

ABSTRACT
The risk of transmission of transmissible spongiform encephalopathies (TSE) between different species has been notoriously unpredictable because the mechanisms of transmission are not fully understood. A transmission barrier between species often prevents infection of a new host with a TSE agent. Nonetheless, some TSE agents are able to cross this barrier and infect new species, with devastating consequences. The host PrPC misfolds during disease pathogenesis and has a major role in controlling the transmission of agents between species, but sequence compatibility between host and agent PrPC does not fully explain host susceptibility. PrPC is posttranslationally modified by the addition of glycan moieties which have an important role in the infectious process. Here, we show in vivo that glycosylation of the host PrPC has a significant impact on the transmission of TSE between different host species. We infected mice carrying different glycosylated forms of PrPC with two human agents (sCJDMM2 and vCJD) and one hamster strain (263K). The absence of glycosylation at both or the first PrPC glycosylation site in the host results in almost complete resistance to disease. The absence of the second site of N-glycan has a dramatic effect on the barrier to transmission between host species, facilitating the transmission of sCJDMM2 to a host normally resistant to this agent. These results highlight glycosylation of PrPC as a key factor in determining the transmission efficiency of TSEs between different species.
IMPORTANCE The risks of transmission of TSE between different species are difficult to predict due to a lack of knowledge over the mechanisms of disease transmission; some strains of TSE are able to cross a species barrier, while others do not. The host protein, PrPC, plays a major role in disease transmission. PrPC undergoes posttranslational glycosylation, and the addition of these glycans may play a role in disease transmission. We infected mice that express different forms of glycosylated PrPC with three different TSE agents. We demonstrate that changing the glycosylation status of the host can have profound effects on disease transmission, changing host susceptibility and incubation times. Our results show that PrPC glycosylation is a key factor in determining risks of TSE transmission between species.
INTRODUCTION
Transmissible spongiform encephalopathies (TSE), or prion diseases, are fatal neurodegenerative diseases that can be sporadic, genetic, or acquired by infection (1). These diseases are characterized by a distinct pathology in the central nervous system (CNS), with neuronal loss, spongiform degeneration, and gliosis (2). Numerous mammalian species are susceptible to infection with TSE agents, such as scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle, Creutzfeldt-Jakob disease (CJD) in humans, and chronic wasting disease (CWD) in cervids.
The host cellular protein PrPC has been shown to have a key role in the transmission of disease (3, 4). During the disease process, PrPC misfolds from the normal conformation to an aberrant form (PrPSc), which is partially resistant to proteases. The prion hypothesis proposes that PrPSc is the infectious agent responsible for disease transmission and that it is able to self-propagate and induce TSE disease in a new host in the apparent absence of any nucleic acid (5).
Transmission of TSE between different species often is limited by a species barrier to infection (6, 7). In experimental models of disease, the species barrier is characterized by an inefficient primary infection with low susceptibility and long incubation times in the new host. Adaptation to the new host then usually occurs in subsequent passages with an increased attack rate and shorter incubation time (6, 8). In naturally occurring TSE, the species barrier prevents transmission of certain agents between different species. However, some agents have been shown to be able to cross this barrier and cause devastating epidemics in a new host. For example, BSE in cattle can be transmitted to humans via the oral route to cause variant CJD (vCJD) (9, 10). BSE also was able to naturally infect a number of different species, such as goats, nyala, kudu, and domestic or captive wild cats (11–13). Understanding how the species barrier is regulated is important, so that the zoonotic potential of a TSE in other animal populations transmitting to humans can be assessed. This is particularly important for newly emergent strains of TSE in both farmed and wild animals (8, 14).
Despite many studies in recent decades, the mechanisms regulating the species barrier to TSE transmission still are elusive. It has been proposed that sequence identity between host and donor PrPC is important to determine the barrier to transmission. In particular, evidence suggests that sequence homology between host PrPC and PrPSc leads to high susceptibility and shorter incubation time, whereas sequence differences between these two proteins can lead to lower susceptibility of the host (6, 15, 16). However, this is not always the case (17–19), and it becomes difficult to predict the transmissibility of a strain in a new recipient based solely on sequence identity between host and donor. It is likely that other factors should be taken in account to understand and predict the species barrier.
PrPC is variably glycosylated at two highly conserved sites (amino acid positions 180 and 196 in mice). N-glycan attachment to these sites results in four glycosylated forms (glycotypes) of PrP, diglycosylated, monoglycosylated at position 180, monoglycosylated at position 196, and unglycosylated. While the ratios of diglycosylated, monoglycosylated, and unglycosylated PrPC remain reasonably constant in uninfected brains, the ratios of PrPSc are highly variable in brains infected with different TSE agents.
In vitro studies have demonstrated that the choice of mutation at the glycosylation sites can have profound effects on PrP trafficking, preventing infection of the resulting mutant mice (20). In order to define the role of glycosylation in the transmission of disease, glycosylation-deficient mice have been produced by three laboratories, and inoculations with a number of TSE agents derived from the same species have been carried out (20–22). Conventional transgenic mice expressing hamster PrP in which the first N-glycan site (T183A) or both the first and second site (T183A, T199A) were disrupted were resistant to hamster-passaged prion strains Sc237 and 139H. Mice devoid of the second site also were resistant to 139H but susceptible to Sc237, with extended incubation periods compared to those of the controls (21). In contrast, transgenic mice expressing a 3F4-tagged murine PrP devoid of the first site (T182N) showed no evidence of resistance to any of the murine agents ME7, 139A, and murine-passaged BSE (301C) (20). Mice lacking the second site (T198A) also were susceptible to these strains with extended incubation times (20). Cancellotti et al. produced three inbred lines of gene-targeted mice in which the first (G1; N180T), second (G2; N196T), or both (G3; N180T and N196T) N-glycan attachment sites were disrupted. The homozygous transgenic mice produce partially or unglycosylated PrPC under the control of the endogenous mouse Prnp promoter (22). Mice lacking the first N-glycan site were susceptible to 79A, but unlike those of Neuendorf et al., they were resistant to ME7. Mice lacking the second site were susceptible to a number of TSE strains, including ME7, 79A, and 301C, similar to those of Neuendorf et al. However, extended incubation periods were observed for all strains in Neuendorf et al. but only in the case of 301C in Tuzi et al. (20, 23). The differences between the lines of mice can be attributed to many variables, including the different point mutations used to disrupt the glycosylation sites, the species of origin of the PrP gene, different gene constructs, the addition of epitopes, copy numbers, and integration sites, and strain differences of the inocula. The gene-targeted mice, however, remove at least some of the variability observed with the conventional lines of mice.
Multiple strains of TSE agents have been identified. Strains differ in their disease characteristics, such as their transmissibility to other host species or the degree of pathology induced in the brain of the host (24–26). These different strains are proposed to result from different conformations of PrPSc (27–29). The ratio of the glycotypes of PrPSc also differs between TSE strains (30, 31). For example, PrPSc associated with vCJD is predominately diglycosylated, whereas that associated with sporadic CJD (sCJD) is mostly monoglycosylated (32). The N-glycans attached to PrP may influence the conformational flexibility of the protein and could influence its misfolding (33, 34). Conformational flexibility may be particularly important during interspecies transmission of TSE, as the infectious agent must adapt to the novel source of PrPC. Indeed, in vitro interspecies misfolding of PrPC induced by PrPSc is inhibited specifically by glycosylation of the protein (35). Moreover, the replication of a given strain may require the misfolding of a precise combination of PrPC glycotypes to replicate the glycoform (36). For example, the replication of a strain in which PrPSc is predominately monoglycosylated may be favored in a host that produces an elevated level of monoglycosylated PrPC.
Here, we have tested the effect of host PrPC glycosylation on the TSE species barrier. This is the first in vivo study of the role of glycosylation of PrPC on the transmission of TSE agents between species. Previous in vitro and in vivo studies to investigate this issue have provided inconsistent results (35, 37, 38) or used mouse-passaged TSE agents (20, 21). Some studies have suggested that the glycosylation of PrPC impedes the transmission between host species (35), whereas others have shown no such effect (20, 38).
We challenged three lines of gene-targeted glycosylation mutants (22) with three nonmurine TSE agents: human vCJD, human sCJD (sCJDMM2), and hamster scrapie 263K. These agents differ in their PrPSc glycoform ratios and their relative transmissibility to wild-type mice (9, 39, 40). Two of the agents used in this study (263K and vCJD) have similar PrPSc glycoform ratios in which PrPSc is predominately diglycosylated. The sCJDMM2 agent has an amino acid sequence identical to that of the vCJD agent and a cleavage site similar to that of proteinase K (PK); however, it has a significantly different PrPSc glycoform ratio, as the PrPSc associated with this agent is predominately monoglycosylated. By comparing the relative transmissibility of these TSE agents to our transgenic models, we have established the importance of host PrPC glycosylation in determining the transmissibility of a TSE strain across a species barrier.
MATERIALS AND METHODS
Preparation of TSE inocula and intracerebral injection.
Inocula were prepared from brain tissue from a patient with pathologically confirmed sCJDMM2 (0.1% [wt/vol] in physiological saline), the National Institute for Biological Standards and Control (NIBSC) variant CJD reference case (0.1% [wt/vol] in physiological saline), and a hamster with clinical scrapie, strain 263K (1% [wt/vol] in physiological saline). Consent for the use of these materials for research was obtained with ethics approval by the Lothian National Health Service Research Ethics Committee (reference no. 2000/4/157). G1 (N180T), G2 (N196T), and G3 (N180T, N196T) transgenic and wild-type mice were genotyped by mismatched PCR as previously described (22) and coded prior to intracerebral (i.c.) injection with 20 μl of inoculum. All groups were age and sex matched.
The second passage was carried out in a similar manner using brain material from selected G2 and wild-type mice showing evidence of TSE vacuolation and/or PrP deposition. A second passage was not carried out in G1 or G3 mice due to extremely low numbers of mice exhibiting TSE vacuolation and/or PrP deposition. Inocula were prepared from mouse brain tissue at 0.1% (wt/vol) in physiological saline, and mice were inoculated via the i.c. route with 20 μl. Animal experiments were approved by The Roslin Institute's Ethical Review Board and were conducted according to the regulations of the 1986 United Kingdom Home Office Animals (Scientific Procedures) Act.
Scoring of clinical TSE disease and pathology.
Mice were scored weekly for clinical signs from 100 days postinoculation (dpi) by operators blind to animal genotype according to a previously established TSE clinical scoring system (41). Mice were scored as unaffected, possibly affected, or definitely affected using standard criteria, including kyphosis, ataxia, paralysis, hyperactivity, urinary incontinence, and weight loss. Any unusual clinical signs were noted. In older animals, signs of aging (kyphosis, weight loss, and reduced activity) were taken into account and were classified as possibly affected due to the similar nature of aging and TSE disease phenotypes. Mice were sacrificed after (i) two consecutive scores of definitely affected, (ii) after receiving scores of definitely affected in 2 out of 3 weeks, or (iii) significant deterioration of condition. Mice with no signs of clinical disease were maintained until approximately 700 dpi, at which point the studies were terminated. Animals in which clinical signs were present without pathological (TSE vacuolation and/or PrP deposition) confirmation were removed from the analysis, as these signs also can be due to other conditions, such as aging, as detailed above. Incubation periods were calculated as the number of days between injection and the clinical endpoint in animals with TSE vacuolation. In the absence of an incubation period, the survival time was calculated in days.
Half brains were fixed in formal saline for 48 h and decontaminated with formic acid, when required, prior to paraffin embedding. Coronal sections were cut and stained with hematoxylin and eosin, and TSE-specific vacuolation was semiquantitatively scored blind to TSE strain and mouse genotype by standard methods (42). Vacuolation profiles were plotted for groups of 6 mice or more. Genotypes of all mice were confirmed postmortem by PCR (22). Early intercurrent deaths (under 200 dpi for the first pass and 100 dpi for the second pass) were excluded from the study.
Biochemical assessment of PrPSc in first-pass mice.
Half brains from mice challenged with either sCJDMM2, vCJD, or 263K were used to prepare a 1% (wt/vol) inocula in physiological saline; from this, PrPSc was extracted using extraction buffer (NP-40, sodium deoxycholate, sodium chloride, Tris [pH 7.4]) and digested with PK (37°C for 1 h, 20 μg/ml). Reactions were terminated by the addition of phenylmethylsulfonyl fluoride. If required, PrPSc then was concentrated by sodium phosphotungstic acid (NaPTA)-mediated precipitation (43). Samples (brain homogenate/NaPTA-concentrated PrPSc) were analyzed for PrPSc by Western blotting using the anti-PrP antibody 8H4 (1/10,000) (44). PrPSc could not be easily detected in vCJD-challenged G2 mice. In order to detect PrPSc in G2 mice, NaPTA-mediated precipitation was carried out on 10% (wt/vol) brain homogenates such that the equivalent of 5 μg of total brain was run per G2 well. Equal PrP signal then was achieved by diluting wild-type PrPSc 10-fold prior to electrophoresis.
Immunohistochemistry.
Coronal brain sections were stained using the 6H4 antibody (1/20,000; Prionics) to detect PrP. Antigen retrieval by autoclaving at 121°C for 15 min and a 5-min formic acid (98%) treatment was used. Sections then were blocked with normal rabbit serum prior to incubation with the primary antibody. Antibody binding was detected with either the catalyzed signal amplification system (Dako) or Vector ABC kit (Vector Laboratories) and visualized with 3,3′-diaminobenzidine chromogen. All sections were counterstained with hematoxylin. In all experiments, normal brain homogenate-inoculated mouse and PrP−/− mouse (NPU, Edinburgh, United Kingdom) sections were used as a negative control (4).
Biochemical assessment of PrPC in uninfected glycosylation-deficient transgenic mice.
Brains from uninfected G1, G2, G3, and wild-type mice were used to prepare 10% (wt/vol) homogenates. An α-tubulin loading control was included to determine variation in the amount of protein being loaded. Samples were analyzed by Western blotting using seven primary anti-PrP antibodies (7A12/epitope, conformation epitope; DE3/epitope, aa146-153; FH10/epitope, aa202-210; AE11/epitope, aa140-145; BH1/epitope, aa143/154; FD12/epitope, unknown at 0.1 μg/ml; and BC6/epitope, aa146-156 at 0.01 mg/ml) (45, 46) and species-specific peroxidase-conjugated AffiniPure antibodies (Stratech) at 0.05 μg/ml diluted in 0.5% blocking solution for 30 min at room temperature. Bound secondary antibody was detected by light emission from SuperSignal West Dura chemiluminescent substrate using a Kodak 440 Image Station with typical exposures of 1, 5, and 10 min. Immunoblots were imaged using the Kodak Image Station and Kodak MI software. Protein bands were selected manually by drawing around them and the sum intensity calculated. A blank region also was selected to measure the background intensity of the blot.
RESULTS
Absence of the first and both PrPC glycosylation sites limits transmission with all three agents.
Infection of G1 and G3 mice with vCJD and sCJDMM2 resulted in no pathologically confirmed clinical disease and no evidence of TSE vacuolation and/or PrP deposition in the brain (Tables 1 and 2). While there were a number of G1 mice (4/14) with TSE vacuolation and/or PrP deposition after 263K infection, again there were no cases of clinical disease or TSE vacuolation and/or PrP deposition in G3 mice (Table 3). Thus, the absence of either the first or both PrPC glycosylation sites in the host appears, particularly in the case of vCJD and sCJDMM2, to restrict replication of the agent within the CNS compared to that of the wild-type mice.
TABLE 1.
Sporadic CJDMM2 inoculation into glycosylation-deficient mice at first and seconda passages
| Mouse line | Inoculum | Incubation timeb (days) ± SEM (range) | Clinical disease (no. positive/total no.) | TSE vac and/or PrP depositionc | Survival time (days) of mice with TSE vac and/or PrPd |
|---|---|---|---|---|---|
| G1 | sCJDMM2 | 0/18 | 0/18 | ||
| G2 | sCJDMM2 | 404 ± 8 (378–430) | 7/20 | 11/20 | 367 ± 50 (245–494) |
| G3 | sCJDMM2 | 0/18 | 0/18 | ||
| Wt | sCJDMM2 | 0/41 | 3/41 | 658 ± 49 (559–707) | |
| G2 | G2sCJDa | 129 ± 3 | 11/11 | 11/11 | |
| Wt | G2sCJDa | 430 ± 20 | 12/12 | 12/12 | |
| G2 | G2sCJDb | 147 ± 3 | 10/11 | 11/11 | |
| Wt | G2sCJDb | 439 ± 22 | 10/10 | 10/10 |
Brain material from two sCJDMM2-inoculated G2 mice with pathological evidence of disease were further passaged into G2 and wild-type mice.
Incubation time for mice with clinical signs and evidence of TSE vacuolation. Studies were maintained to ∼700 dpi for mice with no clinical signs of TSE disease.
TSE vacuolation (vac) and/or PrP deposition were used as evidence of transmission for primary passage due to low numbers of mice with clinical signs and vacuolation present. Limited confirmatory immunohistochemistry for PrP deposition was carried out in the second passage due to high numbers of mice with clinical signs and vacuolation present.
Survival time of mice with no clinical signs and evidence of vacuolation and/or PrP deposition.
TABLE 2.
Variant CJD inoculation into glycosylation-deficient mice at first and seconda passages
| Mouse line | Inoculum | Incubation timeb (days) ± SEM (range) | Clinical disease (no. positive/total no.) | TSE vac and/or PrP depositionc | Survival time (days) of mice with TSE vac and/or PrPd |
|---|---|---|---|---|---|
| G1 | vCJD | 0/17 | 0/17 | ||
| G2 | vCJD | 536 ± 16 (510–600) | 8/18 | 14/18 | 611 ± 29 (505–708) |
| G3 | vCJD | 0/19 | 0/19 | ||
| Wt | vCJD | 477 ± 15 (380–616) | 21/40 | 36/40 | 483 ± 18 (310–616) |
| G2 | G2vCJDa | 283 ± 3 | 10/11 | 11/11 | |
| Wt | G2vCJDa | 192 ± 2 | 12/12 | 12/12 | |
| G2 | G2vCJDb | 315 ± 3 | 11/12 | 12/12 | |
| Wt | G2vCJDb | 193 ± 3 | 12/12 | 12/12 | |
| G2 | WtvCJD | 334 ± 6 | 11/13 | 13/13 | |
| Wt | WtvCJD | 156 ± 1 | 12/12 | 12/12 |
Brain material from two vCJD-inoculated G2 mice and one vCJD inoculated wild-type mouse with pathological evidence of disease were further passaged into G2 and wild-type mice.
Incubation time for mice with clinical signs and evidence of TSE vacuolation. Studies were maintained to ∼700 dpi for mice with no clinical signs of TSE disease.
TSE vacuolation and/or PrP deposition were used as evidence of transmission for primary passage due to low numbers of mice with clinical signs and vacuolation present. Limited confirmatory immunohistochemistry for PrP deposition was carried out in the second passage due to high numbers of mice with clinical signs and vacuolation present.
Survival time of mice with no clinical signs and evidence of TSE vacuolation and/or PrP deposition.
TABLE 3.
263K inoculation into glycosylation-deficient mice at first and second passagesa
| Mouse line | Inoculum | Incubation timeb (days) ± SEM (range) | Clinical disease (no. positive/total no.) | TSE vac and/or PrP depositionc | Survival time (days) of mice with TSE vac and/or PrPd |
|---|---|---|---|---|---|
| G1 | 263K | 0/14 | 4/14 | 579 ± 35 (507–674) | |
| G2 | 263K | 0/18 | 11/18 | 552 ± 18 (461–629) | |
| G3 | 263K | 0/16 | 0/16 | ||
| Wt | 263K | 0/18 | 11/18 | 612 ± 18 (503–698) | |
| G2 | G2–263Ka | 284 ± 7 | 12/12 | 12/12 | |
| Wt | G2–263Ka | 437 ± 32 | 10/12 | 9/12 | |
| G2 | G2–263Kb | 320 ± 7 | 11/11 | 11/11 | |
| Wt | G2/263Kb | 536 ± 6 | 4/10 | 6/10 | 343 ± 61 (282–408) |
| G2 | Wt263K | 365 ± 0 | 11/12 | 12/12 | |
| Wt | Wt263K | 172 ± 5 | 9/10 | 10/10 |
Brain material from two 263K-inoculated G2 mice and one 263K-inoculated wild-type mouse with pathological evidence of disease were further passaged into G2 and wild type mice
Incubation time for mice with clinical signs and evidence of TSE vacuolation. Studies were maintained to ∼700 dpi for mice with no clinical signs of TSE disease.
TSE vacuolation and/or PrP deposition were used as evidence of transmission for primary passage due to low numbers of mice with clinical signs and vacuolation present. Limited confirmatory immunohistochemistry for PrP deposition was carried out in the second passage due to high numbers of mice with clinical signs and vacuolation present.
Survival time of mice with no clinical signs and evidence of TSE vacuolation and/or PrP deposition.
Absence of sugars at the second glycosylation site of PrPC in the host removes the barrier to infection with sCJD.
G2 transgenic mice were susceptible to sCJDMM2, in contrast to the G1, G3, and wild-type mice (Table 1; also see Fig. 2D). Seven out of 20 of the sCJDMM2-challenged G2 transgenic mice developed clinical disease with an average incubation time of 404 ± 8 days (Table 1). Disease status was confirmed by pathology within the brain in which both TSE vacuolation and PrP deposition were observed (Fig. 1A and 2A). PK-resistant PrPSc also was detected by Western blotting in brains of these mice (Fig. 3A). The molecular weight of PrPSc in these samples is consistent with the majority of PrPSc being monoglycosylated, as observed in previous studies using these mice (23). Thus, removal of the second glycosylation site of the host PrPC has rendered the host susceptible to cross-species transmission with sCJDMM2.
FIG 2.

PrP deposition in the brains of wild-type and G2 mice after intracerebral inoculation with sCJDMM2, vCJD, or 263K agent. (A) G2 mouse inoculated with sCJDMM2; (B) G2 mouse inoculated with vCJD; (C) G2 mouse inoculated with 263K; (D) wild-type mouse inoculated with sCJDMM2; (E) wild-type mouse inoculated with vCJD; (F) wild-type mouse inoculated with 263K. Arrows indicate examples of PrP accumulation in the form of plaque-like deposits in panels A and B and examples of fine punctate PrP accumulation in panels C, E, and F. No PrP accumulation was detected in panel D. PrP was detected with 6H4 antibody. dg, dentate gyrus; cc, corpus callosum. Scale bars, 500 μm.
FIG 1.

Lesion profile analysis of wild-type (Wt) and G2 mice after intracerebral inoculation with sCJDMM2 (A), second passage of sCJDMM2 (B), vCJD (C), and second passage of vCJD (D). The second passage was carried out from selected G2 and wild-type mice showing TSE vacuolation and/or PrP. Group size, n ≥ 6 (± standard errors of the means). Gray matter scoring regions are labeled G1 to G9: G1, dorsal medulla; G2, cerebellar cortex; G3, superior colliculus; G4, hypothalamus; G5, thalamus; G6, hippocampus; G7, septum; G8, retrosplenial cortex; G9, cingulate and adjacent motor cortex. White-matter scoring regions are labeled W1 to W3: W1, cerebellar white matter; W2, mesencephalic tegmentum; W3, pyramidal tract.
FIG 3.

Biochemical analysis of PrPSc from the brains of G2 and wild-type (Wt) mice inoculated with sCJDMM2, vCJD, and 263K. (A) PrPSc was isolated from four sCJD-challenged G2 mice, one vCJD-challenged G2 mouse (assayed in duplicate), and one vCJD-challenged wild-type mouse (assayed in duplicate) by standard PK digestion. PrPSc was concentrated by NaPTA precipitation. The amount of PrPSc in the vCJD-challenged G2 sample was too low to detect. Uninfected mice and a 263K-challenged wild-type mouse were included as controls. (B) PrPSc was isolated from vCJD-challenged G2 mice by standard PK digestion followed by NaPTA precipitation. The amount of PrPSc was equalized by dilution following NaPTA precipitation to allow comparison (equivalent of 5 μg of G2 brain and 0.5 to 1 μg of wild-type brain). Uninfected mice were included as controls. (C) PrPSc was isolated from three 263K-challenged G2 mice and three wild-type mice by standard PK digestion. Uninfected mice and a 263K-challenged hamster were included as controls. The different isoforms of PrP are denoted Di (for diglycosylated), Mono (monoglycosylated), and Un (unglycosylated). PrPSc was detected with 8H4.
Brain material from two G2 mice that had developed clinical disease following inoculation with sCJDMM2 (G2-sCJD) was used to challenge G2 and wild-type mice. All challenged G2 mice rapidly developed a clinical disease with remarkably short incubation times of 129 ± 3 days and 147 ± 3 days (Table 1). Wild-type mice that previously were resistant to infection with the sCJDMM2 agent developed a clinical disease after challenge with G2-sCJD brain; however, in this case the incubation time was much longer than that for G2 recipients (430 ± 20 days and 439 ± 22 days) (Table 1).
The pathological signs of disease differed in the two hosts, with the vacuolation profile in G2 mice distinct from that observed in wild-type mice (Fig. 1B). The intensity of TSE vacuolation in the superior colliculus was lower in G2 mice than in wild-type animals infected with G2-sCJD brain material. More intense TSE vacuolation occurred in the cingulate cortex of the G2 mice than in wild-type animals after challenge with G2-sCJD brain material (Fig. 1B). Primary and secondary passage of sCJDMM2 in G2 mice showed a similar trend in vacuolation intensity throughout the brain (Fig. 1A and B). The lack of the second glycosylation site in the host appears to greatly facilitate the infection with sCJD, suggesting that the diglycosylated form of host PrP is important in maintaining the species barrier of a predominantly monoglycosylated strain.
Diglycosylated PrPC in the host influences incubation time in the interspecies transmission of vCJD.
Variant CJD PrPSc has an identical amino acid sequence and PK cleavage pattern by Western blotting similar to that of PrPSc from sCJDMM2 but is predominately diglycosylated compared with the predominantly monoglycosylated sCJDMM2. Moreover, vCJD has been shown to transmit readily to wild-type mice, in contrast to sCJDMM2. Twenty-one out of 40 wild-type mice challenged with vCJD developed clinical disease with an average incubation time of 477 ± 15 days. A similar proportion of G2 mice (8/18) developed clinical disease, with an average incubation time of 536 ± 16 days (Table 2).
The pattern of TSE vacuolation was similar in G2 transgenic and wild-type mice infected with vCJD (Fig. 1C). However, as shown in Fig. 2B and E, there is less PrP deposition in the brains of animals expressing monoglycosylated PrP (G2) than in those of the wild-type mice. Diffuse deposition of PrP was observed in the brains of wild-type mice. In contrast, small plaque-like deposits of PrP were observed in the brains of G2 transgenic mice. In order to detect PrPSc in the G2 mouse brain, NaPTA precipitation of PrPSc was required, followed by equalized PrPSc loading (5 μg of G2 brain versus 1 μg of wild-type brain), indicating that there was less PrPSc in G2 mice than in wild-type mice (Fig. 3B).
To further investigate the effect of the second site on vCJD transmission, we inoculated brain material from two G2 mice (G2-vCJD with monoglycosylated PrPSc) and a wild-type mouse (Wt-vCJD with fully glycosylated PrPSc) previously infected with vCJD into G2 and wild-type recipients. Wild-type mice expressing fully glycosylated PrPC developed clinical disease within 192 ± 2 days and 193 ± 3 days of infection with two independent G2-vCJD isolates. Two groups of G2 mice that were challenged with these isolates developed clinical disease later than the wild-type controls (283 ± 3 days and 315 ± 3) (Table 2). The same pattern also was observed when mice were challenged with the Wt-vCJD brain material; G2 mice developed a clinical disease later (334 ± 6 days) than wild-type mice (156 ± 1 days) (Table 2). Despite changes in incubation times, the pattern of TSE vacuolation appeared to be similar between G2 and wild-type mice and between primary and secondary passages of vCJD (Fig. 1C and D).
Glycosylation compatibility between the hamster strain 263K and host PrPC reduces the species barrier.
Primary transmission was carried out with the hamster TSE strain 263K. PrPSc associated with the 263K agent is predominately diglycosylated. In contrast to vCJD, wild-type mice do not develop a clinical disease with this TSE strain but do show evidence of PK-resistant PrPSc in the brain following inoculation (39, 47). After challenge with 263K, no wild-type or G2 mice exhibited clinical disease, consistent with previous data. However, both G2 and wild-type mice demonstrated pathological signs of a subclinical infection (G2, 11/18; wild type, 11/18) (Table 3 and Fig. 2C and F). PK-resistant PrPSc also was detected in the brains of these mice by Western blotting (Fig. 3C). The PrPSc glycoprofile in these samples is consistent with the majority of PrPSc being monoglycosylated in G2 recipients and fully glycosylated in wild types.
G2 and wild-type mice were challenged by an i.c. route with brain homogenate from two 263K-inoculated G2 mice (G2-263K with monoglycosylated PrPSc) and a 263K-infected wild-type mouse (Wt-263K with fully glycosylated PrPSc). In contrast to the vCJD transmissions, G2 mice had a higher susceptibility to disease after challenge with G2-263K than wild-type mice; all G2 mice succumbed to disease after challenge, whereas 10/12 (with the first G2 brain) and 4/10 (with the second G2 brain) wild-type mice developed clinical signs of TSE disease after G2-263K infection (Table 3). Moreover, G2 mice developed disease more quickly (284 ± 7 days and 320 ± 7 days) than wild-type mice (437 ± 32 days and 536 ± 6 days) after challenge with G2-263K (Table 3). The differences in the attack rate and incubation periods elicited by the two isolates of G2-263K may occur because the two animals from which they were derived were at different stages of subclinical infection; hence, they had different titers of infectivity within their brains. The G2 mice challenged with Wt-263K developed disease later (365 ± 0 days) than wild-type mice (172 ± 5 days) (Table 3). Thus, 263K transmits with similar efficiency in the G2 mice and the wild-type mice in the primary passage, but unlike vCJD on the second passage following G2 transmission, this agent was considerably faster in the G2 host than in the wild-type host.
PrPC expression is reduced in glycosylation-deficient mice.
To ascertain whether the protein levels of PrPC in any of the lines of mice could influence the incubation times in the transmissions described above, we undertook analysis of the PrPC levels in all mouse lines. Immunoblot analysis showed the expected banding patterns for both the transgenic and wild-type mice. Truncated PrPC, designated C1, was observed in all mice and was included in the measurement of total PrPC (selected antibodies are shown in Fig. 4A and B). All of the glycosylation-deficient mice expressed significantly less PrPC than wild-type mice (P < 0.0001). G1 and G2 mice expressed approximately 50% and G3 mice only 32% of that found in wild-type mice, with G3 mice expressing significantly less (P < 0.01) than both G1 and G2 mice. There was no significant difference in total PrP in each mouse line using different antibodies (Fig. 4A and B).
FIG 4.
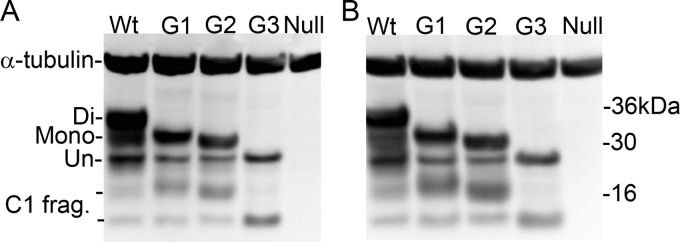
PrPC expression levels in the brains of glycosylation-deficient transgenic mice. Representative Western blots showing the different PrPC isoforms in wild-type (Wt), G1, G2, and G3 mice using BC6 (A) and BH1 (B) antibodies. Western blots underwent densitometry to measure levels of PrPC. α-Tubulin was used as a loading control. The different isoforms of PrPC are denoted Di (for diglycosylated), Mono (monoglycosylated), Un (unglycosylated), and C1 frag. (C1 fragments).
DISCUSSION
Expression of PrPC is known to influence incubation times of a TSE disease, with reduced levels of the protein resulting in longer incubation periods (48). Earlier studies showed conflicting results over whether PrPC expression levels are altered within the glycosylation transgenic mice (22). This most likely was due to the epitope recognition of the antibodies used and detection of only a subset of isoforms. Our expanded studies here, using a range of monoclonal antibodies within the C-terminal and central region of PrPC, are able to detect all isoforms of PrPC, demonstrating that G1, G2, and G3 mice do have lower levels of PrPC expression than wild-type mice. However, while lower levels of PrPC in the G1 and G3 mice may contribute to longer incubation times, the levels observed in these mice are not likely to explain the resistance to TSE disease observed here. Studies have shown that mice heterozygous for PrPC expression and with a level of PrPC expression similar to that of the G1 mice are fully susceptible to TSE disease, albeit with incubation periods of almost twice that of wild-type mice (48–50). Our studies were maintained to approximately 700 dpi, almost twice the incubation period of sCJD in G2 mice, which also show 50% PrPC expression. Thus, factors other than a reduction in PrPC expression level are likely to contribute to the resistance of these mice to TSE disease. While the lower expression of PrPC in G2 mice may contribute to the longer incubation period observed in this model after challenge with vCJD, the G2 mice are more susceptible to infection with the sCJDMM2- and G2-passaged 263K TSE agents despite expressing lower levels of PrPC than wild-type controls. Therefore, this enhanced susceptibility can be directly attributed to the altered glycosylation status of the host.
The monoglycosylated sCJDMM2 agent was transmitted to a normally resistant host (51) by removal of the glycans at PrP residue 196 (as removed in G2 mice). Moreover, sCJDMM2 became adapted in the G2 host and produced very short incubation times on the second pass. The data suggest that the presence of glycans at PrP residue 196 (as present in G1 or wild-type mice) is responsible for the sCJDMM2 transmission barrier; removal of this site may facilitate the interaction between host monoglycosylated PrPC and the infective monoglycosylated PrPSc, allowing replication of the infective agent. This is the first time that glycosylation-deficient transgenic mice have shown an enhanced susceptibility to TSE infection compared to that of wild-type mice. This suggests that glycosylation at the second glycosylation site can protect against transmission both between and within species.
Experimental transmissions from wild-type or G2 mice infected with the 263K strain provide additional evidence that similar glycosylation statuses of host PrPC and the PrPSc in the inoculated strain can greatly accelerate TSE incubation periods. Indeed, the incubation period in G2 recipients was almost half that of wild-type mice after challenge with the G2-263K strain.
In both primary and secondary passages of vCJD, incubation periods were shorter in wild-type mice than in mice in which the second PrPC N-glycan attachment site was disrupted. The shorter incubation periods were observed irrespective of the glycosylation status of the second site in the infecting PrPSc. While these differences in incubation time can be explained on the basis of lower PrPC expression levels in the G2 mice, we cannot discount the possibility that it indicates a preference of this strain for a PrPC diglycosylated host irrespective of the passage history of the strain. This may explain the ability of this agent to infect a large number of host species and its transmissibility across many species barriers.
G1 and G3 mice showed little susceptibility to infection throughout this study. Indeed, these transgenic mice did not develop any pathologically confirmed clinical TSE disease after inoculation with any of the three agents used, although asymptomatic infection in the form of PrP deposition was detected in extremely low numbers. This may be linked to an inability of this particular host PrPC to propagate nonmurine strains; previous experiments performed with a number of mouse-adapted scrapie strains by several routes have highlighted an intrinsic resistance of both G1 and G3 mice to infection (23, 52). Therefore, it is more likely that the resistance observed in G1 and G3 mice in this study is linked to a more general mechanism rather than an effect of the species barrier. Why the absence of the first glycosylation site should lead to such a dramatic loss of host susceptibility may be related to the conversion efficiency of PrPC to PrPSc. Some in vitro conversion assays have previously suggested that glycosylation inhibits the conversion activity (30). However, such in vitro systems have not revealed the complexity of the glycosylation issue observed in these in vivo studies. The resistance observed in the G3 mice likely is related to the absence of the G1 glycosylation. However, G3 mice also show more C1-truncated PrPC upon biochemical analysis than G1, G2, and wild-type mice. Previous in vitro studies have shown that higher levels of C1 PrPC are associated with resistance to TSE infection (53). In addition, G3 mice show the lowest PrPC expression of the three glycosylation mutants and a different PrPC localization (22). All of these factors might contribute to the resistance to TSE infection of this specific line of mice.
The absence of glycans at the second site may alter the biology of PrPC or PrPSc interaction in a very different way than that of the first glycosylation site. A number of biochemical properties and the cellular localization of PrPC in the G2 mice resemble that observed in wild-type and G1 mice (22). However, the presence/absence of carbohydrates in a specific portion of PrPC may influence other characteristics, such as the ultrastructural localization of PrPC (e.g., localization in a different portion of the cell membrane) or its conformation, and this may dictate the different susceptibility to infection of the G2 mice compared to that of the G1 mice.
We have argued that altered glycosylation status of PrPC alters the host susceptibility. An alternative explanation is that the point mutations inserted in order to modify the N-linked glycosylation sites on PrP are the cause of this change (22). Previous transmission studies performed by us (23) and Neuendorf et al. (20) have shown similar results upon primary passage of both ME7 and mouse BSE strains with prolonged incubation periods in mice deficient at the first glycosylation site despite utilizing different amino acid substitutions and expressing different levels of PrP. In addition, Ikeda et al. (54) showed that substitution of Asn residues to abolish glycosylation sites does not prevent conversion of PrPC to PrPSc. In this study, the differences between the wild-type and G2 hosts in susceptibility to primary passage with two human agents, vCJD and sCJDMM2 (characterized by an identical PrPSc sequence and PK cleavage pattern but a different glycoprofile), further argues for the glycosylation status being the main determinant of host susceptibility rather than the change in amino acid sequence.
The deposition of PrP in the brains of G2 mice infected with vCJD differed from that observed in wild-type mice infected with the same agent. First, the total amount of PrP that accumulated by disease endpoint appeared to be lower in G2 mice. This could be due to less PrPC being available for replication, or it could mean that the rates of misfolding, clearance, and/or toxicity of PrP are changed in the absence of glycosylation at the second site. In addition, small PrP aggregates were observed in G2 mice infected with vCJD, in contrast to the diffuse PrP deposition observed in wild-type mice. Large aggregated deposits of PrP also were observed in G2 mice challenged with sCJDMM2. These data suggest that PrPC that lacks the second glycosylation site has altered misfolding or clearance kinetics, which also may have an important effect on host susceptibility.
In summary, we propose that the transmission of TSE agents across different species can be profoundly influenced by posttranslational events in both PrPC and PrPSc. In particular, we have demonstrated that the glycosylation status of host PrPC (55, 56) can dramatically alter cross-species transmission characteristics and likely is important for this protein to act as a receptor for the incoming TSE agent.
On the other hand, the prevalence of certain PrPSc glycotypes in an infectious inoculum may determine its conformation and the ability to interact with the host and cause a TSE infection. This combination may lead to the binding between PrPSc and PrPC occurring through direct interactions between the glycan residues and/or different PrP regions that have been recently suggested to be important for TSE transmission between different species (57) or by interactions with a number of conversion cofactors previously suggested, such as host proteins or nucleic acids (58–60).
The dramatic effects in altered host susceptibility, in particular the resistance of the G1 and G3 mice to infection, suggests this mechanism provides an important focus for blocking the disease process and protecting the infected individual from neurodegeneration.
ACKNOWLEDGMENTS
We acknowledge the excellent technical assistance of Irene McConnell, Val Thomson, Sally Carpenter, Kris Hogan, Gillian Macgregor, Sandra Coupar, Dorothy Kisielewski, and Winggee Liu and the statistical analysis assistance of Jill Sales, BIOSS. We thank Robert Somerville, Wilfred Goldmann, Rona Barron, and Nadia Tuzi for valuable discussions. Antibodies 8H4 and 7A12 were a kind gift of M. S. Sy, University of Cleveland.
This work was supported by the BBSRC and MRC. The NCJDRSU Brain Bank is part of the Edinburgh Brain Bank, which is funded by MRC. F.W. was funded by a Wellcome Trust Ph.D. studentship (069283). K.I. was funded by a BBSRC studentship.
NCJDRSU is supported by the Scottish Government and the Department of Health, England. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health. The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any agency determination or policy.
REFERENCES
- 1.Prusiner SB. 1998. Prions. Proc Natl Acad Sci U S A 95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Will RG. 2003. Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull 66:255–265. doi: 10.1093/bmb/66.1.255. [DOI] [PubMed] [Google Scholar]
- 3.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. 1993. Mice devoid of PrP are resistant to scrapie. Cell 73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 4.Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 1994. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 5.Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 6.Beringue V, Vilotte JL, Laude H. 2008. Prion agent diversity and species barrier. Vet Res 39:47. doi: 10.1051/vetres:2008024. [DOI] [PubMed] [Google Scholar]
- 7.Castilla J, Gonzalez-Romero D, Saa P, Morales R, De Castro J, Soto C. 2008. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 134:757–768. doi: 10.1016/j.cell.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manson JC, Cancellotti E, Hart P, Bishop MT, Barron RM. 2006. The transmissible spongiform encephalopathies: emerging and declining epidemics. Biochem Soc Trans 34:1155–1158. doi: 10.1042/BST0341155. [DOI] [PubMed] [Google Scholar]
- 9.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. 1997. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 10.Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. 1996. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347:921–925. doi: 10.1016/S0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 11.Kirkwood JK, Cunningham AA. 1994. Epidemiological observations on spongiform encephalopathies in captive wild animals in the British Isles. Vet Rec 135:296–303. doi: 10.1136/vr.135.13.296. [DOI] [PubMed] [Google Scholar]
- 12.Vaccari G, Panagiotidis CH, Acin C, Peletto S, Barillet F, Acutis P, Bossers A, Langeveld J, van Keulen L, Sklaviadis T, Badiola JJ, Andreeoletti O, Groschup MH, Agrimi U, Foster J, Goldmann W. 2009. State-of-the-art review of goat TSE in the European Union, with special emphasis on PRNP genetics and epidemiology. Vet Res 40:48. doi: 10.1051/vetres/2009031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H. 1994. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci 343:405–411. doi: 10.1098/rstb.1994.0036. [DOI] [PubMed] [Google Scholar]
- 14.Heisey DM, Mickelsen NA, Schneider JR, Johnson CJ, Langenberg JA, Bochsler PN, Keane DP, Barr DJ. 2010. Chronic wasting disease (CWD) susceptibility of several North American rodents that are sympatric with cervid CWD epidemics. J Virol 84:210–215. doi: 10.1128/JVI.00560-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Browning SR, Mason GL, Seward T, Green M, Eliason GA, Mathiason C, Miller MW, Williams ES, Hoover E, Telling GC. 2004. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol 78:13345–13350. doi: 10.1128/JVI.78.23.13345-13350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott M, Foster D, Mirenda C, Serban D, Coufal F, Walchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, Westaway D, Prusiner SB. 1989. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 17.Cancellotti E, Barron RM, Bishop MT, Hart P, Wiseman F, Manson JC. 2007. The role of host PrP in transmissible spongiform encephalopathies. Biochim Biophys Acta 1772:673–680. doi: 10.1016/j.bbadis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Barron RM, Thomson V, Jamieson E, Melton DW, Ironside J, Will R, Manson JC. 2001. Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO J 20:5070–5078. doi: 10.1093/emboj/20.18.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell'Omo G, Cartoni C, Ingrosso L, Boyle A, Galeno R, Sbriccoli M, Lipp HP, Bruce M, Pocchiari M, Agrimi U. 2006. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2:e12. doi: 10.1371/journal.ppat.0020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neuendorf E, Weber A, Saalmueller A, Schatzl HM, Reifenberg K, Pfaff E, Groschup MH. 2004. Glycosylation deficiency at either one of the two glycan attachment sites of cellular prion protein preserves susceptibility to bovine spongiform encephalopathy and scrapie infections. J Biol Chem 279:53306–53316. doi: 10.1074/jbc.M410796200. [DOI] [PubMed] [Google Scholar]
- 21.DeArmond SJ, Sanchez H, Yehiely F, Qiu Y, Ninchak-Casey A, Daggett V, Camerino AP, Cayetano J, Rogers M, Groth D, Torchia M, Tremblay P, Scott MR, Cohen FE, Prusiner SB. 1997. Selective neuronal targeting in prion disease. Neuron 19:1337–1348. doi: 10.1016/S0896-6273(00)80424-9. [DOI] [PubMed] [Google Scholar]
- 22.Cancellotti E, Wiseman F, Tuzi NL, Baybutt H, Monaghan P, Aitchison L, Simpson J, Manson JC. 2005. Altered glycosylated PrP proteins can have different neuronal trafficking in brain but do not acquire scrapie-like properties. J Biol Chem 280:42909–42918. doi: 10.1074/jbc.M509557200. [DOI] [PubMed] [Google Scholar]
- 23.Tuzi NL, Cancellotti E, Baybutt H, Blackford L, Bradford B, Plinston C, Coghill A, Hart P, Piccardo P, Barron RM, Manson JC. 2008. Host PrP glycosylation: a major factor determining the outcome of prion infection. PLoS Biol 6:e100. doi: 10.1371/journal.pbio.0060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguzzi A, Heikenwalder M, Polymenidou M. 2007. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8:552–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- 25.Bruce ME. 2003. TSE strain variation. Br Med Bull 66:99–108. doi: 10.1093/bmb/66.1.99. [DOI] [PubMed] [Google Scholar]
- 26.Morales R, Abid K, Soto C. 2007. The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta 1772:681–691. doi: 10.1016/j.bbadis.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bessen RA, Marsh RF. 1994. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collinge J, Clarke AR. 2007. A general model of prion strains and their pathogenicity. Science 318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 29.Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergara J, Burton DR, DeArmond SJ, Prusiner SB, Scott MR. 2002. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 34:921–932. doi: 10.1016/S0896-6273(02)00726-2. [DOI] [PubMed] [Google Scholar]
- 30.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. 1996. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 31.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. 1996. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 39:767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 32.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. 1999. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. [PubMed] [Google Scholar]
- 33.Bosques CJ, Imperiali B. 2003. The interplay of glycosylation and disulfide formation influences fibrillization in a prion protein fragment. Proc Natl Acad Sci U S A 100:7593–7598. doi: 10.1073/pnas.1232504100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zuegg J, Gready JE. 2000. Molecular dynamics simulation of human prion protein including both N-linked oligosaccharides and the GPI anchor. Glycobiology 10:959–974. doi: 10.1093/glycob/10.10.959. [DOI] [PubMed] [Google Scholar]
- 35.Priola SA, Lawson VA. 2001. Glycosylation influences cross-species formation of protease-resistant prion protein. EMBO J 20:6692–6699. doi: 10.1093/emboj/20.23.6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khalili-Shirazi A, Quaratino S, Londei M, Summers L, Tayebi M, Clarke AR, Hawke SH, Jackson GS, Collinge J. 2005. Protein conformation significantly influences immune responses to prion protein. J Immunol 174:3256–3263. doi: 10.4049/jimmunol.174.6.3256. [DOI] [PubMed] [Google Scholar]
- 37.Nishina KA, Deleault NR, Mahal SP, Baskakov I, Luhrs T, Riek R, Supattapone S. 2006. The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry 45:14129–14139. doi: 10.1021/bi061526k. [DOI] [PubMed] [Google Scholar]
- 38.Raymond GJ, Hope J, Kocisko DA, Priola SA, Raymond LD, Bossers A, Ironside J, Will RG, Chen SG, Petersen RB, Gambetti P, Rubenstein R, Smits MA, Lansbury PT Jr, Caughey B. 1997. Molecular assessment of the potential transmissibilities of BSE and scrapie to humans. Nature 388:285–288. doi: 10.1038/40876. [DOI] [PubMed] [Google Scholar]
- 39.Kimberlin RH, Walker CA. 1978. Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J Gen Virol 39:487–496. doi: 10.1099/0022-1317-39-3-487. [DOI] [PubMed] [Google Scholar]
- 40.Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, Parchi P, Gambetti P, Will R, Ironside J, Heinrich C, Tremblay P, DeArmond SJ, Prusiner SB. 2003. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci U S A 100:4784–4789. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dickinson AG, Meikle VMH, Fraser H. 1968. Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. J Comp Pathol 78:293–299. doi: 10.1016/0021-9975(68)90005-4. [DOI] [PubMed] [Google Scholar]
- 42.Fraser H, Dickinson AG. 1968. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78:301–311. doi: 10.1016/0021-9975(68)90006-6. [DOI] [PubMed] [Google Scholar]
- 43.Wadsworth JD, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I, Welch J, Stone L, Lloyd SE, Hill AF, Brandner S, Collinge J. 2004. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 306:1793–1796. doi: 10.1126/science.1103932. [DOI] [PubMed] [Google Scholar]
- 44.Zanusso G, Liu D, Ferrari S, Hegyi I, Yin X, Aguzzi A, Hornemann S, Liemann S, Glockshuber R, Manson JC, Brown P, Petersen RB, Gambetti P, Sy MS. 1998. Prion protein expression in different species: analysis with a panel of new mAbs. Proc Natl Acad Sci U S A 95:8812–8816. doi: 10.1073/pnas.95.15.8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCutcheon S, Langeveld JP, Tan BC, Gill AC, de Wolf C, Martin S, Gonzalez L, Alibhai J, Blanco AR, Campbell L, Hunter N, Houston EF. 2014. Prion protein-specific antibodies that detect multiple TSE agents with high sensitivity. PLoS One 9:e91143. doi: 10.1371/journal.pone.0091143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li R, Liu T, Wong BS, Pan T, Morillas M, Swietnicki W, O'Rourke K, Gambetti P, Surewicz WK, Sy MS. 2000. Identification of an epitope in the C terminus of normal prion protein whose expression is modulated by binding events in the N terminus. J Mol Biol 301:567–573. doi: 10.1006/jmbi.2000.3986. [DOI] [PubMed] [Google Scholar]
- 47.Race R, Meade-White K, Raines A, Raymond GJ, Caughey B, Chesebro B. 2002. Subclinical scrapie infection in a resistant species: persistence, replication, and adaptation of infectivity during four passages. J Infect Dis 186(Suppl 2):S166–170. doi: 10.1086/344267. [DOI] [PubMed] [Google Scholar]
- 48.Manson JC, Clarke AR, McBride PA, McConnell I, Hope J. 1994. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 3:331–340. [PubMed] [Google Scholar]
- 49.Bueler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. 1994. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1:19–30. [PMC free article] [PubMed] [Google Scholar]
- 50.Manson J. 1996. Prnp gene dosage, allelic specificity and gene regulation in the transmissible spongiform encephalopathies, p 239–245 InCourt L, Dodet B (ed), Transmissible subacute spongiform encephalopathies: prion diseases. Elsevier, Paris, France. [Google Scholar]
- 51.Bishop MT, Will RG, Manson JC. 2010. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107:12005–12010. doi: 10.1073/pnas.1004688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cancellotti E, Bradford BM, Tuzi NL, Hickey RD, Brown D, Brown KL, Barron RM, Kisielewski D, Piccardo P, Manson JC. 2010. Glycosylation of PrPC determines timing of neuroinvasion and targeting in the brain following transmissible spongiform encephalopathy infection by a peripheral route. J Virol 84:3464–3475. doi: 10.1128/JVI.02374-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis V, Hill AF, Haigh CL, Klug GM, Masters CL, Lawson VA, Collins SJ. 2009. Increased proportions of C1 truncated prion protein protect against cellular M1000 prion infection. J Neuropathol Exp Neurol 68:1125–1135. doi: 10.1097/NEN.0b013e3181b96981. [DOI] [PubMed] [Google Scholar]
- 54.Ikeda S, Kobayashi A, Kitamoto T. 2008. Thr but Asn of the N-glycosylation sites of PrP is indispensable for its misfolding. Biochem Biophys Res Commun 369:1195–1198. doi: 10.1016/j.bbrc.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 55.Somerville RA, Hamilton S, Fernie K. 2005. Transmissible spongiform encephalopathy strain, PrP genotype and brain region all affect the degree of glycosylation of PrPSc. J Gen Virol 86:241–246. doi: 10.1099/vir.0.80251-0. [DOI] [PubMed] [Google Scholar]
- 56.Beringue V, Mallinson G, Kaisar M, Tayebi M, Sattar Z, Jackson G, Anstee D, Collinge J, Hawke S. 2003. Regional heterogeneity of cellular prion protein isoforms in the mouse brain. Brain 126:2065–2073. doi: 10.1093/brain/awg205. [DOI] [PubMed] [Google Scholar]
- 57.Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Fernandez-Borges N, Schwarz P, Castilla J, Wuthrich K, Aguzzi A. 2010. A molecular switch controls interspecies prion disease transmission in mice. J Clin Investig 120:2590–2599. doi: 10.1172/JCI42051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 59.Deleault NR, Lucassen RW, Supattapone S. 2003. RNA molecules stimulate prion protein conversion. Nature 425:717–720. doi: 10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- 60.Graham JF, Agarwal S, Kurian D, Kirby L, Pinheiro TJ, Gill AC. 2010. Low density subcellular fractions enhance disease-specific prion protein misfolding. J Biol Chem 285:9868–9880. doi: 10.1074/jbc.M109.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]