variants by sequencing two different pools of virus sequences within the same plants. The study provides a firm baseline for future studies of plant virus population diversity and dynamics after bottleneck (transmission by vectors, systemic movement within host) or radiation (spread to a new host) events.
MATERIALS AND METHODS
Preparation of infected plant samples.
PVY (strain NTN; isolate NIB V 151)-infected Solanum tuberosum cv. Pentland Squire plants were propagated in stem node culture (in vitro) and transferred to soil. After 3 weeks, whole green parts (leafs and stem) of 60 plantlets were harvested. The leaves and stem of each plant were divided in two pools, one used for the purification of viral particles and the other for small RNA isolation. First, for the isolation of small RNAs, equal amounts of young leaf, old leaf, and stem were sampled from each plant and pooled (amounting in a total of 1 g of plant material). Subsequently, all of the remaining harvested material (16.74 g) from the same plants was combined and used for purification of viral particles. For a detailed scheme of the experiment, see Fig. 1.
FIG 1.

Scheme of the experiment and analysis pipeline of the study.
Purification of PVY particles and isolation and fragmentation of viral RNA.
Viral particles were purified using convective interactive media (CIM) monolithic chromatographic supports as previously described (24). Chromatographic fractions containing purified viruses were pooled and subjected to total RNA isolation using TRIzol LS reagent (Life Technologies, Invitrogen, Carlsbad, CA, USA) by following the manufacturer's instructions. Prior to sequence library preparation, isolated PVY RNA was fragmented using a NEBNext magnesium RNA fragmentation module (NEB, Ipswich, MA, USA). The fragmentation and end repair procedure followed the Illumina directional mRNA-Seq sample preparation guide (15018460, Rev. A, 2010). The size and quantity of purified fragmented viral RNA was assessed with the Agilent 2100 Bioanalyzer using an RNA Pico chip (Agilent Technologies, CA, USA).
Isolation of small RNAs.
Total RNA was isolated from pooled plant material (1 g) using TRIzol reagent (Life Technologies, Invitrogen, Carlsbad, CA, USA) by following the manufacturer's instructions. RNA was separated in 15% urea-polyacrylamide gel. The band corresponding to the size of small RNAs (cloud of the smallest size, ∼10 to 50 nt) was cut from the gel and crushed. Crushed gel pieces were soaked in 700 μl of 2.5 M NaCl with 0.1% β-mercaptoethanol at 4°C overnight. Samples then were spun down, and 700 μl supernatant was transferred to Freeze ‘N Squeeze columns (Bio-Rad, Hercules, CA, USA). The remaining gel pieces were washed with 300 μl of 2.5 M NaCl with 0.1% β-mercaptoethanol, which was added to Freeze ‘N Squeeze columns. The columns were spun at 5,000 rpm for 1 min. Resuspended RNA was cleaned using a 0.5 volume of chloroform, precipitated with 1 volume of isopropanol, and washed with 75% ethanol. Finally, the pellet was air dried and resuspended in 10 μl of RNase-free water.
Library preparation and sequencing.
Illumina TruSeq sequencing libraries were prepared for both fragmented RNA isolated from purified PVY particles (VP) and small RNAs (sRNA) according to the published protocol (26), with the following modifications: (i) reverse transcription reactions were made in a reduced (50%) volume; (ii) in the final PCR enrichment step, a 4× concentration of forward and reverse primers was used. The libraries were size selected in 10% native polyacrylamide gel, and the DNA was purified from the gel using chloroform-isopropanol extraction. The size distribution of purified libraries was inspected on a Caliper LabChip GX using an HS DNA chip (PerkinElmer, Hopkinton, MA, USA). The libraries were sent for sequencing on an Illumina Hiseq2000 platform in single-end 50-nt mode to Fasteris Life Sciences SA (Plan-les-Ouates, Switzerland). In order to confidently discriminate real SNPs in the population from RT, PCR, and sequencing artifacts, we used technical replicates. Two library technical replicates were prepared (from the RT step onward) for each sequence pool, amounting to a total of 4 samples (sRNA1, sRNA2, VP1, and VP2).
Reconstruction of consensus genome sequence.
Subsamples of 500,000 reads were used for the reconstruction of the consensus genome sequence to achieve optimal performance of the algorithms (the performance of de novo assembly algorithms is not optimized for very high sequencing depths; up to ∼100,000× in our case). Velvet 1.2.10 (27) and the CLC Genomics Workbench 6.0 (CLC bio) de novo assembler were compared initially. The latter showed better performance (significantly longer contigs); thus, it was used for all subsequent analyses (using default parameters, with the minimum contig length set to 50 nt). Parameters of assemblies were compared between sRNA and VP pools, and contigs were mapped to reconstructed consensus complete viral genome sequences to define the proportion of genome they cover for each of the samples. The analysis was repeated on 10 subsamples, and the results were plotted using R 3.0.2 (28). The reconstructed PVY complete genome sequence was deposited in GenBank under accession number KM396648.
To define the ratio between viral and host genome sequences, reads from each set first were mapped to the reconstructed PVY genome (KM396648). We removed reads shorter than 15 nt because they could map nonuniquely. Reads that did not map to PVY then were mapped to a published potato genome sequence (International Nucleotide Sequence Database Collaboration [INSDC] assembly no. GCA_000226075.1). CLC Genomics was used for the mapping. The depth of coverage for each position of PVY genome was exported from CLC Genomics and normalized to the sample with the lowest total read count (∼11.67 million reads). The data were visualized as a line plot using R 3.0.2 (28), with lines being smoothed using the smooth.spline function (spar = 0.3).
Analysis of PVY within-plant population structure: SNP calling.
For SNP analysis, a stringent data filtering and trimming strategy was used before mapping to avoid false-negative results: (i) the reads containing ambiguous bases (Ns) were discarded; (ii) for sRNA samples, 21- to 24-nt-long reads were selected, and for VP samples, reads longer than 24 nt (25 to 50 nt) were selected; (iii) test alignments were made and mismatch profiles per read position were inspected (Fig. 2), and then positions <5 and positions >45 were trimmed from VP reads and positions <5 and >21 were trimmed from sRNA reads; and (iv) reads containing nucleotides with a Phred Q score lower than 20 were discarded. Filtered and trimmed reads were aligned to the reference consensus genome sequence obtained by de novo assembly as described above (accession number KM396648). Several mapping algorithms were tested (CLC Genomics Workbench, bowtie, bowtie2, and RazerS3). RazerS3 was selected as the most suitable for our particular case (very short reads of variable length) and was used for the construction of final alignments, allowing no gaps and a minimum 90% identity match. SAMtools view (29) was used to randomly sample the alignments to comparable sequencing depths (the average depth of coverage was then ∼8,000× for each of the samples). The resulting alignments were used for SNP calling using LoFreq (30). LoFreq allows the detection of variants with frequency lower than the average sequencing error rate. It uses Poisson-binomial distribution-based modeling of sequencing errors for each site in the genome separately, considering position-specific Phred quality scores as well as sequencing depth of the position to distinguish legitimate SNPs from sequencing artifacts (30). Thus, error probabilities are computed for each SNP separately without depending on a universal arbitrary frequency cutoff value. However, the algorithmic filtering implemented in LoFreq can account only for the mistakes arising during the last sequencing step of the process. To further eliminate false positives, which could arise during RT or PCR, we used two replicates (from the RT step on) for each of the samples. Thus, SNPs called by LoFreq were filtered, and only the ones occurring in both replicates were considered reliable. This filtering approach allowed us to confidently discriminate real SNPs from the RT, PCR, and sequencing noise, discovering SNPs in the range of 0.06 to 50% (see Fig. 4b) without the use of an arbitrary frequency cutoff value. Moreover, for comparisons between the pools, we used the lofreq.unique script implemented in the program to further filter out the SNPs which did not have sufficient sequencing depth in all of the samples (30).
FIG 2.
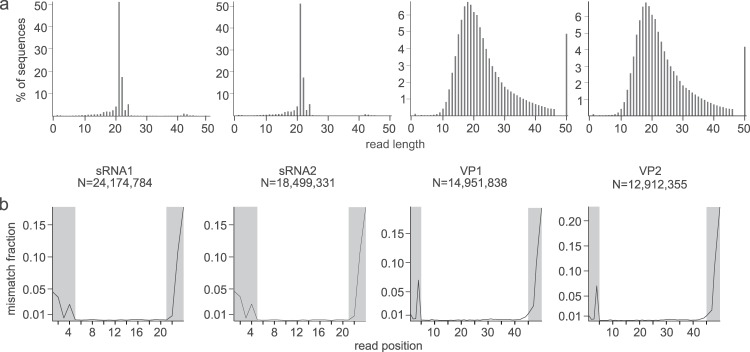
Read length profiles, mismatch fractions per read position, and read trimming. (a) Total number of reads and read length profiles for each of the samples. (b) Relative frequencies of mismatches per position in the read after the test mapping. The higher mismatch frequencies observed at the 5′ and 3′ ends of the reads were trimmed (the positions shaded in gray) and were not used for subsequent SNP calling.
FIG 4.

SNP analysis for VP and sRNA pool. (a) Number of predicted SNPs in two different pools, where the black circle represent SNPs present in the sRNA pool and the gray circle represents SNPs present in the VP pool. The overlap represents SNPs present in both of the pools. SNPs were confirmed in both technical replicates (sRNA1/sRNA2 and VP1/VP2). The numbers in parentheses represent further filtered unique SNPs, which had similar depths in all 4 samples. The SNPs detected in only one of the technical replicates were treated as artifacts and are not depicted here. (b) Violin plots showing the distribution of SNP frequencies in the corresponding pool. The black line indicates median frequency. The white box is the first quartile and the white line is the second quartile of the data. The shaded outer area is a kernel density plot, which describes the distribution of the data. The scale is log10 transformed. The percentages on the lines above violin plots for sRNA and VP unique SNPs denote the upper limits of their distribution. (c to f) Correlation (R is the correlation coefficient) between frequencies of common SNPs for two VP replicates (c), for two sRNA replicates (d), and for common SNPs between the sRNA and VP pool (e). (f) Correlation between ranks of common SNPs between sRNA and the VP pool. The outlined area represents roughly the first 30 ranks (frequencies of higher than 1%), which show higher correlation between the pools.
SNPs were annotated for possible amino acid changes (synonymous/nonsynonymous mutation, consensus amino acid, and variant amino acid) using an in-house-made script. Additionally, the corresponding BLOSUM62 score for each of the SNPs was obtained from the BLOSUM62 matrix (31).
Correlation of SNP frequencies between replicates and between pools was calculated and plotted (see Fig. 4c to f). First, frequencies of SNPs present in both sRNA replicates were compared. Second, the same analysis was made for VP pool replicates. Third, frequencies of SNPs found in both pools (sRNA and VP) were compared. Finally, those SNPs also were sorted by frequency and each SNP was ranked. The ranks of the SNPs in one pool were compared to the ranks of the SNPs in the other pool.
Analysis of PVY within-plant population structure: recombination detection.
For the detection of nonhomologous recombination events, only reads from the VP pool were used. Reads longer than 25 nt first were mapped to the consensus PVY genome and then to the host (potato) reference genome. Reads which did not map to either of them were extracted, and ViReMa (32) was used to detect possible virus-virus nonhomologous recombination events (seed length set to 20 nt, removing PCR duplicates). To further assess the robustness of the results, the same analysis was repeated with longer seed lengths (22 nt and 24 nt). Seed length represents the number of nucleotides used by mapping algorithm for the beginning alignment to the reference. Thus, increasing seed length should increase the specificity but decrease sensitivity of the method. To confirm the reliability of the ViReMa approach for the detection of recombination events, 10 of the most frequent recombination events and 5 other randomly chosen ones were further evaluated. We constructed simulated recombinant sequences, spanning the region of 25 nt before and 25 nt after recombination events detected by ViReMa. The ViReMa input reads then were mapped (using CLC Genomics Workbench) to this simulated sequences. We then visually inspected the mapping for each of the simulated recombinant sequences to ensure there were recombinant reads covering each of the tested recombinant points. Since the sRNAs are too short (21 to 24 nt) to be analyzed for recombination signal using ViReMa or other existing bioinformatics tools, we tried to detect recombination signals by mapping the sRNA sample reads to the same simulated recombinant sequences and then visually inspecting the results.
To search for possible recombination hot spots in the PVY genome, only recombination events supported by two or more nonidentical reads in both replicates were considered. Such a conservative filtering approach significantly reduced the number of the detected recombination points; however, we were able to reliably exclude possible artifacts of the wet-laboratory and analysis procedure. The number of filtered recombination points was counted in each 10-nt-wide window across the PVY genome and visualized as a histogram.
Circos 0.65 (33) was used for the visualization of the results. The square root of relative frequencies of filtered SNPs, their BLOSUM62 scores, and detected recombination events were visualized in a circular plot (see Fig. 5).
FIG 5.

Circular representation of PVY genome with plotted frequencies of SNPs in the sRNA and VP pools, corresponding BLOSUM62 amino acid substitution scores, and recombination points detected by ViReMa (only for the VP pool). (a) BLOSUM62 scores for amino acid changes corresponding to SNPs in the sRNA pool. (b) Square-root-transformed relative frequencies of SNPs detected in the sRNA pool. (c) Square-root-transformed relative frequencies of SNPs detected in the VP pool. (d) BLOSUM62 scores for amino acid changes corresponding to SNPs in the VP pool. (e) Histogram of frequencies of recombination points in 10-nt windows across the PVY genome (scale maximum is 380 events). (f) Visualization of recombination breakpoints detected in the VP pool. The link connects the 3′ and 5′ recombination breakpoint, and its thickness corresponds to the frequency of the event.
Nucleotide sequence accession numbers.
Raw sequencing reads from this experiment have been deposited in the NCBI Sequence Read Archive (SRA) under accession numbers SRR1556759, SRR1556760, SRR1556761, and SRR1556762. The consensus viral genome sequence has been deposited in NCBI GenBank under accession number KM396648.
RESULTS
Illumina deep sequencing resulted in 13 to 24 million reads for four prepared libraries (Fig. 2). The size distribution of sRNA libraries implied excellent enrichment for sRNAs (prominent 21- to 24-nt peak) (Fig. 2a). It was previously shown that the small RNA library preparation could introduce a bias in the frequency of represented sRNAs, likely due to the strong adaptor-sRNA base pairing (34). To compensate for the bias, RNA isolated from VP was fragmented in similarly short fragments (reads of VP libraries range in length from 10 to 50 nt) (Fig. 2). Thus, the similar size of fragments in both types of libraries allowed us an unbiased comparison of the two pools (VP versus sRNA).
Sequencing RNA isolated from purified viral particles enhances reconstruction of consensus viral genome sequence.
De novo assembly of reads obtained by sequencing fragmented RNA from purified viral particles repeatedly (two replicates with 10 500,000-read subsamples) produced one contig encompassing the complete or nearly complete viral genome sequence (9,278 to 9,761 nt) (Fig. 3b). De novo assembly of small RNAs resulted in several shorter contigs (with a maximum length of 611 to 2,543 nt in different subsamples) (Fig. 3b), which did not overlap; the N50 (the parameter used to describe the quality of assembly) was considerably lower for sRNA assemblies (Fig. 3b). The sRNA contigs covered the majority of, but not the complete, viral genome (up to 826 nt not covered by contigs) (Fig. 3c).
FIG 3.

Generation of complete consensus viral genome sequence. De novo assembly quality parameters for 10 subsamples (500,000 reads per subsample). (a) N50; (b) maximum length of contig; (c) total length of viral genome not covered by contigs. Black lines represent average values. (d) The percentage of reads mapped to the viral and host reference genome for the small RNA (sRNA) pool and viral particle (VP) pool. (e) Distribution of sequencing depth across the viral genome for VP and sRNA pools.
In both cases (VP and sRNA), the entire genome was covered when mapping the reads to the reconstructed PVY complete genome sequence, with no zero-coverage regions (Fig. 3e). In VP samples, viral sequences amounted to 89% of the sample, whereas in sRNA samples they amounted to around 54% (Fig. 3d). There was a low level of contamination with background host sequences in VP samples (only 2% of the reads). In sRNA samples, a higher proportion of reads mapped to the host genome (38%), probably mostly representing potato endogenous small RNAs. The remaining reads (8 to 9%; denoted as “other” in Fig. 3d) did not map to either of the genomes and may represent host sequences not sufficiently similar to the published potato genome sequence, which corresponds to Solanum phureja (a closely related diploid potato species), but it also may include sequences of other taxa present in plants and recombinant viral sequences (as confirmed by recombination analysis). The depth of sequencing coverage across the PVY genome was variable but high in both cases. The genome was more uniformly covered in the case of sRNAs, whereas in the case of the VP pool, a notable peak in coverage was observed in the 3′ region of the genome (Fig. 3e).
The mutational landscape is highly similar between VP and sRNA pools.
SNP analysis, following rigorous filtering using replicate data, revealed 103 polymorphic sites for the VP pool and 121 polymorphic sites for the sRNA pool (details, including LoFreq-derived P values, are given in Table SA1 in the supplemental material). The frequencies of variants ranged from 0.06% to 49.86% (Fig. 4b). Eighty-one SNPs were present in both pools. Twenty-two SNPs were found only in the VP pool, and 40 were found only in the sRNA pool. After excluding the sites with uneven coverage across the samples (using the lofreq.unique script), 16 SNPs were confirmed to be unique for the VP pool and 29 for the sRNA pool (Fig. 4a).
The correlation of common SNP frequencies was nearly perfect (R = 0.99) (Fig. 4c and d) between the replicates. The correlation of SNP frequencies for the 81 SNPs present in both pools (VP and sRNA) was strong (R = 0.85) (Fig. 4e). Moreover, when comparing ranks of the SNPs instead of their frequencies, there was even higher interpool correlation for approximately the first 30 ranks, which largely correspond to the SNPs with frequencies higher than ∼1% (Fig. 4f, outlined area).
All of the SNPs which have a frequency of 0.6% or more in the VP pool also were discovered in the sRNA pool. From the opposite perspective, there was a 100% rate of SNP discovery in the VP pool only if this SNP had a frequency of 1.6% or more in the sRNA pool (Fig. 4b).
The SNPs were distributed across the entire genome of PVY, with a possible cold spot in the VPg coding region (Fig. 5b and c). The annotation of SNPs shows larger amounts of nonsimilar amino acid substitutions (lower BLOSUM62 scores) in the P1 and NIb regions of PVY polyprotein (Fig. 5a and d).
Recombinant viral sequences are common in VP pool.
The recombination analysis of the VP pool with the ViReMa algorithm suggested that 0.5 to 3% of reads are recombinant viral sequences (result dependent on the seed length: 20, 22, and 24 nt). However, this represents an extreme underestimation. The short length of reads constituting our libraries (15 to 50 nt) did not allow us to detect the majority of recombination events. According to Routh and Johnson (32), a theoretical maximum efficiency of recombination detection with the ViReMa algorithm can be estimated using the following calculation: there are 49 possible cutting sites in a 50-nt read at which a recombination may occur. With a search seed of 20 nt, recombination events occurring in the first or last 19 cutting sites of the reads will not be detected, leaving 11 possible sites. Therefore, a theoretical maximum efficiency of recombination can be calculated as 11/49 × 100 = 22.4%. Using the same formula, the theoretical maximum efficiency of recombination detection using a seed length of 22 nt is 14%, and using a seed length of 24 nt it is 6%. Correction for theoretical algorithm efficiency was made by dividing the detected number of recombinant viral sequences by the approximate theoretical efficiency of the algorithm. Because our data set contained different read lengths, we could perform only a rough estimation using the most represented read length (50 bp) for the corrections (also being the most conservative one). Thus, considering the theoretical efficiency of recombination detection, we can roughly estimate that the “real” proportion of recombinant sequences amounts to 7 to 15% (Table 1). This is in accordance with the study of Routh and Johnson (32), where, with a similar approach, 8 to 13% of sequences encapsidated in flock house virus (FHV) particles could be annotated as recombinant viral sequences. Detailed results are presented in Table 1.
TABLE 1.
Recombinant reads detected by ViReMa in VP1 and VP2 samples before and after correction for theoretical algorithm efficiency using different analysis settings (seed lengths)
| Parameter | Value for: |
|
|---|---|---|
| VP1 | VP2 | |
| Total no. of reads (>25 nt) | 5,058,459 | 4,413,342 |
| No. of reads mapping to PVY | 4,198,138 | 3,704,865 |
| No. of reads mapping to potato | 9,843 | 8,843 |
| No. of reads for recombination analysis | 850,478 | 699,634 |
| Seed length set to 20 nt | ||
| No. of recombinant reads | 171,191 | 132,950 |
| Proportion (%) of total no. of reads | 3.38 | 3.01 |
| Proportion (%) of total no. of reads, corrected for theoretical algorithm efficiency (/0.22)a | 15.38 | 13.69 |
| No. of reads after filteringb | 2,295 (706 unique) | |
| Seed length set to 22 nt | ||
| No. of recombinant reads | 83,840 | 63,508 |
| Proportion (%) of total no. of reads | 1.66 | 1.44 |
| Proportion (%) of total no. of reads, corrected for theoretical algorithm efficiency (/0.14)a | 11.84 | 10.28 |
| No. of reads after filteringa | 668 (217 unique) | |
| Seed length set to 24 nt | ||
| No. of recombinant reads | 27,757 | 20,458 |
| Proportion (%) of total no. of reads | 0.55 | 0.46 |
| Proportion (%) of total no. of reads, corrected for theoretical algorithm efficiency (/0.06)a | 9.15 | 7.73 |
| No. of reads after filteringb | 38 (16 unique) | |
The correction was made by dividing the number of recombinant reads by theoretical algorithm efficiency, which is given in parentheses.
Recombination events have to be detected in two nonidentical reads in both replicates.
For further analysis, we filtered the detected recombinant events conservatively: only the recombination events confirmed by at least two nonidentical reads in both replicates were considered reliable. This way, we excluded potential experimental and analysis artifacts. With the seed length set to 20 nt, 2,295 (706 unique) filtered nonhomologous recombination events were revealed. Plotting the observed recombination points on the PVY genome revealed a notable hot spot of such events present in the 3′-untranslated region (UTR) and coat protein (CP) region of the genome (Fig. 5e and f). When the specificity of recombination detection was increased by setting seed lengths to 22 nt and 24 nt, the number of detected recombination points decreased to 668 (217 unique) and 38 (16 unique), respectively. Nevertheless, identical hotspots were identified in the viral genome (Fig. 6a). The predicted hotspots (Fig. 5e and f) coincide with the notable peak in the depth of sequencing coverage (Fig. 3e). We tested if the observed pattern could be explained only as a correlation between the number of detected recombination events and the depth of coverage. No notable correlation could be observed (Fig. 6b). Manual inspection of recombination events confirmed the reliability of the ViReMa approach, since all 15 tested simulated recombinant sequences were covered by recombinant reads. No recombinant events could be detected (using the same simulated sequences) within the sRNA pool (see Materials and Methods for details about the analysis).
FIG 6.

Detection of recombination hotspots. (a to c) Recombination points detected by ViReMa (links connect 3′ and 5′ recombination breakpoints) in the PVY genome (black squares designate genes and UTRs) and their frequency in 10-nt-wide windows across the PVY genome (histogram above the genome segments) with seed length set to 20 nt (a), 22 nt (b), and 24 nt (c). Longer seed length increases the specificity and decreases the sensitivity of the method. The number of recombination points stated under each circular diagram represents the number of detected unique recombination events. (b) Number of recombination points (seed length set to 20 nt) within each of the 10-nt-windows across the PVY genome plotted against the corresponding depth of coverage.
DISCUSSION
Through a carefully planned experimental and analysis approach, we compared the diversity of two separate but coexisting viral sequence pools within a plant: virus-derived small RNAs and RNA isolated from purified viral particles. Both of the approaches tested here proved to be efficient for the enrichment of viral nucleic acids from infected plants. However, the isolation of viral particles was shown to be superior when considering the purity and relative amount of viral nucleic acids. It also performed better when the reads were used for de novo reconstruction of the consensus viral genome sequence. Thus, sequencing RNA isolated from purified viral particles is highly efficient when characterizing unknown viruses. The main drawback of this approach could be the length of the purification process. This was, however, greatly reduced in our experimental setup by using a novel purification approach based on CIM monolithic chromatography (24). On the other hand, the sRNA sequencing technique is a more time-efficient and generic approach. It allows us to detect many different viruses without a special protocol modification, and as such it represents a first choice for virus screening or diagnostic usage.
To explore and compare the mutational landscape of within-plant virus populations, we predicted SNPs present in both of the investigated pools. Using a stringent filtering approach, we were able to reliably detect variants with frequency as low as 0.06%, which is the highest resolution, obtained in whole-genome studies of plant virus populations to date. Confirmation of identified variants in two independent pools of sequences provides additional reliability for investigating the diversity of viral populations. We showed that the SNPs of two distinct viral sequence pools overlap greatly. All of the mutations with frequency higher than 1.6% were detected in both pools, implicating a highly conserved mutational landscape between VP and sRNA pools. Nevertheless, both of the pools contained some unique SNPs, which in all of the cases had low frequencies (up to 1.6% in the sRNA pool, up to 0.6% in the VP pool).
The occurrence of these pool-unique SNPs can be explained by several scenarios. First, it could be a result of a genetic drift in localized virus populations within a plant. Even though special care was taken to ensure representative sampling for both of the pools (Fig. 1), it is likely that mutations from a very recent and/or localized replication event would not occur in both of the pools. Moreover, in the case of sRNAs, unique SNPs could arise as random mutations through viral genome replication (dsRNA) but may later be negatively selected during viral genome encapsidation. Second, they also could arise through errors introduced by host RNA-dependent RNA polymerase during the generation of secondary vsiRNAs (18). The possibility of technical artifacts should not be completely excluded, because the extremely low input concentrations of the current protocols do not allow us to exactly equilibrate the amount of molecules between the pools before the ligation reaction. This could result in a dropout of low-frequency variants in some of the libraries. Nevertheless, since we observed unique SNPs in both of the pools and the frequencies of SNPs between technical replicates and between the pools have a good correlation, it is not likely that observed unique SNPs would arise solely as a technical artifact of unequilibrated molecule counts.
Several studies reported prominent hotspots and cold spots in depths of vsiRNAs coverage across the viral genome (35, 36). Different explanations have been suggested for these observations, including biological explanations, e.g., connecting the hotspots with predicted secondary structures in viral genomes (23, 36), as well as library preparation-connected ligation biases (34). In other cases (37, 38) such patterns were not observed. In our study, vsiRNAs were uniformly distributed across the PVY genome without prominent coverage hotspots or cold spots (Fig. 3e). In addition, the evenness of coverage was greater for sRNA samples than for chemically fragmented viral RNA (Fig. 3e). The pattern of vsiRNA coverage is specific for different plant-virus systems and should be evaluated in each specific case. However, one can speculate that even if notable cold/hotspots in coverage are observed, they do not greatly affect the relative frequency of detected variants but only reduce/enhance the resolution in such regions of the viral genome. Moreover, such a variation could be taken into account, when comparing the samples, filtering the positions with uneven coverage across the samples.
Recently, some studies have shown that recombination events should be approximately as common as mutations in the evolution of RNA viruses (32, 39, 40). A recent study (41) illustrated this phenomenon in the case of tobacco etch virus, a member of the same (Potyvirus) genus as PVY. We used an algorithm (32) which allowed us to detect nonhomologous recombinant reads in the VP pool. The reads in the sRNA pool were too short to be included in the analyses. The results of our study implicated that roughly 7 to 15% of the VP reads obtained in this experiment represent recombinant viral sequences originating from nonhomologous recombination events. This is congruent with the data obtained for flock house virus (FHV), where a similar methodology was used (32). A relatively high proportion of nonhomologous recombinant sequences in virions highlights an important but rather understudied part of the variability in virus populations. Further studies should be made that employ an approach which would allow the straightforward distinction of in vivo recombinants from artifactual recombinations that may have been introduced through library preparation. Here, we have used a conservative approach to reliably distinguish in planta recombination events from possible artifactual recombinations by stringently filtering the recombination events based on their occurrence in both of the sequenced technical replicates. This allowed us to perform a high-resolution mapping of nonhomologous recombination events on the viral genome, which indicated hot spots for recombination events in the 3′-untranslated region of the PVY genome as well as in the 5′ and 3′ ends of the coat protein gene. The mechanism and relevance behind this interesting pattern should be further explored.
In the potato-PVY system, both of the sequence pools investigated allowed reliable determination of low-frequency SNPs in virus populations. We showed that sequencing virus-derived small RNAs captures a highly detailed picture of the within-plant virus mutational landscape compared to the RNA packed in viral particles. The deep resolution obtained with each of the approaches would allow us to track in detail the fluctuations of variant frequencies in population studies of plant virus evolution and emergence. Both tested methods have their advantages and disadvantages; small RNA sequencing would allow an easier, quicker, and more generic method to explore within-plant virus population structure, whereas deep sequencing of RNA isolated from viral particles provides an additional insight into the recombination events, which are an important but often overlooked source of diversity in viral populations. The presented comparison can serve as a firm baseline for the employment of both methods in within-plant virus population studies.
Supplementary Material
ACKNOWLEDGMENTS
The work was supported by the Slovenian Research Agency through grant L4-5525. D.K. is a recipient of a Ph.D. research grant from the Slovenian Research Agency.
We thank Lidija Matičič and Polona Kogovšek for help with the preparation of plant material and virus isolates.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03685-14.
REFERENCES
- 1.Domingo E, Sheldon J, Perales C. 2012. Viral quasispecies evolution. Microbiol Mol Biol Rev 76:159–216. doi: 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McElroy K, Thomas T, Luciani F. 2014. Deep sequencing of evolving pathogen populations: applications, errors, and bioinformatic solutions. Microb Inform Exp 4:1. doi: 10.1186/2042-5783-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright CF, Morelli MJ, Thébaud G, Knowles NJ, Herzyk P, Paton DJ, Haydon DT, King DP. 2011. Beyond the consensus: dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J Virol 85:2266–2275. doi: 10.1128/JVI.01396-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Routh A, Domitrovic T, Johnson JE. 2012. Host RNAs, including transposons, are encapsidated by a eukaryotic single-stranded RNA virus. Proc Natl Acad Sci U S A 109:1907–1912. doi: 10.1073/pnas.1116168109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morelli MJ, Wright CF, Knowles NJ, Juleff N, Paton DJ, King DP, Haydon DT. 2013. Evolution of foot-and-mouth disease virus intra-sample sequence diversity during serial transmission in bovine hosts. Vet Res 44:1–15. doi: 10.1186/1297-9716-44-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohshima K, Akaishi S, Kajiyama H, Koga R, Gibbs AJ. 2010. Evolutionary trajectory of turnip mosaic virus populations adapting to a new host. J Gen Virol 91:788–801. doi: 10.1099/vir.0.016055-0. [DOI] [PubMed] [Google Scholar]
- 7.Kim T, Youn MY, Min BE, Choi SH, Kim M, Ryu KH. 2005. Molecular analysis of quasispecies of Kyuri green mottle mosaic virus. Virus Res 110:161–167. doi: 10.1016/j.virusres.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Acosta-Leal R, Bryan BK, Rush CM. 2010. Host effect on the genetic diversification of beet necrotic yellow vein virus single-plant populations. Phytopathology 100:1204–1212. doi: 10.1094/PHYTO-04-10-0103. [DOI] [PubMed] [Google Scholar]
- 9.Ali A, Roossinck MJ. 2010. Genetic bottlenecks during systemic movement of Cucumber mosaic virus vary in different host plants. Virology 404:279–283. doi: 10.1016/j.virol.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 10.Schneider W, Roossinck M. 2001. Genetic diversity in RNA virus quasispecies is controlled by host-virus interactions. J Virol 75:6566–6571. doi: 10.1128/JVI.75.14.6566-6571.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martínez F, Lafforgue G, Morelli MJ, González-Candelas F, Chua N-H, Daròs J-A, Elena SF. 2012. Ultradeep sequencing analysis of population dynamics of virus escape mutants in RNAi-mediated resistant plants. Mol Biol Evol 29:3297–3307. doi: 10.1093/molbev/mss135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabre F, Montarry J, Coville J, Senoussi R, Simon V, Moury B. 2012. Modelling the evolutionary dynamics of viruses within their hosts: a case study using high-throughput sequencing. PLoS Pathog 8:e1002654. doi: 10.1371/journal.ppat.1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montarry J, Doumayrou J, Simon V, Moury B. 2011. Genetic background matters: a plant-virus gene-for-gene interaction is strongly influenced by genetic contexts. Mol Plant Pathol 12:911–920. doi: 10.1111/j.1364-3703.2011.00724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morroni M, Jacquemond M, Tepfer M. 2013. Deep sequencing of recombinant virus populations in transgenic and nontransgenic plants infected with cucumber mosaic virus. Mol Plant Microbe Interact 26:801–811. doi: 10.1094/MPMI-02-13-0057-R. [DOI] [PubMed] [Google Scholar]
- 15.Tugume AK, Mukasa SB, Kalkkinen N, Valkonen JPT. 2010. Recombination and selection pressure in the ipomovirus sweet potato mild mottle virus (Potyviridae) in wild species and cultivated sweet potato in the centre of evolution in East Africa. J Gen Virol 91:1092–1108. doi: 10.1099/vir.0.016089-0. [DOI] [PubMed] [Google Scholar]
- 16.Yu X-Q, Jia J-L, Zhang C-L, Li X-D, Wang Y-J. 2010. Phylogenetic analyses of an isolate obtained from potato in 1985 revealed potato virus X was introduced to China via multiple events. Virus Genes 40:447–451. doi: 10.1007/s11262-010-0468-5. [DOI] [PubMed] [Google Scholar]
- 17.Satya VR, DiCarlo J. 2014. Edge effects in calling variants from targeted amplicon sequencing. BMC Genomics 15:1073. doi: 10.1186/1471-2164-15-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Llave C. 2010. Virus-derived small interfering RNAs at the core of plant-virus interactions. Trends Plant Sci 15:701–707. doi: 10.1016/j.tplants.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 19.Kreuze JF, Perez A, Untiveros M, Quispe D, Fuentes S, Barker I, Simon R. 2009. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology 388:1–7. doi: 10.1016/j.virol.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 20.Wu Q, Luo Y, Lu R, Lau N, Lai EC, Li W-X, Ding S-W. 2010. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc Natl Acad Sci U S A 107:1606–1611. doi: 10.1073/pnas.0911353107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seguin J, Rajeswaran R, Malpica-López N, Martin RR, Kasschau K, Dolja VV, Otten P, Farinelli L, Pooggin MM. 2014. De novo reconstruction of consensus master genomes of plant RNA and DNA viruses from siRNAs. PLoS One 9:e88513. doi: 10.1371/journal.pone.0088513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steyer A, Gutiérrez-Aguire I, Kolenc M, Koren S, Kutnjak D, Pokorn M, Poljšak-Prijatelj M, Racki N, Ravnikar M, Sagadin M, Fratnik Steyer A, Toplak N. 2013. High similarity of novel orthoreovirus detected in a child hospitalized with acute gastroenteritis to mammalian orthoreoviruses found in bats in Europe. J Clin Microbiol 51:3818–3825. doi: 10.1128/JCM.01531-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Candresse T, Filloux D, Muhire B, Julian C, Galzi S, Fort G, Bernardo P, Daugrois J-H, Fernandez E, Martin DP, Varsani A, Roumagnac P. 2014. Appearances can be deceptive: revealing a hidden viral infection with deep sequencing in a plant quarantine context. PLoS One 9:e102945. doi: 10.1371/journal.pone.0102945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rupar M, Ravnikar M, Tušek-Žnidarič M, Kramberger P, Glais L, Gutiérrez-Aguirre I. 2013. Fast purification of the filamentous Potato virus Y using monolithic chromatographic supports. J Chromatogr A 1272:33–40. doi: 10.1016/j.chroma.2012.11.058. [DOI] [PubMed] [Google Scholar]
- 25.Scholthof K-BG, Adkins S, Czosnek H, Palukaitis P, Jacquot E, Hohn T, Hohn B, Saunders K, Candresse T, Ahlquist P, Hemenway C, Foster GD. 2011. Top 10 plant viruses in molecular plant pathology. Mol Plant Pathol 12:938–954. doi: 10.1111/j.1364-3703.2011.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y-R, Zheng Y, Liu B, Zhong S, Giovannoni J, Fei Z. 2012. A cost-effective method for Illumina small RNA-Seq library preparation using T4 RNA ligase 1 adenylated adapters. Plant Methods 8:41. doi: 10.1186/1746-4811-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Core Team R. 2013. R: a language and environment for statistical computing. 3.0.2. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 29.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. 2012. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Routh A, Johnson JE. 2014. Discovery of functional genomic motifs in viruses with ViReMa–a virus recombination mapper–for analysis of next-generation sequencing data. Nucleic Acids Res 42:e11. doi: 10.1093/nar/gkt916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra Ma. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sorefan K, Pais H, Hall AE, Kozomara A, Griffiths-Jones S, Moulton V, Dalmay T. 2012. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence 3:4. doi: 10.1186/1758-907X-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, Gao S, Hernandez AG, Wechter WP, Fei Z, Ling K-S. 2012. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS One 7:e37127. doi: 10.1371/journal.pone.0037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aregger M, Borah BK, Seguin J, Rajeswaran R, Gubaeva EG, Zvereva AS, Windels D, Vazquez F, Blevins T, Farinelli L, Pooggin MM. 2012. Primary and secondary siRNAs in geminivirus-induced gene silencing. PLoS Pathog 8:e1002941. doi: 10.1371/journal.ppat.1002941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Cheng X, Wu X, Wang A, Wu X. 2014. Characterization of complete genome and small RNA profile of pagoda yellow mosaic associated virus, a novel badnavirus in China. Virus Res 188:103–108. doi: 10.1016/j.virusres.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Fuentes S, Heider B, Tasso RC, Romero E, Zum Felde T, Kreuze JF. 2012. Complete genome sequence of a potyvirus infecting yam beans (Pachyrhizus spp.) in Peru. Arch Virol 157:773–776. doi: 10.1007/s00705-011-1214-6. [DOI] [PubMed] [Google Scholar]
- 39.Routh A, Ordoukhanian P, Johnson JE. 2012. Nucleotide-resolution profiling of RNA recombination in the encapsidated genome of a eukaryotic RNA virus by next-generation sequencing. J Mol Biol 424:257–269. doi: 10.1016/j.jmb.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Froissart R, Roze D, Uzest M, Galibert L, Blanc S, Michalakis Y. 2005. Recombination every day: abundant recombination in a virus during a single multi-cellular host infection. PLoS Biol 3:e89. doi: 10.1371/journal.pbio.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tromas N, Zwart MP, Poulain M, Elena SF. 2014. Estimation of the in vivo recombination rate for a plant RNA virus. J Gen Virol 95:724–732. doi: 10.1099/vir.0.060822-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.