

ABSTRACT
Hepatitis C virus (HCV) entry into host cells is a complex process requiring multiple host factors, including claudin-1 (CLDN1). Safe and effective therapeutic entry inhibitors need to be developed. We isolated a human hepatic Huh7.5.1-derived cell mutant that is nonpermissive to HCV, and comparative microarray analysis showed that the mutant was CLDN1 defective. Four hybridomas were obtained, which produced monoclonal antibodies (MAbs) that interacted with the parental Huh7.5.1 cell but not with the CLDN1-defective mutant. All MAbs produced by these hybridomas specifically bound to human CLDN1 with a very high affinity and prevented HCV infection of Huh7.5.1 cells in a dose-dependent manner, without apparent cytotoxicity. Two selected MAbs also inhibited HCV infection of human liver-chimeric mice without significant adverse effects. CLDN1 may be a potential target to prevent HCV infection in vivo. Anti-CLDN1 MAbs may hence be promising candidates as novel anti-HCV agents.
IMPORTANCE Safe and effective therapeutic entry inhibitors against hepatitis C virus (HCV) are very useful for combination therapies with other anti-HCV drugs, such as direct-acting antivirals. In this study, we first showed an effective strategy for developing functional monoclonal antibodies (MAbs) against extracellular domains of a multimembrane-spanning target protein, claudin-1 (CLDN1), by using parental cells expressing the intact target membrane protein and target-defective cells. The established MAbs against CLDN1, which had a very high affinity for intact CLDN1, efficiently inhibited in vitro and in vivo HCV infections. These anti-CLDN1 MAbs are promising leads for novel entry inhibitors against HCV.
INTRODUCTION
Worldwide, 170 million people are infected with hepatitis C virus (HCV), which is a major cause of liver cirrhosis and hepatocellular carcinoma. Thus, overcoming HCV infection is an important global health care issue (1). HCV is an enveloped, positive-sense, single-stranded RNA virus in the Flaviviridae family (2). Recent clinical research using direct-acting antivirals that target HCV enzymes, such as sofosbuvir and simeprevir, has provided new insights into combination therapy with inhibitors of multiple targets (3–5).
Preventing viral entry into hepatocytes is an attractive target for anti-HCV agents, but strategies for preventing HCV entry into host cells are clinically unavailable (6). Host factors involved in initiating infection include heparan sulfate (7), low-density lipoprotein receptor (8), CD81 (9), scavenger receptor class B type I (SRBI) (10), claudin-1 (CLDN1) (11), occludin (12, 13), epidermal growth factor receptor (EGFR) (14), and Niemann-Pick C1-like 1 (15). Among these, CLDN1 is considered a potent target because it is essential for HCV entry into cells via interaction with CD81 and for cell-to-cell HCV transmission (16, 17). Anti-CLDN1 antibodies (Abs) that inhibit HCV infection in vitro were reported by Baumert et al. (18, 19) and Hötzel et al. (20), but a CLDN1 binder that prevents HCV infection in vivo has not yet been developed.
In this study, we showed that CLDN1 is a promising anti-HCV target based on genetic approaches using hepatic cell mutants defective in HCV infection. We developed a unique method for screening CLDN1 binding and established novel anti-human CLDN1 (anti-hCLDN1) monoclonal Abs (MAbs) that prevent in vitro and in vivo HCV infections, without apparent adverse effects.
MATERIALS AND METHODS
Cells and plasmid construction.
Human hepatoma Huh-7.5.1 cells (21) were subcloned by limiting dilution, and a highly HCV-JFH1-permissive subclonal cell line, Huh-7.5.1-8 (22), was used. Huh-7.5.1-derived cells and human hepatoma HepG2 cells were maintained as described previously (22). The pcDNA3.1/Hyg-hCLDN1 expression vector was prepared by insertion of hCLDN1 cDNA into the KpnI/NotI-digested pcDNA3.1-Hyg vector (Life Technologies Corp.). Huh-7.5.1-derived S7-A cells that stably expressed hCLDN1 (S7-A/hCLDN1 cells) were established by the following procedure. The pcDNA3.1/Hyg-hCLDN1 vector was transfected into S7-A cells by use of FuGENE6 transfection reagent (Roche Diagnostics), and hygromycin-resistant clones were selected and cloned by limiting dilution. Huh7.5.1-8 cells that expressed green fluorescent protein (GFP) in the nucleus (Huh7.5.1-8/GFP-Nuc cells) were established via the transfection of pAcFP1-Nuc (TaKaRa Bio Inc.) into Huh7.5.1-8 cells. Human embryonic kidney 293T cells and human fibrosarcoma HT1080 cells were obtained from the ATCC (Manassas, VA) and the Japanese Collection of Research Bioresources (Osaka, Japan), respectively. These cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin G, and 100 μg/ml streptomycin sulfate. The N-terminal FLAG-tagged CLDN1 and CLDN4 expression vectors, composed of tagged genes inserted into pcDNA3.1(+), were prepared using PCR to amplify the tagged genes. Various FLAG-tagged CLDN1 vectors with point mutations were constructed using a KODplus mutagenesis kit (Toyobo Co. Ltd., Osaka, Japan). These FLAG-tagged CLDN1 vectors were transiently introduced into 293T cells by use of X-tremeGENE HP DNA transfection reagent (Roche Diagnostics). Mouse CLDN1 and human CLDN1, -2, -4, -5, -6, -7, and -9 cDNAs were generated via PCR, using primer pairs specific to each CLDN (23). The resultant cDNAs were cloned into pcDNA3.1(−) (Invitrogen, CA). The CLDN expression vectors were then introduced into HT1080 cells, and G418-resistant clones were selected, resulting in the isolation of cells that stably expressed each CLDN (23).
Mice.
Autoimmune BXSB mice were purchased from Japan SCL. For HCV infection studies, human liver-chimeric mice (24) were used as described previously (25). The procedures were approved by the Animal Ethics Committee of PhoenixBio Co., Ltd. All the animal experiments were performed according to the guidelines of Osaka University.
Isolation and characterization of Huh7.5.1-derived cell mutants resistant to HCV.
Since Huh7.5.1 cells showed a pronounced cytopathic effect about 10 days after infection with large amounts of our cell-cultured infectious HCV-JFH1 (HCVcc) stock (see “In vitro HCV infection,” below), we tried to isolate cell mutants that survived after HCV infection (HCV-resistant cells). Huh7.5.1 cells were seeded at 5 × 105 cells in 10-cm dishes and infected on the next day with HCVcc (HCV core content, 0.2 nmol/liter) at a multiplicity of infection (MOI) of >10. After 2 weeks, surviving cells were reinfected with HCVcc (HCV core content, 0.2 nmol/liter) and cultured for another 2 weeks. Each cell colony was picked and recloned by limiting dilution. To further isolate HCV-resistant cells not defective in CD81, we changed and added some steps for screening. Huh7.5.1 cells were seeded at 1 × 106 cells in 10-cm dishes and infected on the next day with HCVcc (HCV core content, 0.2 nmol/liter) at an MOI of >10. After 8 days, surviving cells were transfected with the pcDNA3.1-hCD81 vector and cultured for 1.5 days. Transfected cells were then reinfected with HCVcc (HCV core content, 0.2 nmol/liter) and cultured for 6 days in the normal medium and for another 25 days in the normal medium containing 1 mg/ml G418 to concentrate CD81-expressing cells. Each cell colony was picked and recloned by limiting dilution. The reason for using transiently CD81-transfected cells, not stably CD81-expressing cells, for HCV infection screening is that treatment of host cells with selection drugs, such as G418, showed less HCV production and less cytopathic effect under our culture conditions.
In vitro HCV infection.
HCV-JFH1 (26) in cell culture (HCVcc) was prepared from culture supernatants of Huh7.5.1 cells that had been transfected with in vitro-transcribed HCV-JFH1 RNA (22), passaged a few times using Huh7.5.1-8 cells, and used to infect Huh7.5.1-derived cells (27). During infection, cells were incubated with the virus at an MOI of 1 for 2 h at 37°C.
Transcriptome analysis.
Total RNA was purified from cells by use of an RNeasy Mini Kit (Qiagen K.K.). Comprehensive RNA microarray analyses were performed using a NimbleGen Human Oligo 72K chip (Roche Diagnostics K.K.). Data were analyzed using ArrayStar software (DNAStar Inc., WI).
Isolation of mouse anti-hCLDN1 MAb.
Six-week-old male BXSB mice were immunized with a eukaryotic expression vector that encoded hCLDN1 by use of proprietary Genovac technology (Genovac GmbH) (18, 23). The lymphocytes were removed 7 days after the final immunization and fused with P3-UI cells in the presence of polyethylene glycol 1000. A conditioned medium of hybridomas was screened by flow cytometry, based on the ability to bind Huh7.5.1-8 cells but not CLDN1-defective cells. After cloning by limiting dilution, four stable hybridomas were obtained. Immunoglobulin classes/subclasses of clones were determined using a mouse immunoglobulin isotyping enzyme-linked immunosorbent assay (ELISA) kit (BD Biosciences). A rat MAb recognizing extracellular domains of CLDN4 was prepared as described previously (23).
Flow cytometric analysis.
To analyze MAb binding to CLDNs, cells were detached and incubated with MAbs (2 to 10 μg/ml), followed by secondary treatment with phycoerythrin-, fluorescein-, or Alexa 488-conjugated goat anti-mouse IgG. MAb-bound cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences) (22).
Immunofluorescence analysis.
Cells were stained with anti-HCV core MAb and 4′,6-diamidino-2-phenylindole (DAPI) as described previously (22, 28). In brief, cells cultured on collagen-coated glass coverslips in a 24-well plate were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) for 30 min. After washing with PBS containing 30 mM glycine, the cells were permeabilized with PBS containing 0.1% Triton X-100 for 10 min, blocked with 5% (wt/vol) skim milk in PBS for 30 min, and incubated with a 1:500 dilution of mouse anti-HCV core protein MAb (2H9) followed by a 1:500 dilution of Alexa 488-conjugated goat anti-mouse IgG (Life Technologies) and DAPI. To detect CLDN1, permeabilized and blocked cells were incubated with a 1:200 dilution of rabbit anti-CLDN1 polyclonal Abs (pAbs) (Life Technologies Corp.) followed by a 1:200 dilution of Alexa 488-conjugated goat anti-rabbit IgG. Stained cells were analyzed using a confocal laser scanning microscope (Axiovert 100M; Carl Zeiss).
qRT-PCR analysis.
Total RNA was isolated from cells by using a Blood/Cultured Cell total RNA Mini Kit (Favorgen Biotech Corp., Pingtung City, Taiwan). Cellular contents of viral RNA were determined by one-step quantitative real-time reverse transcription-PCR (qRT-PCR) with RNA-direct real-time PCR master mix (Toyobo Co. Ltd., Osaka, Japan), using specific primers and a TaqMan probe as described previously (22). The cellular mRNA contents of CD81, CLDN1, CLDN6, CLDN9, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were determined by one-step qRT-PCR with RNA-direct SYBR green real-time PCR master mix (Toyobo Co. Ltd., Osaka, Japan), using the following specific primers: 5′-AAGCAGTTCTATGACCAGGCCCTAC-3′ and 5′-TGAGGTGGTCAAAGCAGTCAGTG-3′ for CD81, 5′-GCACATACCTTCATGTGGCTCAG-3′ and 5′-TGGAACAGAGCACAAACATGTCA-3′ for CLDN1, 5′-AATGCAGATCCTGGGAGTCGTC-3′ and 5′-ACGATGCTGTTGCCGATGAA-3′ for CLDN6, 5′-TTCGACCTTGGCCTGATGAC-3′ and 5′-CTGCAGCCAGGTGTAGCTTG-3′ for CLDN9, and 5′-GCACCGTCAAGGCTGAGAAC-3′ and 5′-TGGTGAAGACGCCAGTGGA-3′ for GAPDH. PCR was performed in a LightCycler real-time PCR system (F. Hoffmann-La Roche AG Konzern-Hauptsitz, Basel, Switzerland), and the PCR conditions were as follows: 90°C for 1 min, 60°C for 20 min, 95°C for 30 s, and 45 cycles of 95°C for 0 s and 60°C for 45 s.
HCVpp infection.
HCV pseudoparticles (HCVpp) were generated as described previously (29), using glycoprotein (HCV E1 and E2)-expressing vectors (JFH1 [genotype 2a] and TH [genotype 1b]) (30).
For infection assay (22), Huh7.5.1-8 cells were infected with HCVpp for 3 h at room temperature. The medium was replaced by fresh medium, and cells were cultured for an additional 2 days at 37°C. Luciferase activities of cell lysates were measured using a luciferase reporter assay kit (PicaGene; Toyo Ink).
TJ function assays.
To examine tight junction (TJ) integrity in the monolayers, we observed the paracellular diffusion of solutes from the bile-canalicular (BC) lumen into the basolateral medium (19). In brief, on day 0, HepG2 cells were seeded at 105 cells in 24-well plates. On day 5, the cell monolayers were treated with or without 0.1 μg of tumor necrosis factor alpha (TNF-α), 5 μg of anti-CLDN1 MAbs, or control mouse IgG and cultured for 1 h at 37°C. Cells were then stained with 5 mM 5-chloromethylfluorescein diacetate (CMFDA) at 37°C for 10 min, and the numbers of BC dot-like structures were counted by fluorescence microscopy.
In vivo HCV infection experiments.
Human liver-chimeric PXB mice (24) were generated by transplanting 106 human primary hepatocytes (BD Gentest inducible-qualified human cryohepatocytes; HF284, lot 195; BD Biosciences) into the spleens of 2- to 3-week-old uPA/SCID mice as previously described (25). Five weeks after transplantation, mouse serum was analyzed for the presence of human albumin by use of ELISA. Animals with human albumin levels of >1 mg/ml were considered to be successfully engrafted and used for the infection studies. PXB mice were injected intraperitoneally with anti-hCLDN1 MAb (3A2 or 7A5) or a control Ab at 30 mg/kg of body weight (4 mice/group). Eight hours after Ab administration, mice were inoculated intravenously with HCV-HCR6 (104 copies) (genotype 1b; accession no. AY045702), which was obtained from HCV-HCR6-infected PXB mice. One copy corresponds to 0.185 IU. Since the total blood volume of a mouse is approximately 80 ml/kg of body weight (31), 104 HCV RNA copies/mouse corresponds to ∼1.7 × 104 HCV RNA copies/ml (∼3 × 103 IU/ml) of serum. Mice were injected intraperitoneally with 3A2 or 7A5 at 20, 10, and 10 mg/kg on days 3, 7, and 10, respectively. Sera were collected from mice on days 0, 3, 7, 10, 14, 21, 28, 35, and 42 and used for analyses.
HCV measurement in mouse serum.
HCV RNA in mouse serum was quantified using TaqMan EZ RT-PCR core reagent (Life Technologies Corp.) and an ABI Prism 7500 sequence detector system (Life Technologies Corp.). The lower quantification limit of the assay was 4.0 × 104 copies/ml.
Human albumin, ALT, and AST measurements in mouse serum.
The human albumin level in mouse serum was measured using an LX Alb-II kit (Eiken Co., Ltd.). Serum alanine aminotransferase (ALT) and aspartate transaminase (AST) levels were measured using a Transaminase-CII kit (Wako Pure Chemical).
Anti-hCLDN1 MAb measurements in mouse serum.
Anti-hCLDN1 MAb 3A2 and 7A5 levels in mouse serum were determined by a modified method based on that described by Meuleman et al. (31). We used Huh7.5.1-8 cells instead of Huh7.5 cells, purified anti-hCLDN1 MAbs 3A2 and 7A5 as standards, and a 1:1,000 dilution of Alexa 488-conjugated goat anti-mouse IgG(H+L) (Life Technologies Corp.) as the secondary antibody. Mouse sera and antibodies were diluted with PBS containing 2% fetal bovine serum.
Statistical analysis.
The drug concentration providing 50% inhibition (IC50) was calculated by using the “log (inhibitor) versus normalized response-variable slope” equation of the nonlinear regression model included in GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Statistical analysis was performed with Student's t test, using the GraphPad calculator (http://www.graphpad.com/quickcalcs/); differences with P values of <0.05 were considered statistically significant.
Cell ELISA.
Cell ELISA was performed as follows. Huh7.5.1-8 cells seeded at 1.5 × 104 cells/well in 96-well plates were cultured for 2 days at 37°C and fixed with 100 μl of 3.7% formaldehyde-PBS for 1 h at room temperature. After washing with PBS containing 30 mM glycine, cells were blocked with 100 μl of 5% (wt/vol) skim milk in TBS-T (Tris-buffered saline containing 0.1% [vol/vol] Tween 20) for 30 min and then incubated with 5-fold serial dilutions of 100 μl of anti-CLDN1 MAbs or control IgG (2.5 μg/ml) for 2 h, followed by a 1:5,000 dilution of 100 μl of horseradish peroxidase-conjugated AffiniPure goat anti-mouse IgG(H+L) (Jackson ImmunoResearch Laboratories, Inc.) for 1 h at room temperature. The plates were washed 3 times with TBS-T between incubations. To detect antibody binding to the cells, o-phenylenediamine dihydrochloride and hydrogen peroxide were used as substrates. The absorbance at 492 nm for each well was determined on an OpsysMR plate reader (Dynex Technologies Inc.). The half-saturating concentrations (apparent Kd values [antibody dissociation constants]) were determined as described previously (32).
Other methods.
The HCV core protein level in the culture supernatant, which indicates the level of secreted virus, was quantified by ELISA using an Ortho HCV antigen ELISA kit (Ortho-Clinical Diagnostics, Inc.). Immunoblot analyses were performed using the NuPAGE system (Life Technologies Corp.) as described previously (22, 27).
Microarray data accession number.
Microarray data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-2589.
RESULTS
Isolation of Huh7.5.1-derived cell mutants resistant to HCV.
To understand the overall HCV life cycle, host factors involved in HCV infection should be elucidated. Genetic approaches to isolate host cell mutants defective in HCV infection are powerful strategies for identifying genes essential for the HCV life cycle. We isolated cell mutants that survived after HCV infection (HCV-resistant cells) because some of these mutants are expected to be defective in host factors essential for HCV infection. In the first trial, we cloned >20 lines of surviving cells which were not permissive to HCV, except for one persistently infected clone. Biochemical analyses of these HCV-nonpermissive cells showed that they were all CD81 defective and that transient expression of human CD81 in these cells recovered their capacity for HCV infection. Results for representative clones, i.e., 751r-1 and 751r-2 cells, are shown in Fig. 1, indicating that CD81 is essential for HCV infection, as described previously (9, 33). To isolate other HCV-resistant cells, multicopy CD81 expression plasmids were introduced into Huh7.5.1 cells before secondary HCVcc infection during the screening. Various cell mutants were established again after cloning. Among them, three clones, S7-A, -B, and -C, were characterized in the following experiments.
FIG 1.
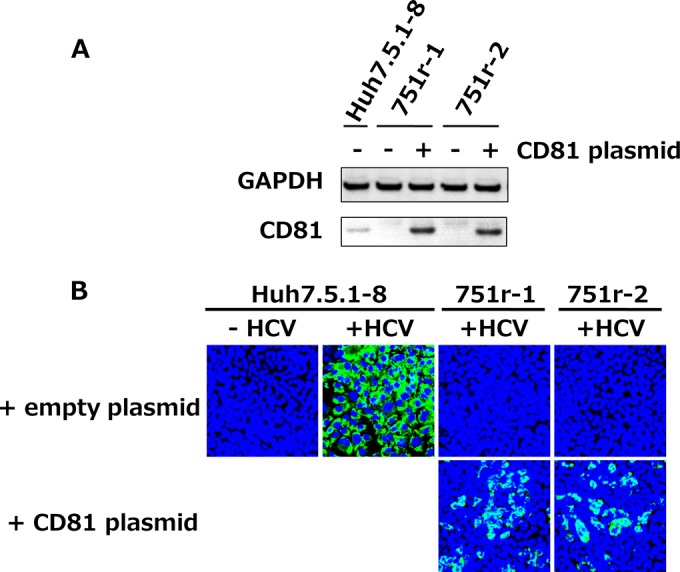
Most cell clones with resistance to HCV were CD81 defective and HCV nonpermissive. Huh7.5.1-derived cell clones (751r-1 and 751r-2) with resistance to HCV, which were isolated by the screen described in Materials and Methods, were transfected with the pcDNA3.1-hCD81 vector and cultured for 2 days. (A) Cell lysates were subjected to immunoblotting to detect the GAPDH and CD81 proteins. (B) Transfected cells and control Huh7.5.1-8 cells were infected with HCVcc and stained with anti-HCV core protein (green) and DAPI (blue) at 3 dpi.
Characterization of Huh7.5.1-derived CLDN1-defective cell clones.
S7-A, -B, and -C cells were not permissive to HCVcc (Fig. 2A), indicating that they were defective in host factors essential for HCV infection. We then performed comparative microarray analyses of Huh7.5.1-8 and S7-A cells. The scatterplot as well as qRT-PCR analysis clearly showed that CLDN1 expression was defective in S7-A cells (Fig. 2B and Table 1), consistent with the immunohistochemistry data (Fig. 2E). Immunoblot analyses also showed that there was no CLDN1 protein in S7-A, -B, and -C cells and that CD81 was enhanced in these cells because of the transfection of the CD81 expression vector during screening (Fig. 2C). Similar levels of CLDN6 mRNA were expressed in both Huh7.5.1 and S7-A cells, which were about 1/5 the levels of CLDN1 (Table 1), suggesting that HCV-JFH1 cannot infect Huh7.5.1-derived cells via CLDN6. Other host factors involved in HCV entry, such as SRBI and occludin, were detected in S7-A, -B, and -C cells (Fig. 2C). When the hCLDN1 expression vector was transiently transfected into S7-A, -B, and -C cells, the permissiveness to HCV infection was recovered in transfected cells (Fig. 2D). S7-A cells stably expressing hCLDN1 (S7-A/hCLDN1 cells) (Fig. 2E) were permissive to HCVcc (Fig. 2F) and released significant amounts of HCV particles (Fig. 2G), indicating that all stages of the HCV life cycle were restored by expression of CLDN1 in S7-A cells. These observations demonstrate that HCV resistance of S7-A, -B, and -C cells was due to their deficiency of CLDN1 expression and that CLDN1 is essential for HCV infection, as described previously (11). We also examined the role of CLDN1 in cell-to-cell transmission by using CLDN1-defective S7-A cells. HCV-preinfected Huh7.5.1-8/GFP-Nuc cells with a GFP nuclear marker were mixed with naive Huh7.5.1-8, S7-A, or CD81-defective 751r-1 cells. After culture for 5.5 days, HCV-infected cells were observed by immunostaining of HCV core proteins. S7-A cells with blue nuclei were never infected by HCV, while one or two 751r-1 cells adjacent to Huh7.5.1-8/GFP-Nuc cells were infected (Fig. 2H). To confirm the CD81 defect in 751r-1 cells, we examined the amounts of CD81 mRNA in Huh7.5.1-8 and 751r-1 cells by qRT-PCR. The level in Huh7.5.1-8 cells was (1.12 ± 0.11) × 105 copies/μg total RNA, whereas the level in 751r-1 cells was <1/104 (<10 copies/μg total RNA) of that in Huh7.5.1-8 cells (under the limit of detection). Note that cell-to-cell transmission in 751r-1 cells is quite unlikely to be mediated by the very minimal amounts of CD81 on the surfaces of 751r-1 cells (Fig. 2H). These results indicate that cell-to-cell transmission of HCV can occur at a low efficiency without the presence of CD81, but CLDN1 seems to be absolutely essential, as previously reported (17).
FIG 2.

Isolation and characterization of Huh7.5.1-derived CLDN1-defective clones. (A) Huh7.5.1-derived cell clones, i.e., S7-A, -B, and -C, and control Huh7.5.1-8 cells were infected with HCVcc. At 3 dpi, cells were stained with anti-HCV core protein (green) and DAPI (blue). (B) Scatterplot showing the results of the RNA microarray experiments (Huh7.5.1-8 versus S7-A cells). Each data point represents a single gene. The arrow indicates CLDN1. (C) Each cell lysate was subjected to immunoblotting to detect CD81, SRBI, CLDN1, occludin, and GAPDH. (D) Each cell was transfected with pcDNA3.1/Hygro (empty vector) or pcDNA3.1/Hygro-hCLDN1 (CLDN1 vector). After 2 days, transfected cells were infected with HCVcc and stained with anti-HCV core protein (green) and DAPI (blue) at 3 dpi. The transfection efficiency was usually ∼30% when we observed it 2 days after transfection. (E) Huh7.5.1-8 cells, S7-A cells, and S7-A cells that stably expressed human CLDN1 (S7-A/hCLDN1) were stained with anti-CLDN1 pAb (red) and observed by confocal fluorescence microscopy. (F) Each cell culture was infected with HCVcc and stained with anti-HCV core protein (green) and DAPI (blue) at 3.5 dpi. (G) HCV core contents in the culture supernatants from the different cell types were quantitated by ELISA at 3.5 dpi. (H) Huh7.5.1-8/GFP-Nuc cells with green nuclear staining were infected with HCVcc and cultured for 1.5 days. HCV-infected Huh7.5.1-8/GFP-Nuc cells were mixed with naive Huh7.5.1-8, S7-A, or 751r-1 cells, at a cell number ratio of 1:10 in each case, and plated onto a 24-well plate. After 5.5 days, cells were stained with anti-HCV core protein (red) and DAPI (blue). Arrows indicate HCV-infected 751r-1 cells.
TABLE 1.
Expression levels of CLDN1, CLDN6, CLDN9, and GAPDH mRNAs in Huh7.5.1-8 cells and S7-A cells
| Cell line | No. of copies/μg total RNA (n = 3)a |
|||
|---|---|---|---|---|
| CLDN1 mRNA | CLDN6 mRNA | CLDN9 mRNA | GAPDH mRNA | |
| Huh7.5.1-8 | (5.8 ± 1.3) × 105 | (1.1 ± 0.0) × 105 | (4.6 ± 1.0) × 101 | (1.2 ± 0.1) × 108 |
| S7-A | <102 | (1.1 ± 0.3) × 105 | (4.3 ± 0.7) × 101 | (1.3 ± 0.2) × 108 |
The limit of detection was 102 copies of mRNA.
Isolation of MAbs against extracellular domains of hCLDN1.
These studies using CLDN1-defective mutants, together with previous studies (11, 17–19), indicate that CLDN1 is a promising target for anti-HCV agents; therefore, we tried to establish MAbs against extracellular domains of hCLDN1. An effective screening system is crucial for developing functional Abs that prevent HCV entry into hepatocytes; thus, it was very important to screen MAbs recognizing “intact” extracellular domains of CLDN1 in hepatocytes. For screening, we used Huh7.5.1-8 cells that expressed intact CLDN1 and CLDN1-defective S7-A cells that we established in this study. We used fluorescence-activated cell sorting (FACS) analysis to screen hybridomas producing MAbs that interacted with Huh7.5.1-8 cells but not S7-A cells (Fig. 3A). Autoimmune BXSB mice were immunized with an hCLDN1 expression vector, resulting in the isolation of four hybridomas producing MAbs (2C1, 3A2, 5F2, and 7A5) that bound to Huh7.5.1-8 cells but not S7-A cells (Fig. 3B).
FIG 3.

Establishment of mouse anti-hCLDN1 MAbs. (A) Strategy for MAb development against extracellular domains of hCLDN1. s.c., subcutaneous. (B) Huh7.5.1-8 (black histograms) or S7-A (white histograms) cells were incubated with the conditioned medium from each hybridoma clone and treated with phycoerythrin-conjugated secondary Abs. Stained cells were analyzed by flow cytometry. (C) 293T cells were transiently transfected with pcDNA3.1(+)-FLAG-hCLDN1 (FLAG-hCLDN1) or pcDNA3.1(+)-FLAG-hCLDN4 (FLAG-hCLDN4). After 2 days, cells were stained with anti-hCLDN1 MAbs or an anti-CLDN4 MAb and analyzed by flow cytometry. Gray and white patterns are for vehicle- and MAb-treated cells, respectively. (D) Huh7.5.1-8 (upper) and S7-A (lower) cells were incubated with anti-hCLDN1 MAbs or polyclonal Abs (pAbs) that recognized the cytosolic domain of CLDN1 and then treated with Alexa 488-conjugated secondary Abs. Stained cells were observed by confocal microscopy. (E) Huh7.5.1-8 or S7-A cell lysates were subjected to immunoblotting with anti-hCLDN1 MAbs, anti-CLDN1 pAbs, or an anti-GAPDH MAb.
Characterization of MAbs that recognized extracellular domains of CLDN1.
Isotype analyses of these MAbs showed that clones 2C1 and 3A2 were IgG2b, 5F2 was IgG2a, and 7A5 was IgG1. To confirm whether these MAbs recognized hCLDN1, HEK293T cells were transiently transfected with a FLAG-hCLDN1 or FLAG-hCLDN4 vector and stained with these MAbs or an anti-CLDN4 MAb. All MAbs bound to FLAG-CLDN1-expressing cells but not FLAG-CLDN4-expressing cells, to which an anti-CLDN4 MAb bound (Fig. 3C). We also investigated the specificity of these MAbs by using mouse CLDN1-expressing L cells and hCLDN1, -2, -4, -5, -6, -7, and -9-expressing HT1080 cells. All clones reacted with hCLDN1-expressing cells, but they did not react with cells expressing mouse CLDN1 and other hCLDNs (Fig. 4). These results showed that all MAbs specifically recognized extracellular domains of hCLDN1.
FIG 4.

Established mouse MAbs specifically recognized human CLDN1. HT1080 cells that expressed human CLDN1, CLDN2, CLDN4, CLDN5, CLDN6, CLDN7, or CLDN9 and mouse CLDN1-expressing L cells were incubated with anti-CLDN1 MAbs, i.e., 2C1, 3A2, 5F2, and 7A5, at 5 μg/ml and then treated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG. Gray and white patterns indicate vehicle- and Ab-treated cells, respectively.
Based on immunohistochemical analyses under nonpermeabilized conditions, we observed that cell-cell contact sites in Huh7.5.1-8 cells were mainly stained with these anti-hCLDN1 MAbs, while those in CLDN1-defective S7-A cells were not (Fig. 3D), suggesting that all anti-hCLDN1 MAbs can recognize intact extracellular domains of CLDN1, which are mainly localized to TJs in cell monolayers. Additionally, the immunoblot analysis showed that only 7A5 reacted with hCLDN1 (Fig. 3E), indicating that 7A5 recognized both intact and denatured forms of hCLDN1, while the other clones (2C1, 3A2, and 5F2) reacted only with the intact form.
The 7A5 clone recognized the primary structure of hCLDN1; thus, we determined the 7A5 epitope on hCLDN1 by mutational analyses. Five amino acid residues in two extracellular loops (EL1 and EL2) of CLDN1 differ between the human and mouse CLDN1 proteins (R31, M46, and S74 in EL1 and M152 and V155 in EL2 of hCLDN1) (Fig. 5A). Replacement of these hCLDN1 amino acids with those of mouse CLDN1 (R31K, M46I, S74N, M152L, and V155I) eliminated the 7A5-hCLDN1 reaction (Fig. 5B, FLAG-hCLDN1-EL-m panels; also see Fig. 4). These results suggest that the critical epitopes of 7A5 are probably located among these five amino acids. Next, amino acids in EL1 or EL2 of hCLDN1 were replaced with those in EL1 or EL2 of mouse CLDN1 (FLAG-hCLDN1-EL1-m and FLAG-hCLDN1-EL2-m constructs, respectively). The 7A5 clone bound to cells that expressed FLAG-hCLDN1-EL1-m but not to those that expressed FLAG-hCLDN1-EL2-m, for which cell surface expression was verified using the 2C1 clone (Fig. 5B), suggesting that 7A5 recognizes only the EL2 domain of hCLDN1. Further investigations showed that the binding of 7A5 to FLAG-hCLDN1 was preserved by the replacement of valine with isoleucine at position 155 (V155I) but not by the replacement of methionine with leucine at position 152 (M152L) (Fig. 5B). Consistent with this, the 7A5 clone recognized FLAG-hCLDN1-V155I but not FLAG-hCLDN1-M152L, according to immunoblot analysis (Fig. 5C). These results indicate that the methionine at position 152 (M152) in EL2 is a critical residue during the 7A5-hCLDN1 interaction.
FIG 5.

Analysis of anti-hCLDN1 epitope on hCLDN1. (A) Schematic structure of CLDN1 and homologies of the first (EL1) and second (EL2) extracellular loops of human and mouse CLDN1 proteins. (B and C) 293T cells were transiently transfected without a vector (mock) or with the FLAG-hCLDN1, FLAG-hCLDN1-EL-m, FLAG-hCLDN1-EL1-m, FLAG-hCLDN1-EL2-m, FLAG-hCLDN1-M152L, or FLAG-hCLDN1-V155I expression vector, as indicated. (B) Flow cytometric analyses were performed using anti-hCLDN1 MAbs (7A5, 2C1, 3A2, and 5F2). Each cell culture was incubated with 2 μg/ml of anti-hCLDN1 MAb (white histograms) or control IgG (gray histograms), and Ab binding was detected using Alexa 488-conjugated goat anti-mouse IgG(H+L). (C) The cell lysates were subjected to immunoblotting with anti-FLAG, anti-hCLDN1 (7A5), and anti-GAPDH.
We determined the amino acid sequences of the VH and VL regions of MAbs (data not shown). All MAbs had different sequences, but the homologies between 2C1 and 3A2 were quite high (93% and 97% similarity in the VH and VL regions, respectively), and those between 5F2 and 7A5 were also high (88% and 95% similarity in the VH and VL regions, respectively), reflecting the similar binding characteristics of these MAbs (Fig. 5B).
We also examined the binding properties of the MAbs for Huh7.5.1-8 cells (Fig. 6). The apparent Kd values were all in the picomolar range (95 [2C1], 124 [3A2], 228 [5F2], and 201 [7A5] pM), indicating that these MAbs bind with very high affinity to intact CLDN1 on Huh7.5.1-8 cells.
FIG 6.
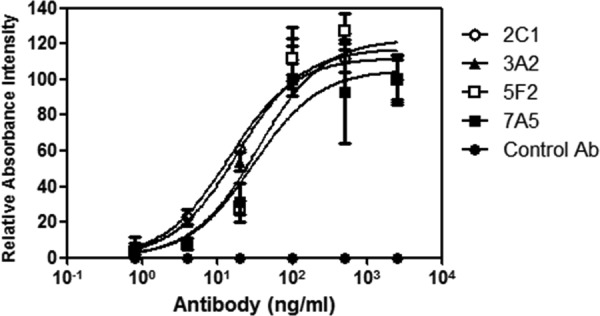
Measurement of anti-CLDN1 MAb binding to cellular CLDN1 by cell ELISA. After fixation, Huh7.5.1-8 cells in 96-well plates were incubated with serial dilutions of each anti-CLDN1 MAb (2C1, 3A2, 5F2, and 7A5) or control IgG, followed by horseradish peroxidase-conjugated second antibody. The binding of each antibody to cellular CLDN1 was measured as described in Materials and Methods.
Anti-hCLDN1 MAbs inhibited HCV infection in vitro.
To investigate whether anti-hCLDN1 MAbs could prevent HCV infection, Huh7.5.1-8 cells were infected with HCVcc in the presence or absence of each CLDN1 MAb. HCV infection was assessed using an ELISA for the HCV core protein in the conditioned medium at 4 days postinfection (dpi). Treatment of cells with each MAb decreased the amount of the HCV core protein in supernatants in a dose-dependent manner, indicating that these MAbs strongly inhibited HCV infection (Fig. 7A). MAb levels that resulted in a 50% inhibition of HCV production were 0.19 (2C1 and 3A2), 0.23 (7A5), and 5.8 (5F2) μg/ml, which seemed to correlate well with the binding affinities of MAbs for cellular CLDN1 (Fig. 6). Quantification of the HCV RNA by qRT-PCR (Fig. 7B) and immunoblot analysis of HCV proteins (Fig. 7C) in infected cells also detected similar inhibition rates of HCV infection by these MAbs.
FIG 7.

Inhibition of HCV infection by anti-hCLDN1 MAbs in vitro. Huh7.5.1-8 cells were seeded at 5 × 104 cells in 48-well plates and pretreated with the indicated amount of anti-hCLDN1 MAb or control IgG (control Ab) for 1 h at room temperature and then were subjected to the following procedures. (A to C) Cells were infected with HCVcc (MOI = 1) for 2 h at room temperature and cultured for 4 days in the presence of the indicated amounts (0.1 to 5 μg/well) of MAbs (400 μl/well). (A) Culture supernatants were collected, and levels of HCV core proteins were quantified by ELISA. (B) The cellular total RNA was extracted, and HCV RNA contents were quantified by qRT-PCR. The dotted line represents the average level in HCV-uninfected cells. In panels A and B, values are expressed as percentages, and data represent the means ± standard deviations (SD) (n = 3). (C) Cell lysates were subjected to immunoblotting of HCV NS3, core, and GAPDH proteins. (D and E) Cells were then infected with HCVpp (genotype 2a [D] or genotype 1b [E]) for 3 h at room temperature and cultured for 2 days in the presence of the indicated amounts of each anti-hCLDN1 MAb. Luciferase activities in cell lysates were measured using a luminometer. Values are expressed as percentages. Data in each graph represent the means ± SD (n = 3). RLU, relative light units.
HCVpp infection is a valid model of HCV entry into cells. Thus, we investigated the effects of anti-hCLDN1 MAbs on HCVpp (genotypes 2a and 1b) entry into Huh7.5.1-8 cells. The treatment of cells with control IgG (10 μg/ml) did not reduce HCVpp infection. In contrast, treatment with each anti-hCLDN1 MAb decreased the entry of both genotypes of HCVpp (2a [Fig. 7D] and 1b [Fig. 7E]) in a dose-dependent manner. MAb levels that resulted in a 50% inhibition of HCVpp entry were 0.2 (2C1 and 3A2), 1.2 (7A5), and 6.5 (5F2) μg/ml for genotype 2a and 0.4 (2C1 and 3A2), 0.9 (7A5), and 5 (5F2) μg/ml for genotype 1b, which are comparable between genotypes. From these findings, anti-hCLDN1 MAbs may be promising candidates for the development of HCV entry inhibitors.
Anti-hCLDN1 MAbs did not affect TJ functions in cultured cells.
CLDN1 is a TJ protein and has important roles in the regulation of cell polarity and barrier functions. Thus, we examined whether anti-hCLDN1 MAbs affected CLDN1 functions in TJs. First, the cellular CLDN1 distribution was tested after treating Huh7.5.1-8 cells with 5 μg of anti-hCLDN1 MAbs for 4 days. Even after the long treatments with MAbs, CLDN1 was detected at cell-cell contact sites (Fig. 8A), suggesting that CLDN1 localization to cell-cell contact sites, probably TJs, was not affected by anti-hCLDN1 MAbs. We next examined whether the binding of anti-hCLDN1 MAbs to polarized HepG2 cells perturbed TJ integrity. CMFDA retention in the BC lumen was comparable in polarized HepG2 cells treated with anti-hCLDN1 MAbs, control IgG, or PBS, while it was decreased in TNF-α-treated cells (Fig. 8B), suggesting that all anti-hCLDN1 MAbs had no effects on TJ integrity (barrier function).
FIG 8.
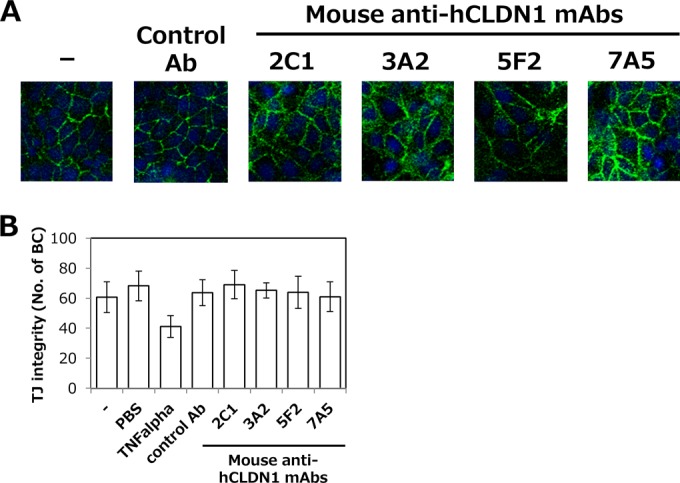
Anti-CLDN1 MAbs did not affect TJ functions in cultured cells. (A) Huh7.5.1-8 cells were treated with or without 5 μg of anti-hCLDN1 MAbs or control IgG (control Ab) and cultured for 4 days at 37°C. Cells were fixed, permeabilized, and stained with anti-CLDN1 pAbs, which recognize the cytosolic domain of CLDN1, as described in Materials and Methods. (B) HepG2 cells were treated with or without TNF-α, anti-hCLDN1 MAbs, or control IgG (control Ab) and incubated for 1 h at 37°C. Cells were then stained with CMFDA, and numbers of BCs were counted by fluorescence microscopy. Data represent the means ± SD (n = 3).
Anti-hCLDN1 MAbs prevented HCV infection in vivo.
We tested whether MAb administration inhibited HCV infection in vivo by using human liver-chimeric mice, a well-known animal model of HCV infection (24, 34–36). We focused on clones 3A2 and 7A5 in the in vivo HCV infection analysis, because these two MAbs recognized different epitopes and substantially inhibited HCV infection in vitro (Fig. 5 and 7). Chimeric mice were administered 3A2 or 7A5 intraperitoneally, at 30, 20, 10, and 10 mg/kg on days 0, 3, 7, and 10, respectively. On day 0, 8 h after MAb administration, these mice were challenged with HCV (genotype 1b) (Fig. 9A). All mice that received the control Ab were HCV positive on day 14. Serum HCV RNA levels in three mice reached a plateau that exceeded 107 copies/ml on day 21, while the level in the 4th mouse (control IgG-1) also reached a plateau of about 107 copies/ml, on day 28 (Fig. 9B). Three mice that received 3A2 were HCV negative even after day 42, while the 4th mouse that received 3A2 (3A2-4) had a very low serum HCV RNA level, near the detection limit, until day 14. Three mice that received 7A5 exhibited an apparent increase in delay of serum HCV RNA levels compared with mice that received the control Ab, while the 4th mouse that received 7A5 (7A5-1) was HCV negative even after day 42. These results show that anti-hCLDN1 clones 3A2 and 7A5, especially 3A2, substantially prevent HCV infection in vivo.
FIG 9.

Inhibition of HCV infection by anti-hCLDN1 MAbs in vivo. (A) In vivo HCV infection procedure using human liver-chimeric mice. These mice were treated with control IgG or an anti-hCLDN1 MAb, i.e., 3A2 or 7A5, using the indicated amounts, via intraperitoneal (i.p.) injection. HCV (genotype 1b; 104 copies) was used for infections in these experiments. (B) HCV RNA contents of sera were determined by qRT-PCR at the indicated time points. The dotted line indicates the detection limit. The arrows show the times of injection of MAbs. P values were 0.019, 0.121, 0.069, 0.043, and 0.044 for 3A2 and 0.019, 0.123, 0.219, 0.045, and 0.064 for 7A5 at days 7, 14, 21, 28, and 35, respectively, when the undetected values were considered to be 2 × 104 copies/ml. (C to F) Characteristics of MAb-treated chimeric mice, including body weight (C) and human albumin (D), AST (E), and ALT (F) levels in sera.
To investigate why some MAb-treated mice showed a breakthrough in viremia at different time points, we determined the serum MAb levels at 3 and 10 days post-HCV inoculation (Table 2). Interestingly, the serum MAb levels in 3A2-treated mice at day 3 were very low (<2 μg/ml), and those at day 10 were under the limit of detection; nevertheless, three of four 3A2-treated mice did not show HCV breakthrough (Fig. 9B). Although we have to investigate the distribution and metabolism of the 3A2 MAb in animals, we speculate that this MAb may completely block HCV entry through its higher affinity for human liver CLDN1. On the other hand, the 7A5 MAb was detected significantly in sera of 7A5-treated mice at days 3 and 10, except in that of the 7A5-3 mouse, in which the level was extremely low (<0.21 μg/ml) at day 3: injection of the MAb may have been missed, or the MAb was efficiently excreted or metabolized by an unknown mechanism. Consistent with the results, the 7A5-3 mouse showed an early breakthrough in viremia (Fig. 9B). The 7A5-1 mouse showed the highest concentration of 7A5 MAb in serum at days 3 and 10 (Table 2) and did not show HCV breakthrough (Fig. 9B). Thus, serum 7A5 levels seemed to be reversely correlated with serum HCV RNA levels.
TABLE 2.
Levels of anti-CLDN1 MAbs in sera of human liver-chimeric mice in in vivo HCV infection experiments
| Mouse no. | Antibody level in serum (μg/ml)a |
|
|---|---|---|
| Day 3 | Day 10 | |
| 3A2-1 | 1.67 ± 0.10 | <0.085 |
| 3A2-2 | 1.13 ± 0.01 | <0.085 |
| 3A2-3 | 0.15 ± 0.07 | <0.085 |
| 3A2-4 | 1.85 ± 0.23 | <0.085 |
| 7A5-1 | 282.6 ± 20.9 | 110.3 ± 8.3 |
| 7A5-2 | 224.7 ± 8.7 | 95.7 ± 5.9 |
| 7A5-3 | <0.21 | 14.0 ± 0.3 |
| 7A5-4 | 105.3 ± 3.4 | 12.6 ± 0.3 |
Data represent the means ± SD (n = 4). The limit of detection was 0.085 for the 3A2-treated mice and 0.21 for the 7A5-treated mice.
To monitor the toxic effects of anti-hCLDN1 MAbs, we measured the body weights and analyzed human albumin, AST, and ALT levels in sera of MAb-treated mice. No significant changes in body weight or human albumin, AST, and ALT levels were observed in mice that received control Abs or anti-hCLDN1 MAbs (Fig. 9C to F and Table 3). In particular, the constant human albumin levels detected in anti-hCLDN1 MAb-treated mice during the experimental period indicated that there were no dropouts of human hepatocytes and no defects in human hepatocyte functions of chimeric mice. Additionally, there were no apparent changes in body weight or AST and ALT levels in the anti-hCLDN1 MAb-treated chimeric mice which were not infected with HCV (data not shown). These data suggest that anti-hCLDN1 MAbs 3A2 and 7A5 do not have significant cytotoxic effects on human hepatocytes at the MAb doses used.
TABLE 3.
ALT and AST levels in sera of human liver-chimeric mice in in vivo HCV infection experiments
| Enzyme measured and antibody used | Enzyme level (U/liter)a (P value) |
||||||
|---|---|---|---|---|---|---|---|
| Day 0 | Day 7 | Day 14 | Day 21 | Day 28 | Day 35 | Day 42 | |
| ALT | |||||||
| Control | 101 ± 10 | 110 ± 15 | 172 ± 76 | 201 ± 68 | 267 ± 91 | 299 ± 193 | 299 ± 232 |
| 3A2 | 97 ± 14 (0.7402) | 116 ± 33 (0.6355) | 172 ± 76 (0.6541) | 201 ± 68 (0.8063) | 331 ± 156 (0.4561) | 385 ± 151 (0.6500) | 519 ± 113 (0.2372) |
| 7A5 | 98 ± 22 (0.9524) | 106 ± 37 (0.9421) | 156 ± 77 (0.7707) | 213 ± 96 (0.6927) | 376 ± 128 (0.1965) | 524 ± 93 (0.1340) | 541 ± 163 (0.1975) |
| AST | |||||||
| Control | 147 ± 79 | 112 ± 16 | 137 ± 35 | 126 ± 29 | 200 ± 28 | 219 ± 116 | 229 ± 121 |
| 3A2 | 126 ± 8 (0.4295) | 193 ± 35 (0.2870) | 147 ± 54 (0.6889) | 153 ± 31 (0.2588) | 238 ± 77 (0.3157) | 234 ± 38 (0.9152) | 393 ± 75 (0.0856) |
| 7A5 | 173 ± 114 (0.6588) | 144 ± 69 (0.2486) | 133 ± 54 (0.9822) | 167 ± 50 (0.1805) | 249 ± 69 (0.1853) | 353 ± 57 (0.1268) | 318 ± 102 (0.1268) |
Data represent the means ± SD (n = 4).
DISCUSSION
We applied genetic approaches to host cell mutants to identify host factors essential for the HCV life cycle. We isolated numerous HCV-resistant cell mutants, including those defective in CD81 or CLDN1 (Fig. 1 and 2), which are known host factors involved in HCV entry (9, 11). Using these mutants, we confirmed that CD81 and CLDN1 are essential for HCV infection in hepatocytes (Fig. 1 and 2).
HCV entry into cells is a multistep process involving host receptors, such as CD81, SRBI, CLDN1, and occludin (9-11, 13). These receptors may be promising targets for novel antiviral treatments (6). For example, Abs recognizing extracellular regions of these receptors are lead molecules for development of HCV entry inhibitors. Anti-CD81, -SRBI, and -CLDN1 Abs have been created to inhibit HCV infection in vitro (18, 31, 37). Anti-CD81 and -SRBI Abs can suppress HCV infection in vivo (31, 37, 38). It is unknown whether CLDN1 is a critical receptor for HCV infection in vivo. Here we established a unique screening system for CLDN1 binders by using CLDN1-defective cells (Fig. 3A), and we developed four novel anti-hCLDN1 MAbs: 2C1, 3A2, 5F2, and 7A5 (Fig. 3). 3A2 and 7A5 prevented HCV infection in vitro and in vivo (Fig. 7 and 9). Since we can now use genome editing strategies, such as the CRISPR/Cas9 system, to establish specific gene knockout mutant cells easily, this Ab screening method using parental cells expressing the intact target membrane protein and the target-defective cells would be a useful strategy for developing functional MAbs against extracellular domains of intact multimembrane-spanning proteins.
There are two major differences between the methods used by Fofana et al. to develop anti-CLDN1 MAbs (18) and those used in our study (Fig. 3A). Fofana et al. used rats as immunized animals, while we used autoimmune BXSB mice. They used CLDN1-overexpressing cells for hybridoma screening, while we used intact CLDN1-bearing Huh7.5.1 and CLDN1-defective S7-A cells. These differences may have resulted in the different characteristics of the two MAb groups. MAbs developed by Fofana et al. recognized the conserved motif W30-GLW51-C54-C64 and the I33, Y35, and D38 residues in EL1 of hCLDN1 (18). In contrast, our MAb 7A5 recognized M152 in EL2 (Fig. 5). Three other MAbs recognized structures with three-dimensional conformations; thus, the real epitopes cannot be defined until the cocrystal structures are solved in the future. However, our preliminary flow cytometric observations based on mutational analyses showed that the binding strengths of 2C1, 3A2, and 5F2 to the mouse-type EL1 or EL2 mutants were significantly low or undetectable (Fig. 5), suggesting that these clones may recognize three-dimensional conformations of EL1 and EL2. The rat anti-CLDN1 MAbs by Fofana et al. resulted in a >90% inhibition of HCV infection in vitro at 10 to 50 μg/ml (18), whereas our mouse MAbs had similar inhibitory effects at only 0.3 to 8 μg/ml (Fig. 7). The binding affinities of rat MAbs for cellular CLDN1 were in the nanomolar range (2 to 9 nM) (18), whereas those of our mouse MAbs were in the picomolar range (100 to 230 pM) (Fig. 6). At least two of our anti-hCLDN1 MAbs, 3A2 and 7A5, significantly inhibited HCV infection in vivo (Fig. 9). Thus, the tight association between our MAbs and domains that included intact EL2 of hCLDN1 may have resulted in higher in vitro and successful in vivo anti-HCV activities. 3A2 exhibited a more efficient inhibition of HCV infection in vivo and in vitro than that by 7A5 (Fig. 7 and 9), possibly because of differences in their epitopes, i.e., 7A5 recognizes EL2, while 3A2 may recognize EL1 and EL2. The crystal structure of mouse CLDN15 was reported very recently (39). The transmembrane four-helix bundle of CLDN15 is tightly packed; thus, N-terminal EL1 and C-terminal EL2 are located in close proximity. CLDN1 may have a similar conformation, because homology modeling of the CLDN family indicates that the conformation of the four-helix bundle is highly conserved. A previous study showed that the first extracellular loop of CLDN1 is required for HCV entry (11). In this study, anti-CLDN1 MAbs 5F2 and 7A5, recognizing the second extracellular loop of CLDN1 (Fig. 5B and C), could also prevent HCV infection (Fig. 7). We now think that binding of the 5F2 and 7A5 MAbs to the second extracellular loop of CLDN1 is likely to mask the first extracellular loop as well as the second one and to block HCV entry, since MAbs are ∼5-fold larger than the CLDN1 molecule, and both extracellular loops of CLDN1 might be located very closely on the membranes, like those of CLDN15 (39).
CLDN has pivotal roles in the physiological barrier that regulates the free movement of solutes via the paracellular space (40). Hepatocytes are highly polarized cells, and their plasma membranes are separated into apical-canalicular and basolateral-sinusoidal domains by TJs (41). TJs are physiological barriers that prevent HCV entry into host cells, and TJ disruption increases HCV entry (42). Thus, adverse effects may occur in the liver if anti-hCLDN1 MAbs disrupt TJ seals. However, MAb treatment prevented HCV entry without affecting CLDN1 localization at cell-cell contact sites in Huh7.5.1-8 cells (Fig. 8A). Importantly, MAb treatment did not have significant adverse effects on human liver function at the doses used (Fig. 9D to F and Table 3). CLDN1 is expressed on apical and basolateral surfaces of hepatocytes in normal liver tissue (43), but the presence of TJs (42) or CLDN1 localization at TJs (11) is not critical for HCV infection. Therefore, nonjunctional CLDN1 may be a promising target for HCV therapy. The treatment of cells with anti-hCLDN1 MAbs for a long period resulted in CLDN1 localization to cell-cell contact sites and dot-like localization to the cell surface near cell-cell contact sites (Fig. 8A). Under these conditions, nonjunctional CLDN1, which may be involved in HCV infection, may be trapped by MAbs.
In conclusion, we developed a novel Ab screening system using CLDN1-defective cells and successfully created mouse anti-hCLDN1 MAbs that strongly inhibited HCV infection in vitro and in vivo, without apparent adverse effects, thereby providing new insights into the future development of CLDN1-targeting anti-HCV agents, although further investigations, including in vivo HCV infection experiments with other genotypes of the virus, are needed. These agents may be useful in inhibiting primary infections, such as those after liver transplantation, and in combination therapies with other anti-HCV drugs, such as direct-acting antivirals.
ACKNOWLEDGMENTS
We thank all members of our laboratory for their useful comments. We thank F. V. Chisari (The Scripps Research Institute, La Jolla, CA) for donating Huh7.5.1 cells. We thank Y. Matsuura (Osaka University, Osaka, Japan) for the gift of the pcDNA3.1-hCD81 vector. We thank Ryo Abe and Jo Chiba (Tokyo University of Science, Chiba, Japan), Masahiro Nishijima (Showa Pharmaceutical University), and Minoru Tada and Akiko Ishii-Watabe (National Institute of Health Sciences, Tokyo, Japan) for their helpful comments and support.
This research was supported by a Health and Labor Sciences Research Grant from the Ministry of Health, Labor, and Welfare of Japan; Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (grant 23590104 to M.F. and grant 24390042 to M.K.); the Adaptable and Seamless Technology Transfer Program through Target-Driven R&D, Japan Science and Technology Agency; the Platform for Drug Discovery, Informatics, and Structural Life Science of the Ministry of Education, Culture, Sports, Science, and Technology, Japan; the Takeda Science Foundation (to M.K. and K.H.); and the Nakatomi Foundation.
Except for Kohki Endo, we declare that there are no conflicts of interest. Kohki Endo is an employee of Wako Pure Chemical Industries, Ltd.
REFERENCES
- 1.El-Serag HB. 2012. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142:1264–1273. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray CL, Rice CM. 2011. Turning hepatitis C into a real virus. Annu Rev Microbiol 65:307–327. doi: 10.1146/annurev-micro-090110-102954. [DOI] [PubMed] [Google Scholar]
- 3.McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J, Nyberg LM, Lee WM, Ghalib RH, Schiff ER, Galati JS, Bacon BR, Davis MN, Mukhopadhyay P, Koury K, Noviello S, Pedicone LD, Brass CA, Albrecht JK, Sulkowski MS. 2009. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med 361:580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- 4.Poordad F, McCone J Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawitz E, Sulkowski MS, Ghalib R, Rodriguez-Torres M, Younossi ZM, Corregidor A, DeJesus E, Pearlman B, Rabinovitz M, Gitlin N, Lim JK, Pockros PJ, Scott JD, Fevery B, Lambrecht T, Ouwerkerk-Mahadevan S, Callewaert K, Symonds WT, Picchio G, Lindsay KL, Beumont M, Jacobson IM. 2014. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet 384:1756–1765. doi: 10.1016/S0140-6736(14)61036-9. [DOI] [PubMed] [Google Scholar]
- 6.Zeisel MB, Lupberger J, Fofana I, Baumert TF. 2013. Host-targeting agents for prevention and treatment of chronic hepatitis C—perspectives and challenges. J Hepatol 58:375–384. doi: 10.1016/j.jhep.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 7.Barth H, Schnober EK, Zhang F, Linhardt RJ, Depla E, Boson B, Cosset FL, Patel AH, Blum HE, Baumert TF. 2006. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J Virol 80:10579–10590. doi: 10.1128/JVI.00941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monazahian M, Bohme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R. 1999. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J Med Virol 57:223–229. [DOI] [PubMed] [Google Scholar]
- 9.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. [DOI] [PubMed] [Google Scholar]
- 10.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 12.Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T. 2009. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol 83:2011–2014. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris HJ, Davis C, Mullins JG, Hu K, Goodall M, Farquhar MJ, Mee CJ, McCaffrey K, Young S, Drummer H, Balfe P, McKeating JA. 2010. Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem 285:21092–21102. doi: 10.1074/jbc.M110.104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, McKeating JA. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24. doi: 10.1002/hep.21959. [DOI] [PubMed] [Google Scholar]
- 18.Fofana I, Krieger SE, Grunert F, Glauben S, Xiao F, Fafi-Kremer S, Soulier E, Royer C, Thumann C, Mee CJ, McKeating JA, Dragic T, Pessaux P, Stoll-Keller F, Schuster C, Thompson J, Baumert TF. 2010. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 139:953–964, 964.e1–964.e4. doi: 10.1053/j.gastro.2010.05.073. [DOI] [PubMed] [Google Scholar]
- 19.Krieger SE, Zeisel MB, Davis C, Thumann C, Harris HJ, Schnober EK, Mee C, Soulier E, Royer C, Lambotin M, Grunert F, Dao Thi VL, Dreux M, Cosset FL, McKeating JA, Schuster C, Baumert TF. 2010. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology 51:1144–1157. doi: 10.1002/hep.23445. [DOI] [PubMed] [Google Scholar]
- 20.Hötzel I, Chiang V, Diao J, Pantua H, Maun HR, Kapadia SB. 2011. Efficient production of antibodies against a mammalian integral membrane protein by phage display. Protein Eng Des Sel 24:679–689. doi: 10.1093/protein/gzr039. [DOI] [PubMed] [Google Scholar]
- 21.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shirasago Y, Sekizuka T, Saito K, Suzuki T, Wakita T, Hanada K, Kuroda M, Abe R, Fukasawa M. Isolation and characterization of a Huh.7.5.1-derived cell clone highly permissive to hepatitis C virus. J Infect Dis, in press. doi: 10.7883/yoken.JJID.2014.231. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Iida M, Tada M, Watari A, Kawahigashi Y, Kimura Y, Yamashita T, Ishii-Watabe A, Uno T, Fukasawa M, Kuniyasu H, Yagi K, Kondoh M. 2014. Development of an anti-claudin-3 and -4 bispecific monoclonal antibody for cancer diagnosis and therapy. J Pharmacol Exp Ther 351:206–213. doi: 10.1124/jpet.114.216911. [DOI] [PubMed] [Google Scholar]
- 24.Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, Addison WR, Fischer KP, Churchill TA, Lakey JR, Tyrrell DL, Kneteman NM. 2001. Hepatitis C virus replication in mice with chimeric human livers. Nat Med 7:927–933. doi: 10.1038/90968. [DOI] [PubMed] [Google Scholar]
- 25.Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, Tachibana A, Soeno Y, Asahina K, Hino H, Asahara T, Yokoi T, Furukawa T, Yoshizato K. 2004. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am J Pathol 165:901–912. doi: 10.1016/S0002-9440(10)63352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saito K, Shirasago Y, Suzuki T, Aizaki H, Hanada K, Wakita T, Nishijima M, Fukasawa M. 2015. Targeting cellular squalene synthase, an enzyme essential for cholesterol biosynthesis, is a potential antiviral strategy against hepatitis C virus. J Virol 89:2220–2232. doi: 10.1128/JVI.03385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nitahara-Kasahara Y, Fukasawa M, Shinkai-Ouchi F, Sato S, Suzuki T, Murakami K, Wakita T, Hanada K, Miyamura T, Nishijima M. 2009. Cellular vimentin content regulates the protein level of hepatitis C virus core protein and the hepatitis C virus production in cultured cells. Virology 383:319–327. doi: 10.1016/j.virol.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Murakami Y, Fukasawa M, Kaneko Y, Suzuki T, Wakita T, Fukazawa H. 2013. Selective estrogen receptor modulators inhibit hepatitis C virus infection at multiple steps of the virus life cycle. Microbes Infect 15:45–55. doi: 10.1016/j.micinf.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Wakita T, Wands JR. 1994. Specific inhibition of hepatitis C virus expression by antisense oligodeoxynucleotides. In vitro model for selection of target sequence. J Biol Chem 269:14205–14210. [PubMed] [Google Scholar]
- 31.Meuleman P, Hesselgesser J, Paulson M, Vanwolleghem T, Desombere I, Reiser H, Leroux-Roels G. 2008. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 48:1761–1768. doi: 10.1002/hep.22547. [DOI] [PubMed] [Google Scholar]
- 32.Catanese MT, Graziani R, von Hahn T, Moreau M, Huby T, Paonessa G, Santini C, Luzzago A, Rice CM, Cortese R, Vitelli A, Nicosia A. 2007. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J Virol 81:8063–8071. doi: 10.1128/JVI.00193-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akazawa D, Date T, Morikawa K, Murayama A, Miyamoto M, Kaga M, Barth H, Baumert TF, Dubuisson J, Wakita T. 2007. CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J Virol 81:5036–5045. doi: 10.1128/JVI.01573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bukh J, Meuleman P, Tellier R, Engle RE, Feinstone SM, Eder G, Satterfield WC, Govindarajan S, Krawczynski K, Miller RH, Leroux-Roels G, Purcell RH. 2010. Challenge pools of hepatitis C virus genotypes 1–6 prototype strains: replication fitness and pathogenicity in chimpanzees and human liver-chimeric mouse models. J Infect Dis 201:1381–1389. doi: 10.1086/651579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, Rice CM. 2006. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci U S A 103:3805–3809. doi: 10.1073/pnas.0511218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meunier JC, Engle RE, Faulk K, Zhao M, Bartosch B, Alter H, Emerson SU, Cosset FL, Purcell RH, Bukh J. 2005. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc Natl Acad Sci U S A 102:4560–4565. doi: 10.1073/pnas.0501275102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meuleman P, Catanese MT, Verhoye L, Desombere I, Farhoudi A, Jones CT, Sheahan T, Grzyb K, Cortese R, Rice CM, Leroux-Roels G, Nicosia A. 2012. A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology 55:364–372. doi: 10.1002/hep.24692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vercauteren K, Van Den Eede N, Mesalam AA, Belouzard S, Catanese MT, Bankwitz D, Wong-Staal F, Cortese R, Dubuisson J, Rice CM, Pietschmann T, Leroux-Roels G, Nicosia A, Meuleman P. 2014. Successful anti-scavenger receptor class B type I (SR-BI) monoclonal antibody therapy in humanized mice after challenge with HCV variants with in vitro resistance to SR-BI-targeting agents. Hepatology 60:1508–1518. doi: 10.1002/hep.27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, Dohmae N, Tsukita S, Nureki O, Fujiyoshi Y. 2014. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 344:304–307. doi: 10.1126/science.1248571. [DOI] [PubMed] [Google Scholar]
- 40.Furuse M, Tsukita S. 2006. Claudins in occluding junctions of humans and flies. Trends Cell Biol 16:181–188. doi: 10.1016/j.tcb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Boyer JL. 2004. The maintenance and generation of membrane polarity in hepatocytes. Hepatology 39:892–899. doi: 10.1002/hep.20039. [DOI] [PubMed] [Google Scholar]
- 42.Mee CJ, Grove J, Harris HJ, Hu K, Balfe P, McKeating JA. 2008. Effect of cell polarization on hepatitis C virus entry. J Virol 82:461–470. doi: 10.1128/JVI.01894-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reynolds GM, Harris HJ, Jennings A, Hu K, Grove J, Lalor PF, Adams DH, Balfe P, Hubscher SG, McKeating JA. 2008. Hepatitis C virus receptor expression in normal and diseased liver tissue. Hepatology 47:418–427. doi: 10.1002/hep.22028. [DOI] [PubMed] [Google Scholar]