

ABSTRACT
The retinoblastoma (Rb) tumor suppressor controls cell cycle, DNA damage, apoptotic, and metabolic pathways. DNA tumor virus oncoproteins reduce Rb function by either inducing Rb degradation or physically disrupting complexes between Rb and its myriad binding proteins. Human cytomegalovirus (HCMV), a betaherpesvirus being investigated for potential roles in human cancers, encodes multiple lytic-phase proteins that inactivate Rb in distinct ways, leading to the hypothesis that reduced Rb levels and/or activity would benefit HCMV lytic infection. Paradoxically, we found that Rb knockdown prior to infection, whether transient or constitutive, impaired HCMV lytic infection at multiple stages, notably viral DNA replication, late protein expression, and infectious virion production. The existence of differentially modified forms of Rb, the temporally and functionally distinct means by which HCMV proteins interact with Rb, and the necessity of Rb for efficient HCMV lytic replication combine to highlight the complex relationship between the virus and this critical tumor suppressor.
IMPORTANCE Initial work examining viral protein modulation of cell cycle progression and oncogenic transformation revealed that these proteins inactivated the function of cellular tumor suppressor proteins. However, subsequent work, including experiments described here using human cytomegalovirus, demonstrate a more nuanced interaction that includes the necessity of cellular tumor suppressors for efficient viral replication. Understanding the positive impacts that cellular tumor suppressors have on viral infections may reveal new activities of these well-studied yet incompletely understood proteins. The basis for oncolytic viral therapy is the selective replication of viruses in transformed cells in which tumor suppressor function may be compromised. Understanding how tumor suppressors support viral infections may allow for the generation of modified oncolytic viruses with greater selective tumor cell replication and killing.
INTRODUCTION
The retinoblastoma (Rb) protein is a tumor suppressor (1, 2). Loss of both Rb alleles predisposes patients to the development of cancer (3). Rb, through its association with more than 200 other cellular proteins (4), controls pathways that regulate cell cycle progression, DNA repair, apoptosis, and energy metabolism, all of which are intimately involved in oncogenic transformation and tumor cell survival (5–7). Most, if not all, human tumors have defects (mutations) in one or more components of the pathways controlled by Rb (8).
The unphosphorylated or hypophosphorylated form of Rb is generally considered the active form of the protein (9). Hypophosphorylated Rb interacts with many cellular proteins, including a critical association with the E2F family of transcription factors (10). E2F transcription factors control the expression of many genes required for cell cycle progression, and Rb binding inhibits E2F-dependent transcription (11). Rb binding to E2F protects cells from untimely progression through the cell cycle and prevents E2F-mediated oncogenic transformation (12, 13). During normal cell cycle progression, a series of cellular cyclin-dependent kinases (Cdks) phosphorylate Rb, converting it into a fully phosphorylated form, termed hyperphosphorylated Rb. This form is considered inactive (14), although it may retain some unrecognized function (15). Hyperphosphorylated Rb no longer binds E2F and thus permits E2F-dependent transcription and cell cycle progression (10). Recently, Cdk-dependent monophosphorylation of Rb has been reported (16), but the physiological relevance of this is unknown. Rb can also be acetylated, methylated, SUMOylated, ubiquitinated, and phosphorylated on non-Cdk-mediated sites in response to stimuli that may activate non-cell-cycle-associated functions of Rb (17).
In addition to being a tumor suppressor, Rb might also be a “virus suppressor,” at least for the DNA tumor virus human papillomavirus (HPV). The HPV E7 protein binds to Rb and induces its proteasomal degradation (18, 19). E7 proteins unable to bind or degrade Rb are unable to support productive papillomavirus replication (20, 21). However, as E7 Rb-binding-deficient mutants have other defects (22), it is premature to conclude the inability to degrade Rb is the only reason for the observed defects in the viral life cycle. Unfortunately, the role of Rb during HPV infection remains unclear due to difficulties in studying productive HPV replication in vitro (through organotypic raft cultures) and the justifiable focus on the essential role of E7-mediated inactivation of Rb during HPV-induced cellular transformation and human cancers.
Adenovirus, another DNA tumor virus, encodes the E1A protein that binds Rb and disrupts its complexes with E2F (23). E1A mutants unable to bind Rb display only modest defects in viral replication (24). Therefore, Rb does not appear to be a suppressor of adenovirus. In fact, during adenoviral infection, Rb-E2F1 complexes selectively remain intact (25), and recently E1A-Rb complexes were demonstrated to suppress the transcription of genes with antiviral functions in adenovirus-infected cells (26). Thus, Rb might contribute to adenovirus infection in a positive way, although this remains to be determined through knockdown or knockout studies.
Human cytomegalovirus (HCMV) is a DNA virus that is being explored as a potential cofactor for human cancers, most notably glioblastoma brain tumors (27–30). One possible mechanism by which HCMV could contribute to cellular transformation is manipulation of the cell cycle through inactivation of Rb (28). HCMV has multiple, potentially semiredundant ways in which to modulate Rb protein function during lytic infection. The viral pp71 protein is delivered to cells by infecting virions where it binds to and induces the proteasomal degradation of hypophosphorylated Rb (31, 32). This pp71-mediated degradation occurs only within the first hours of infection, and Rb reaccumulates later during infection (33). The biological relevance of this degradation remains unclear, as a pp71 mutant virus unable to induce Rb degradation supports wild-type levels of lytic infection in fibroblasts (34). The viral UL97 protein is a viral cyclin-dependent kinase (v-Cdk) that phosphorylates Rb on several Cdk target sites (33, 35). A mutant virus lacking UL97 fails to phosphorylate Rb and has a growth defect, and it should also fail to phosphorylate other known cellular and viral targets (36). Mutants unable to phosphorylate Rb but retaining kinase activity have not been reported. The viral IE1 and IE2 proteins have also been reported to activate E2F-responsive promoters (37–39), though mechanisms have not been explored and evidence for Rb inactivation by IE1 and IE2 during viral infection is lacking.
With at least two and perhaps as many as four ways to inactivate the Rb protein, we suspected that Rb would suppress HCMV infection. However, we found that Rb knockdown, either transiently by small interfering RNA (siRNA) transfection or constitutively through short hairpin RNA (shRNA) expression, impaired HCMV lytic replication in fibroblasts. HCMV viral DNA replication and late gene expression were decreased, and as many as 10-fold-fewer infectious virions were produced. We conclude that the Rb protein is required for efficient HCMV replication and that the multiple ways in which HCMV interacts with Rb represent viral attempts not to completely eliminate Rb function but to modulate it in temporally and likely functionally distinct ways.
MATERIALS AND METHODS
Cells and viruses.
Primary normal human dermal fibroblasts (NHDFs) (Clonetics) used for transient experiments and to derive the stable knockdown populations were cultured in Dulbecco's modified Eagle medium (DMEM) (Invitrogen and Sigma). THP-1 cells (ATCC TIB-202) were cultured in RPMI 1640 medium (Invitrogen). Both DMEM and RPMI 1640 medium were supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Sigma), and 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.292 mg/ml glutamine (PSG) (Invitrogen) to generate complete media. Transduction with shRNA retroviruses was performed as previously described (40). Transduced cells were maintained in complete media containing 1 μg/ml puromycin (Sigma) and used for experiments until passage 16. Asynchronous cells were plated at a subconfluent density and cultured in complete media for 24 h before harvesting or infection. To arrest cells by serum starvation, cells were plated at 1 × 104 cells/cm2, and 24 h after plating, cells were washed twice with Dulbecco's phosphate-buffered saline (DPBS) (Invitrogen) and then maintained in DMEM containing 0.1% FBS supplemented with PSG (0.1% FBS DMEM+PSG) (low-serum medium) for 48 h before harvesting or infection. Confluent cells were fed complete media every 2 days for 144 h after plating before beginning contact inhibition cell experiments. All infections used HCMV-AD169 or an AD169 derivative expressing IE2 fused to green fluorescent protein (AD169 IE2-GFP, 41) or GFP-expressing HCMV strain FIX (42). Multiplicities of infection (MOIs) were calculated for each cell line by counting an extra plated well. Cells were infected in minimal volumes for 60 min, inocula were removed, and media were replaced. Viral titers were measured by standard plaque assay on nontransduced fibroblasts.
RNA interference.
The pSUPER retro puro Scr shRNA (retro stands for retroviral, puro stands for puromycin, and Scr stands for scrambled) was a gift from John Gurdon (Addgene plasmid 30520) (43), and the Rb2 and Rb3 plasmids were previously described (RB-sh2 and RB-sh3 [44]). Transient knockdown was achieved by transfecting cells with 80 pg/cell of small interfering oligonucleotides (small interfering RNA [siRNA]) from Dharmacon (scrambled control [Scr], catalog no. D-001810-01-20; siRNA targeting the coding region of the Rb mRNA [siRb1], 5′-GAA CAG GAG UGC ACG GAU AUU-3′) using Lonza nucleofection reagents (VPI-1002; Lonza) according to the manufacturer's instructions.
Inhibitors and antibodies.
Valproic acid (VPA) (1 mM) (Sigma) dissolved in water was added to THP-1 cells 3 h prior to infection and maintained throughout the infection. Antibodies used in these experiments that are available from commercial sources are as follows: anti-Rb (4H1; Cell Signaling; catalog no. 554136; BD Pharmingen), anti-p107 (C-18; Santa Cruz), anti-cyclin E (C-19; Santa Cruz), anti-tubulin (clone DM 1A; Sigma), and anti-UL44 (10D8; Virusys). Monoclonal antibodies against pp71 (2H10-9), IE1 (1B12), and pp150 (CMV127) have been described previously (45). Infrared (IR) dye 680- and 800-conjugated secondary antibodies (Li-Cor) were used for Western blotting, and Alexa Fluor 488-conjugated secondary antibody (catalog no. A-11017; Molecular Probes; Thermo Fisher Scientific) was used for immunofluorescence.
Western blots.
Equal cell numbers were lysed in 1% SDS and 2% β-mercaptoethanol and boiled for 20 min prior to separation by SDS-PAGE and transfer to nitrocellulose membranes (Whatman). Five percent bovine serum albumin (BSA) in Tris-buffered saline with Tween 20 (TBST [46]) was used to block membranes and incubate primary and secondary antibodies. Membranes were washed in TBST and imaged with the Odyssey Fc imager (Li-Cor). Images were generated, and bands were quantitated with the Image Studio version 2.1.10 software (Li-Cor).
Flow cytometry.
After trypsinization, cells were harvested by low-speed centrifugation, washed twice with PBS, and fixed with 70% ethanol at −20°C. Cells were then stained with propidium iodide (PI) (Sigma) 1 h prior to analysis on a FACSCalibur flow cytometer (BD Biosciences). Data were analyzed with ModFit LT software (Verity Software House).
Quantitative real-time PCR.
Total DNA was extracted from equal numbers of cells using a genomic DNA minikit (IB47202; IBI). DNA was quantitated using iTaq universal SYBR green supermix (catalog no. 172-5124; Bio-Rad) on an Applied Biosystems 7900HT instrument. Viral DNA was amplified with IE1 primers (47), and cellular genomes were measured with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers (48).
Indirect immunofluorescence.
Cells were plated on glass coverslips and serum starved for 48 h prior to infection. After infection, cells were fixed with 1% paraformaldehyde in PBS and processed as previously described (46). Stained cells were visualized with a Nikon Ti-Eclipse inverted wide-field microscope, and images were collected with a CoolSnap HQ camera and Nikon NIS Elements software (v 4.00.03). At least 400 nuclei were counted per condition.
RESULTS
Rb knockdown in primary human fibroblasts inhibits HCMV lytic infection.
To determine whether Rb helps or hinders HCMV replication, we employed RNA interference to knock down the levels of the Rb protein prior to infection. Initial experiments utilized transient transfection of a duplexed siRNA targeting the coding region of the Rb mRNA (siRb1) or a scrambled control (siScr). Western blot analysis indicated that siRb1 was effective at reducing the steady-state level of the Rb protein, as well as its activity, as evidenced by the increased steady-state levels of the proteins encoded by the Rb-repressed, E2F-responsive genes p107 and cyclin E (Fig. 1A). Transient Rb knockdown cells produced fewer infectious virions than the scrambled controls after infections at either low (Fig. 1B) or high (Fig. 1C) multiplicities of infection (MOIs) with the laboratory-adapted AD169 strain of HCMV. We conclude that transient Rb knockdown in fibroblasts inhibits HCMV productive lytic replication.
FIG 1.
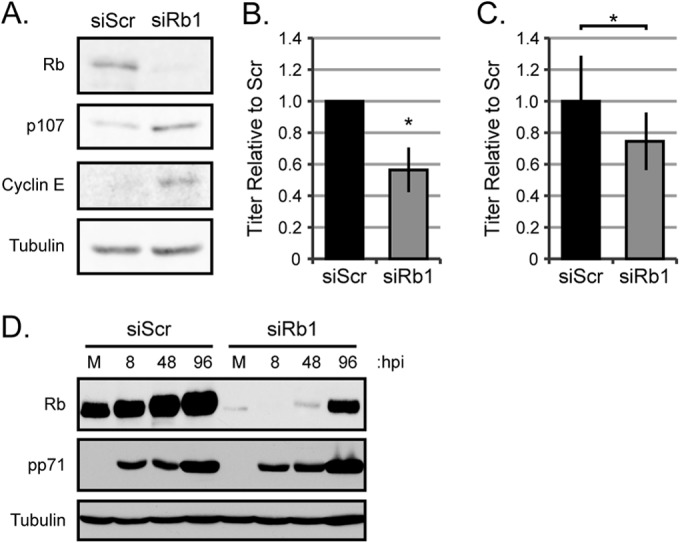
Transient reduction of steady-state Rb protein levels decreases HCMV virion production. (A) Primary fibroblasts were electroporated to deliver scrambled siRNA (siScr) or Rb-targeting siRNA (siRb1) to cells. Twenty-four hours after transfection, cells were serum starved, and equal amounts of protein lysates were analyzed by Western blotting with the indicated antibodies. (B) Serum-starved siScr- or siRb1-transfected cells were infected with AD169 at an MOI of 0.1. Combined cell-free and cell-associated virus was collected 6 days postinfection, and the titers of virus were determined by standard plaque assay. The data are mean titers normalized to the siScr titer ± standard deviation (error bar) from 6 biological replicates. Statistical significance was determined by a permutation test of titers before normalization (*, P < 0.04). (C) Serum-starved siScr- or siRb1-transfected cells were infected with AD169 at an MOI of 1. Combined cell-free and cell-associated virus was collected 4 days postinfection, and the titers of virus were determined by standard plaque assay. The data are mean titers normalized to siScr titer ± standard deviations from 5 biological replicates. Statistical significance was determined by a permutation test prior to normalization (*, P < 0.025). (D) Serum-starved siScr- or siRb1-transfected cells were mock infected (M) or infected with AD169 at an MOI of 1 and harvested at the indicated time (hours postinfection [hpi]). Equal amounts of protein lysates were analyzed by Western blotting with the indicated antibodies.
We suspected the modest effect after transient knockdown might be due to Rb protein reaccumulation during the lengthy HCMV productive replication cycle, which we frequently observed (Fig. 1D), compounded by an inability to selectively infect only transfected cells. Therefore, we stably decreased Rb protein steady-state levels by transducing cells with a retroviral vector expressing an shRNA to Rb or a scrambled control sequence. The retroviral vector also encodes the gene for resistance to puromycin, which allowed us to generate and maintain a population of transduced cells by selecting for resistance to puromycin. We used a different sequence complementary to the Rb coding sequence for the stable knockdowns (Rb2) compared to the transient knockdowns (siRb1) to control for off target effects inherent with RNA interference approaches. shRNA-Rb2 was effective at reducing the steady-state level of the Rb protein, as well as its activity, as evidenced by the increased steady-state levels of E2F-responsive genes (Fig. 2A). Similar to the transient-knockdown experiments, the stable Rb knockdown cells produced fewer infectious virions than the scrambled controls after infections at either low (Fig. 2B) or high (Fig. 2C) MOI HCMV-AD169 infections. A clinical strain of HCMV (FIX) also showed decreased replication in stable Rb knockdown cells (Fig. 2D). Rb did not reaccumulate during any of these experiments (Fig. 2E), likely accounting for the increased magnitude of the observed negative effect on HCMV replication compared to transient knockdown, where Rb did reaccumulate. We conclude that stable Rb knockdown in fibroblasts inhibits HCMV productive lytic replication. Due to the similar but more durable effect of stable compared to transient knockdown, we used the stable knockdown cells for experiments to determine at what stage(s) Rb enhances HCMV infection.
FIG 2.
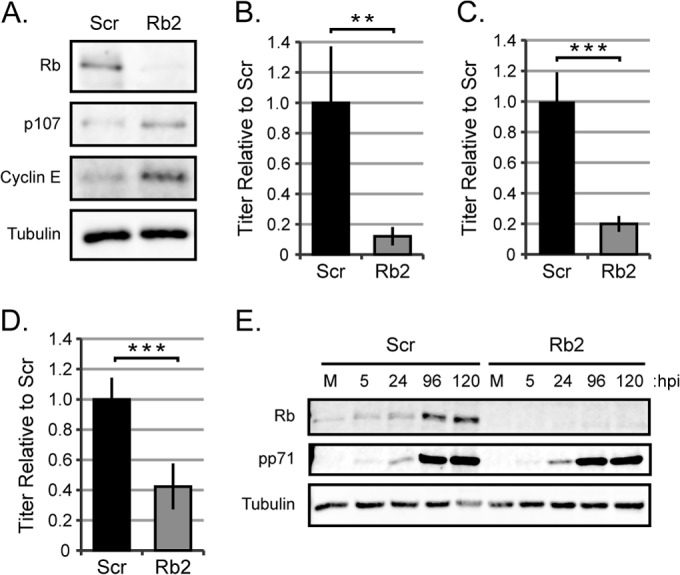
Stable Rb knockdown reduces the production of HCMV virions. (A) Equal amounts of protein lysates from subconfluent, serum-starved primary fibroblasts transduced with a retrovirus expressing a scrambled shRNA sequence (Scr) or an shRNA targeting Rb mRNA (Rb2) were analyzed by Western blotting with the indicated antibodies. (B to D) Serum-starved Scr or Rb2 cells were infected with AD169 at an MOI of 0.1 (B) or 1 (C) or with HCMV strain FIX labeled with GFP (FIX-GFP) at an MOI of 1 (D). Combined cell-free and cell-associated virus was collected 4 days postinfection, and the titers of virus were determined by standard plaque assay. The data are mean titers normalized to the Scr titer ± standard deviations from three biological replicates with technical triplicates. Statistical significance was determined by a permutation test prior to normalization (**, P < 0.01; ***, P < 0.001). (E) Serum-starved Scr or Rb2 cells were mock infected (M) or infected with AD169 at an MOI of 0.1 and harvested at the indicated hours postinfection (hpi). Equal amounts of protein lysates were analyzed by Western blotting with the indicated antibodies.
HCMV does not initiate lytic replication during the DNA synthesis (S) phase of the cell cycle, and Rb loss can compromise the restriction of cell entry into the S phase under certain conditions (49, 50). Thus, we examined whether the observed HCMV replication defect could be due to unscheduled S-phase entry following Rb knockdown in our primary fibroblasts. Our stable Rb knockdown cells, as well as scrambled controls, retained the ability to growth arrest in response to serum starvation as well as upon contact inhibition (Fig. 3A). Furthermore, the DNA content profiles, which are indicative of cell cycle stage, for Rb knockdown and scrambled controls, were indistinguishable when cells were either growing asynchronously (Fig. 3B) or grown to confluence (Fig. 3C) or starved for serum (Fig. 3D). The DNA content profile also revealed that the Rb knockdown and scrambled control cells maintain an intact G1/S restriction point, as approximately 90% of the serum-starved or contact-arrested cells were in the G1 phase. Finally, HCMV produced fewer infectious virions than scrambled controls in stable Rb knockdown cells that were serum starved (Fig. 2B), asynchronously growing (Fig. 3E), or confluent (Fig. 3F). We conclude that our population of stable Rb knockdown primary fibroblasts has growth and synchronization characteristics indistinguishable from those of their Rb-expressing counterparts and that the defect in HCMV replication observed in the Rb knockdown cells does not result from cell cycle perturbations secondary to Rb knockdown.
FIG 3.

Cell cycle defects are not responsible for the decrease in virions produced by Rb knockdown cells. (A) Scr or Rb2 cells were plated at 1 × 104 cells/cm2 and serum starved at 24 h after plating or maintained in complete media. Cell density was measured after trypsinization by counting with a hemocytometer. Each data point represents the mean cell density ± standard deviation from three biological replicates. (B to D) Scr or Rb2 cells were plated at a subconfluent density and harvested 24 h later as an asynchronous population (B), fed every 48 h for 6 days until contact inhibited (C), or serum starved for 48 h (D) before harvesting for analysis of PI staining by flow cytometry. A representative histogram from three biological replicates for each condition is shown. (E and F) Scr or Rb2 cells were plated at a subconfluent density and 24 h later were infected in complete media with AD169 at an MOI of 1 (E) or were fed every 48 h for 6 days and then infected in complete media by AD169 at an MOI of 1 (F). Combined cell-free and cell-associated virus was collected 4 days postinfection, and the titers of virus were determined by standard plaque assay. The data are mean titers normalized to the Scr titer ± standard deviations from three biological replicates with technical triplicates. Statistical significance was determined by a permutation test prior to normalization (***, P < 0.001).
Rb knockdown reduces entry but not IE1 or UL44 protein accumulation.
We systematically interrogated landmark steps during the HCMV lytic replication cycle (entry, immediate early [IE] gene expression, early gene expression, viral DNA replication, and late gene expression) to decipher how these stages were enhanced by the presence of Rb. As an initial assay for viral entry, we analyzed the steady-state levels of the viral pp71 tegument protein that is delivered to cells by infecting virions. Western blot analyses revealed a small, but noticeable and reproducible, reduction in the level of pp71 protein in Rb knockdown cells at 6 h postinfection (Fig. 4A). Quantitation of multiple experiments normalized to tubulin levels detected a 32% decrease in pp71 levels in Rb knockdown cells (Fig. 4B). We further analyzed entry by quantitating viral genome delivery to cells at both high (Fig. 4C) and low (Fig. 4D) MOIs. Again, we found an approximately 20% decrease in viral genomes delivered to Rb knockdown cells compared to scrambled controls. The simplest explanation for these results is that viral entry is less efficient in the absence of Rb, although we cannot rule out effects on the stability of the pp71 protein or the viral genomic DNA. As pp71 and viral genomes appear to traffic independently in cells (40), this defect seems to be at an early stage of viral entry.
FIG 4.
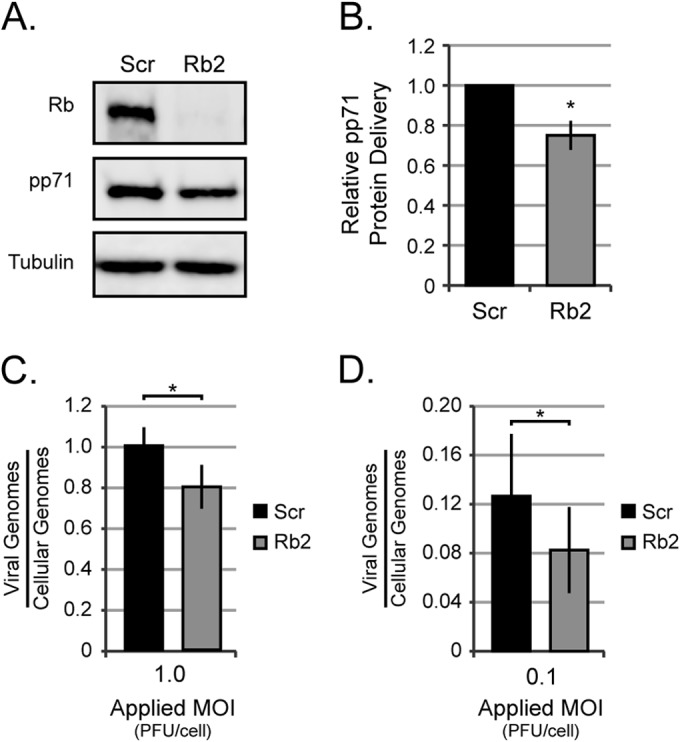
Rb knockdown reduces the ability of HCMV to enter cells. (A) Serum-starved Scr or Rb2 cells were infected with AD169 at an MOI of 1, and equal amounts of protein from lysates harvested between 4 and 6 h postinfection were analyzed by Western blotting with the indicated antibodies. (B) The data are mean values of tegument-delivered pp71 relative to tubulin and normalized to Scr ± standard deviations from five biological replicates. Statistical significance was determined by Student's t test prior to normalization (*, P < 0.02). (C and D) Serum-starved Scr or Rb cells were infected with AD169 at an MOI of 1 (C) or an MOI of 0.1 (D) and harvested at 6 h postinfection. Viral and cellular genomes were then quantitated by qPCR. The data are mean numbers of viral genomes relative to those of cellular genomes ± standard deviations from four biological replicates for each MOI. Statistical significance was determined by Student's t test prior to normalization (*, P < 0.04).
The small effect on entry, while reproducible and statistically significant, may not have biological relevance because we found no difference in the steady-state accumulation of the IE1 protein in Rb knockdown cells compared to scrambled controls by Western blotting (Fig. 5A). Quantitation of IE1 protein normalized to tubulin levels across multiple experiments revealed no difference in IE1 protein expression (Fig. 5B). Furthermore, there was no difference in the number of scrambled or Rb knockdown cells that became IE1 positive (as detected by indirect immunofluorescence microscopy) after HCMV infection at three different MOIs (Fig. 5C). Viral early gene expression follows IE gene expression. By Western blotting, we observed similar steady-state levels of the early protein UL44 in Rb knockdown and scrambled control fibroblasts (Fig. 5D). Quantitation over multiple experiments actually revealed a small increase in UL44 protein at 72 h postinfection in the absence of Rb (Fig. 5E). Taking all the data together, we conclude that pre-IE or IE defects are not responsible for the HCMV infection defect observed in Rb knockdown cells and that Rb is not required for efficient early protein accumulation in HCMV-infected fibroblasts.
FIG 5.

Immediate early genes are not reduced in Rb knockdown cells. (A) Serum-starved Scr or Rb2 cells were infected with AD169 at an MOI of 1, and equal amounts of protein lysates from 6 h postinfection were analyzed by Western blotting with the indicated antibodies. (B) The data are mean values of IE1 protein relative to tubulin and normalized to Scr ± standard deviations from five biological replicates. Student's t test prior to normalization determined there was no statistical difference between Scr and Rb2 cells (P > 0.51). (C) Scr or Rb2 cells plated on coverslips were serum starved and infected with AD169 at the indicated MOIs. Twenty-four hours after infection, cells were fixed and stained with an antibody against IE1 and imaged by indirect fluorescence microscopy. The data are the mean percentage of cells staining positive for IE1 ± standard deviation from three biological replicates. Student's t test determined that there was no statistical difference between Scr and Rb2 cells (P > 0.2 for all MOIs). (D) Serum-starved Scr or Rb2 cells were mock infected (M) or infected with AD169 at an MOI of 1, and equal amounts of protein lysates harvested at the indicated hours postinfection were analyzed by Western blotting. (E) The data are mean values of UL44 protein relative to tubulin and normalized to Scr ± standard deviations from four biological replicates. Statistical significance was determined by Student's t test prior to normalization (*, P < 0.04).
Interestingly, nonquantitative Western blotting revealed a slight delay in IE1 and IE2 protein accumulation in cells infected with a virus expressing a pp71 point mutant (C219G) that fails to degrade Rb (34). Perhaps Rb has a modest negative effect on IE gene expression that is relieved by pp71-mediated degradation. Rb knockdown would also relieve this slight restriction, improving IE gene expression and thus allow equivalent levels of IE1 protein accumulation (Fig. 5A) despite the minor defect in viral entry observed in Rb knockdown cells (Fig. 4).
Rb does not suppress viral IE gene expression during latency.
The potential for a negative effect of Rb on IE gene expression during a lytic infection prompted us to ask whether or not Rb knockdown had any effect on the ability of HCMV to suppress IE gene expression during the establishment of latency. Daxx, another pp71 substrate, silences viral IE gene expression at the start of a lytic infection unless it is degraded by pp71 (46). Daxx also silences IE gene expression during the establishment of latency in undifferentiated cells (51). Daxx is not degraded during latency because when HCMV enters the undifferentiated myeloid cell types where latency is established, pp71 does not enter the nucleus where its substrates (Daxx and Rb) reside (51). Knockdown of Daxx, or small-molecule inhibition of histone deacetylases with valproic acid (VPA), permits IE gene expression upon infection of THP-1 cells, a commonly used model for in vitro, experimental latency (40).
We stably decreased Rb steady-state protein levels in THP-1 cells by transducing them with retroviruses that express either shRNA-Rb2 or shRNA-Rb3 (each targeting unique sequences in the coding region of the Rb mRNA) and selected for puromycin-resistant populations. While both shRNAs were effective at reducing Rb levels, neither was able to permit IE1 expression in the absence of VPA (Fig. 6). Furthermore, even in the absence of Rb, IE gene expression remained VPA responsive in THP-1 cells (Fig. 6). We conclude that Rb has no demonstrable or significant role in silencing viral IE gene expression during the establishment of latency.
FIG 6.
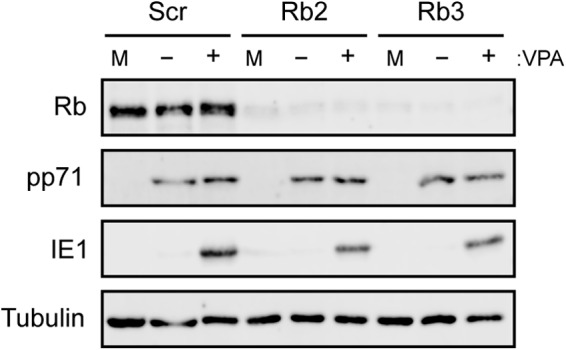
HCMV efficiently silences IE1 protein expression during a latent infection in cells with reduced levels of Rb. THP-1 cells in which Scr, Rb2, and Rb3 had been knocked down were mock treated or pretreated with valproic acid (VPA) 3 h prior to infection with AD169 IE2-GFP at an MOI of 1. Eighteen hours postinfection, mock-infected (M) cells and infected cells treated with VPA (+) or not treated with VPA (−) were harvested, and equal amounts of protein lysates were analyzed by Western blotting with the indicated antibodies. Data are representative of two biological replicates.
Viral DNA accumulates to lower levels in Rb knockdown cells.
Since Rb does not appear to have a biological impact on the establishment of latency or early events in a lytic infection, we next examined viral DNA replication during lytic infection in the Rb knockdown fibroblasts. Using quantitative PCR (qPCR), we quantitated viral genome amplification in scrambled and Rb knockdown cells to determine whether Rb knockdown impaired viral DNA replication. We determined the amount of viral DNA encoding the IE1 gene relative to the amount of cellular DNA encoding the GAPDH gene at a time prior to DNA replication (6 h postinfection [hpi]) (Fig. 7A) and then again after DNA replication (72 hpi) (Fig. 7B). This allowed us to determine a value for viral DNA amplification (Fig. 7C) that would be independent of the observed defect in genome delivery (Fig. 4C and D and 7A). Using this approach, we observed a 20% decrease in viral DNA replication in Rb knockdown cells compared to control cells.
FIG 7.

Viral DNA accumulates to lower levels in cells with reduced levels of Rb. (A to C) Serum-starved Scr and Rb2 cells were infected with AD169 at an MOI of 1. (D to F) Serum-starved Scr cells were infected with AD169 at an MOI of 1, and Rb2 cells were infected with AD169 at an MOI of 1.2 to achieve equal genome entry. (G and H) Serum-starved Scr cells were infected with AD169 at an MOI of 1, and Rb2 cells were infected with AD169 at an MOI of 1.6 to achieve a minimum of 1.2-fold-higher genome entry in the Rb knockdown cells. Infections were harvested at 6 h (A, D, and G) and 72 h (B, E, and H) postinfection. DNA was isolated, and the levels of viral and cellular genomes were measured by qPCR. The horizontal bars represent the mean measured quantities of viral genomes/cellular genomes from a minimum of four biological replicates, and the symbols represent data points from the individual experiments. The data in panels C, F, and I are the mean fold increase in viral genomes (viral genomes/cellular genomes at 72 hpi divided by viral genomes/cellular genomes at 6 hpi) ± standard deviation from a minimum of four biological replicates. Statistical significance was determined by Student's t test (*, P < 0.05; **, P < 0.01).
Even though our experimental approach should eliminate the mathematical problem resulting from decreased viral entry into Rb knockdown cells, it may not mitigate any biological issue associated with this phenomenon. Therefore, we repeated the experiments with an increased dosage of virus to the Rb knockdown cells so that we could achieve either equivalent (Fig. 7D, E, and F) or higher (Fig. 7G, H, and I) genome entry levels into Rb knockdown cells compared to scrambled controls. Despite the increased input genome levels, we still detected a reproducible and statistically significant defect in viral genome amplification in Rb knockdown cells compared to scrambled controls. Likewise, infection of Rb knockdown cells at higher applied MOI (as assayed by increased genome levels relative to scrambled control cells [Fig. 8A]) still resulted in a defect in infectious progeny production (Fig. 8B). We conclude that Rb is required for efficient viral genome replication during HCMV lytic replication in fibroblasts.
FIG 8.
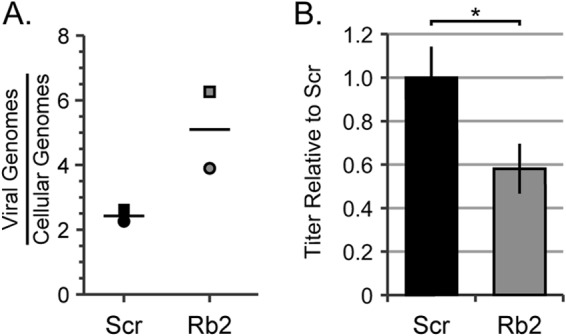
Overcoming the entry defect does not rescue viral replication in Rb knockdown cells. (A) Serum-starved Scr cells were infected with AD169 at an MOI of 1, and Rb2 cells were infected with AD169 at an MOI of 1.6. DNA was isolated at 6 h postinfection, and the levels of viral and cellular genomes were measured by qPCR. The horizontal bars represent the mean measured quantities of viral genomes/cellular genomes from two biological replicates, and the symbols represent data points from individual experiments. (B) Serum-starved Scr or Rb2 cells were infected with AD169 at an MOI of 1 and 1.6, respectively. Combined cell-free and cell-associated virus was collected 4 days postinfection, and the titers of virus were determined by standard plaque assay. The data are mean titers normalized to the siScr titer ± standard deviations from two biological replicates. Statistical significance was determined by a parametric test before normalization (*, P < 0.04).
The late protein pp150 accumulates to lower levels in Rb knockdown cells.
Viral late gene expression follows DNA replication. As expected based on the defect in viral DNA synthesis in the absence of Rb (Fig. 7), Western blotting revealed a decrease in the steady-state level of the true late UL32 gene product pp150 in Rb knockdown cells compared to scrambled control cells (Fig. 9A). Quantitation over multiple experiments revealed a 39% defect that was both reproducible and statistically significant (Fig. 9B). We conclude that Rb is required for efficient late protein accumulation in HCMV-infected fibroblasts. Overall, we conclude that Rb contributes in a positive way to multiple steps during HCMV infection, promoting efficient lytic replication.
FIG 9.
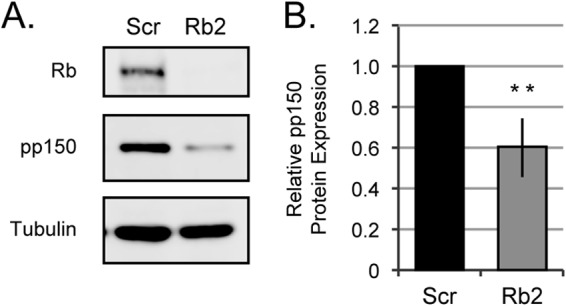
Rb knockdown cells accumulate lower levels of late proteins. (A) Serum-starved Scr or Rb2 cells were mock infected or infected with AD169 at an MOI of 1, and equal amounts of protein lysates harvested at the indicated hours postinfection were analyzed by Western blotting. (B) The data are mean values of pp150 protein relative to tubulin and normalized to Scr ± standard deviations from five biological replicates. Statistical significance was determined by Student's t test prior to normalization (**, P < 0.01).
DISCUSSION
We detected multiple steps at which Rb deficiency decreased parameters of HCMV infection. HCMV entered Rb knockdown cells less efficiently, but it is unlikely this is a direct effect, as Rb localizes to the nucleus and mitochondria but not at the sites of HCMV entry (52, 53). The decrease in entry may result from the myriad transcriptional changes that likely result from knocking down a widely acting corepressor like Rb. Rb-mediated transcriptional repression of IE1 expression may explain why IE1 protein accumulation is not defective in Rb knockdown cells despite decreased viral genome delivery (Fig. 4 and 5). This may also explain why the pp71-C219G virus, which eliminates the ability of pp71 to degrade Rb, displays an apparent delay in IE protein accumulation (34). HCMV may utilize tegument-delivered pp71 to degrade Rb prior to viral IE gene expression to ensure a robust lytic infection. However, this effect appears to be insignificant, at least in fibroblasts in vitro, as the pp71-C219G virus displays no overt growth phenotype (34).
Rb is considered an inhibitor of both cellular and viral DNA replication through repression of the E2F-responsive genes that participate in nucleotide metabolism and DNA synthesis (11). Our data indicate that Rb is not inhibitory for HCMV viral DNA replication but actually enhances this process. Whether Rb accomplishes this directly by physically interacting with HCMV genomes or indirectly through transcriptional modulation of viral or cellular gene expression remains to be determined. In HCMV-infected cells, Rb reaccumulates after its degradation by pp71 at pre-IE times, but it is subsequently phosphorylated by the HCMV UL97 v-Cdk at times when viral DNA is synthesized (33). While UL97 clearly facilitates HCMV viral DNA replication (54), it appears to do so independently of Rb phosphorylation (C. V. Kuny and R. F. Kalejta, submitted for publication). Likewise, it remains unknown whether the hypo-, mono-, or hyperphosphorylated form of Rb is responsible for inducing efficient HCMV DNA synthesis. Furthermore, it is currently unclear whether the observed decreases in late gene expression in the absence of Rb result from a direct or indirect effect on late gene transcription or translation or are secondary to the observed defect on viral DNA replication.
Rb promoting HCMV infection may seem counterintuitive because this virus and the small DNA tumor viruses encode multiple proteins reported to inactivate Rb. However, the term “inactivate” may not accurately describe the effects of these viral proteins on Rb. While certain functions of Rb will clearly be inhibited, other activities may be retained in the presence of these viral proteins. We previously described an HCMV mutant that was wild type except for an engineered ability to induce Rb degradation by expressing the papillomavirus E7 protein from an early/late HCMV promoter (55). This mutant HCMV with E7 virus displayed a growth defect, while a similar HCMV virus expressing an E7 mutant that is unable to bind and degrade Rb had no growth defect (55). This is consistent with the knockdown data presented here that Rb promotes efficient HCMV infection and suggests that inactivation by degradation is not synonymous with hyperphosphorylation.
Multiple proteins target Rb during HCMV infection, but infection is inefficient at multiple steps in its absence. Analogous findings have been reported for p53, the other major cellular tumor suppressor. At least three viral proteins, IE1, IE2, and UL44, have been reported to inactivate p53 function (56–58). Furthermore, p53 is sequestered within viral replication compartments in infected cells (59). In p53-null cells, HCMV replicates to decreased titers and shows defects at multiple stages of the viral life cycle, including DNA replication and late gene expression (60, 61), similar to what is observed here in Rb knockdown cells. Clearly, HCMV does not simply inactivate these tumor suppressors but modifies their function to stimulate its replication.
The proteins that induce Rb degradation, HPV E7 and HCMV pp71, target only the hypophosphorylated form, and not the hyperphosphorylated form (19, 31). While this inactivates the S-phase restriction function of hypophosphorylated Rb, it would not necessarily inactivate any function that a more-phosphorylated form of Rb might possess. Analogously, although adenovirus E1A associates with hypophosphorylated Rb and disrupts its complexes with certain E2F proteins to inactivate the S-phase restriction, Rb remains able to interact with E2F1 in the presence of E1A, perhaps preventing E2F1-induced apoptosis during adenovirus infection (25). Additionally, recent experiments clearly demonstrate that E1A-bound Rb does not remain inert, but rather E1A directs the Rb-mediated transcriptional repression of a modified set of cellular genes (26). Many of these genes have annotated antiviral functions, indicating that, as is the case for HCMV, Rb may very well enhance adenovirus infection (26).
Deciphering how HCMV might use Rb to its advantage is complicated because at least four viral proteins (pp71, IE1, IE2, and UL97) have been demonstrated to physically and/or functionally interact with the Rb/E2F pathway (31–33, 37–39). Furthermore, an additional 14 HCMV proteins (US16, US22, US26, US29, UL20, UL23, UL24, UL25, UL29, UL69, UL77, and UL93 in all strains and UL111a and UL128 in clinical strains) encode a canonical Rb-binding LXCX(D/E) motif (62). While any one or more of these proteins may be a functional partner with Rb, none are known to play direct roles in the processes shown here to be deficient in the absence of Rb (viral DNA replication and late gene expression), although genetic ablation of UL69 shows a similar phenotype (63). UL77 and UL93 play roles in DNA encapsidation (64, 65), provocatively suggesting that Rb may participate in another viral process critical for HCMV replication not directly assayed here. No doubt, the interaction between HCMV and Rb is multifaceted and complex.
The vast majority of research on the interactions between viruses and Rb has focused on oncogenic transformation events (66). Such explorations have been instrumental in defining the Rb-E2F pathway and its role in both normal cell cycle progression and cancer. However, oncogenicity is not a central part of the infectious cycle of these viruses, and the actual role of viral targeting of Rb remains, for the most part, undefined. Thus, despite intensive study using viral Rb-inactivating proteins to illuminate the contribution of the Rb pathway to tumor suppression and oncogenesis, we still have a rudimentary comprehension of the role of Rb in productive viral replication. Understanding how viruses manipulate the function of Rb to their benefit may reveal novel functions of this fascinating tumor suppressor.
ACKNOWLEDGMENTS
We thank Phil Balandyk for expert technical assistance, Paul Lambert and Hiroyuki Sakai for shRNA retroviral plasmids, Nathan Sherer for sharing equipment, and Emily Albright for comments on the manuscript.
This work was supported by National Institutes of Health grant AI080675 to R.F.K. H.R.V. was supported by training grant T32 CA009135.
REFERENCES
- 1.Viatour P, Sage J. 2011. Newly identified aspects of tumor suppression by RB. Dis Model Mech 4:581–585. doi: 10.1242/dmm.008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Fiore R, D'Anneo A, Tesoriere G, Vento R. 2013. RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol 228:1676–1687. doi: 10.1002/jcp.24329. [DOI] [PubMed] [Google Scholar]
- 3.Horowitz JM, Park SH, Bogenmann E, Cheng JC, Yandell DW, Kaye FJ, Minna JD, Dryja TP, Weinberg RA. 1990. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci U S A 87:2775–2779. doi: 10.1073/pnas.87.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris EJ, Dyson NJ. 2001. Retinoblastoma protein partners. Adv Cancer Res 82:1–54. doi: 10.1016/S0065-230X(01)82001-7. [DOI] [PubMed] [Google Scholar]
- 5.Dick FA, Rubin SM. 2013. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicolay BN, Dyson NJ. 2013. The multiple connections between pRB and cell metabolism. Curr Opin Cell Biol 25:735–740. doi: 10.1016/j.ceb.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Sherr CJ, McCormick F. 2002. The RB and p53 pathways in cancer. Cancer Cell 2:103–112. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 9.Rubin SM. 2013. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci 38:12–19. doi: 10.1016/j.tibs.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. 1999. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98:859–869. doi: 10.1016/S0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 11.Trimarchi JM, Lees JA. 2002. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 12.Johnson DG. 2000. The paradox of E2F1: oncogene and tumor suppressor gene. Mol Carcinog 27:151–157. doi:. [DOI] [PubMed] [Google Scholar]
- 13.Johnson DG, Cress WD, Jakoi L, Nevins JR. 1994. Oncogenic capacity of the E2F1 gene. Proc Natl Acad Sci U S A 91:12823–12827. doi: 10.1073/pnas.91.26.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke JR, Hura GL, Rubin SM. 2012. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev 26:1156–1166. doi: 10.1101/gad.189837.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrientes S, Cooke C, Goodrich DW. 2000. Glutamic acid mutagenesis of retinoblastoma protein phosphorylation sites has diverse effects on function. Oncogene 19:562–570. doi: 10.1038/sj.onc.1203332. [DOI] [PubMed] [Google Scholar]
- 16.Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. 2014. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 3:e02872. doi: 10.7554/eLife.02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macdonald JI, Dick FA. 2012. Posttranslational modifications of the retinoblastoma tumor suppressor protein as determinants of function. Genes Cancer 3:619–633. doi: 10.1177/1947601912473305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huh K, Zhou X, Hayakawa H, Cho J-Y, Libermann TA, Jin J, Harper JW, Munger K. 2007. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol 81:9737–9747. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyer SN, Wazer DE, Band V. 1996. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res 56:4620–4624. [PubMed] [Google Scholar]
- 20.Collins AS, Nakahara T, Do A, Lambert PF. 2005. Interactions with pocket proteins contribute to the role of human papillomavirus type 16 E7 in the papillomavirus life cycle. J Virol 79:14769–14780. doi: 10.1128/JVI.79.23.14769-14780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bodily JM, Mehta KPM, Cruz L, Meyers C, Laimins LA. 2011. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J Virol 85:8852–8862. doi: 10.1128/JVI.00664-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roman A, Munger K. 2013. The papillomavirus E7 proteins. Virology 445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bagchi S, Raychaudhuri P, Nevins JR. 1990. Adenovirus E1A proteins can dissociate heteromeric complexes involving the E2F transcription factor: a novel mechanism for E1A trans-activation. Cell 62:659–669. doi: 10.1016/0092-8674(90)90112-R. [DOI] [PubMed] [Google Scholar]
- 24.Sauthoff H, Pipiya T, Heitner S, Chen S, Bleck B, Reibman J, Chang W, Norman RG, Rom WN, Hay JG. 2004. Impact of E1a modifications on tumor-selective adenoviral replication and toxicity. Mol Ther 10:749–757. doi: 10.1016/j.ymthe.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Seifried LA, Talluri S, Cecchini M, Julian LM, Mymryk JS, Dick FA. 2008. pRB-E2F1 complexes are resistant to adenovirus E1A-mediated disruption. J Virol 82:4511–4520. doi: 10.1128/JVI.02713-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrari R, Gou D, Jawdekar G, Johnson SA, Nava M, Su T, Yousef AF, Zemke NR, Pellegrini M, Kurdistani SK, Berk AJ. 2014. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 16:663–676. doi: 10.1016/j.chom.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cinatl J, Scholz M, Kotchetkov R, Vogel J-U, Doerr HW. 2004. Molecular mechanisms of the modulatory effects of HCMV infection in tumor cell biology. Trends Mol Med 10:19–23. doi: 10.1016/j.molmed.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Soroceanu L, Cobbs CS. 2011. Is HCMV a tumor promoter? Virus Res 157:193–203. doi: 10.1016/j.virusres.2010.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barami K. 2010. Oncomodulatory mechanisms of human cytomegalovirus in gliomas. J Clin Neurosci 17:819–823. doi: 10.1016/j.jocn.2009.10.040. [DOI] [PubMed] [Google Scholar]
- 30.Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. 2012. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J Virol 86:854–864. doi: 10.1128/JVI.06097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23:1885–1895. doi: 10.1128/MCB.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalejta RF, Shenk T. 2003. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc Natl Acad Sci U S A 100:3263–3268. doi: 10.1073/pnas.0538058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320:797–799. doi: 10.1126/science.1152095. [DOI] [PubMed] [Google Scholar]
- 34.Cantrell SR, Bresnahan WA. 2005. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J Virol 79:7792–7802. doi: 10.1128/JVI.79.12.7792-7802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prichard MN, Sztul E, Daily SL, Perry AL, Frederick SL, Gill RB, Hartline CB, Streblow DN, Varnum SM, Smith RD, Kern ER. 2008. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J Virol 82:5054–5067. doi: 10.1128/JVI.02174-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacob T, Van den Broeke C, Favoreel HW. 2011. Viral serine/threonine protein kinases. J Virol 85:1158–1173. doi: 10.1128/JVI.01369-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poma EE, Kowalik TF, Zhu L, Sinclair JH, Huang ES. 1996. The human cytomegalovirus IE1-72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J Virol 70:7867–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiebusch L, Asmar J, Uecker R, Hagemeier C. 2003. Human cytomegalovirus immediate-early protein 2 (IE2)-mediated activation of cyclin E is cell-cycle-independent and forces S-phase entry in IE2-arrested cells. J Gen Virol 84:51–60. doi: 10.1099/vir.0.18702-0. [DOI] [PubMed] [Google Scholar]
- 39.Song Y-J, Stinski MF. 2002. Effect of the human cytomegalovirus IE86 protein on expression of E2F-responsive genes: a DNA microarray analysis. Proc Natl Acad Sci U S A 99:2836–2841. doi: 10.1073/pnas.052010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81:9109–9120. doi: 10.1128/JVI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanchez V, Clark CL, Yen JY, Dwarakanath R, Spector DH. 2002. Viable human cytomegalovirus recombinant virus with an internal deletion of the IE2 86 gene affects late stages of viral replication. J Virol 76:2973–2989. doi: 10.1128/JVI.76.6.2973-2989.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasque V, Gillich A, Garrett N, Gurdon JB. 2011. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J 30:2373–2387. doi: 10.1038/emboj.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ueno T, Sasaki K, Yoshida S, Kajitani N, Satsuka A, Nakamura H, Sakai H. 2006. Molecular mechanisms of hyperplasia induction by human papillomavirus E7. Oncogene 25:4155–4164. doi: 10.1038/sj.onc.1209445. [DOI] [PubMed] [Google Scholar]
- 45.Nowak B, Sullivan C, Sarnow P, Thomas R, Bricout F, Nicolas JC, Fleckenstein B, Levine AJ. 1984. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132:325–338. doi: 10.1016/0042-6822(84)90039-4. [DOI] [PubMed] [Google Scholar]
- 46.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang J, Saffert RT, Kalejta RF. 2011. Elongin B-mediated epigenetic alteration of viral chromatin correlates with efficient human cytomegalovirus gene expression and replication. mBio 2(2):e00023-11. doi: 10.1128/mBio.00023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Juckem LK, Boehme KW, Feire AL, Compton T. 2008. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol 180:4965–4977. doi: 10.4049/jimmunol.180.7.4965. [DOI] [PubMed] [Google Scholar]
- 49.Salvant BS, Fortunato EA, Spector DH. 1998. Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol 72:3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sage J, Miller AL, Pérez-Mancera PA, Wysocki JM, Jacks T. 2003. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 51.Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol 84:5594–5604. doi: 10.1128/JVI.00348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, Walensky LD, Lees JA. 2013. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev 27:1003–1015. doi: 10.1101/gad.211326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Antonucci LA, Egger JV, Krucher NA. 2014. Phosphorylation of the Retinoblastoma protein (Rb) on serine-807 is required for association with Bax. Cell Cycle 13:3611–3617. doi: 10.4161/15384101.2014.964093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolf DG, Courcelle CT, Prichard MN, Mocarski ES. 2001. Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc Natl Acad Sci U S A 98:1895–1900. doi: 10.1073/pnas.98.4.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamil JP, Hume AJ, Jurak I, Münger K, Kalejta RF, Coen DM. 2009. Human papillomavirus 16 E7 inactivator of retinoblastoma family proteins complements human cytomegalovirus lacking UL97 protein kinase. Proc Natl Acad Sci U S A 106:16823–16828. doi: 10.1073/pnas.0901521106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang E-S, Zhang Z, Cai H, Huang DY, Huong S-M, Cha C-Y, Huang E-S. 2009. Human cytomegalovirus IE1-72 protein interacts with p53 and inhibits p53-dependent transactivation by a mechanism different from that of IE2-86 protein. J Virol 83:12388–12398. doi: 10.1128/JVI.00304-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finkel T, Epstein SE. 1994. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 265:391–394. doi: 10.1126/science.8023160. [DOI] [PubMed] [Google Scholar]
- 58.Kwon Y, Kim M-N, Young Choi E, Heon Kim J, Hwang E-S, Cha C-Y. 2012. Inhibition of p53 transcriptional activity by human cytomegalovirus UL44. Microbiol Immunol 56:324–331. doi: 10.1111/j.1348-0421.2012.00446.x. [DOI] [PubMed] [Google Scholar]
- 59.Fortunato EA, Spector DH. 1998. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J Virol 72:2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. 2006. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol 80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hannemann H, Rosenke K, O'Dowd JM, Fortunato EA. 2009. The presence of p53 influences the expression of multiple human cytomegalovirus genes at early times postinfection. J Virol 83:4316–4325. doi: 10.1128/JVI.02075-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chellappan S, Kraus VB, Kroger B, Munger K, Howley PM, Phelps WC, Nevins JR. 1992. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci U S A 89:4549–4553. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hayashi ML, Blankenship C, Shenk T. 2000. Human cytomegalovirus UL69 protein is required for efficient accumulation of infected cells in the G1 phase of the cell cycle. Proc Natl Acad Sci U S A 97:2692–2696. doi: 10.1073/pnas.050587597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toropova K, Huffman JB, Homa FL, Conway JF. 2011. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J Virol 85:7513–7522. doi: 10.1128/JVI.00837-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bellacchio E, Paggi MG. 2013. Understanding the targeting of the RB family proteins by viral oncoproteins to defeat their oncogenic machinery. J Cell Physiol 228:285–291. doi: 10.1002/jcp.24137. [DOI] [PubMed] [Google Scholar]