

ABSTRACT
Human cytomegalovirus (HCMV) is a major cause of birth defects that include severe neurological deficits, hearing and vision loss, and intrauterine growth restriction. Viral infection of the placenta leads to development of avascular villi, edema, and hypoxia associated with symptomatic congenital infection. Studies of primary cytotrophoblasts (CTBs) revealed that HCMV infection impedes terminal stages of differentiation and invasion by various molecular mechanisms. We recently discovered that HCMV arrests earlier stages involving development of human trophoblast progenitor cells (TBPCs), which give rise to the mature cell types of chorionic villi—syncytiotrophoblasts on the surfaces of floating villi and invasive CTBs that remodel the uterine vasculature. Here, we show that viral proteins are present in TBPCs of the chorion in cases of symptomatic congenital infection. In vitro studies revealed that HCMV replicates in continuously self-renewing TBPC lines derived from the chorion and alters expression and subcellular localization of proteins required for cell cycle progression, pluripotency, and early differentiation. In addition, treatment with a human monoclonal antibody to HCMV glycoprotein B rescues differentiation capacity, and thus, TBPCs have potential utility for evaluation of the efficacies of novel antiviral antibodies in protecting and restoring placental development. Our results suggest that HCMV replicates in TBPCs in the chorion in vivo, interfering with the earliest steps in the growth of new villi, contributing to virus transmission and impairing compensatory development. In cases of congenital infection, reduced responsiveness of the placenta to hypoxia limits the transport of substances from maternal blood and contributes to fetal growth restriction.
IMPORTANCE Human cytomegalovirus (HCMV) is a leading cause of birth defects in the United States. Congenital infection can result in permanent neurological defects, mental retardation, hearing loss, visual impairment, and pregnancy complications, including intrauterine growth restriction, preterm delivery, and stillbirth. Currently, there is neither a vaccine nor any approved treatment for congenital HCMV infection during gestation. The molecular mechanisms underlying structural deficiencies in the placenta that undermine fetal development are poorly understood. Here we report that HCMV replicates in trophoblast progenitor cells (TBPCs)—precursors of the mature placental cells, syncytiotrophoblasts and cytotrophoblasts, in chorionic villi—in clinical cases of congenital infection. Virus replication in TBPCs in vitro dysregulates key proteins required for self-renewal and differentiation and inhibits normal division and development into mature placental cells. Our findings provide insights into the underlying molecular mechanisms by which HCMV replication interferes with placental maturation and transport functions.
INTRODUCTION
Human cytomegalovirus (HCMV) is the most common cause of congenital viral infection in the United States. Each year, at least 40,000 babies are born with congenital infection, resulting in about 400 deaths and leaving 4,000 to 8,000 children with permanent neurological complications, such as hearing loss, visual impairment, and mental retardation (1, 2). HCMV infection is also associated with stillbirth, preterm delivery, and intrauterine growth restriction (IUGR) (3–9), which are risk factors for perinatal and lifetime morbidity (10), including cardiovascular disease (11, 12). There are more cases of permanent disability from congenital HCMV infection than from other, better known congenital conditions, such as Down syndrome, fetal alcohol syndrome, and neural tube defects (13, 14). The burden to families and the economic costs to society of congenital HCMV infection are immense, with direct annual costs of more than one billion dollars (15). Despite its public health significance, however, the specific molecular and cellular basis of HCMV's effects on the placenta and fetus and the reasons why clinical outcomes vary are poorly understood. Although direct fetal infection is involved in severe cases of neuropathology, infection of the placenta—with attendant effects on its development and function leading to an hypoxic environment (16–19)—can result in IUGR and stillbirth (20–22).
Models used to uncover the molecular mechanisms of HCMV pathogenesis in the human placenta have focused on the terminal stages of trophoblast differentiation and have been limited to primary cytotrophoblasts (CTBs), chorionic villous explants, and transformed trophoblast cell lines. In CTBs, HCMV replication reduces expression of the differentiation markers integrin α1β1, integrin αVβ3, and major histocompatibility complex (MHC) class I protein HLA-G (23) and reduces both the expression and activity of matrix metalloproteinase-9 (MMP-9) (24), which degrades the extracellular matrix (25), thereby impairing the ability of CTBs to differentiate and invade the uterine vasculature. Infected CTBs increase production of the immunosuppressive cytokines interleukin-10 (IL-10) and cytomegalovirus IL-10 (cmvIL-10), which further reduce invasiveness (24). HCMV replication activates the peroxisome proliferator-activated receptor γ (PPARγ), which also compromises CTB functions (26, 27). Together, these results suggest that HCMV infection reduces CTB differentiation and invasion in utero, required to ensure an adequate blood supply to the placenta and fetus (23, 24, 26–30). Using human placental villous explants (23, 31) and xenografts implanted into SCID-hu mice (32), we reported that infection with a pathogenic HCMV strain impairs villous growth. Although these studies provided important clues to the mechanisms by which HCMV infection impairs placental development and function, they did not provide an understanding of how and whether infection affects earlier stages of trophoblast development.
We previously reported that placentas from infants with symptomatic congenital infection and poor outcome exhibit signs of hypoxia (17, 18). Passive immunization with hyperimmune globulin, a pooled immunoglobulin preparation from donors with high-avidity anti-HCMV antibodies, was shown to reduce viral replication and enable compensatory growth of chorionic villi, increasing the placental surface area perfused by maternal blood (17, 33) and, for some babies, improving the outcome (34–38). In addition, we recently found that placentas from cases of IUGR with primary maternal infection contain unusual clusters of extravillous CTBs, indicating that infection may have effects that block differentiation and fusion into syncytiotrophoblasts (STBs) (18). These observations suggested impaired development of the trophoblast lineages at stages earlier than previously indicated by studies of CTBs in culture (23).
Chorionic villi, the functional units of the placenta, grow from the chorion, where trophoblast progenitor cells (TBPCs) reside (39) (see Fig. 1A and B). Cells in the chorion coexpress trophoblast markers and proteins whose homologs in mice mark pluripotent progenitors, suggesting that placental growth is supported by progenitors in the chorion throughout gestation (39). Lines of continuously self-renewing TBPCs were established from cells that expressed these markers isolated from the chorion. These cells express the CTB marker cytokeratin 7 and transcriptional regulators required for trophoblast fate specification and/or differentiation of the major lineages in mice, including Eomes (Eomesodermin), geminin, GCM1 (glial cells missing homolog 1) and GATA4. They also express the neonatal Fc receptor (FcRn) for maternal IgG transport and passive immunity of the fetus (40, 41). When TBPCs are cultured under conditions that trigger differentiation, the cells aggregate and form spheres containing mononuclear and multinuclear cells. These spheres upregulate HLA-G, a marker of differentiating/invading CTBs, and increase expression of human placental lactogen and secretion of human chorionic gonadotropin (hCG)—two hormones secreted from STBs (see Fig. 1C). In addition, TBPCs show high invasive capacity and can be used to study early steps in trophoblast differentiation.
FIG 1.
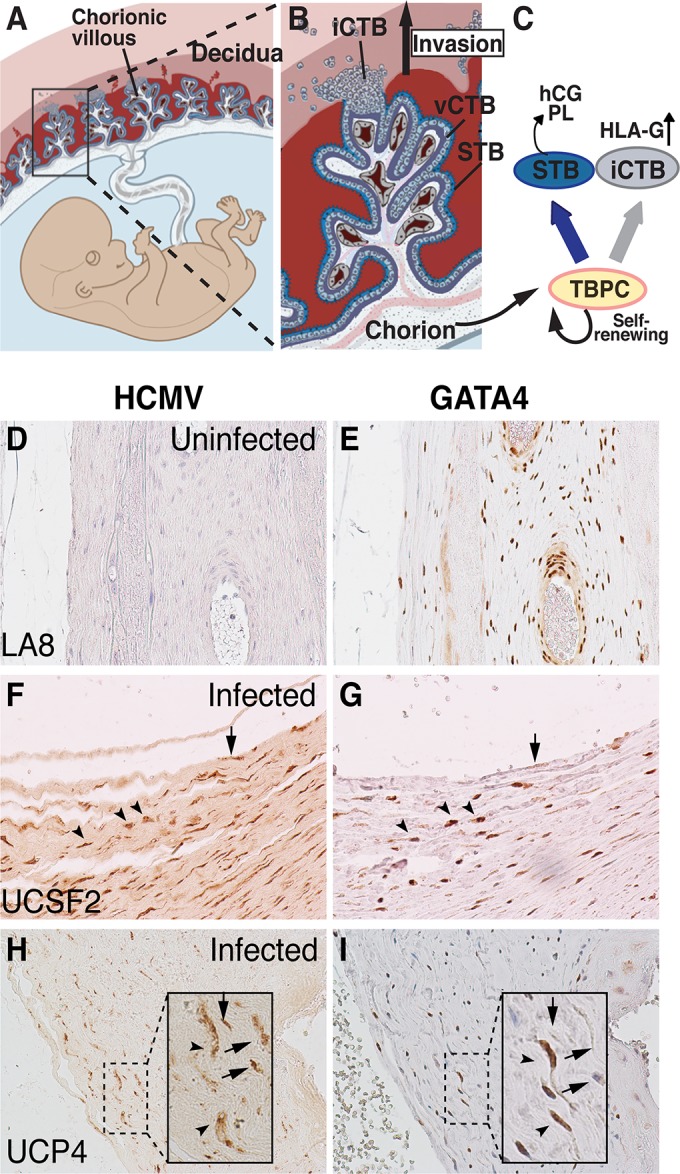
Anatomy of the human placenta. (A and B) Chorionic villi, the functional units of the placenta, consist of STBs and villous CTBs (vCTBs), which terminally differentiate into invasive CTBs (iCTBs). Adapted from Maltepe et al. (65). (C) TBPCs from chorion differentiate into STBs and iCTBs. hCG, human chorionic gonadotropin; PL, placental lactogen. (D to I) HCMV proteins detected in cells of chorion from placentas of babies with congenital infection. Immunohistochemical staining for HCMV proteins (D, F, and H) and GATA4 (E, G, and I) in adjacent sections of an uninfected placenta (D and E) and two placentas from cases of symptomatic congenital infection (F to I). HCMV protein-positive cells (F and H) had lower levels of GATA4 expression (arrows), which labels TBPCs (G and I) in the chorion. Some HCMV protein-positive cells still express GATA4 (arrowheads).
Since TBPCs give rise to the mature cells of the human placenta, impaired proliferation and function would be expected to lead to hypoxia and developmental defects in the fetus, such as IUGR. Here we show that HCMV-infected cell proteins are present in TBPCs in the chorion in cases of symptomatic congenital infection. We then use the established TBPC cell lines to investigate the impact of viral replication on self-renewal and generation of the mature placental cell types.
MATERIALS AND METHODS
Cell culture, virus strains, and HCMV infection in vitro.
The University of California's Committee on Human Research approved this study. TBPC lines isolated from human chorionic membranes (7.3 and 15.6 weeks of gestation) were cultured on gelatin-coated plates in Dulbecco's modified Eagle's medium with nutrient mixture F-12 (DMEM/F12) supplemented with 10 ng/ml basic fibroblast growth factor (bFGF) (R&D Systems, Minneapolis, MN), 10 μM SB431542 (Tocris Biosciences, Minneapolis, MN) and 10% fetal bovine serum (FBS) (39). TBPCs—100% positive by immunostaining for cytokeratin with the rat monoclonal antibody 7D3 (42) and GATA4—were used up to passage 18. The cells were infected with the pathogenic clinical HCMV strain VR1814 (43) at a multiplicity of infection (MOI) of 0.01 to 1. VR1814 virus stocks were propagated in human umbilical vein endothelial cells at low passage; high-titer stocks were produced by one passage in human foreskin fibroblasts (HF). Since clinical HCMV strains are highly cell associated (44), infected cells were pelleted by centrifugation, and supernatant was removed. The cell pellet was sonicated in 1% FBS-containing DMEM, and cell debris was removed by centrifugation (45).
Serological and other reagents.
The following antibodies were purchased: rabbit polyclonal antibodies to GATA4, Hand1, HMGA2 (high-mobility group AT-hook 2), and hCG (Abcam, Cambridge, MA); goat polyclonal antibodies to GATA3 and GATA4 (R&D Systems); rabbit polyclonal antibodies to geminin (Santa Cruz Biotechnology, Dallas, TX); anti-PPARγ rabbit monoclonal antibodies (clones C26H12 and 81B8; Cell Signaling Technology, Danvers, MA) and rabbit polyclonal antibody (Proteintech, Chicago, IL); mouse monoclonal antibody to human cytokeratin 7 (clone OV-TL 12/30; Dako, Carpinteria, CA); and anti-actin mouse monoclonal antibody (Sigma-Aldrich, St. Louis, MO). Anti-human/rat neonatal Fc receptor (FcRn) α-chain rabbit polyclonal antibody (46) was a generous gift from Neil E. Simister, and rat monoclonal anti-human cytokeratin (clone 7D3) was a gift from Susan Fisher (42). The following mouse monoclonal antibodies to HCMV ICPs (infected cell proteins), generated by the Pereira lab (47, 48), were also used: CH112-2 to glycoprotein B (UL55); CH19 to pp28 (UL99); CH160 to immediate early (IE) nuclear proteins (IE1 and IE2, UL122, and UL123) and CH443 to IE1 (UL123). We also used M23, a mouse monoclonal antibody to UL112 and -113 (49, 50), and a cocktail of mouse monoclonal antibodies directed against HCMV ICPs (MAB8121 containing clones 8B1.2, 1G5.2, and 2D4.2; Millipore, Billerica, MA). TRL345, a human IgG1 monoclonal antibody (MAb) against the conserved AD-2 epitope of HCMV gB (51), and control IgG1 MAb Synagis, which is reactive with respiratory syncytial virus (52), were provided by Trellis Bioscience, LLC (Menlo Park, CA).
Immunofluorescence staining.
Cells grown on coverslips were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 or ice-cold methanol. Nonspecific antibody binding to the viral Fc receptor was blocked using normal human serum (53). For double immunostaining, cells were simultaneously incubated with primary antibodies from different species and secondary antibodies labeled with fluorescein isothiocyanate (FITC) or rhodamine red-X (RRX). Nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). Alternatively, the coverslips were incubated with primary antibodies against cellular proteins overnight, followed by incubation with secondary antibodies, then stained with antibodies to HCMV proteins. Images were obtained using a Nikon Eclipse 50i microscope equipped with a Spot 7.4 Slider camera (Diagnostic Instruments) controlled by Spot advanced software.
Immunoblot analysis.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing a protease inhibitor cocktail (Thermo Scientific, Rockford, IL) and clarified by centrifugation. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to Hybond membranes (GE Healthcare Life Sciences, Pittsburgh, PA), and blocked for 2 h in phosphate-buffered saline (PBS) containing 5% skim milk and 0.05% Tween 20. After incubation with primary antibody for 16 h at 4°C and then with a peroxidase-conjugated secondary antibody for 1 h, blots were developed with WesternBright enhanced chemiluminescence (ECL) or Quantum horseradish peroxidase (HRP) substrate (Advansta, Menlo Park, CA).
Immunohistochemical analysis. (i) Placental samples.
Placental biopsy specimens were obtained from patients delivering at Long Hospital, University of California, San Francisco, with informed written consent and institutional review board approval. Biopsy specimens (1 central biopsy specimen and 4 peripheral biopsy specimens) were fixed in 10% formalin and embedded in paraffin. Serial 5-μm-thick sections were either stained with hematoxylin and eosin or immunostained (16, 54). Additional biopsy specimens were obtained from the standing collection in the Department of Pathology, University of California, San Francisco.
(ii) Immunostaining of cellular and viral antigens.
Slides were deparaffinized using Clear-Rite 3 (Thermo Scientific) and rehydrated in a decreasing ethanol concentration series. Antigen retrieval was performed for each antigen as described below, followed by blocking with 1 to 2% normal horse serum (for staining with mouse monoclonal antibodies) or normal rabbit serum (for staining with goat polyclonal antibodies) for 30 min to overnight (18). Sections were incubated with primary antibody overnight at 4°C, washed, and processed for color development using Vectastain avidin-biotin complex (ABC) horseradish peroxidase kit (mouse or goat; Vector Laboratories) according to the manufacturer's instructions, followed by color development with a diaminobenzidine (DAB) substrate kit (Abcam). Slides were counterstained with hematoxylin (Sigma-Aldrich), dehydrated, and mounted using Vectamount AQ (Vector Laboratories). The antibodies used for immunohistochemistry were mouse monoclonal anti-HCMV blend (MAB8121, which react with IE, early, and late antigens; Millipore) and goat polyclonal anti-GATA4 (Abcam). Antigen retrieval was performed for each antibody as follows: for HCMV ICPs, sections were incubated with 0.4% pepsin (Sigma-Aldrich) in 0.01 N HCl (30 min, 37°C); for GATA4, slides were heat treated in 10 mM sodium citrate (pH 6.0) in a 2100 Retriever pressure cooker (Diatome, Hatfield, PA) using its automated pressurized heat cycle (∼15 min) and cooled for 2 h. Images were taken on a Nikon TS100 inverted microscope equipped with a Nikon DS-F12 camera controlled by Nikon NIS-Elements F4 software.
Differentiation of TBPCs.
Mock-infected control and infected TBPCs were dissociated into single cells with Accutase (Stemcell Technologies, Vancouver, BC, Canada), and live cells were counted using trypan blue exclusion. A total of 2 × 105 cells (live cells) were plated on undiluted Matrigel-coated 24-well plates in differentiation medium consisting of KnockOut DMEM (Invitrogen, Grand Island, NY) supplemented with KnockOut serum replacement (Invitrogen), 40 ng/ml epidermal growth factor (EGF), 10 ng/ml fibroblast growth factor 4 (FGF4) (R&D Systems), and 10% FBS and cultured for up to 3 days (39). When cells were infected at higher MOIs and/or maintained for longer periods before dissociation, infected cells did not form any spheres but became apoptotic. Thus, we chose infection at an MOI of 1 and culture for 4 days for further differentiation assays. In some experiments, mock-infected control and infected cells were cultured in media containing ganciclovir (450 μM) (55) prior to differentiation. To evaluate the efficacy of anti-gB MAb TRL345 (51), TBPCs were infected, and 20 μg/ml of the antibody was added 1 day after infection. Cells were further cultured 3 or 4 days, after which differentiation assays were performed. Since infection at a lower MOI has only minimal effects on sphere formation and infection at a higher MOI is not physiologically relevant, we chose infection at an MOI of 0.01 to 0.1 (56). We noticed that infectivity was slightly different depending on passage number, i.e., higher-passage cells had lower infectivity, especially at a low MOI. The isotype-matched negative-control antibody Synagis (52) was included in these experiments.
In vitro cell invasion assays.
Cell invasion assays were performed as reported with minor modifications (24, 39). Accutase-dissociated mock-infected control and infected TBPCs (4 days postinfection [p.i.]; MOI of 1) (5,000 cells) were plated on undiluted Matrigel-coated Transwell polycarbonate filters (8-μm pores; Corning Costar, Tewksbury, MA) in differentiation medium. After 72 h, filters were fixed and stained for CTB-specific cytokeratin with 7D3 antibody. Nuclei and cytokeratin-positive cells that migrated to the underside of the filters were counted. Each condition was tested in duplicate, and the experiments were performed 3 times.
Image and statistical analyses.
Fluorescence intensities of the immunofluorescence staining of geminin, HMGA2 and GATA4 were quantified using NIH ImageJ software. Three to five images (magnification of ×200) from randomly selected areas were taken at constant settings from at least 3 independent experiments. Within each image, signal intensities were measured for each nucleus using the integrated density function of ImageJ. A total of 100 to 600 measurements were made for each experimental condition within each experiment. The statistical significance of differences between the means within experiments was determined using the Mann-Whitney rank sum test with the Real Statistic Resource Pack Add In for Excel. P values of less than 0.05 were considered significant. Data from multiple experiments were not combined, as statistical significance was easily achieved in all individual experiments in comparisons between infected and mock-infected cells and between infected and infected, ganciclovir-treated cells. For presentation in the charts in Fig. 2, fluorescence intensities of ganciclovir-treated mock-infected control, infected cells, and infected, ganciclovir-treated cells were normalized to the mean intensities of the mock-infected control in the same experiment.
FIG 2.

HCMV infection of TBPCs alters expression levels of key regulatory proteins. (A) TBPCs express trophoblast-specific cytokeratins (CK) and neonatal Fc receptor (FcRn). Original magnification, ×200. (B) TBPCs were mock infected (control) or infected with HCMV strain VR1814 and fixed at 5 days p.i. The cells were immunostained for specific regulatory proteins (green) and HCMV pp28 (red) and counterstained with DAPI (blue). Colocalization of green and blue signals (turquoise) and of red and green signals (yellow) is shown. Original magnification, ×200. Bar = 20 μm. (C) Fluorescence intensities of geminin, HMGA2, and GATA4 were quantified using NIH ImageJ software. In each experiment, the mean intensities of ganciclovir (GCV)-treated uninfected control cells, infected cells, and infected, GCV-treated cells were normalized to the mean intensities of the control cells. Graphs show representative relative fluorescence intensity (mean ± SE) from 1 of 3 independent experiments. Significant differences were found by the Mann-Whitney rank sum test and indicated as follows: *, P < 0.0001 for mock-infected control cells and infected cells; #, P < 0.0001 for infected cells and infected, GCV-treated cells; and +, P < 0.0001 for GCV-treated mock-infected control cells and GCV-treated, infected cells. Significant differences indicated here for one experiment were found in all experiments. (D) Immunoblotting of key regulatory proteins. Cell lysates from control or infected TBPCs at 2 and 5 days (d) p.i. were immunoblotted with antibodies to TBPC regulatory proteins and actin (loading control).
To obtain sphere diameter values, entire wells (24-well plate) for each condition (e.g., mock-infected control in the presence and absence of ganciclovir, infection in the presence and absence of ganciclovir or antibodies) were photographed using a Nikon TS100 inverted microscope equipped with a Nikon DS-F12 camera controlled by Nikon NIS-Elements F4 software, and sphere diameters were manually analyzed. Each condition was tested in duplicate, and each experiment was performed 4 times. The statistical significance of differences between conditions was determined within each experiment using the Mann-Whitney rank sum test (described above). Although experiments were repeated, each individual experiment provided sufficient statistical power to achieve significance in comparisons, so data from multiple experiments were not combined. The figures show the results of representative experiments and indicate P values for two-way comparisons. P values of less than 0.05 were considered significant.
For invasion assays, data are expressed as means ± standard errors (SE). Student's t test was used to analyze the difference in invasion between mock-infected control and infected cells and between infected and infected, ganciclovir-treated cells. A P value of < 0.05 was considered significant.
Ethics statement.
The Institutional Review Board of the University of California, San Francisco—the Human Research Protection Program Committee on Human Research—approved the collection and use of human tissue samples, including the identification and use of sample UCP4, obtained from the existing collection of the Department of Pathology. Samples were obtained only from adults who provided written informed consent.
RESULTS
HCMV infects trophoblast progenitor cells in the chorion of the placenta in utero.
We recently published evidence that congenital HCMV infection interferes with CTB differentiation and STB formation in the placenta (18), suggesting that the virus affects differentiation of the trophoblast lineages at stages earlier than previously demonstrated using primary CTBs in culture (23). As chorionic villi develop from progenitor cells in the chorion, we hypothesized that HCMV replicates in these cells (TBPCs), leading to impaired differentiation at early stages of the development of villi (Fig. 1A to C). To determine whether TBPC infection occurs in utero, we immunostained placentas from cases of confirmed congenital infection and healthy pregnancies using a cocktail of antibodies against HCMV antigens (IE, early, and late) and GATA4, a marker of TBPCs not expressed in CTBs (39). We observed viral antigens in cells weakly expressing GATA4 (Fig. 1F to I, arrow) in the chorions of cases of symptomatic congenital infection, but not in control placentas (Fig. 1D and E) or when isotype control antibodies were used (data not shown). In addition, chorions from cases of congenital infection tended to contain 2- to 3-fold-fewer GATA4-positive cells compared with control placentas, suggesting that infection decreases the number of TBPCs. These results indicated that HCMV replicates in TBPCs and could interfere with the development of chorionic villi (Fig. 1C). This offers an explanation why placentas from cases of congenital infection treated with hyperimmune globulin to reduce virus replication contained more immature chorionic villi than placentas from cases of untreated infection and healthy subjects (17). We and others reported that HCMV replicates in decidual cells and glandular epithelial cells (16, 18, 57), suggesting that infection spreads to the adjacent chorion, reducing the number of TBPCs available to differentiate into CTBs and STBs and undermining development of new chorionic villi in response to hypoxic conditions (17).
HCMV infection of TBPCs alters expression of proteins that control differentiation.
We recently reported that TBPCs from first- and second-trimester placentas are permissive for HCMV replication and that the development of spheres is reduced in vitro (i.e., support the full viral life cycle and produce progeny virions) (56). To explore the molecular mechanisms by which HCMV infection impairs trophoblast differentiation, we used lines of continuously self-renewing TBPCs established previously (39, 56). For these experiments, we routinely confirmed that the cells expressed cytokeratin, neonatal Fc receptor (FcRn) (Fig. 2A) and GATA4 (Fig. 2B), markers not expressed in human placental fibroblasts (data not shown). TBPCs plated on gelatin were infected with the pathogenic HCMV clinical strain VR1814. We analyzed expression of proteins with potential roles in self-renewal and differentiation (referred to as TBPC regulatory proteins) based on their known functions in mouse trophoblast progenitor cells and human embryonic stem cells (ESCs). These proteins included the regulators of pluripotency and cell cycle progression geminin (58, 59) and HMGA2 (high-mobility group AT-hook 2) (60) and the differentiation factors GATA3 (61–63), GATA4 (64), and Hand1 (65).
VR1814-infected TBPCs and mock-infected controls were immunostained (Fig. 2B and 3), and extracts were analyzed by immunoblotting at 2 and 5 days postinfection (p.i.) (i.e., early and late) (Fig. 2D). At 5 days p.i., geminin was upregulated (Fig. 2B), with a significant increase in fluorescence intensity (Fig. 2C), which could prevent TBPC differentiation. Geminin is involved in the maintenance of the undifferentiated state of neural progenitor cells, and overexpression of geminin prevents neuronal differentiation (66–68). HMGA2 (Fig. 2B and C) and SOX2, a component of the core pluripotency factor network in ESCs (69, 70) (data not shown), were downregulated in infected cells, suggesting reduced capacity for self-renewal and pluripotency. GATA4, a differentiation-related transcription factor, was significantly decreased in comparison to the control (Fig. 2B and C), which could inhibit differentiation, as demonstrated in human ESCs (71). In some cells, GATA4 disappeared from nuclei (Fig. 2B, blue dashed line), and in other cells, GATA4 accumulated in small spots in nuclei (Fig. 2B, white arrowheads). Altered expression of geminin and SOX2 was observed at early time points after infection (data not shown), but downregulation of HMGA2 and GATA4 was more pronounced at late time points. Immunoblot analysis confirmed upregulation of geminin and downregulation of HMGA2 and GATA4, especially at later time points (Fig. 2D).
FIG 3.

HCMV infection of TBPCs alters the subcellular localization of differentiation transcription factors GATA3 and Hand1. TBPCs were mock infected (control) or infected with VR1814 and fixed at 5 days p.i. Cells were immunostained for specific regulatory proteins (green) and HCMV pp28 (red) and counterstained with DAPI (blue). Colocalization of green and blue signals (turquoise) and of red and green signals (yellow) is shown. Nuclei (blue dashed lines) and pp28 (red dashed lines) are indicated. Original magnification, ×200. Bar = 20 μm.
Interestingly, infection caused changes in the subcellular localization of two other differentiation-related transcription factors, GATA3 and Hand1. In uninfected cells, these proteins are found in the nucleus; in infected cells, these proteins were found diffusely in the cytoplasm and at the periphery of the cytoplasmic virion assembly compartment (cVAC), with both proteins colocalizing with gB (not shown) and pp28 (Fig. 3) (nuclei [light blue dashed lines]; pp28 [red dashed lines]). Loss of nuclear localization of these transcription factors could further reduce the capacity of TBPCs to differentiate. In addition, immunoblot analyses revealed additional bands of GATA3 at late time points and increased levels of Hand1 (Fig. 2D), and these changes were similar between first- and second-trimester TBPCs (56).
Viral gene expression is required for the altered expression and localization of TBPC regulatory proteins.
Our results indicated that altered expression of TBPC regulatory proteins was more pronounced at 5 days p.i. (Fig. 2 and 3). We therefore asked whether late viral genes are required for these changes in infected cells. For these experiments, TBPCs were infected in the presence of ganciclovir, an inhibitor of the viral DNA polymerase. Ganciclovir-treated, infected cells showed punctate nuclear localization of UL112/113 proteins, early gene products, in contrast to accumulation in large nuclear inclusion bodies (49, 50) (Fig. 4A), which indicates the absence of viral DNA replication (50). Moreover, these cells did not express the late gene product pp28, further confirming the absence of viral DNA replication. Ganciclovir treatment was not able to prevent geminin upregulation (Fig. 2C and 4B) or SOX2 downregulation (data not shown) in infected cells, indicating that these events are mediated through IE or early genes. In contrast, ganciclovir treatment was able to block the changes in HMGA2 (completely) and GATA4 (partially) (Fig. 2C), also confirmed by immunoblot analysis (Fig. 4B), indicating that these changes require viral DNA replication. As expected, GATA3 and Hand1 failed to accumulate in discrete cytoplasmic compartments or in the cVAC. Instead, the bulk of the GATA3 signal remained nuclear, and Hand1 staining was diffuse in both the cytoplasm and nuclei (Fig. 4C). As cVAC formation is blocked when infection is carried out in the presence of inhibitors of viral DNA replication, it is likely that alterations in subcellular localization of regulatory proteins may be linked to cVAC formation. Note there was no effect of expression of these proteins in uninfected, ganciclovir-treated controls (Fig. 2C and 4B and C). Altogether, these results suggest that HCMV DNA replication is required for interference with TBPC self-renewal capacity and differentiation.
FIG 4.

Effect of ganciclovir treatment on expression of TBPC regulatory proteins. (A) TBPCs were infected with VR1814 with ganciclovir (GCV) (+) or without GCV (−), fixed at 5 days p.i., and immunostained for viral proteins UL112/113 and pp28. Original magnification, ×200. Bar = 20 μm. (B) Immunoblot of TBPC regulatory proteins (geminin, HMGA2, and GATA4) in uninfected controls and infected cells with GCV. (C) Immunofluorescence analysis of GATA3 and Hand1 in uninfected controls and infected cells with ganciclovir. Cellular proteins (green), HCMV IE1 (red), and DAPI (blue) are indicated. Colocalization of red and green signals (yellow) and of red, green, and blue signals (white) are indicated. Original magnification, ×200. Bar = 20 μm.
HCMV infection inhibits TBPC differentiation.
Having found that changes in differentiation factor expression and localization require viral DNA replication, we asked whether infection of TBPCs inhibits their differentiation, as we reported for primary CTBs (23). For these experiments, TBPCs were infected with HCMV strain VR1814 and cultured for 4 days for further differentiation assays (56). Control cells formed large, dense aggregates by 3 days after plating on Matrigel (range of mean diameters ± SE among all experiments was 328 ± 12 to 346 ± 10 μm) (Fig. 5A), indicating differentiation (39). In contrast, VR1814-infected cells formed significantly smaller and looser spheres with fewer cells (112 ± 3 to 168 ± 4 μm) (Fig. 5A). When infected TBPC monolayers were cultured with ganciclovir and then differentiated, larger aggregates formed (163 ± 3 to 256 ± 7 μm) (Fig. 5A). Although sphere diameters varied in infected cells in each experiment, they were significantly different between control and infected cells (P < 0.0001) and between infected and ganciclovir-treated infected cells (P < 0.0001) in all experiments. Minimal effects were observed in ganciclovir-treated, mock-infected controls compared to untreated controls (315 ± 13 to 322 ± 10 μm) (Fig. 5A). The histogram of sphere diameters for the infected cells was skewed toward smaller spheres (Fig. 5B), further confirming that infected TBPCs impair sphere formation in parallel with altered expression of differentiation factors.
FIG 5.

HCMV infection reduces aggregation and invasion of differentiating TBPCs and alters proteins that function during differentiation. (A) Sphere formation was inhibited by VR1814 infection. Original magnification, ×10. (B) Representative histograms of sphere diameters from 1 of 4 individual experiments. For each condition, 100 to 1,200 spheres were counted. The mean sphere diameters are indicated by the black arrows. The mean sphere diameter ± SE values were as follows: 328 ± 12 μm for the mock-infected control cells, 315 ± 13 μm for the ganciclovir-treated, mock-infected control cells (Control + GCV), 168 ± 4 μm for the VR1814-infected cells (VR1814 inf.), and 256 ± 7 μm for the infected, ganciclovir-treated cells (VR1814 inf. + GCV). P values for Mann-Whitney comparisons between infected and mock-infected control cells and between infected and ganciclovir-treated, infected cells were below 0.0001 in all experiments, indicating statistical significance. (C) Invasion assays with uninfected control (Cont.) cells alone and with ganciclovir (GCV) and infected cells alone and with ganciclovir. Results are presented as means (± SE) from two to five experiments done in triplicate. Significant differences in the percentage of cells crossing the filters for infected and mock-infected controls are indicated by asterisks as follows: *, P < 0.05; **, P < 0.001. A significant difference between the infected cells and infected, GCV-treated cells is indicated by the pound symbol (#) (P < 0.001). (D) Immunoblotting of key proteins in differentiated TBPCs. Control and infected cells with ganciclovir (4 days p.i.) were differentiated and cultured for 3 days. Cell lysates from differentiated control (Cont.) cells and differentiated infected TBPCs with or without GCV were immunoblotted with antibodies to PPARγ, hCG, and actin (loading control). hCG-H, hyperglycosylated hCG.
To measure the invasive capacity of TBPCs, mock-infected control and infected cells at 4 days p.i. were plated in differentiation medium on Matrigel-coated Transwell filters, and invasion assays were performed by counting the numbers of cells that traversed the pores. Significantly fewer infected TBPCs traversed the Transwell filters, whereas ganciclovir treatment partially restored invasiveness (Fig. 5C). Interestingly, even when invasion assays were performed at 2 days p.i., the invasiveness of infected cells was significantly impaired (∼60% reduction compared with control) (data not shown).
Activation of PPARγ inhibits CTB invasion (27, 72) and MMP-9 activity (73) and inversely controls the expression of transcripts for the α and β subunits of hCG and total hCG secretion (27, 74). hCG is mainly secreted from STBs and further stimulates STB formation. Having found impaired sphere formation and decreased invasion by infected TBPCs, we analyzed PPARγ and hCG expression in differentiated cells. Immunoblot analysis showed that PPARγ expression increased (Fig. 5D), in accord with decreased invasiveness (Fig. 5C). Moreover, hCG expression decreased, and a hyperglycosylated form of hCG was produced (Fig. 5D), suggesting reduced STB formation. Hyperglycosylated hCG has low bioactivity and increases in mid-trimester Down syndrome pregnancies, resulting in defects in trophoblast differentiation into STBs (75, 76). In ganciclovir-treated, infected cells, hCG levels were restored, but expression levels of PPARγ and hyperglycosylated hCG were comparable to those of infected cells (Fig. 5D). Since invasive capacity is inhibited early after infection and partially restored by ganciclovir treatment, even while PPARγ levels remain high, there may be PPARγ-independent effects on invasive capacity that depend on viral replication. Taken together, these results further confirm that infection impairs TBPC differentiation.
Anti-gB monoclonal antibody restores the capacity for self-renewal and differentiation.
We recently showed that a human neutralizing monoclonal antibody (MAb) to HCMV gB, but not a MAb to UL131A of the pentamer (gH/gL/UL128-131A), potently inhibits viral entry into TBPCs (99% reduction at 10 μg/ml) and enables the cells to differentiate and form spheres (56). HCMV hyperimmune globulin also exhibits neutralizing activity, although it is less potent (∼42% reduction at 10 μg/ml and ∼81% reduction at 100 μg/ml) (56); most neutralizing activity in hyperimmune globulin is attributed to anti-pentameric complex antibodies (77). To determine whether anti-gB treatment following infection restores TBPC functions, cells were infected, then treated with a human MAb to gB (TRL345) (20 μg/ml) 1 day after infection, and cultured for another 3 days. Immunoblot analysis showed that at 3 days after anti-gB treatment following infection, the HMGA2 level was comparable to the control level, and GATA4 expression was partially restored (Fig. 6A). In contrast, an isotype-matched negative-control MAb, Synagis, which lacks HCMV neutralizing activity and fails to block virus entry into TBPCs (56), had no effect on protein expression. These cells were then stimulated to differentiate by plating on Matrigel in differentiation medium. Anti-gB treatment markedly shifted the distribution of TBPC sphere diameters toward that characteristic of the uninfected state (Fig. 6B). Mean sphere diameters (mean diameter ± SE) ranged from 328 ± 12 to 414 ± 13 μm for control cells, 155 ± 2 to 257 ± 52 μm for infected cells, and 226 ± 6 to 292 ± 7 μm for anti-gB-treated cells, suggesting that anti-gB rescues the TBPC phenotype and the capacity for differentiation. In all these comparisons within experiments, differences were statistically significant. Negative-control antibody treatment had minimal effects on sphere formation (162 ± 2 to 242 ± 5 μm) compared to untreated, infected cells. In addition, cells receiving anti-gB treatment formed greater numbers of spheres (1.5- to 4-fold) than did control cells (data not shown), which could explain the increased numbers of villi observed in placentas from mothers treated with hyperimmune globulin (17).
FIG 6.
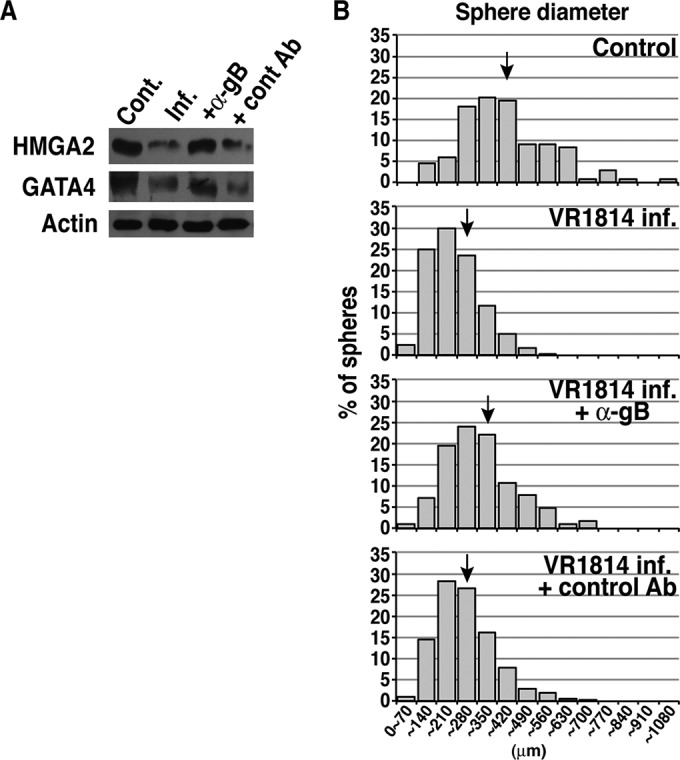
Anti-gB monoclonal antibody restores the capacity for self-renewal and differentiation. Anti-gB (TLR345) (20 μg/ml) was added at 1 day after infection, and cells were further cultured for 3 days, after which differentiation assays were performed. (A) Immunoblotting of key regulatory proteins prior to differentiation. Cell lysates from control (Cont.) or infected (Inf.) TBPCs with or without anti-gB (α-gB) or control antibody (cont Ab) (Synagis) were immunoblotted with antibodies to HMGA2, GATA4, and actin (loading control). (B) Representative histograms of sphere diameters from 1 of 4 independent experiments. For each condition, 100 to 1,200 spheres were counted. The mean sphere diameters are indicated by the black arrows. The mean sphere diameter ± SE values were as follows: 378 ± 13 μm for the mock-infected control cells, 257 ± 52 μm for VR1814-infected cells (VR1814 inf.), 292 ± 5 μm for infected cells with anti-gB treatment (VR1814 inf. + α-gB), and 242 ± 5 μm for infected cells with negative-control MAb (Synagis) treatment (VR1814 inf. + control Ab). The P values for Mann-Whitney comparisons between infected and mock-infected control cells and between infected and ganciclovir-treated, infected cells were below 0.0001 in all experiments, indicating statistical significance.
DISCUSSION
Placental development is central to the maintenance of a healthy pregnancy. The placenta functions at the interface between mother and fetus, preventing rejection of the fetal hemiallograft, enabling respiratory gas exchange, nutrient and IgG transport, and secretion of peptide and steroid hormones with autocrine functions. Our recent studies have suggested that infection could impair the early stages of trophoblast differentiation in cases of IUGR with primary maternal infection (18). Here, we provide the first evidence that cases of congenital HCMV infection involve viral infection of TBPCs in the chorion in utero, which could reduce the number and functional capability of TBPCs, as shown by the reduction in both cell number and GATA4 expression (Fig. 1). Experiments using TBPC lines showed that viral replication impairs the ability to self-renew, maintain an undifferentiated state, as indicated by reduced HMGA2 and SOX2 expression, and differentiate to form the mature trophoblast cell types, as indicated by impaired sphere formation, reduced hCG production, and increased PPARγ expression (Fig. 2 to 5). Reduced TBPC numbers in the chorion could correlate with decreased HMGA2 expression, which reduces stem cell numbers and self-renewal potential when it occurs in neural and hematopoietic stem cells (78, 79). We also found that infected TBPCs fail to migrate, as determined by wound healing assays in vitro (data not shown). Reduced migration and villous formation could explain why placentas from cases of symptomatic congenital infection have reduced compensatory development, in contrast to recipients of hyperimmune globulin, which suppresses viral replication (17, 33, 80, 81).
Currently, there are no approved therapeutics for congenital HCMV infection due to concerns over the toxicity and teratogenicity of available antiviral drugs (reviewed in references 82 and 83). A major reason for the lack of treatments is poor understanding of the molecular mechanisms that lead to placental injury and reduced developmental capacity. There are no appropriate animal models for studying congenital HCMV infection that recapitulate human disease and birth defects, and experiments on human tissues pose significant obstacles. In nonrandomized clinical studies, treatment of pregnant women with hyperimmune globulin was reported to improve pregnancy outcome in several studies (34, 35, 38, 84) with limited effect in one study (37). As novel therapeutics, human MAbs specific for epitopes on HCMV proteins have been developed using technological advancements in human B-cell cloning (51, 85, 86). TBPC phenotype and differentiation capacity were rescued by anti-gB MAb treatment (Fig. 6) and increased the numbers of spheres, indicating that TBPCs could model the increased villus numbers observed in placentas from hyperimmune globulin treatment (17). In addition, a human MAb to gH had potent HCMV neutralizing activity in TBPCs (T. Tabata and L. Pereira, unpublished data). TBPCs have self-renewal capacity and can be maintained indefinitely and serve as a consistent (genetic and proliferative) source for trophoblast differentiation. In addition, they allow studies of viral effects on trophoblast proliferation and early stage differentiation, which are required for continuous growth of the placenta throughout pregnancy. Thus, human TBPCs have potential utility for evaluation of the efficacies of novel antiviral antibodies in protecting and restoring placental development. Such studies have not been possible using primary CTBs and chorionic villous explants, models previously used, which do not support long periods of viral replication and can be used to study only the terminal steps of differentiation.
HCMV can establish latency in human ESCs (87), pluripotent cells that display unlimited self-renewal in an undifferentiated state. ESCs, derived from the inner cell mass, differentiate into all embryonic tissues, including the germ line, but have lost the ability to form trophoblast derivatives of the placenta. In contrast, progenitors are more lineage committed than stem cells and differentiate into their specific target cells in tissues. TBPCs (56) and human neural progenitor cells from fetal brains (88, 89) are fully permissive for HCMV replication. Human induced pluripotent stem cells (iPS) are not permissive for infection, whereas their derivatives, neural progenitor cells, are fully permissive for infection and alter expression of genes related to neural metabolism and differentiation (90). We found that HCMV reduces stem cell self-renewal factors HMGA2 and SOX2 (60, 69, 70) (Fig. 2). Infected TBPCs upregulate geminin, which coordinates proliferation and differentiation with multiple roles in development and homeostasis. Downregulation of geminin occurs around the time of cell cycle exit (91, 92) and is required to initiate differentiation into STBs and invasive CTBs, especially during the first and second trimesters (93). Infection of fibroblasts synchronized in G0 or S inhibits G1/S progression in several ways, including upregulation of geminin and downregulation of HMGA2 mRNA (94–97). Thus, changes in the levels of geminin and HMGA2 arrest the cell cycle (98, 99) and inhibit host cell DNA replication (100, 101), preventing TBPC differentiation. Since progenitors/stem cells have unique cell cycles and expression profiles of cell cycle regulators linked to the activities of factors that enable self-renewal (58, 102, 103), we are currently examining the expression of cell cycle regulators that could affect TBPC self-renewal.
For the most part, the inhibitory effects of HCMV infection on TBPC differentiation require late viral gene expression, which parallels findings for neural progenitor cells, where late viral gene products were found to inhibit differentiation (55, 88, 104). However, upregulation of geminin and downregulation of SOX2 occur before viral DNA replication. Overexpression of geminin prevents neuronal differentiation (67). SOX2 is required for mouse trophoblast formation (105), and expression levels are high in human ESCs during differentiation (106). Furthermore, overexpression of SOX2 enhances the differentiation of human mesenchymal stem cells and mouse ESCs (107, 108). Thus, inhibition of viral DNA replication only partially restored differentiation capacity (Fig. 5).
HCMV infection causes a dramatic reorganization of components of the secretory pathway, including the endoplasmic reticulum, Golgi complex, and trafficking vesicles, to facilitate cVAC formation and virion egress (109–113). In infected TBPCs, GATA3 and Hand1 colocalize with pp28 and accumulate in the cVAC (Fig. 3) (109–111), but ganciclovir treatment prevents the relocalization of GATA3 and Hand1 (Fig. 4C). Thus, cVAC formation could reroute GATA3 and Hand1 and prevent their localization to the nucleus, leading to loss of function of these proteins. Loss of GATA3 from the nuclei of infected TBPCs could further downregulate trophoblast-specific genes, including placental lactogen, secreted by STBs (61–63). With regard to GATA4, downregulation in human ESCs significantly inhibits differentiation, as shown by inhibition of embryoid body (sphere) formation (71). Hand1, a member of the basic helix-loop-helix (bHLH) transcription factor family, is essential for adequate differentiation of mouse trophoblasts (114), promotes cell cycle exit in mouse trophoblast stem cells (115), and is critical for trophoblast giant cell formation, the equivalent of the human invasive CTBs (65). Thus, altered localization of Hand1 in infected TBPCs could further impair differentiation/invasion.
Unlike fibroblasts, TBPCs and their derivatives, CTBs, cooperate as they differentiate, increasing cell contact during sphere formation and aggregation (23, 39). Although HCMV infected about 30% of CTBs in our previous reports, the levels of cell-matrix and cell-cell adhesion molecules were dramatically altered, impairing the ability to differentiate and invade the extracellular matrix (23, 116). In addition, increased secretion of both the IL-10 and cmvIL-10 reduce the expression and activity of MMP-9, further contributing to defective CTB invasiveness (24). A vivid illustration of the impact of infection on intercellular communication is the impairment of CTB outgrowth in villous explants, leading to formation of truncated, spindly cell columns, even though only a fraction of these CTBs were infected (32). Interestingly, we found that infection at a low MOI induced considerable changes in TBPC functions, indicated by decreased expression of HMGA2 and GATA4 and reduced size of spheres (Fig. 6). Ongoing studies should better define the intercellular signals and cell-matrix interactions involved in the sphere formation process.
Here we have shown that infection inhibits the developmental pathways for both CTBs and STBs. Infection of differentiated TBPCs increases expression of PPARγ, which correlates with impaired invasion (26, 27) and decreases hCG production, indicating defects in the formation of invasive CTBs and STBs, respectively (Fig. 5C and D). PPARγ is also involved in the pathophysiology of IUGR and preeclampsia (117). Strong transactivation of PPARγ during early pregnancy could reduce CTB invasiveness, one explanation for the pathophysiology of preeclampsia (72, 118–120). In addition, PPARγ is a regulator of fatty acid storage and glucose metabolism. The placental transfer of fatty acids during pregnancy is important for adequate fetal growth and development, and imbalances adversely affect development and could lead to IUGR (121). These observations suggest that the clinical use of thiazolidinediones (PPARγ inhibitors) to treat gestational diabetes mellitus (3 to 6% of pregnancies) could have unanticipated detrimental effects (122).
STBs play a major role in placental functions throughout pregnancy by synthesizing steroid and peptide hormones necessary for fetal development. Among the hormones produced by STBs is hCG, which stimulates progesterone production and promotes fusion and STB formation in an autocrine fashion (123). Disturbances in STB maturation and functions are found in numerous clinical conditions, including preeclampsia, IUGR, trisomy 21 (reviewed in reference 124), and congenital HCMV infection (18). Infected TBPCs reduce hCG expression and increase levels of hyperglycosylated hCG, with lower bioactivity (75, 76) (Fig. 5D) contributing to impaired development and functions of the mature cell types.
In conclusion, we have reported for the first time that HCMV infects TBPCs in the chorion in utero, and using continuously self-renewing TBPC lines, we have shown that viral replication interferes with essential progenitor cell properties, including self-renewal and lineage allocation, and could explain dysregulation of placental development. Furthermore, IUGR occurs in the absence of fetal infection, suggesting that negative outcomes can result from placental pathology alone (125). Our results shed light on novel molecular mechanisms that underlie IUGR and further define the constellation of developmental defects associated with congenital infection, thereby providing insights that could lead to more effective treatment.
ACKNOWLEDGMENTS
We thank members of Susan Fisher's laboratory for thoughtful discussions, Gabrielle Rizzuto for providing human tissue samples from the Department of Pathology collection, and Nancy Hills for advice on statistical analysis.
This work was supported by grants from the National Institutes of Health (RO1AI046657 and R56AI101130 to L.P., R21 HD061890 to T.T., and R44AI102396 to L.M.K.), a postdoctoral fellowship of the Deutsche Forschungsgemeinschaft DFG (ZY110/1-1 to M.Z.), and a scholarship award in Herpesvirus Research from the Japan Herpesvirus Infection Forum (M.T.). We also thank the Eunice Kennedy Shriver NICHD cooperative agreement as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research.
REFERENCES
- 1.Ross SA, Boppana SB. 2005. Congenital cytomegalovirus infection: outcome and diagnosis. Semin Pediatr Infect Dis 16:44–49. doi: 10.1053/j.spid.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Cannon MJ, Davis KF. 2005. Washing our hands of the congenital cytomegalovirus disease epidemic. BMC Public Health 5:70. doi: 10.1186/1471-2458-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagy A, Endreffy E, Streitman K, Pinter S, Pusztai R. 2004. Incidence and outcome of congenital cytomegalovirus infection in selected groups of preterm and full-term neonates under intensive care. In Vivo 18:819–823. [PubMed] [Google Scholar]
- 4.Panhani S, Heinonen KM. 1994. Screening for congenital cytomegalovirus infection among preterm infants born before the 34th gestational week in Finland. Scand J Infect Dis 26:375–378. doi: 10.3109/00365549409008607. [DOI] [PubMed] [Google Scholar]
- 5.Perlman JM, Argyle C. 1992. Lethal cytomegalovirus infection in preterm infants: clinical, radiological, and neuropathological findings. Ann Neurol 31:64–68. doi: 10.1002/ana.410310112. [DOI] [PubMed] [Google Scholar]
- 6.Turner KM, Lee HC, Boppana SB, Carlo WA, Randolph DA. 2014. Incidence and impact of CMV infection in very low birth weight infants. Pediatrics 133:e609–e615. doi: 10.1542/peds.2013-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto AY, Mussi-Pinhata MM, Cristina P, Pinto G, Moraes Figueiredo LT, Jorge SM. 2001. Congenital cytomegalovirus infection in preterm and full-term newborn infants from a population with a high seroprevalence rate. Pediatr Infect Dis J 20:188–192. doi: 10.1097/00006454-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 8.Iwasenko JM, Howard J, Arbuckle S, Graf N, Hall B, Craig ME, Rawlinson WD. 2011. Human cytomegalovirus infection is detected frequently in stillbirths and is associated with fetal thrombotic vasculopathy. J Infect Dis 203:1526–1533. doi: 10.1093/infdis/jir121. [DOI] [PubMed] [Google Scholar]
- 9.Syridou G, Spanakis N, Konstantinidou A, Piperaki ET, Kafetzis D, Patsouris E, Antsaklis A, Tsakris A. 2008. Detection of cytomegalovirus, parvovirus B19 and herpes simplex viruses in cases of intrauterine fetal death: association with pathological findings. J Med Virol 80:1776–1782. doi: 10.1002/jmv.21293. [DOI] [PubMed] [Google Scholar]
- 10.Bristow BN, O'Keefe KA, Shafir SC, Sorvillo FJ. 2011. Congenital cytomegalovirus mortality in the United States, 1990-2006. PLoS Negl Trop Dis 5:e1140. doi: 10.1371/journal.pntd.0001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barker DJ, Bull AR, Osmond C, Simmonds SJ. 1990. Fetal and placental size and risk of hypertension in adult life. BMJ 301:259–262. doi: 10.1136/bmj.301.6746.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barker DJ. 1999. Fetal origins of cardiovascular disease. Ann Med 31(Suppl 1):S3–S6. [PubMed] [Google Scholar]
- 13.Fowler KB, Stagno S, Pass RF. 2003. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA 289:1008–1011. doi: 10.1001/jama.289.8.1008. [DOI] [PubMed] [Google Scholar]
- 14.Ross DS, Victor M, Sumartojo E, Cannon MJ. 2008. Women's knowledge of congenital cytomegalovirus: results from the 2005 HealthStyles survey. J Womens Health (Larchmt) 17:849–858. doi: 10.1089/jwh.2007.0523. [DOI] [PubMed] [Google Scholar]
- 15.Griffiths PD. 2002. Strategies to prevent CMV infection in the neonate. Semin Neonatol 7:293–299. doi: 10.1016/S1084-2756(02)90123-5. [DOI] [PubMed] [Google Scholar]
- 16.Pereira L, Maidji E, McDonagh S, Genbacev O, Fisher S. 2003. Human cytomegalovirus transmission from the uterus to the placenta correlates with the presence of pathogenic bacteria and maternal immunity. J Virol 77:13301–13314. doi: 10.1128/JVI.77.24.13301-13314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maidji E, Nigro G, Tabata T, McDonagh S, Nozawa N, Shiboski S, Muci S, Anceschi MM, Aziz N, Adler SP, Pereira L. 2010. Antibody treatment promotes compensation for human cytomegalovirus-induced pathogenesis and a hypoxia-like condition in placentas with congenital infection. Am J Pathol 177:1298–1310. doi: 10.2353/ajpath.2010.091210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereira L, Petitt M, Fong A, Tsuge M, Tabata T, Fang-Hoover J, Maidji E, Zydek M, Zhou Y, Inoue N, Logahvi S, Pepkowitz S, Kuuvar L, Ogunyemi D. 2014. Intrauterine growth restriction caused by underlying congenital cytomegalovirus infection. J Infect Dis 209:1573–1584. doi: 10.1093/infdis/jiu019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabrielli L, Bonasoni MP, Santini D, Piccirilli G, Chiereghin A, Petrisli E, Dolcetti R, Guerra B, Piccioli M, Lanari M, Landini MP, Lazzarotto T. 2012. Congenital cytomegalovirus infection: patterns of fetal brain damage. Clin Microbiol Infect 18:E419–E427. doi: 10.1111/j.1469-0691.2012.03983.x. [DOI] [PubMed] [Google Scholar]
- 20.Demmler GJ. 1996. Congenital cytomegalovirus infection and disease. Adv Pediatr Infect Dis 11:135–162. [PubMed] [Google Scholar]
- 21.Grammatikopoulou I, Lambropoulou M, Chatzaki E, Deftereou TE, Lambropoulou V, Simopoulou M, Papadopoulos E, Galazios G, Dimitriou T, Petrou A, Papadopoulos N. 2012. Molecular diagnosis of CMV infection in fetal aborted tissues in the region of Thrace. Clin Exp Obstet Gynecol 39:96–102. [PubMed] [Google Scholar]
- 22.Tanaka K, Yamada H, Minami M, Kataoka S, Numazaki K, Minakami H, Tsutsumi H. 2006. Screening for vaginal shedding of cytomegalovirus in healthy pregnant women using real-time PCR: correlation of CMV in the vagina and adverse outcome of pregnancy. J Med Virol 78:757–759. doi: 10.1002/jmv.20619. [DOI] [PubMed] [Google Scholar]
- 23.Fisher S, Genbacev O, Maidji E, Pereira L. 2000. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J Virol 74:6808–6820. doi: 10.1128/JVI.74.15.6808-6820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto-Tabata T, McDonagh S, Chang HT, Fisher S, Pereira L. 2004. Human cytomegalovirus interleukin-10 downregulates matrix metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J Virol 78:2831–2840. doi: 10.1128/JVI.78.6.2831-2840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Librach CL, Werb Z, Fitzgerald ML, Chiu K, Corwin NM, Esteves RA, Grobelny D, Galardy R, Damsky CH, Fisher SJ. 1991. 92-kD type IV collagenase mediates invasion of human cytotrophoblasts. J Cell Biol 113:437–449. doi: 10.1083/jcb.113.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fournier T, Guibourdenche J, Handschuh K, Tsatsaris V, Rauwel B, Davrinche C, Evain-Brion D. 2011. PPARgamma and human trophoblast differentiation. J Reprod Immunol 90:41–49. doi: 10.1016/j.jri.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Rauwel B, Mariame B, Martin H, Nielsen R, Allart S, Pipy B, Mandrup S, Devignes MD, Evain-Brion D, Fournier T, Davrinche C. 2010. Activation of peroxisome proliferator-activated receptor gamma by human cytomegalovirus for de novo replication impairs migration and invasiveness of cytotrophoblasts from early placentas. J Virol 84:2946–2954. doi: 10.1128/JVI.01779-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan G, Hemmings DG, Yurochko AD, Guilbert LJ. 2002. Human cytomegalovirus-caused damage to placental trophoblasts mediated by immediate-early gene-induced tumor necrosis factor-alpha. Am J Pathol 161:1371–1381. doi: 10.1016/S0002-9440(10)64413-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maidji E, Percivalle E, Gerna G, Fisher S, Pereira L. 2002. Transmission of human cytomegalovirus from infected uterine microvascular endothelial cells to differentiating/invasive placental cytotrophoblasts. Virology 304:53–69. doi: 10.1006/viro.2002.1661. [DOI] [PubMed] [Google Scholar]
- 30.Angelova M, Zwezdaryk K, Ferris M, Shan B, Morris CA, Sullivan DE. 2012. Human cytomegalovirus infection dysregulates the canonical Wnt/beta-catenin signaling pathway. PLoS Pathog 8:e1002959. doi: 10.1371/journal.ppat.1002959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maidji E, Genbacev O, Chang HT, Pereira L. 2007. Developmental regulation of human cytomegalovirus receptors in cytotrophoblasts correlates with distinct replication sites in the placenta. J Virol 81:4701–4712. doi: 10.1128/JVI.02748-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabata T, Petitt M, Fang-Hoover J, Rivera J, Nozawa N, Shiboski S, Inoue N, Pereira L. 2012. Cytomegalovirus impairs cytotrophoblast-induced lymphangiogenesis and vascular remodeling in an in vivo human placentation model. Am J Pathol 181:1540–1559. doi: 10.1016/j.ajpath.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varani S, Frascaroli G, Landini MP, Soderberg-Naucler C. 2009. Human cytomegalovirus targets different subsets of antigen-presenting cells with pathological consequences for host immunity: implications for immunosuppression, chronic inflammation and autoimmunity. Rev Med Virol 19:131–145. doi: 10.1002/rmv.609. [DOI] [PubMed] [Google Scholar]
- 34.Nigro G, Adler SP, La Torre R, Best AM. 2005. Passive immunization during pregnancy for congenital cytomegalovirus infection. N Engl J Med 353:1350–1362. doi: 10.1056/NEJMoa043337. [DOI] [PubMed] [Google Scholar]
- 35.Nigro G, Adler SP, Parruti G, Anceschi MM, Coclite E, Pezone I, Di Renzo GC. 2012. Immunoglobulin therapy of fetal cytomegalovirus infection occurring in the first half of pregnancy–a case-control study of the outcome in children. J Infect Dis 205:215–227. doi: 10.1093/infdis/jir718. [DOI] [PubMed] [Google Scholar]
- 36.Visentin S, Manara R, Milanese L, Da Roit A, Forner G, Salviato E, Citton V, Magno FM, Orzan E, Morando C, Cusinato R, Mengoli C, Palu G, Ermani M, Rinaldi R, Cosmi E, Gussetti N. 2012. Early primary cytomegalovirus infection in pregnancy: maternal hyperimmunoglobulin therapy improves outcomes among infants at 1 year of age. Clin Infect Dis 55:497–503. doi: 10.1093/cid/cis423. [DOI] [PubMed] [Google Scholar]
- 37.Revello MG, Lazzarotto T, Guerra B, Spinillo A, Ferrazzi E, Kustermann A, Guaschino S, Vergani P, Todros T, Frusca T, Arossa A, Furione M, Rognoni V, Rizzo N, Gabrielli L, Klersy C, Gerna G. 2014. A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N Engl J Med 370:1316–1326. doi: 10.1056/NEJMoa1310214. [DOI] [PubMed] [Google Scholar]
- 38.Buxmann H, Stackelberg OM, Schlosser RL, Enders G, Gonser M, Meyer-Wittkopf M, Hamprecht K, Enders M. 2012. Use of cytomegalovirus hyperimmunoglobulin for prevention of congenital cytomegalovirus disease: a retrospective analysis. J Perinat Med 40:439–446. doi: 10.1515/jpm-2011-0257. [DOI] [PubMed] [Google Scholar]
- 39.Genbacev O, Donne M, Kapidzic M, Gormley M, Lamb J, Gilmore J, Larocque N, Goldfien G, Zdravkovic T, McMaster MT, Fisher SJ. 2011. Establishment of human trophoblast progenitor cell lines from the chorion. Stem Cells 29:1427–1436. doi: 10.1002/stem.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Firan M, Bawdon R, Radu C, Ober RJ, Eaken D, Antohe F, Ghetie V, Ward ES. 2001. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int Immunol 13:993–1002. doi: 10.1093/intimm/13.8.993. [DOI] [PubMed] [Google Scholar]
- 41.Story CM, Mikulska JE, Simister NE. 1994. A major histocompatibility complex class I-like Fc receptor cloned from human placenta: possible role in transfer of immunoglobulin G from mother to fetus. J Exp Med 180:2377–2381. doi: 10.1084/jem.180.6.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Damsky CH, Fitzgerald ML, Fisher SJ. 1992. Distribution patterns of extracellular matrix components and adhesion receptors are intricately modulated during first trimester cytotrophoblast differentiation along the invasive pathway, in vivo. J Clin Invest 89:210–222. doi: 10.1172/JCI115565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Revello MG, Baldanti F, Percivalle E, Sarasini A, De-Giuli L, Genini E, Lilleri D, Labo N, Gerna G. 2001. In vitro selection of human cytomegalovirus variants unable to transfer virus and virus products from infected cells to polymorphonuclear leukocytes and to grow in endothelial cells. J Gen Virol 82:1429–1438. [DOI] [PubMed] [Google Scholar]
- 44.Frascaroli G, Sinzger C. 2014. Distinct properties of human cytomegalovirus strains and the appropriate choice of strains for particular studies. Methods Mol Biol 1119:29–46. doi: 10.1007/978-1-62703-788-4_3. [DOI] [PubMed] [Google Scholar]
- 45.Patrone M, Secchi M, Fiorina L, Ierardi M, Milanesi G, Gallina A. 2005. Human cytomegalovirus UL130 protein promotes endothelial cell infection through a producer cell modification of the virion. J Virol 79:8361–8373. doi: 10.1128/JVI.79.13.8361-8373.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCarthy KM, Yoong Y, Simister NE. 2000. Bidirectional transcytosis of IgG by the rat neonatal Fc receptor expressed in a rat kidney cell line: a system to study protein transport across epithelia. J Cell Sci 113:1277–1285. [DOI] [PubMed] [Google Scholar]
- 47.Pereira L, Hoffman M, Tatsuno M, Dondero D. 1984. Polymorphism of human cytomegalovirus glycoproteins characterized by monoclonal antibodies. Virology 139:73–86. doi: 10.1016/0042-6822(84)90331-3. [DOI] [PubMed] [Google Scholar]
- 48.Dondero DV, Pereira L. 1990. Monoclonal antibody production. American Public Health Association, Washington, DC. [Google Scholar]
- 49.Yamamoto T, Suzuki S, Radsak K, Hirai K. 1998. The UL112/113 gene products of human cytomegalovirus which colocalize with viral DNA in infected cell nuclei are related to efficient viral DNA replication. Virus Res 56:107–114. doi: 10.1016/S0168-1702(98)00032-X. [DOI] [PubMed] [Google Scholar]
- 50.Iwayama S, Yamamoto T, Furuya T, Kobayashi R, Ikuta K, Hirai K. 1994. Intracellular localization and DNA-binding activity of a class of viral early phosphoproteins in human fibroblasts infected with human cytomegalovirus (Towne strain). J Gen Virol 75:3309–3318. doi: 10.1099/0022-1317-75-12-3309. [DOI] [PubMed] [Google Scholar]
- 51.McCutcheon KM, Gray J, Chen NY, Liu K, Park M, Ellsworth S, Tripp RA, Tompkins SM, Johnson SK, Samet S, Pereira L, Kauvar LM. 2014. Multiplexed screening of natural humoral immunity identifies antibodies at fine specificity for complex and dynamic viral targets. mAbs 6:460–473. doi: 10.4161/mabs.27760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The IMpact-RSV Study Group.1998. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 102:531–537. doi: 10.1542/peds.102.3.531. [DOI] [PubMed] [Google Scholar]
- 53.Lilley BN, Ploegh HL, Tirabassi RS. 2001. Human cytomegalovirus open reading frame TRL11/IRL11 encodes an immunoglobulin G Fc-binding protein. J Virol 75:11218–11221. doi: 10.1128/JVI.75.22.11218-11221.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDonagh S, Maidji E, Chang HT, Pereira L. 2006. Patterns of human cytomegalovirus infection in term placentas: a preliminary analysis. J Clin Virol 35:210–215. doi: 10.1016/j.jcv.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 55.Luo MH, Hannemann H, Kulkarni AS, Schwartz PH, O'Dowd JM, Fortunato EA. 2010. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J Virol 84:3528–3541. doi: 10.1128/JVI.02161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zydek M, Petitt M, Fang-Hoover J, Adler B, Kauvar LM, Pereira L, Tabata T. 2014. HCMV infection of human trophoblast progenitor cells of the placenta is neutralized by a human monoclonal antibody to glycoprotein B and not by antibodies to the pentamer complex. Viruses 6:1346–1364. doi: 10.3390/v6031346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weisblum Y, Panet A, Zakay-Rones Z, Haimov-Kochman R, Goldman-Wohl D, Ariel I, Falk H, Natanson-Yaron S, Goldberg MD, Gilad R, Lurain NS, Greenfield C, Yagel S, Wolf DG. 2011. Modeling of human cytomegalovirus maternal-fetal transmission in a novel decidual organ culture. J Virol 85:13204–13213. doi: 10.1128/JVI.05749-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang VS, Carter SA, Hyland SJ, Tachibana-Konwalski K, Laskey RA, Gonzalez MA. 2011. Geminin escapes degradation in G1 of mouse pluripotent cells and mediates the expression of Oct4, Sox2, and Nanog. Curr Biol 21:692–699. doi: 10.1016/j.cub.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tabrizi GA, Bose K, Reimann Y, Kessel M. 2013. Geminin is required for the maintenance of pluripotency. PLoS One 8:e73826. doi: 10.1371/journal.pone.0073826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfannkuche K, Summer H, Li O, Hescheler J, Droge P. 2009. The high mobility group protein HMGA2: a co-regulator of chromatin structure and pluripotency in stem cells? Stem Cell Rev 5:224–230. doi: 10.1007/s12015-009-9078-9. [DOI] [PubMed] [Google Scholar]
- 61.Ray S, Dutta D, Rumi MA, Kent LN, Soares MJ, Paul S. 2009. Context-dependent function of regulatory elements and a switch in chromatin occupancy between GATA3 and GATA2 regulate Gata2 transcription during trophoblast differentiation. J Biol Chem 284:4978–4988. doi: 10.1074/jbc.M807329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma GT, Roth ME, Groskopf JC, Tsai FY, Orkin SH, Grosveld F, Engel JD, Linzer DI. 1997. GATA-2 and GATA-3 regulate trophoblast-specific gene expression in vivo. Development 124:907–914. [DOI] [PubMed] [Google Scholar]
- 63.Ng YK, George KM, Engel JD, Linzer DI. 1994. GATA factor activity is required for the trophoblast-specific transcriptional regulation of the mouse placental lactogen I gene. Development 120:3257–3266. [DOI] [PubMed] [Google Scholar]
- 64.Fujikura J, Yamato E, Yonemura S, Hosoda K, Masui S, Nakao K, Miyazaki Ji J, Niwa H. 2002. Differentiation of embryonic stem cells is induced by GATA factors. Genes Dev 16:784–789. doi: 10.1101/gad.968802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maltepe E, Bakardjiev AI, Fisher SJ. 2010. The placenta: transcriptional, epigenetic, and physiological integration during development. J Clin Invest 120:1016–1025. doi: 10.1172/JCI41211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seo S, Herr A, Lim JW, Richardson GA, Richardson H, Kroll KL. 2005. Geminin regulates neuronal differentiation by antagonizing Brg1 activity. Genes Dev 19:1723–1734. doi: 10.1101/gad.1319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seo S, Richardson GA, Kroll KL. 2005. The SWI/SNF chromatin remodeling protein Brg1 is required for vertebrate neurogenesis and mediates transactivation of Ngn and NeuroD. Development 132:105–115. doi: 10.1242/dev.01548. [DOI] [PubMed] [Google Scholar]
- 68.Hough SR, Clements I, Welch PJ, Wiederholt KA. 2006. Differentiation of mouse embryonic stem cells after RNA interference-mediated silencing of OCT4 and Nanog. Stem Cells 24:1467–1475. doi: 10.1634/stemcells.2005-0475. [DOI] [PubMed] [Google Scholar]
- 69.Adachi K, Suemori H, Yasuda SY, Nakatsuji N, Kawase E. 2010. Role of SOX2 in maintaining pluripotency of human embryonic stem cells. Genes Cells 15:455–470. doi: 10.1111/j.1365-2443.2010.01400.x. [DOI] [PubMed] [Google Scholar]
- 70.Fong H, Hohenstein KA, Donovan PJ. 2008. Regulation of self-renewal and pluripotency by Sox2 in human embryonic stem cells. Stem Cells 26:1931–1938. doi: 10.1634/stemcells.2007-1002. [DOI] [PubMed] [Google Scholar]
- 71.Huang HN, Chen SY, Hwang SM, Yu CC, Su MW, Mai W, Wang HW, Cheng WC, Schuyler SC, Ma N, Lu FL, Lu J. 2014. miR-200c and GATA binding protein 4 regulate human embryonic stem cell renewal and differentiation. Stem Cell Res 12:338–353. doi: 10.1016/j.scr.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 72.Fournier T, Therond P, Handschuh K, Tsatsaris V, Evain-Brion D. 2008. PPARgamma and early human placental development. Curr Med Chem 15:3011–3024. doi: 10.2174/092986708786848677. [DOI] [PubMed] [Google Scholar]
- 73.Hetzel M, Walcher D, Grub M, Bach H, Hombach V, Marx N. 2003. Inhibition of MMP-9 expression by PPARgamma activators in human bronchial epithelial cells. Thorax 58:778–783. doi: 10.1136/thorax.58.9.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Handschuh K, Guibourdenche J, Tsatsaris V, Guesnon M, Laurendeau I, Evain-Brion D, Fournier T. 2007. Human chorionic gonadotropin produced by the invasive trophoblast but not the villous trophoblast promotes cell invasion and is down-regulated by peroxisome proliferator-activated receptor-gamma. Endocrinology 148:5011–5019. doi: 10.1210/en.2007-0286. [DOI] [PubMed] [Google Scholar]
- 75.Cole LA, Khanlian SA. 2007. Hyperglycosylated hCG: a variant with separate biological functions to regular hCG. Mol Cell Endocrinol 260-262:228–236. doi: 10.1016/j.mce.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 76.Frendo JL, Guibourdenche J, Pidoux G, Vidaud M, Luton D, Giovangrandi Y, Porquet D, Muller F, Evain-Brion D. 2004. Trophoblast production of a weakly bioactive human chorionic gonadotropin in trisomy 21-affected pregnancy. J Clin Endocrinol Metab 89:727–732. doi: 10.1210/jc.2009-030668.. [DOI] [PubMed] [Google Scholar]
- 77.Fouts AE, Chan P, Stephan JP, Vandlen R, Feierbach B. 2012. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J Virol 86:7444–7447. doi: 10.1128/JVI.00467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nishino J, Kim I, Chada K, Morrison SJ. 2008. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf expression. Cell 135:227–239. doi: 10.1016/j.cell.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Copley MR, Babovic S, Benz C, Knapp DJ, Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, Mader H, Kuchenbauer F, Humphries RK, Eaves CJ. 2013. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol 15:916–925. doi: 10.1038/ncb2783. [DOI] [PubMed] [Google Scholar]
- 80.La Torre R, Nigro G, Mazzocco M, Best AM, Adler SP. 2006. Placental enlargement in women with primary maternal cytomegalovirus infection is associated with fetal and neonatal disease. Clin Infect Dis 43:994–1000. doi: 10.1086/507634. [DOI] [PubMed] [Google Scholar]
- 81.Adler SP, Nigro G, Pereira L. 2007. Recent advances in the prevention and treatment of congenital cytomegalovirus infections. Semin Perinatol 31:10–18. doi: 10.1053/j.semperi.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 82.Mercorelli B, Sinigalia E, Loregian A, Palu G. 2008. Human cytomegalovirus DNA replication: antiviral targets and drugs. Rev Med Virol 18:177–210. doi: 10.1002/rmv.558. [DOI] [PubMed] [Google Scholar]
- 83.Benoist G, Leruez-Ville M, Magny JF, Jacquemard F, Salomon LJ, Ville Y. 2013. Management of pregnancies with confirmed cytomegalovirus fetal infection. Fetal Diagn Ther 33:203–214. doi: 10.1159/000342752. [DOI] [PubMed] [Google Scholar]
- 84.Nigro G, La Torre R, Pentimalli H, Taverna P, Lituania M, de Tejada BM, Adler SP. 2008. Regression of fetal cerebral abnormalities by primary cytomegalovirus infection following hyperimmunoglobulin therapy. Prenatal Diagn 28:512–517. doi: 10.1002/pd.2013. [DOI] [PubMed] [Google Scholar]
- 85.Macagno A, Bernasconi NL, Vanzetta F, Dander E, Sarasini A, Revello MG, Gerna G, Sallusto F, Lanzavecchia A. 2010. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol 84:1005–1013. doi: 10.1128/JVI.01809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kauvar LM, Liu K, Park M, DeChene N, Stephenson R, Tenorio E, Ellsworth SL, Tabata T, Petitt M, Tsuge M, Fang-Hoover J, Adler SP, Cui X, McVoy MA, Pereira L. 2015. A high-affinity native human antibody neutralizes human cytomegalovirus infection of diverse cell types. Antimicrob Agents Chemother 59:1558–1568. doi: 10.1128/AAC.04295-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Penkert RR, Kalejta RF. 2013. Human embryonic stem cell lines model experimental human cytomegalovirus latency. mBio 4(3):e00298-13. doi: 10.1128/mBio.00298-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Odeberg J, Wolmer N, Falci S, Westgren M, Seiger A, Soderberg-Naucler C. 2006. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol 80:8929–8939. doi: 10.1128/JVI.00676-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luo MH, Schwartz PH, Fortunato EA. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol 82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.D'Aiuto L, Di Maio R, Heath B, Raimondi G, Milosevic J, Watson AM, Bamne M, Parks WT, Yang L, Lin B, Miki T, Mich-Basso JD, Arav-Boger R, Sibille E, Sabunciyan S, Yolken R, Nimgaonkar V. 2012. Human induced pluripotent stem cell-derived models to investigate human cytomegalovirus infection in neural cells. PLoS One 7:e49700. doi: 10.1371/journal.pone.0049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seo S, Kroll KL. 2006. Geminin's double life: chromatin connections that regulate transcription at the transition from proliferation to differentiation. Cell Cycle 5:374–379. doi: 10.4161/cc.5.4.2438. [DOI] [PubMed] [Google Scholar]
- 92.Xouri G, Lygerou Z, Nishitani H, Pachnis V, Nurse P, Taraviras S. 2004. Cdt1 and geminin are down-regulated upon cell cycle exit and are over-expressed in cancer-derived cell lines. Eur J Biochem 271:3368–3378. doi: 10.1111/j.1432-1033.2004.04271.x. [DOI] [PubMed] [Google Scholar]
- 93.Genbacev O, McMaster MT, Fisher SJ. 2000. A repertoire of cell cycle regulators whose expression is coordinated with human cytotrophoblast differentiation. Am J Pathol 157:1337–1351. doi: 10.1016/S0002-9440(10)64648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Biswas N, Sanchez V, Spector DH. 2003. Human cytomegalovirus infection leads to accumulation of geminin and inhibition of the licensing of cellular DNA replication. J Virol 77:2369–2376. doi: 10.1128/JVI.77.4.2369-2376.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fortunato EA, Sanchez V, Yen JY, Spector DH. 2002. Infection of cells with human cytomegalovirus during S phase results in a blockade to immediate-early gene expression that can be overcome by inhibition of the proteasome. J Virol 76:5369–5379. doi: 10.1128/JVI.76.11.5369-5379.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Salvant BS, Fortunato EA, Spector DH. 1998. Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol 72:3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shlapobersky M, Sanders R, Clark C, Spector DH. 2006. Repression of HMGA2 gene expression by human cytomegalovirus involves the IE2 86-kilodalton protein and is necessary for efficient viral replication and inhibition of cyclin A transcription. J Virol 80:9951–9961. doi: 10.1128/JVI.01300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. 2000. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 290:2309–2312. doi: 10.1126/science.290.5500.2309. [DOI] [PubMed] [Google Scholar]
- 99.Tada S, Li A, Maiorano D, Mechali M, Blow JJ. 2001. Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat Cell Biol 3:107–113. doi: 10.1038/35055000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hodgson B, Li A, Tada S, Blow JJ. 2002. Geminin becomes activated as an inhibitor of Cdt1/RLF-B following nuclear import. Curr Biol 12:678–683. doi: 10.1016/S0960-9822(02)00778-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee C, Hong B, Choi JM, Kim Y, Watanabe S, Ishimi Y, Enomoto T, Tada S, Cho Y. 2004. Structural basis for inhibition of the replication licensing factor Cdt1 by geminin. Nature 430:913–917. doi: 10.1038/nature02813. [DOI] [PubMed] [Google Scholar]
- 102.Boheler KR. 2009. Stem cell pluripotency: a cellular trait that depends on transcription factors, chromatin state and a checkpoint deficient cell cycle. J Cell Physiol 221:10–17. doi: 10.1002/jcp.21866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mantel C, Guo Y, Lee MR, Kim MK, Han MK, Shibayama H, Fukuda S, Yoder MC, Pelus LM, Kim KS, Broxmeyer HE. 2007. Checkpoint-apoptosis uncoupling in human and mouse embryonic stem cells: a source of karyotpic instability. Blood 109:4518–4527. doi: 10.1182/blood-2006-10-054247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Odeberg J, Wolmer N, Falci S, Westgren M, Sundtrom E, Seiger A, Soderberg-Naucler C. 2007. Late human cytomegalovirus (HCMV) proteins inhibit differentiation of human neural precursor cells into astrocytes. J Neurosci Res 85:583–593. doi: 10.1002/jnr.21144. [DOI] [PubMed] [Google Scholar]
- 105.Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. 2003. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kallas A, Pook M, Trei A, Maimets T. 2014. SOX2 is regulated differently from NANOG and OCT4 in human embryonic stem cells during early differentiation initiated with sodium butyrate. Stem Cells Int 2014:298163. doi: 10.1155/2014/298163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Han SM, Han SH, Coh YR, Jang G, Chan Ra J, Kang SK, Lee HW, Youn HY. 2014. Enhanced proliferation and differentiation of Oct4- and Sox2-overexpressing human adipose tissue mesenchymal stem cells. Exp Mol Med 46:e101. doi: 10.1038/emm.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kopp JL, Ormsbee BD, Desler M, Rizzino A. 2008. Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells 26:903–911. doi: 10.1634/stemcells.2007-0951. [DOI] [PubMed] [Google Scholar]
- 109.Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol 85:5864–5879. doi: 10.1128/JVI.00155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol 74:975–986. doi: 10.1128/JVI.74.2.975-986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J Virol 83:11421–11428. doi: 10.1128/JVI.00762-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alwine JC. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog 8:e1002878. doi: 10.1371/journal.ppat.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Scott IC, Anson-Cartwright L, Riley P, Reda D, Cross JC. 2000. The HAND1 basic helix-loop-helix transcription factor regulates trophoblast differentiation via multiple mechanisms. Mol Cell Biol 20:530–541. doi: 10.1128/MCB.20.2.530-541.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hughes M, Dobric N, Scott IC, Su L, Starovic M, St-Pierre B, Egan SE, Kingdom JC, Cross JC. 2004. The Hand1, Stra13 and Gcm1 transcription factors override FGF signaling to promote terminal differentiation of trophoblast stem cells. Dev Biol 271:26–37. doi: 10.1016/j.ydbio.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 116.Tabata T, McDonagh S, Kawakatsu H, Pereira L. 2007. Cytotrophoblasts infected with a pathogenic human cytomegalovirus strain dysregulate cell-matrix and cell-cell adhesion molecules: a quantitative analysis. Placenta 28:527–537. doi: 10.1016/j.placenta.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 117.Holdsworth-Carson SJ, Lim R, Mitton A, Whitehead C, Rice GE, Permezel M, Lappas M. 2010. Peroxisome proliferator-activated receptors are altered in pathologies of the human placenta: gestational diabetes mellitus, intrauterine growth restriction and preeclampsia. Placenta 31:222–229. doi: 10.1016/j.placenta.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 118.Fournier T, Pavan L, Tarrade A, Schoonjans K, Auwerx J, Rochette-Egly C, Evain-Brion D. 2002. The role of PPAR-gamma/RXR-alpha heterodimers in the regulation of human trophoblast invasion. Ann N Y Acad Sci 973:26–30. doi: 10.1111/j.1749-6632.2002.tb04601.x. [DOI] [PubMed] [Google Scholar]
- 119.Schild RL, Schaiff WT, Carlson MG, Cronbach EJ, Nelson DM, Sadovsky Y. 2002. The activity of PPARgamma in primary human trophoblasts is enhanced by oxidized lipids. J Clin Endocrinol Metab 87:1105–1110. doi: 10.1210/jcem.87.3.8284. [DOI] [PubMed] [Google Scholar]
- 120.Fournier T, Handschuh K, Tsatsaris V, Guibourdenche J, Evain-Brion D. 2008. Role of nuclear receptors and their ligands in human trophoblast invasion. J Reprod Immunol 77:161–170. doi: 10.1016/j.jri.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 121.Gil-Sanchez A, Demmelmair H, Parrilla JJ, Koletzko B, Larque E. 2011. Mechanisms involved in the selective transfer of long chain polyunsaturated fatty acids to the fetus. Front Genet 2:57. doi: 10.3389/fgene.2011.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knox CA, Delaney JA, Winterstein AG. 2014. Anti-diabetic drug utilization of pregnant diabetic women in US managed care. BMC Pregnancy Childbirth 14:28. doi: 10.1186/1471-2393-14-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cole LA. 2010. Biological functions of hCG and hCG-related molecules. Reprod Biol Endocrinol 8:102. doi: 10.1186/1477-7827-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pidoux G, Gerbaud P, Cocquebert M, Segond N, Badet J, Fournier T, Guibourdenche J, Evain-Brion D. 2012. Human trophoblast fusion and differentiation: lessons from trisomy 21 placenta. Placenta 33(Suppl):S81–S86. doi: 10.1016/j.placenta.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 125.Benirschke K, Kaufmann P. 2000. Pathology of the human placenta, 4th ed Springer, New York, NY. [Google Scholar]