

ABSTRACT
Orf virus (ORFV) OV20.0L is an ortholog of vaccinia virus (VACV) gene E3L. The function of VACV E3 protein as a virulence factor is well studied, but OV20.0 has received less attention. Here we show that like VACV E3L, OV20.0L encodes two proteins, a full-length protein and a shorter form (sh20). The shorter sh20 is an N-terminally truncated OV20.0 isoform generated when a downstream AUG codon is used for initiating translation. These isoforms differed in cellular localization, with full-length OV20.0 and sh20 found throughout the cell and predominantly in the cytoplasm, respectively. Nonetheless, both OV20.0 isoforms were able to bind double-stranded RNA (dsRNA)-activated protein kinase (PKR) and dsRNA. Moreover, both isoforms strongly inhibited PKR activation as shown by decreased phosphorylation of the translation initiation factor eIF2α subunit and protection of Sindbis virus infection against the activity of interferon (IFN). In spite of this apparent conservation of function in vitro, a recombinant ORFV that was able to express only the sh20 isoform was attenuated in a mouse model.
IMPORTANCE The OV20.0 protein of orf virus (ORFV) has two isoforms and contributes to virulence, but the roles of the two forms are not known. This study shows that the shorter isoform (sh20) arises due to use of a downstream initiation codon and is amino-terminally truncated. The sh20 form also differs in expression kinetics and cellular localization from full-length OV20.0. Similar to the full-length isoform, sh20 is able to bind dsRNA and PKR, inactivate PKR, and thus act as an antagonist of the interferon response in vitro. In vivo, however, wild-type OV20.0 could not be replaced with sh20 alone without a loss of virulence, suggesting that the functions of the isoforms are not simply redundant.
INTRODUCTION
Orf virus (ORFV), a member of the Parapoxvirus genus and the Poxviridae family, is the causative agent of contagious ecthyma in sheep, goats, and other ruminants. The disease is characterized by the development of pustular lesions around the nostrils and mouth with a high incidence rate and a low mortality rate in healthy adult animals. In contrast, infection in immunosuppressed animals or in lambs may be fatal (1). ORFV is also of concern as a source of zoonotic infection because it can cause cutaneous lesions in humans in contact with infected animals. Persistent infection with ORFV can be observed in goats and sheep, and while the severity of lesions is reduced compared with that seen in primary infection, this persistence suggests that the virus is able to evade host immunity (2–4). In line with this observation, ORFV has been shown to encode several proteins that modulate the host response to infection. These include viral homologues of ovine cytokines, such as vascular endothelial growth factor, interleukin-10 (IL-10), and a granulocyte-macrophage colony-stimulating factor (GM-CSF)-inhibiting protein, as well as an apoptosis inhibitor (5–7). ORFV also antagonizes interferon (IFN) signaling, and this is done by the product of the gene OV20.0L. As a result of the concerted expression of these genes, ORFV is able to promote its replication even while under host immune attack (8).
The double-stranded RNA (dsRNA)-dependent protein kinase (PKR) pathway is an important mechanism in innate sensing and response to virus infection in mammalian cells (9). PKR itself consists of two N-terminal dsRNA binding domains (DRBD) and a C-terminal kinase catalytic domain. The protein is activated by binding of dsRNA to the DRBD, so that dimerization followed by stimulation of transautophosphorylation of PKR occurs (10–12). PKR activation leads to phosphorylation of a number of different substrates that have diverse antiviral effects. These include activation of the transcription factors NF-κB and interferon regulatory factor 3 (IRF3), known to promote proinflammatory responses (13). In addition, caspase activation and the induction of apoptosis have also been observed when PKR is activated by certain stimuli (14). Finally, activation of PKR contributes to an inhibition of protein synthesis, which occurs via phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α), which plays a vital role in translational control in viral infection (15). The E3 protein (here abbreviated as VVE3) of vaccinia virus (VACV) competes with PKR for dsRNA binding and interacts directly with PKR itself to block activation of this pathway (5, 16). These activities result in the maintenance of protein synthesis and an inhibition of apoptosis, which favor viral replication and survival. A similar strategy has also been described for other viral proteins, for instance, the reovirus σ3 protein, the influenza virus NS1 protein, and the human cytomegalovirus TRS1 protein, which can circumvent the shutoff of protein synthesis due to the phosphorylation of the elF-2α translation initiation factor mediated by activated PKR (17–20).
VVE3 is encoded by the E3L gene, which has orthologs in many chordopoxviruses, including members of the Orthopoxvirus, Leporipoxvirus, Capripoxvirus, Suipoxvirus, Yatapoxvirus, and Parapoxvirus genera, including OV20.0L of ORFV (21, 22). The VACV E3L gene encodes two isoforms of VVE3 with molecular masses of 25 and 20 kDa that arise due to leaky scanning of the ribosome leading to the use of two alternate initiation codons (5). Current knowledge of E3 structure and function is based largely on the longest form of VVE3, which comprises approximately190 amino acids and is a crucial factor in VACV host range and virulence (21, 22). This VVE3 form contains two nucleic acid binding domains (BD): an N-terminal Z-DNA-BD (residues 4 to 72) and a C-terminal dsRNA-BD (residues 117 to 182) (23, 24). In addition, VVE3 physically interacts with PKR via a domain near the N terminus (16).
OV20.0, the ORFV ortholog of VVE3, is relatively poorly studied. The amino acid sequence of OV20.0 has low overall identity with VVE3 (Fig. 1A) but retains predicted functional motifs at the N- and C-terminal ends (6). The dsRNA binding ability of OV20.0 has been pinpointed by electrophoretic mobility shift assays (EMSA) using recombinant fusion proteins (6). However, dsRNA binding ability in the course of ORFV infection has not been examined. Haig et al. demonstrated that OV20.0 (also referred to as the OVIFNR gene product) inhibits PKR activation and overexpression of OV20.0 is able to protect an unrelated virus infection from the antiviral effects of both type I and type II IFN in cultures of ovine fibroblasts (25). A study of recombinant VACV expressing a series of the chimeric VVE3-OV20.0 proteins has indicated that the N-terminal, but not C-terminal (including the dsRNA binding), domain of OV20.0 is able to complement the relevant function of VVE3 (26). This suggests that OV20.0 may interact with dsRNA via a mechanism that is distinct from that of VVE3. Furthermore, OV20.0 is able to rescue the IFN-sensitive and restricted host range phenotypes of E3-deficient VACV only in cultured cells, but such rescue does not occur in animal models (26). Hence, the precise mechanism of how OV20.0 modulates the host immune pathway remains unclear, and while OV20.0 shares some properties with VVE3, the two proteins are not entirely functionally equivalent.
FIG 1.

Sequence analysis and expression of OV20.0L of ORFV. (A) Sequence alignment of the E3L orthologs of VACV, ORFV, and goat pox virus. Markings include the predicted NLS (red frame) and conserved binding motifs that directly interact with Z-DNA (16) (blue dashed boxes). The predicted initiating methionine (M) of sh20 is indicated by a red asterisk; dashes indicate gaps in the alignments. (B) The sequences of the OV20.0L gene of ORFV (viral OV20.0) and three constructs used in this study are shown. BamHI was the communal site for insertion of the OV20.0L DNA into the vector. In constructs Kozak 20.0-eGFP and sh20-eGFP, the Kozak consensus sequence (CCACCATGG) was inserted at the upstream region of initial codon ATG. (C) Expression of the OV20.0 isoforms in ORFV-infected cells. Goat fibroblast cells were mock infected (lane1) or infected by ORFV at an MOI of 1, and total cell lysate was harvested at 12, 24, and 36 h postinfection (hpi). (D) OV20.0 and sh20 expression in cells. Human embryonic cells (293T) were transfected with plasmids designed to express wild-type OV20.0L (OV20.0-eGFP), full-length OV20.0 only (Kozak 20-eGFP), or sh20 only (sh20-eGFP) in the left blot. In the right blot, plasmids expressing wild-type OV20.0 (OV20.0-TAP) and a second construct expressing only full-length OV20.0 (Δ3ATG-TAP) were used. Total cell lysates were collected after 24 h, resolved by SDS-PAGE, and detected using an OV20.0-specific antibody on Western blots.
In this study, we found that like VACV E3L, OV20.0L encodes two isoforms. Next, the origin of the two isoforms was explored as well as several aspects of the mechanisms underlying their function. Specifically, the role that OV20.0 plays in the inhibition of PKR signaling was analyzed with a focus on understanding the equivalence of the two OV20.0 isoforms in vitro and in vivo.
MATERIALS AND METHODS
Cell maintenance.
Human embryonic kidney cell line 293T and Vero cells were propagated in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Life Technologies Corporation, Carlsbad, CA, USA) with 10% fetal calf serum (FCS; HyClone, Logan, UT, USA) and 1% penicillin-streptomycin (Gibco BRL). Human A549, goat primary testis (GT), and goat fibroblast cells were grown in RPMI medium (Gibco BRL) supplemented with 10% FCS (HyClone, Logan, UT, USA) and 1% penicillin-streptomycin (Gibco BRL). These cells were cultured at 37°C with 5% CO2.
Viruses and infection.
Wild-type ORFV (strain Hoping) (27) was cultivated in goat primary testis cells, and its titer was determined by standard plaque assay. Recombinant Sindbis virus (SINV) containing an enhanced green fluorescent protein (eGFP) expression cassette inserted between the capsid and E3 genes of Sindbis virus (28) was a gift from Lih-Hwa Hwang (Graduate Institute of Microbiology and Immunology, National Yang Ming University, Taipei, Taiwan). For infection, 80% confluent cell monolayers were incubated with ORFV at the indicated multiplicity of infection (MOI) in infection medium (DMEM without FCS). After allowing 1 h for viral adsorption, the infection medium was replaced by fresh DMEM with 2% FCS, and vessels were returned to 37°C with 5% CO2.
Construction of plasmids.
A series of plasmids were generated to express various recombinant VVE3 (or OV20.0) proteins in an Escherichia coli system. VVE3, OV20.0, and PKR genes were amplified by their respective designed primer sets (vaccinia E3L-F, 5′-AAGGATCCCATATGTCTAAAATCTATATCGACG-3′, and vaccinia E3L-R, 5′-AAGCGGCCGCCTCGAGGAATCTAATGATGACGTAACC-3′; Orf-OV20.0L-F, 5′-ATACGCCCATATGGCCTGCGAGTGC-3′, and Orf-OV20.0L-R, 5′-CGGGATAAGTCGACGAAGCTGATGCCG-3′; PKR-F, 5′-CCGCTAGCATGGCTGGTGATCTTTCAG-3′, and PKR-R, 5′-CGCTCGAGACATGTGTGTCGTTC-3′) based on the sequences published in GenBank; the accession numbers for OV20.0L and vaccinia virus E3L are ABY41266 and AAA02759, respectively. All fragments were produced under the same PCR conditions: initial denaturation at 95°C (5 min), followed by 35 cycles of denaturation (95°C, 30 s), annealing (55°C, 45 s), and extension (72°C, 45 s), and a final extension (72°C, 7 min). PCR products were digested by corresponding restriction enzymes introduced in primers (underlined sequences) and ligated into pET24a, a prokaryotic protein expression vector; the resulting plasmids were named pET24a-VVE3, pET24a-OVE3, and pET24a-PKR. The construct expressing short-form OVE3, designated pET24a-sh20, was constructed by self-ligation of the fragment of pET24a OVE3 linearized with NdeI and NcoI, followed by a Klenow enzyme reaction and blunt-end ligation. A similar strategy was used to generate pET24a-VVE3ΔC expressing C-terminally truncated VV E3: pET24a-VVE3 was digested by AatII and XhoI enzymes followed by blunt-end self-ligation.
Several plasmids were constructed for transient transfection of various E3 ortholog proteins in mammalian cells; the sequences surrounding the putative translation start sites are shown in Fig. 1B. DNA fragments of eGFP, OV20.0L, sh20, OV20.0L flanked with Kozak consensus sequences, and human PKR were obtained from templates (pEGFP-C1, pET24a-OV20.0, pET24a-sh20, pET24a-Kozak-OV20, and pET24a-PKR, respectively) by PCR using the following primer sets: GFP-NotI-F, 5′-AAGCGGCCGCAATGGCTAGCAAAG-3′, and GFP-XbaI-R, 5′-CGGCTATCTAGATGTACAGTTCATC-3′; Orf OV20.0L-Kozak-BamH I-F, 5′-TTTGGATCCGCCACCATGGCCTGCGAGTG-3′, Orf OV20.0L-BamH I-F, 5′-CTTGGATCCTTATGGCCTGCGAGTG-3′, Orf OV20.0L-NotI-R, 5′-GCTTGCGGCCGCTAAGAAGCTGATG-3′, and Orf sh20-BamH I-F, 5′-TTTGGATCCGCCACCATGGAGACTGAGG-3′; PKR-NheI-F, 5′-CCGCTAGCATGGCTGGTGATCTTTCAG-3′, and PKR-HA-XbaI-R, 5′-GGTCTAGATTAAGCATAATCTGGAACATCATATGGATAACATGTGTGTCGTTCATTTTTCTC-3′, respectively. The sequence of the hemagglutinin (HA) tag, written in italics, was introduced in the reverse primer to generate PKR with the HA tag fused at the 3′ terminus. The PCR amplification was conducted by initial denaturation at 95°C (5 min), followed by 35 thermal cycles of denaturation (95°C, 30 s), annealing (55°C, 60 s), and extension (68°C, 60 s), followed by a final extension (68°C, 7 min). The resulting PCR products were digested with restriction enzymes (as indicated by underlining in primers) and ligated with pcDNA3.1 (+) cleaved with enzymes corresponding to those used in insert fragment preparations. The resulting plasmids were designated pcDNA3.1-eGFP, pcDNA3.1-Kozak-OV20.0-eGFP, pcDNA3.1-OV20.0-eGFP, pcDNA3.1-sh20-eGFP, and pcDNA3.1-PKR-HA.
pTAP-OV20.0-Δ3ATG was generated to change the third ATG codon (ATG→ATT) via a site-directed mutagenesis kit (QuikChange Site-Directed mutagenesis; Stratagene Corp., CA, USA) on the plasmid pTAP-OV20.0, which contains the full-length OV20.0L gene in the pNTAP-C vector (Agilent Technologies).
Expression and purification of recombinant proteins.
Recombinant proteins were expressed in E. coli strain BL21(DE3) with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) induction for 4 h at 37°C. Bacterial pellets were collected and dissolved in binding buffer (1/10 of culture volume) without denaturing agent (0.01 M imidazole, 0. 5 M NaCl, 0.05 M Tris-HCl, 200 μg/ml lysozyme) followed by sonication. The protein in soluble fraction used for assays was obtained by centrifugation at full speed for 10 min at 4°C. All the proteins were expressed as fusion proteins with the six-histidine tag (His tag) at the C terminus, which allows further purification by metal affinity chromatography, using chelating Sepharose Fast Flow (GE Healthcare) according to the manufacturer's instructions. The high concentration of imidazole was removed from purified protein by a series of dialysis against phosphate-buffered saline (PBS) (0.02 M phosphate, 0.15 M NaCl) at 4°C. The Bradford method (Bio-Rad) was used to determine protein concentrations.
Poly(I·C) pulldown assay.
For poly(I·C) pulldown assays, first, poly(C)-coated agarose (Sigma-Aldrich) was mixed with 2 volumes of 2-mg/ml poly(I) (Sigma-Aldrich) prepared in buffer containing 50 mM Tris (pH 7.0) and 150 mM NaCl and incubated at 4°C for overnight with gentle rocking. The poly(I·C) agarose beads were collected by centrifugation at 800 × g, washed with 50 mM Tris (pH 7.0)–150 mM NaCl, and resuspended in binding buffer A (50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, and 1% Nonidet P-40) as a 10% final slurry. For dsRNA binding assay, poly(I·C) beads were combined with 1 μg of recombinant OV20.0 or sh20 proteins of binding buffer A (in 50 μl of reaction mixture) and incubated at 4°C for 1 h. The poly(I·C) beads were collected by centrifugation at 800 × g and washed twice with buffer C (10 mM Tris-HCl [pH 7.8], 6 mM MgCl2, 80 mM KCl, 2 mM dithiothreitol, 250 mM sucrose, and 0.1 mM EDTA). After the final wash procedure, beads were collected by centrifugation and resuspended in 3× SDS sample dye.
The orf virus-infected cell lysate containing OV20.0 isoforms used for dsRNA binding assay was prepared as follows: goat fibroblast cells were infected with orf virus at an MOI of 1. At 24 h postinfection (hpi), cells were trypsinized and pelleted by centrifugation at 800 rpm. The cell pellets were then resuspended in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.9], 150 mM NaCl, 0.1% SDS, 1.0% NP-40, 0.5% sodium deoxycholate) and kept on ice for 10 min, followed by centrifugation (11,000 × g).
ELISA-based dsRNA binding assay.
For the enzyme-linked immunosorbent assay (ELISA), two strands of RNAs (5′-AGACCCAAGCUGGCGGGCGAAUUGGAGCUCCACCGCGGUGGC-biotin; 5′-GCCACCGCGGUGGAGCUCCAAUUCGCCCGCCAGCUUGGGUCU, synthesized by Invitrogen) were resuspended in Tris-EDTA (TE) buffer (10 mM Tris, 1 mM EDTA [pH 8.0]). Equal molarity amounts of each RNA strand were mixed in annealing buffer (10 mM Tris [pH 8.0], 20 mM NaCl), heated to 90°C in a water bath for 5 min, and gradually cooled down to room temperature. The biotin-dsRNA was incubated with 1 μg of VVE3, OV20.0, or sh20 proteins (all recombinant proteins fused with His tag) at room temperature for 1 h in PBS. The mixture of biotin-dsRNA and E3 (or OV20.0) proteins was then added straight, or with 20, 100, or 500 ng poly(I·C) (Sigma-Aldrich) as competitor, into a 5% bovine serum albumin (BSA)-blocked 96-well microplate coated with streptavidin (Thermo Fisher Scientific). Engagement of the dsRNA complex with the streptavidin-coated plate was carried out for 1 h at room temperature. For detection of the presence of E3 (or OV20.0) proteins in the RNA complex, 1:5,000-diluted His-specific antibody (AbD Serotec) was added in the well for 1 h, followed by a wash procedure, and incubated with 1:10,000-diluted horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Jackson) for 1 h. The signal was detected by chromogenic substrate (tetramethylbenzidine [TMB]) for 10 min and terminated by addition of 50 μl H2SO4. The optical density (OD) of each well was read at 450 nm with reduction at 630 nm using a microplate reader (Tecan). Between all steps, wells were washed by PBST, i.e., PBS (pH 7.4) containing 0.1% (vol/vol) Tween 20, three times.
Western blot analysis.
Proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then electrophoretically transferred to a polyvinylidene difluoride (PVDF) membrane, followed by immunoblotting with diluted antibodies, such as polyclonal anti-OV20.0 sera (1:2,000) generated from mice immunized with purified OV20.0 recombinant protein, anti-eIF2α antibody (1:2,000; 2103s; Cell Signaling), anti-eIF2α-p antibody (1:1,000; ab32157; Abcam), anti-PKR antibody (1:2,000; ab32052; Abcam), anti-PKR-p antibody (1:1,000; ab32036; Abcam), or anti-His antibody (1:5,000; GTX115045; Gene Tex), anti-actin antibody (1:5,000; GTX26276; Gene Tex), and anti-eGFP sera (1:5,000) from mice immunized with purified eGFP recombinant protein. Goat anti-rabbit or mouse IgG-conjugated HRP (1:10,000-fold diluted; Jackson Laboratory) was incubated with membranes for 1 h at room temperature. After an extensive wash with PBST, the PVDF membrane was treated by an enzyme-linked chemiluminescence system (ECL; Amersham, GE Healthcare), and the signal was detected by chemiluminescence. Several PBST washes were conducted between all steps.
Transfection and immunofluorescence assay.
Transfection of plasmids for transient expression was done with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. In brief, cells were seeded in the plate 1 day before transfection. For observing OV20.0 cellular distribution, 0.8 μg of constructs expressing OV20.0-eGFP was transfected individually to cells (seeded in a 24-well plate). At 24 h posttransfection, cells were fixed with 4% paraformaldehyde for 15 min and then washed twice with PBS. To visualize the nuclei, cells were stained with 4,6-diamidino-2-phenylindole (DAPI) for 10 min and mounted onto slides, followed by fluorescence microscopy. Cell lysates for coimmunoprecipitation and determination of PKR and eIF2α phosphorylation levels were prepared by the same transfection procedures.
Detection of PKR and eIF2α phosphorylation following poly(I·C) stimulation.
Constructs expressing full-length OV20.0 (designated Kozak OV20-eGFP), sh20-eGFP, or eGFP (control) were individually transfected into 293T cells. After 24 h of transfection, cells were treated with poly(I·C) by transfection. Total cell lysate was collected at the indicated times for detection of basal and phosphorylation forms of PKR and eIF2α by Western blotting. Actin was used as a control in immunoblotting.
Coimmunoprecipitation of PKR and OV20.0.
For the in vitro coimmunoprecipitation assay, 1 μg of OV20.0 or sh20 recombinant proteins was mixed with 1 μg of PKR protein in PBS as a binding buffer and incubated at 30°C, and a small aliquot was taken as input control. After 1 h of incubation, specific antibody was added into the tube. After a 1-h interaction time, protein A-agarose (Millipore) was blended with the mixture for another hour. Immunocomplex-protein A beads were harvested by centrifugation at 2,000 rpm for 5 min. Supernatant containing unbound proteins was kept for further analysis. After successive PBS washes, the agarose beads were resuspended in SDS sample dye and analyzed by Western blotting.
For immunoprecipitation of proteins expressed in mammalian cells, at 24 h posttransfection, cell pellets were resuspended in RIPA buffer and incubated on ice for 10 min followed by centrifugation at full speed. Supernatant, as crude cell lysate, was mixed with anti-PKR antibody at 30°C for 1 h. Protein A-agarose beads were then added into the reaction mixture, which was incubated for 1 h. Following thorough wash procedures with TNTG (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% Triton X-100, 10% glycerol), the protein bound with agarose beads was harvested for Western blot analysis.
RNA extraction and real-time PCR.
At 24 h after Kozak20, sh20, or eGFP transfection, cells were further transfected with 1 μg/ml of poly(I·C) for 4 h. Total RNA was then extracted by the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was synthesized by using oligo(dT) primers. The primer sets for cytokines and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and the conditions for PCR were the same as described in a previous report (13). Real-time PCR was performed using LightCycler 480 SYBR green I Master (Roche) on a LightCycler 480 instrument (Roche). The expression level of GAPDH was used as an internal control to establish standardization among samples. Data were normalized to the geometric mean of GAPDH and analyzed by the 2−ΔΔCT method.
Construction of transfer vectors for generating recombinant orf viruses.
For the ease of screening recombinant orf virus, the eGFP coding region was cloned into the transfer vector. To do so, initially, two strands of oligonucleotides for vaccinia virus H5 promoter (VACVH5) (sequences, 5′-GGATCCAATAAATACTAATAAAGAGCGAAGTTAAATTGTGTTGGGAGTTCTTGGAAACATAAATAAAAGTTAAAAAGCTAGC-3′) were synthesized, annealed, and trimmed with BamHI and NheI (the sequences of enzyme recognition [underlined] were introduced in the oligonucleotides). The coding region of eGFP was amplified from pEGFP-N (Clontech) by PCR. The DNA containing eGFP sequences trimmed with NheI and XbaI and the VACVH5 fragment were cloned into pUC19 linearized with BamHI and XbaI, resulting in plasmid pUC19-VACVH5 eGFP. Upstream and downstream regions of the OV20.0L gene that serve as flanking sequences for homologous recombination were separately amplified by two primer sets. The sequences of the primers used for amplification are as follows: for the upstream region, F-up-HindIII primer, 5′-TATAAGCTTCGGGCGATGGACGAACACCG-3′, and R-up-SalI primer, 5′-TAAGTCGACTGTGGCGTATTTACGGGGCGTG-3′; for the downstream region, F-down-BamHI primer, 5′-AAGGATCCTTTTTATCTCGCGCCGGCCACCG, and R-down-EcoRI primer, 5′-TTGAATTCTGGTGTACAGCGCGGAGGCCATC 3′). The PCR products containing upstream and downstream regions were sequentially cloned into the pUC19-VACVH5 eGFP plasmid cut with SalI/HindIII and EcoRI/BamHI restriction enzymes, generating plasmid pUC19-flank E3-VACVH5eGFP.
To construct the transfer vector for sh20 orf virus, DNA containing sh20 sequences was amplified by PCR (with primers F-sh20-SalI, 5′-TCGTCGACATGGAGACTGAGGCTG-3′, and R-sh20-XbaI, 5′-CCTCTAGATTACTTATCGTCGTCATCCTTGTAATCGAAGCTGATGCCGCAGTTGTTGATG-3′) and was then cloned into pUC19-flank OV20.0-VACVH5eGFP cut with XbaI/SalI. For OV20.0-FLAG recombinant virus, a fragment containing OV20.0L sequences with fusion of FLAG tag at its C terminus was amplified by PCR using degenerated primers F-OV20.0-HindIII, 5′-GGAAGCTTAAATACGCCACAATGGCCTGCGAG-3′, and R-OV20.0-FLAG-SalI, 5′-CCGTCGACTTACTTATCGTCGTCATCCTTGTAATCGAAGCTGATGCCGCAGTTGTTGATG-3′. The PCR fragment was then cloned into plasmid pUC19-flank 20.0-VACVH5eGFP linearized with SalI/HindIII to replace the DNA of the upstream region.
A revertant virus, of which the sh20-eGFP expression cassette in sh20-GFP orf virus was replaced by the wild-type OV20.0L gene, was also made. To do so, a set of primers, i.e., a 5′ primer (5′-CATCGCGTGTTTGGGCTTTC-3′) and a 3′ primer (5′-ATGCCGATGATGATCCAGAC-3′), was used to amplify the OV20.0L gene containing its up- and downstream sequences from wild-type orf virus DNA. Then, the PCR product was cloned into the TA plasmid, which serves as the transfer vector for creating revertant orf virus.
Generation of recombinant orf viruses.
Initially, 293T cells were infected with wild-type orf virus at an MOI of 0.05. At 1 hpi, infected cells were transfected with the transfer plasmids specific for generating recombinant orf virus, namely, FLAG-OV20.0-GFP orf, FLAG-sh20-GFP orf virus, or revertant OV20.0 orf virus (sh20-Rev). Three days postinfection (p.i.), cells were harvested, followed by sonication. The cell lysate containing orf virus was 10-fold serially diluted and inoculated into goat fibroblast cells. The infected cell monolayer was overlaid with 0.6% agarose gel, and cell morphology was observed by fluorescent microscopy daily. Pure recombinant sh20-GFP orf virus was isolated after three rounds of plaque purification. The homogeneity of recombinant viruses in each run of plaque purification was initially determined by PCR using primers forward rOrf E3-5′-CATCGCGTGTTTGGGCTTTC-3′ and reverse rOrf E3-5′-ATGCCGATGATGATCCAGAC-3′. The genotype of purified recombinant virus was further confirmed by Southern blotting using a DNA probe targeting specific segments from viral DNA cut with XhoI restriction enzyme (data not shown). The OV20.0 protein expression profile in recombinant virus-infected cell lysates was confirmed by Western blot analysis (data not shown).
Pathogenicity of orf viruses in the mouse model.
Groups of 6-week-old female BALB/c mice (five per group) were inoculated with 107 PFU of orf virus or with PBS (as negative control) in the internal face of the ear scraped by sandpaper as described previously (29). The protocol was approved by the IACUC of National Chung-Hsing University (approval number 101-40). Mice were weighed daily for 5 days postinfection, and the weight change was calculated. Clinical signs, including erythema, exudate, macule, papule, vesicle, pustule, and scab, were recorded daily and scored according to the guideline whereby each symptom registers one point toward a cumulative score. Blood was taken from each mouse at 5 days p.i. for determining the concentrations of IL-6, tumor necrosis factor alpha (TNF-α), and IFN-γ in serum by ELISA. At 5 days p.i., mice were sacrificed and ear samples were collected followed by sonication in 1 ml DMEM. The supernatants were inoculated onto monolayers of goat fibroblast cells for determination of virus loads by standard plaque assay.
Statistical analysis.
Data are shown as means ± standard errors of the means (SEM). Student's t test was used to evaluate significant differences. The results of analysis were conducted by using the GraphPad statistical package analysis tool (GraphPad Software, San Diego, CA). A P value of <0.05 indicates a statistically significant difference; in all figures, single, double, and triple asterisks indicate P values of <0.05, <0.01, and <0.001, respectively.
RESULTS
Two OV20.0 isoforms are expressed during infection.
We began by comparing OV20.0 with its orthologs from goatpox virus (GPV; a capripoxvirus) and VACV. OV20.0 shares 31% amino acid identity with VACV E3, but motifs for the Z-DNA binding domains, a nuclear localization signal (NLS), and the dsRNA binding domain are recognizable across these E3L orthologs, despite some variations (Fig. 1A). Next, we characterized OV20.0L expression during infection of goat fibroblasts with ORFV by Western blotting (Fig. 1C). A product of the expected mobility for full-length OV20.0 was observed starting from 12 h postinfection (hpi) with a peak at 24 hpi before it declined by 36 hpi. In addition, a second, shorter protein (called sh20) was detected, also appearing first at 12 hpi but continuing to build up through 36 hpi. The apparent existence of two isoforms of OV20.0 was not entirely unexpected because VACV E3 is expressed as two isoforms, possibly due to leaky scanning and initiation on a downstream AUG codon (30). To explore this possibility, several expression constructs of OV20.0 were made and tested in transient transfections. These constructs were (i) wild-type OV20.0 with a C-terminal eGFP fusion (OV20.0-eGFP), (ii) OV20.0-eGFP with a Kozak consensus sequence introduced in the optimal location ahead of the first ATG (Kozak20-eGFP), and (iii) OV20.0-eGFP truncated such that it starts with the downstream ATG anticipated to give rise to sh20 (sh20-eGFP). Both the full-length and smaller isoforms were found in lysates from cells transfected with OV20.0-eGFP, whereas only the larger and smaller isoforms were found where the Kozak20-eGFP and sh20-eGFP constructs were transfected, respectively (Fig. 1D, left). This finding was extended further, using tandem affinity purification (TAP) fusions in case eGFP or other vector elements might be affecting the result (Fig. 1D, right). Again, both isoforms were detected for wild-type OV20.0. However, when a modification was made to ablate the putative downstream initiating codon (change from AUG to AUU), sh20 was no longer detectable. Taken together, these data show that OV20.0L encodes two protein isoforms that are expressed during ORFV infection and suggest that these arise due to the use of alternate start codons during translation.
Cellular distribution of OV20.0 and sh20 in mammalian cells.
Since the deduced sequence of an NLS is not present in sh20 (Fig. 1A), we investigated whether the localization of this isoform differed from that of full-length OV20.0. To do this, we took advantage of Kozak20-eGFP as a comparison because this construct expresses only the full-length OV20.0. Cells transfected with Kozak20-eGFP contained fluorescence predominantly in the nucleus, with only a subset of cells having subnuclear fluorescence. This contrasted with sh20-eGFP, in which the fluorescence was largely in the cytoplasm (Fig. 2A). To detect the cellular distribution of sh20 in ORFV-infected cells, recombinant ORFVs were made, expressing either wild-type OV20.0 or sh20. In these viruses, OV20.0L was replaced with two expression cassettes, the first expressing eGFP from a vaccinia virus H5 promoter and the second expressing the sh20 isoform or wild-type OV20.0 under its native promoter. These viruses were named sh20-GFP and OV20.0-GFP. The results from infected cells are consistent with those obtained from transiently transfected cells (Fig. 2B). These observations were supported by fractionation and immunoblot analyses that found that the proteins expressed by Kozak20 and sh20 were mainly in the nucleus and cytoplasmic fractions, respectively (Fig. 2C and D). These locations mirrored those of the relevant isoform expressed from wild-type OV20.0L.
FIG 2.

Cellular distribution of OV20.0 proteins. (A) Vero cells were transfected with Kozak 20 and sh20-eGFP plasmids and after 24 h were stained with DAPI and examined by fluorescence microscopy. (B) Cells were infected at an MOI of 1 with recombinant ORFV OV20.0L-FLAG-eGFP (OV20-GFP virus) or sh20-FLAG-eGFP (sh20-GFP) for 48 h before detection of FLAG by immunostaining and fluorescence microscopy. (C) The cellular distribution of OV20.0 isoforms was analyzed by immunoblotting. Lysates of Vero cells transfected with plasmids expressing eGFP fused to wild-type OV20.0 (WT OV20.0), Kozak-OV20.0 (Kozak20), or short-form OV20.0 (sh20) were divided into nuclear (N) and cytoplasmic (C) fractions before separation by SDS-PAGE and detection on Western blots using anti-OV20.0 antibody. The nuclear protein TATA box binding protein (TBP) and two cytoplasmic proteins, GAPDH and eIF4H, were used as controls for nuclear and cytoplasmic fractions, respectively. (D) The intensity of full-length OV20.0-eGFP and sh20-eGFP expressed in these transfectants was measured, and the ratios of nuclear to cytoplasmic proteins were plotted.
Evaluating the dsRNA binding ability of OV20.0 and sh20.
A major function for OV20.0 is binding of dsRNA. To facilitate the investigation of this function in vitro, the OV20.0 isoforms were expressed in E. coli and purified via nickel-nitrilotriacetic acid (Ni-NTA) chromatography (Fig. 3A). VACV E3 and VVE3ΔC (the C-terminally truncated VACV E3 without the dsRNA binding domain) were also produced to be used as positive and negative controls, respectively. First, both OV20.0 isoforms were shown to bind poly(I·C) (a synthetic dsRNA) beads in a pulldown assay, remaining in the pellet fraction after intensive wash procedures, unlike VVE3ΔC (Fig. 3B). Second, in a competitive assay in which both OV20.0 isoforms were simultaneously incubated with poly(I·C) beads, the dsRNA binding capability of full-length 20.0 and sh20 were found to be similar (Fig. 3C). Third, these results were confirmed using an ELISA-based method in which the OD450 values reflected the amount of recombinant protein bound to dsRNA. VVE3 and both isoforms of OV20.0 bound dsRNA to a similar extent, and in each case this binding was significantly better than that of VVE3ΔC (Fig. 3D). Fourth, the specificity of the ELISA-based dsRNA binding assay was confirmed using free poly(I·C) as a competitor that was shown to decrease the OD450 in a dose-dependent manner for all proteins except for VVE3ΔC (Fig. 3E). Finally, poly(I·C-coated beads were able to pull down both isoforms of OV20.0 in lysates from infected cells (Fig. 3F). Together, these findings indicate that both the full-length OV20.0 and sh20 are able to bind dsRNA.
FIG 3.

Interaction of double-stranded RNA with OV20.0 proteins. (A) Recombinant His-tagged proteins, including OV20.0, sh20, VACV E3 protein (VVE3), and VACV E3 with a dsRNA binding motif deletion (VVE3ΔC), were expressed and purified. (B) Full-length OV20.0, sh20, or VVE3ΔC (negative control for dsRNA binding) were incubated with poly(I·C) beads for 1 h. After centrifugation to collect any protein bound to beads, the supernatant (S) was kept before the pellet was washed two times (W1 and W2) before being collected (P). Proteins in each part were detected using an anti-His antibody on Western blots. (C) For analysis of the relative dsRNA binding ability between OV20.0 and sh20 proteins, equal amounts of the two proteins were incubated in the same tube with binding buffer for 1 h at 4°C, followed by fractionation, wash steps, and Western blotting as described above. (D) Quantification of the dsRNA binding ability of various E3 orthologous proteins by ELISA. One microgram of purified recombinant proteins as indicated was incubated with biotin-conjugated dsRNA beads for 1 h. The protein-dsRNA mixtures were then subjected to quantification by ELISA as described in Materials and Methods. Each protein was normalized against the negative control, and the value for the VVE3 protein was set to 1.0. Each bar represents the mean (±SE) of three independent repeats. (E) Competition of poly(I·C) with dsRNA binding with E3 orthologs. Poly(I·C) amounts as indicated were incubated with protein mixtures and biotin-conjugated dsRNA beads for 1 h, and bound protein was detected by ELISA as described for panel D. (F) Interaction of viral OV20.0 isoforms with dsRNA in orf virus-infected cells. Goat fibroblast cells were infected with ORFV at an MOI of 5 for 24 h, cell lysates were harvested, and dsRNA-binding activity was detected by poly(I·C) pulldown assay.
Wild-type OV20.0 directly interacts with PKR.
Inhibition of PKR activation mediated by OV20.0 has been demonstrated in an in vitro system, but the mechanism was not determined (25). VACV E3 inhibits PKR activation by sequestering the dsRNA substrate and via a direct interaction (5, 16, 31). For this reason, we tested whether wild-type OV20.0 also interacts directly with PKR using coimmunoprecipitation of recombinant proteins in vitro and from lysates from transfected mammalian cells and ORFV-infected cells. Recombinant OV20.0 was able to be precipitated using an anti-PKR antibody protein after a 1-h incubation with human PKR (Fig. 4A, top), unlike the negative-control thioredoxin (Fig. 4A, bottom). This indicates that OV20.0 interacts with PKR in the absence of other proteins. Consistent with this, OV20.0 but not eGFP coimmunoprecipitated with human PKR in lysates of 293T cells transfected with constructs expressing these proteins (Fig. 4B). Finally, anti-PKR was able to precipitate OV20.0 in lysates of human A549 cells transfected with a PKR expression plasmid and infected with ORFV at an MOI of 1, demonstrating that this interaction can also occur during infection (Fig. 4C).
FIG 4.

Interaction of OV20.0 with cellular PKR protein. Binding of OV20.0 protein with PKR was determined by immunoprecipitation of recombinant proteins in vitro (A), after transfection of 293T cells (B), and after ORFV infection (C). (A) Purified recombinant His-tagged human PKR was incubated with His-tagged OV20.0 (top) or recombinant thioredoxin protein (Thx, bottom) for 1 h before immunoprecipitation with rabbit anti-PKR antibody. Samples of the input (as shown) and from the nonprecipitated (IP-PKR, lane 2), the final wash (IP-PKR, lane 3, top panel only), and the anti-PKR precipitated fraction (IP-PKR, lane 4) were analyzed by Western blotting (WB) using an anti-His tag antibody to detect all proteins. (B) Human 293T cells were cotransfected with plasmids expressing HA-tagged human PKR and OV20.0-FLAG (left) or eGFP (right) as a negative control. Total cell lysates were immunoprecipitated with rabbit anti-PKR antibody, and various fractions were detected by immunoblotting with anti-OV20.0 (left), anti-eGFP (right), and anti-PKR (top section for both blots). Fractions were input control (lane 1), unbound fraction (lane 2), the supernatant from the final wash (lane 3), and precipitate (lane 4). (C) Human A549 cells were transfected with a plasmid expressing PKR-HA and infected with ORFV at an MOI of 1 for 24 h. The cell lysate was immunoprecipitated using rabbit anti-PKR antibody, and various fractions were detected on a Western blot using anti-PKR (top) and anti-OV20.0 (bottom). Fractions were as described for panel B.
sh20 directly interacts with PKR.
Next, we wondered whether sh20 might also directly interact with PKR. Using a cell-free system similar to the one described above, recombinant sh20 was found to interact with PKR protein (Fig. 5A). To confirm this interaction in mammalian cells, plasmids expressing (i) Kozac20-eGFP, (ii) sh20-eGFP, or (iii) eGFP were cotransfected with a PKR expression construct and cell lysates were immunoprecipitated with anti-PKR. Both isoforms of OV20.0, but not eGFP alone, were precipitated with PKR (Fig. 5B); however, we noted that the full-length OV20.0 was more efficient than sh20 in this assay.
FIG 5.
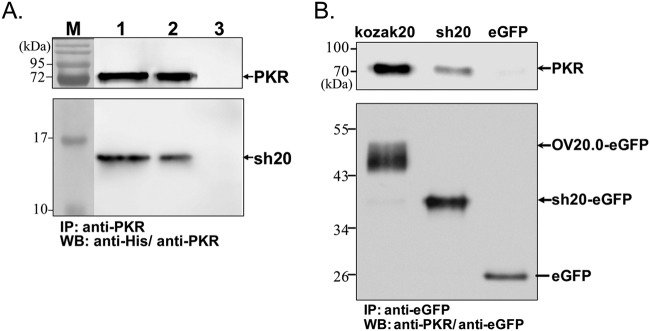
Interaction of OV20.0 isoforms with PKR. The interaction of sh20 with PKR was determined in vitro (A) and in transfected cells, where it was compared with the full-length OV20.0 (B). (A) Purified recombinant His-tagged human PKR was incubated with His-tagged sh20 for 1 h before immunoprecipitation with rabbit anti-PKR antibody. Samples of the input (lane 1) and from the anti-PKR precipitated (lane 2) and supernatant (lane 3) fractions were analyzed by Western blotting (WB) using an anti-His tag antibody to detect all proteins. (B) Full-length OV20.0-eGFP (Kozak20), sh20-eGFP (sh20), and eGFP were transiently expressed in human 293T cells with human PKR. Total cell lysates were immunoprecipitated with anti-eGFP, and the resulting fractions were analyzed on Western blots probed with anti-human PKR (top) and anti-eGFP (bottom).
OV20.0 and sh20 inhibit phosphorylation of PKR and its substrate eIF2α.
To probe the effect of OV20.0 on PKR activation and its downstream signaling, 293T cells were transfected with plasmids expressing OV20.0 or eGFP followed by poly(I·C) stimulation for 0.5, 1, 2, or 4 h. In cells expressing eGFP, the level of PKR phosphorylation was first detected 1 h after poly(I·C) stimulation and was substantially increased at the two later time points (Fig. 6A, top). In contrast, OV20.0 completely blocked the activation of PKR at 1 and 2 h after poly(I·C) treatment. Furthermore, the inhibition of PKR activation by OV20.0 led to greatly reduced eIF2α phosphorylation compared with that of the eGFP-transfected cells (Fig. 6A, bottom). To examine whether both isoforms inhibited PKR activation, transfections were repeated using plasmids expressing OV20.0, Kozak20 (referred to as K20), or sh20, and PKR phosphorylation was determined 4 h after poly(I·C) stimulation. PKR activation after poly(I·C) treatment was observed in both the mock-transfected and eGFP-expressing cells, but this phosphorylation was strongly reduced by all the OV20.0 constructs (Fig. 6B).
FIG 6.
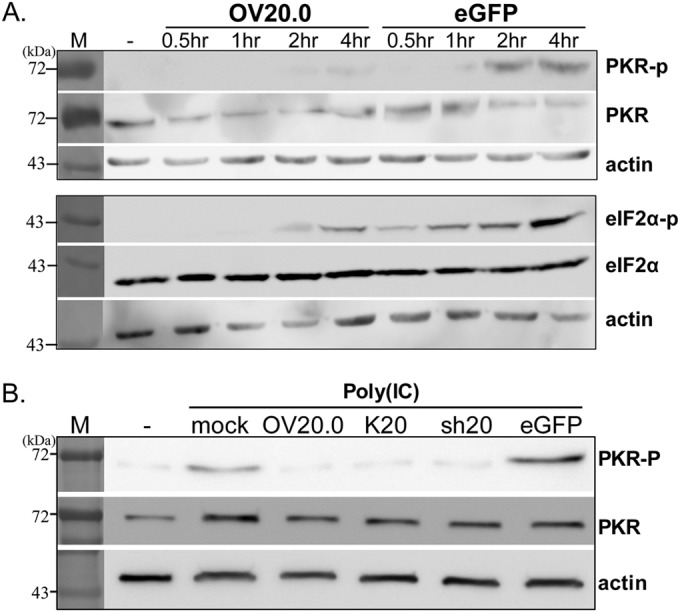
Inhibition of eIF2α phosphorylation by OV20.0 proteins via a PKR-dependent mechanism. (A) Human 293T cells were transfected with plasmids expressing the wild-type (OV20.0) with a FLAG tag or eGFP. One microgram of synthetic dsRNA (poly(I·C) was transfected at 24 h posttransfection. Cells were collected at the times indicated after addition of poly(I·C), and their proteins were separated by SDS-PAGE followed by Western blotting using antiserum specific to the unphosphorylated and phosphorylated forms of eIF2α and PKR. β-Actin is presented as a loading control. (B) Human 293T cells were transfected with plasmids expressing the wild-type OV20.0L (OV20.0), OV20.0L flanked with a Kozak sequence (K20), the short-form OV20.0 (sh20), or eGFP or were left untransfected (mock). At 24 h posttransfection, 1 μg of synthetic dsRNA [poly(I·C] was then transfected into 293T cells for 4 h. −, control lanes.
OV20.0 and sh20 inhibit production of antiviral cytokines.
Inhibition of PKR is known to block the cytokine response to virus infection (13, 32). To test whether both OV20.0 isoforms could inhibit the release of antiviral cytokines, supernatants from OV20.0 isoform-expressing cells stimulated with poly(I·C) were tested for their ability to inhibit Sindbis virus (SINV) infection. SINV infection is highly sensitive to IFN (28), and we used a recombinant SINV expressing eGFP to allow easy monitoring of infection. In these experiments, all the supernatants reduced SINV infection compared to the mock-treated culture. However, based on both the intensity of fluorescence and the number of eGFP+ cells, SINV infection was less inhibited when cells were treated with supernatant from poly(I·C)-stimulated cells expressing full-length OV20.0 or sh20 than with the control (Fig. 7A). This was confirmed by immunoblotting, which showed greater eGFP expression in the SINV-infected cultures pretreated with supernatants from both OV20.0-expressing cells than in those treated with the control (Fig. 7B). To explore the cytokines that might be inhibited by OV20.0, transcription of IFN-β and TNF-α was measured in cells transfected with OV20.0 constructs before poly(I·C) stimulation. Expression of IFN-β was reduced in the presence of both OV20.0 isoforms, compared with eGFP-expressing control cells (Fig. 7C). However, TNF-α expression was reduced only by full-length OV20.0. Taken together, these findings demonstrate that both OV20.0 isoforms inhibit antiviral defenses.
FIG 7.

Induction of an antiviral state by both full-length OV20.0 and sh20 proteins. 293T cells were transfected with plasmids expressing full-length OV20.0 (Kozak20), short-form OV20.0 (sh20), or eGFP proteins. After a 24-h posttransfection, the cells were further transfected with 1 μg/ml poly(I·C) for 4 h. The supernatant was transferred to another well and incubated with 293T cells for 8 h, followed by infection with recombinant Sindbis virus (SINV) expressing eGFP (at an MOI of 0.5) for 24 h. The infection with SINV was measured by fluorescence microscopy; the image under “Mock” shows infection with SINV in cells without any treatment (A). The eGFP expression level was also measured by Western blotting with eGFP antibody (B). The RNA levels of various cytokines (TNF-α and IFN-β) in K20, sh20, and eGFP plasmid-transfected cells followed by poly(I·C) treatment were monitored by real-time reverse transcription-PCR. The relative expression level of TNF-α and IFN-β to GAPDH (serves as internal control) was plotted (C).
Attenuation of virulence in mice infected with recombinant orf virus expressing sh20.
The C terminus of vaccinia virus E3 confers resistance to IFN and is well characterized, but recently a separate role in the inhibition of IFN was found for the N terminus of E3 in mice and primary cells (33). Our study thus far has demonstrated that full-length OV20.0 and sh20 function similarly in vitro, but it remains important to determine if they are equivalent in vivo.
To this end, a revertant of sh20-GFP in which the H5-eGFP-sh20 cassettes were replaced with the original OV20.0L sequences (sh20-Rev), in comparison with the other two recombinant viruses (OV20.0-GFP and sh20-GFP), was also made. The growth of these viruses in goat fibroblast cells was indistinguishable from that of the parent ORFV over 24 h (Fig. 8A), demonstrating that neither insertion of the eGFP cassette nor the expression of sh20 instead of wild-type OV20.0 alters replication in vitro.
FIG 8.

Characterization of the recombinant orf virus bearing sh20. In addition to OV20-GFP and the sh20-GFP recombinant orf viruses, a revertant virus (sh20-Rev) was also made by replacement of the sh20L-eGFP expression cassette with the wild-type OV20.0L-coding region. (A) The growth kinetics of these three recombinant viruses, OV20-GFP, sh20-GFP, and sh20-Rev viruses, were compared with that of wild-type orf virus (WT). Pathogenicity and the regulation of the immune reaction by wild-type orf virus (WT) and recombinant sh20-GFP and sh20-Rev orf viruses were characterized using a mouse model. The weight loss of the mice infected with the various orf viruses or with PBS (as mock control) was monitored from the day of infection as indicated; asterisks indicate (as detailed in Materials and Methods) the significant difference observed between mice infected with OV20-GFP virus and those infected with sh20-GFP virus (B). (C) Cumulative lesion scores were recorded. (D) Orf viruses in mice were isolated from the region of inoculation, and the virus titer of each mouse was determined by standard plaque assay. The average titer of the virus was then plotted. (E) On the fifth day after infection, the mice were bled and sacrificed. The levels of cytokines (IL-6, TNF-α, and IFN-γ) were detected by ELISA.
To test whether sh20 could substitute for wild-type OV20.0, BALB/c mice were infected with sh20-GFP, OV20.0-GFP, and sh20-Rev by scarification of the ear. As shown in Fig. 8B, mice infected with the ORFV expressing wild-type OV20.0 lost more weight than those that received sh2-GFP or PBS (Fig. 8B). This was statistically significant on days 2, 3, and 4 (Fig. 8B). Similarly, infection with the sh20-GFP virus caused less severe lesions than OV20.0-GFP and sh20-Rev, as indicated by the significantly lower cumulative score of clinical signs (Fig. 8C). To determine if this difference in pathogenesis was related to increases in virus growth in vivo, ears were removed from groups of mice on day 5 after infection and virus loads were measured by plaque assay. Similar amounts of virus were recovered from mice infected by OV20.0-GFP and sh20-Rev, but in comparison, titers of sh20-GFP were reduced by over 3 orders of magnitude (Fig. 8D). Finally to examine the role of OV20.0 isoforms in immune modulation, the levels of IFN-γ, IL-6, and TNF-α were measured in the sera of infected mice at 5 days p.i. The amounts of all cytokines were similar in mice infected with OV20.0-GFP and sh20-Rev, but in comparison with these groups, those infected with sh20-GFP had much higher levels in their sera (Fig. 8E). In comparison with what was seen in mock-infected mice (PBS), IL-6 and IFN-γ were not suppressed by sh20-GFP infection and TNF-α was strongly stimulated. Taken together, our findings from this mouse model demonstrate that the presence of the N terminus of the OV20.0 plays an important role in ORFV virulence, most likely due to a role in restraining the release of proinflammatory cytokines.
DISCUSSION
This study presents the first detailed analysis of OV20.0 that takes into account the two isoforms, separately analyzes their functions in vitro, and demonstrates a crucial role for the full-length isoform in vivo.
Leaky scanning by ribosomes is used by several viruses to generate multiple proteins from a single mRNA transcript (34, 35). For instance, the S1 genome segment of avian reovirus has three sequential open reading frames, which partially overlap each other, and two of the three proteins, namely, p10 and p17, are produced from different translation initiation sites (35). Another example is human papillomavirus (HPV) type 16, which produces the E7 protein from an E6/E7 bicistronic mRNA by leaky scanning (34). The present investigation demonstrated that this mechanism is most likely used by ORFV to produce two isoforms of OV20.0, as is the case with E3, the ortholog from vaccinia virus (26). This conclusion is supported by our data from assays using two different mutations. First, the shorter sh20 isoform was predicted to be produced when the ribosome scans through two AUGs and initiates translation at the third AUG (codon 80), which has an intermediate Kozak sequence (CCCAUGG, where underlining indicates the consensus sequence) (36). Consistent with this, mutation of this third AUG to AUU abolished sh20 expression (Fig. 1D, lane Δ3ATG). Second, in contrast to this third possible site for initiation, the first AUG has a poorer context (Fig. 1B). Creation of a full Kozak consensus sequence around this first AUG also eliminates the sh20 isoform. Finally, we also noted that a faint band between full-length OV20.0 and sh20 was observed, and we speculate that this form might result when translation starts at the second AUG (codon 43), which is also in a poor Kozak context. In contrast to our findings in mammalian cells, only one form of the OV20.0 protein was produced from His-tagged OV20L in a prokaryotic expression system. This result was expected because the expression vector contains a built-in ribosome binding site (consensus Shine-Dalgarno sequence AGGAGG) at the 5′ end of the OV20.0L gene to ensure that the first ATG will be used to initiate translation.
Viruses have evolved numerous ways of counteracting PKR activation. Human cytomegalovirus (HCMV) TRS1 protein directly binds with PKR, and interaction of TRS1 directs PKR to the nucleus, where PKR may be sequestered from its substrate (19, 37, 38). Alternatively, some viruses interfere with PKR signaling by competing with dsRNA, one of the substrates that induce PKR activation (39–41). The NS1 protein of influenza virus possesses dsRNA binding ability and causes inhibition of PKR phosphorylation and therefore also transphosphorylation of eIF2α, ultimately leading to the IFN-α/β signaling pathway being disarmed (42). The VACV E3 protein interacts with the αβββα structure in the N terminus of PKR as well as the substrate domain at the C terminus of PKR (16, 43, 44). The present study shows that OV20.0 protein is able to block PKR activity by several means. Both isoforms were shown to bind dsRNA, which would inhibit PKR activation, as well as to interact directly with PKR itself. Nevertheless, whether dsRNA bridging could enhance the interaction of OV20.0 with PKR requires further investigation. One of the consequences of PKR activation is phosphorylation of eIF2α, which brings a halt to the initiation of translation of both cellular and viral RNAs (45, 46). VACV encodes two proteins that interferes with PKR signaling, E3 as noted already and K3. K3 shares 33% identity with eIF2α and mimics the eIF2α structure to interfere with the binding of PKR to its authentic substrate (47). Thus far, OV20.0 is the only ORFV protein that inhibits PKR, and this virus lacks a K3 ortholog (21). While OV20.0 and sh20 are able to curb the activation of PKR following poly(I·C) stimulation, resulting in reduced eIF2α phosphorylation, the lack of a K3 ortholog means that OFRV is more reliant on this mechanism to counteract the host IFN response than some other poxviruses.
It is worth noting that in contrast with the full-length OV20.0 protein, a large proportion of sh20 remains in the cytoplasm in transfected cells (Fig. 2A) and also in ORFV-infected cells (Fig. 2B). It is possible that the different cellular distributions are due to the putative NLS in the full-length OV20.0 and its absence in the N-truncated sh20 protein. An NLS motif has been reported in the N terminus of VACV E3 (48). As indicated by underlining in Fig. 1A, residues 41 to 45 (KREVNK) of VACV E3 show some resemblance to a degenerate NLS motif (K K/R X K/R) (49), and this is partially conserved in OV20.0 (KHEANR). However, some sh20 was found in the nucleus, even though this form lacks this sequence, suggesting that OV20.0 has an additional mechanism for nuclear localization. This is reminiscent of myxoma virus M029, a functional ortholog of OV20.0 and E3, which like sh20 is missing around 80 amino acids from the N terminus (50). All poxviruses replicate and transcribe their genomes exclusively in the cytoplasm, which is then the main site of viral dsRNA production. Therefore, a cytoplasmic location for sh20 might increase the possibility for sh20 to interact with dsRNA and PKR. The one clear difference between the functions of the isoforms in vitro was the failure of sh20 to inhibit TNF-α transcription. It is tempting to speculate that this function of full-length OV20.0 might be due to the putative Z-DNA binding domain that is largely conserved compared with E3 and is missing from sh20. The Z-DNA binding domain of E3 is important for VACV pathogenicity (51) through the suppression of proinflammatory signal transduction (52–54). If so, nuclear localization of full-length OV20.0 could be important to allow interaction with cytokine promoters. Again, there are further precedents from other viruses: the W protein of Nipah virus is localized to the nucleus and suppresses the activation of promoters responding to the stimulation of the Toll-like receptor 3 (TLR3) by dsRNA (55). Another property of VACV E3 related to its localization is that it covalently conjugates with small ubiquitin-like modifiers, such as SUMO-1 or SUMO-2 via a SUMO-interacting motif (SIM), and colocalizes with SUMO-1 to subnuclear domains (56). This interaction with SUMO proteins reduces transcriptional transactivation of p53-regulated genes by E3. Interestingly, OV20.0 was also found in punctate subnuclear foci (Fig. 2A). Further, a comparison of E3 and OV20.0 amino acid sequences found that the conserved lysine residues involved in SUMO modification and the putative SIM domains are conserved between these proteins (56). To elucidate the function of OV20.0 in the nucleus, several issues are critical and require further investigation; these include whether localization of OV20.0 in the subnuclear domains is related to SUMO-1 modification, what possible effect changes in SUMO modification brought by OV20.0 can have on virus replication, and whether the interaction of OV20.0 with protein(s) in the nucleus contributes to its effect on innate antiviral defense.
With the exception of the inhibition of TNF-α noted above, OV20.0 and sh20 appeared to be largely functionally redundant in vitro. This was supported by the generation and testing in vitro of a recombinant ORFV expressing only the sh20 isoform of OV20.0 and suitable control viruses. To extend the study to an in vivo setting, we used a recently described model for ORFV infection in which the ears of mice are scarified with a needle and a drop of virus is added to mimic the natural route of infection (29). These studies demonstrated a very profound role for the full-length OV20.0 for ORFV replication in vivo and pathogenesis. Continuous weight loss was observed in mice infected with recombinant ORFV expressing wild-type OV20.0 (OV20.0-GFP and sh20-Rev), whereas mice infected with sh20-GFP virus began to regain weight at 2 days p.i. Further, sh20-GFP was less able to replicate in mouse ears, with virus loads being under 3 log10 lower than control viruses by 5 days p.i. This is a very rapid difference that suggests a role for full-length OV20.0 in inhibiting very early innate responses to ORFV infection. In line with this, the production of proinflammatory cytokines IL-6 and TNF-α was greatly increased in the sera of mice infected with sh20-GFP compared with that in the sera of mice receiving the control recombinant viruses. These cytokines were also higher in sh20-GFP-infected mice than in mock-infected mice, whereas IL-6 and TNF-α were reduced or unaltered, respectively, by OV20.0-GFP infection. IFN-γ was reduced in the sera of OV20.0-GFP-infected mice, but sh20-GFP was unable to lower the levels of this cytokine. All of this evidence points to an important role for the N-terminal sequences of OV20.0 in modulating production of multiple inflammatory and antiviral cytokines. This is supported by the demonstration that full-length OV20.0, but not sh20, was able to inhibit TNF-α transcription after poly(I·C) stimulation of cells in culture. A virus deficient in OV20.0 would be useful to define the contribution of OV20.0 in PRFV virulence in animals. However, loss of OV20.0 seems detrimental to ORFV in that we were not able to generate an OV20.0 null ORFV that was free of parent virus. We speculate that OV20.0 is the only inhibitor of IFN signaling in ORFV and therefore is essential for growth of the virus and plays an irreplaceable role in counteracting cell-intrinsic antiviral responses.
In conclusion, we have characterized the two isoforms of OV20.0, finding that they perform very comparably in their ability to bind dsRNA and directly interact with PKR, leading to inactivation of this pathway in assays in vitro. In contrast, the sh20 isoform is unable to control production of other cytokines, such as TNF, and this might lead to a requirement for the full-length OV20.0 to maintain ORFV replication in the face of a full host response in vivo.
ACKNOWLEDGMENTS
We thank L. H. Hwang, in the Institute of Microbiology and Immunology, National Yang-Ming University, Taiwan, and L. S. Tiley, in the Department of Veterinary Medicine, University of Cambridge, United Kingdom, for providing Sindbis-GFP virus and vaccinia virus, respectively.
This project was supported by the National Science Council of Taiwan (101-2321-B-005-005, 102–2321-B-005-001, and 103–2321-B-005-001).
REFERENCES
- 1.Gumbrell RC, McGregor DA. 1997. Outbreak of severe fatal orf in lambs. Vet Rec 141:150–151. doi: 10.1136/vr.141.6.150. [DOI] [PubMed] [Google Scholar]
- 2.Pye D. 1990. Vaccination of sheep with cell culture grown orf virus. Aust Vet J 67:182–186. doi: 10.1111/j.1751-0813.1990.tb07751.x. [DOI] [PubMed] [Google Scholar]
- 3.Haig DM, McInnes C, Deane D, Reid H, Mercer A. 1997. The immune and inflammatory response to orf virus. Comp Immunol Microbiol Infect Dis 20:197–204. doi: 10.1016/S0147-9571(96)00045-8. [DOI] [PubMed] [Google Scholar]
- 4.Deane D, McInnes CJ, Percival A, Wood A, Thomson J, Lear A, Gilray J, Fleming S, Mercer A, Haig D. 2000. Orf virus encodes a novel secreted protein inhibitor of granulocyte-macrophage colony-stimulating factor and interleukin-2. J Virol 74:1313–1320. doi: 10.1128/JVI.74.3.1313-1320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang HW, Watson JC, Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A 89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McInnes CJ, Wood AR, Mercer AA. 1998. Orf virus encodes a homolog of the vaccinia virus interferon-resistance gene E3L. Virus Genes 17:107–115. doi: 10.1023/A:1026431704679. [DOI] [PubMed] [Google Scholar]
- 7.Westphal D, Ledgerwood EC, Hibma MH, Fleming SB, Whelan EM, Mercer AA. 2007. A novel Bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J Virol 81:7178–7188. doi: 10.1128/JVI.00404-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buttner M, Rziha HJ. 2002. Parapoxviruses: from the lesion to the viral genome. J Vet Med B Infect Dis Vet Public Health 49:7–16. doi: 10.1046/j.1439-0450.2002.00539.x. [DOI] [PubMed] [Google Scholar]
- 9.Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG. 1990. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 62:379–390. doi: 10.1016/0092-8674(90)90374-N. [DOI] [PubMed] [Google Scholar]
- 10.Saunders LR, Barber GN. 2003. The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J 17:961–983. doi: 10.1096/fj.02-0958rev. [DOI] [PubMed] [Google Scholar]
- 11.Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE. 2005. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 12.Dar AC, Dever TE, Sicheri F. 2005. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 13.Myskiw C, Arsenio J, van Bruggen R, Deschambault Y, Cao J. 2009. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathways. J Virol 83:6757–6768. doi: 10.1128/JVI.02570-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang P, Jacobs BL, Samuel CE. 2008. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J Virol 82:840–848. doi: 10.1128/JVI.01891-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farrell PJ, Balkow K, Hunt T, Jackson RJ, Trachsel H. 1977. Phosphorylation of initiation factor elF-2 and the control of reticulocyte protein synthesis. Cell 11:187–200. doi: 10.1016/0092-8674(77)90330-0. [DOI] [PubMed] [Google Scholar]
- 16.Romano PR, Zhang F, Tan SL, Garcia-Barrio MT, Katze MG, Dever TE, Hinnebusch AG. 1998. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol Cell Biol 18:7304–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu Y, Wambach M, Katze MG, Krug RM. 1995. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology 214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 18.Jacobs BL, Langland JO. 1998. Reovirus sigma 3 protein: dsRNA binding and inhibition of RNA-activated protein kinase. Curr Top Microbiol Immunol 233:185–196. [DOI] [PubMed] [Google Scholar]
- 19.Child SJ, Hakki M, De Niro KL, Geballe AP. 2004. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol 78:197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hakki M, Geballe AP. 2005. Double-stranded RNA binding by human cytomegalovirus pTRS1. J Virol 79:7311–7318. doi: 10.1128/JVI.79.12.7311-7318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bratke KA, McLysaght A, Rothenburg S. 2013. A survey of host range genes in poxvirus genomes. Infect Genet Evol 14:406–425. doi: 10.1016/j.meegid.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haller SL, Peng C, McFadden G, Rothenburg S. 2014. Poxviruses and the evolution of host range and virulence. Infect Genet Evol 21:15–40. doi: 10.1016/j.meegid.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang HW, Jacobs BL. 1993. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology 194:537–547. doi: 10.1006/viro.1993.1292. [DOI] [PubMed] [Google Scholar]
- 24.Kwon JA, Rich A. 2005. Biological function of the vaccinia virus Z-DNA-binding protein E3L: gene transactivation and antiapoptotic activity in HeLa cells. Proc Natl Acad Sci U S A 102:12759–12764. doi: 10.1073/pnas.0506011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haig DM, McInnes CJ, Thomson J, Wood A, Bunyan K, Mercer A. 1998. The orf virus OV20.0L gene product is involved in interferon resistance and inhibits an interferon-inducible, double-stranded RNA-dependent kinase. Immunology 93:335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vijaysri S, Talasela L, Mercer AA, McInnes CJ, Jacobs BL, Langland JO. 2003. The Orf virus E3L homologue is able to complement deletion of the vaccinia virus E3L gene in vitro but not in vivo. Virology 314:305–314. doi: 10.1016/S0042-6822(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 27.Chan KW, Yang CH, Lin JW, Wang HC, Lin FY, Kuo ST, Wong ML, Hsu WL. 2009. Phylogenetic analysis of parapoxviruses and the C-terminal heterogeneity of viral ATPase proteins. Gene 432:44–53. doi: 10.1016/j.gene.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 28.Huang PY, Guo JH, Hwang LH. 2012. Oncolytic Sindbis virus targets tumors defective in the interferon response and induces significant bystander antitumor immunity in vivo. Mol Ther 20:298–305. doi: 10.1038/mt.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cargnelutti JF, Masuda EK, Martins M, Diel DG, Rock DL, Weiblen R, Flores EF. 2011. Virological and clinico-pathological features of orf virus infection in experimentally infected rabbits and mice. Microb Pathog 50:56–62. doi: 10.1016/j.micpath.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Yuwen H, Cox JH, Yewdell JW, Bennink JR, Moss B. 1993. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3L gene. Virology 195:732–744. doi: 10.1006/viro.1993.1424. [DOI] [PubMed] [Google Scholar]
- 31.Langland JO, Jacobs BL. 2004. Inhibition of PKR by vaccinia virus: role of the N- and C-terminal domains of E3L. Virology 324:419–429. doi: 10.1016/j.virol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Gilfoy FD, Mason PW. 2007. West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J Virol 81:11148–11158. doi: 10.1128/JVI.00446-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White SD, Jacobs BL. 2012. The amino terminus of the vaccinia virus E3 protein is necessary to inhibit the interferon response. J Virol 86:5895–5904. doi: 10.1128/JVI.06889-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stacey SN, Jordan D, Williamson AJ, Brown M, Coote JH, Arrand JR. 2000. Leaky scanning is the predominant mechanism for translation of human papillomavirus type 16 E7 oncoprotein from E6/E7 bicistronic mRNA. J Virol 74:7284–7297. doi: 10.1128/JVI.74.16.7284-7297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Racine T, Barry C, Roy K, Dawe SJ, Shmulevitz M, Duncan R. 2007. Leaky scanning and scanning-independent ribosome migration on the tricistronic S1 mRNA of avian reovirus. J Biol Chem 282:25613–25622. doi: 10.1074/jbc.M703708200. [DOI] [PubMed] [Google Scholar]
- 36.Kozak M. 1987. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res 15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hakki M, Marshall EE, De Niro KL, Geballe AP. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol 80:11817–11826. doi: 10.1128/JVI.00957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Child SJ, Geballe AP. 2009. Binding and relocalization of protein kinase R by murine cytomegalovirus. J Virol 83:1790–1799. doi: 10.1128/JVI.01484-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu S, Kaufman RJ. 1997. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem 272:1291–1296. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- 40.Tian B, Mathews MB. 2001. Functional characterization of and cooperation between the double-stranded RNA-binding motifs of the protein kinase PKR. J Biol Chem 276:9936–9944. doi: 10.1074/jbc.M007328200. [DOI] [PubMed] [Google Scholar]
- 41.Zhang F, Romano PR, Nagamura-Inoue T, Tian B, Dever TE, Mathews MB, Ozato K, Hinnebusch AG. 2001. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem 276:24946–24958. doi: 10.1074/jbc.M102108200. [DOI] [PubMed] [Google Scholar]
- 42.Dauber B, Schneider J, Wolff T. 2006. Double-stranded RNA binding of influenza B virus nonstructural NS1 protein inhibits protein kinase R but is not essential to antagonize production of alpha/beta interferon. J Virol 80:11667–11677. doi: 10.1128/JVI.01142-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharp TV, Moonan F, Romashko A, Joshi B, Barber GN, Jagus R. 1998. The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: implications for PKR autoregulation. Virology 250:302–315. doi: 10.1006/viro.1998.9365. [DOI] [PubMed] [Google Scholar]
- 44.Katze MG, Wambach M, Wong ML, Garfinkel M, Meurs E, Chong K, Williams BR, Hovanessian AG, Barber GN. 1991. Functional expression and RNA binding analysis of the interferon-induced, double-stranded RNA-activated, 68,000-Mr protein kinase in a cell-free system. Mol Cell Biol 11:5497–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang HY, Wek RC. 2005. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem 280:14189–14202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- 46.Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. 2004. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dar AC, Sicheri F. 2002. X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol Cell 10:295–305. doi: 10.1016/S1097-2765(02)00590-7. [DOI] [PubMed] [Google Scholar]
- 48.Brandt TA, Jacobs BL. 2001. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J Virol 75:850–856. doi: 10.1128/JVI.75.2.850-856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chelsky D, Ralph R, Jonak G. 1989. Sequence requirements for synthetic peptide-mediated translocation to the nucleus. Mol Cell Biol 9:2487–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rahman MM, Liu J, Chan WM, Rothenburg S, McFadden G. 2013. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog 9:e1003465. doi: 10.1371/journal.ppat.1003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, Jacobs BL, Rich A. 2003. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc Natl Acad Sci U S A 100:6974–6979. doi: 10.1073/pnas.0431131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aminev AG, Amineva SP, Palmenberg AC. 2003. Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res 95:59–73. doi: 10.1016/S0168-1702(03)00163-1. [DOI] [PubMed] [Google Scholar]
- 53.Langland JO, Kash JC, Carter V, Thomas MJ, Katze MG, Jacobs BL. 2006. Suppression of proinflammatory signal transduction and gene expression by the dual nucleic acid binding domains of the vaccinia virus E3L proteins. J Virol 80:10083–10095. doi: 10.1128/JVI.00607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol 77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaw ML, Cardenas WB, Zamarin D, Palese P, Basler CF. 2005. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J Virol 79:6078–6088. doi: 10.1128/JVI.79.10.6078-6088.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gonzalez-Santamaria J, Campagna M, Garcia MA, Marcos-Villar L, Gonzalez D, Gallego P, Lopitz-Otsoa F, Guerra S, Rodriguez MS, Esteban M, Rivas C. 2011. Regulation of vaccinia virus E3 protein by small ubiquitin-like modifier proteins. J Virol 85:12890–12900. doi: 10.1128/JVI.05628-11. [DOI] [PMC free article] [PubMed] [Google Scholar]