

ABSTRACT
Due to a scarcity of immunocompetent animal models for viral hepatitis, little is known about the early innate immune responses in the liver. In various hepatotoxic models, both pro- and anti-inflammatory activities of recruited monocytes have been described. In this study, we compared the effect of liver inflammation induced by the Toll-like receptor 4 ligand lipopolysaccharide (LPS) with that of a persistent virus, lymphocytic choriomeningitis virus (LCMV) clone 13, on early innate intrahepatic immune responses in mice. LCMV infection induces a remarkable influx of inflammatory monocytes in the liver within 24 h, accompanied by increased transcript levels of several proinflammatory cytokines and chemokines in whole liver. Importantly, while a single LPS injection results in similar recruitment of inflammatory monocytes to the liver, the functional properties of the infiltrating cells are dramatically different in response to LPS versus LCMV infection. In fact, intrahepatic inflammatory monocytes are skewed toward a secretory phenotype with impaired phagocytosis in LCMV-induced liver inflammation but exhibit increased endocytic capacity after LPS challenge. In contrast, F4/80high-Kupffer cells retain their steady-state endocytic functions upon LCMV infection. Strikingly, the gene expression levels of inflammatory monocytes dramatically change upon LCMV exposure and resemble those of Kupffer cells. Since inflammatory monocytes outnumber Kupffer cells 24 h after LCMV infection, it is highly likely that inflammatory monocytes contribute to the intrahepatic inflammatory response during the early phase of infection. Our findings are instrumental in understanding the early immunological events during virus-induced liver disease and point toward inflammatory monocytes as potential target cells for future treatment options in viral hepatitis.
IMPORTANCE Insights into how the immune system deals with hepatitis B virus (HBV) and HCV are scarce due to the lack of adequate animal model systems. This knowledge is, however, crucial to developing new antiviral strategies aimed at eradicating these chronic infections. We model virus-host interactions during the initial phase of liver inflammation 24 h after inoculating mice with LCMV. We show that infected Kupffer cells are rapidly outnumbered by infiltrating inflammatory monocytes, which secrete proinflammatory cytokines but are less phagocytic. Nevertheless, these recruited inflammatory monocytes start to resemble Kupffer cells on a transcript level. The specificity of these cellular changes for virus-induced liver inflammation is corroborated by demonstrating opposite functions of monocytes after LPS challenge. Overall, this demonstrates the enormous functional and genetic plasticity of infiltrating monocytes and identifies them as an important target cell for future treatment regimens.
INTRODUCTION
Viral hepatitis, predominantly caused by the hepatitis B and C viruses (HBV and HCV, respectively), is a global health burden (1, 2). Although clearance of HBV and HCV infection is executed by multiple epitope-specific adaptive CD4+ T, CD8+ T, and B cell responses (3–6), these responses are dependent on and shaped by the early immunological events provided by innate immune cells in the liver (6, 7). Since immunological studies of virus-induced hepatitis in human are difficult to perform (reviewed in references 8 and 9), infections of mice with lymphocytic choriomeningitis virus (LCMV) have demonstrated to be a valid model system to examine intrahepatic antiviral immunity (reviewed in reference 10). Although mice persistently infected with LCMV exhibit altered innate responses to subsequent Toll-like receptor (TLR) stimulations and secondary infections (11, 12), the overall and intrahepatic alterations of the innate immune system during early LCMV infections have been less studied.
Monocytes survey the body for inflammatory foci and are therefore among the first innate immune cells to respond to infection. They are equipped with chemokine and adhesion receptors to mediate migration to the site of infection or inflammation, upon which they can further differentiate into tissue macrophages and dendritic cells (13). Depending on the nature of the inflammatory agent and the organ system involved, monocytes can exert both a proinflammatory and an anti-inflammatory role. They have the ability to produce tumor necrosis factor (TNF) and inducible nitric oxide synthase (iNOS) (14–16), to carry microbial antigens to local lymph nodes (17), and to present antigens to T cells (18–20). Alternatively, monocytes may differentiate into anti-inflammatory macrophages (16) or suppress proliferation and production of cytokines by T cells (21), suggesting their role in maintaining homeostasis. In Listeria monocytogenes, Mycobacterium tuberculosis, and Toxoplasma gondii mouse models, for example, monocyte migration from bone marrow results in resistance to infection (15, 22–24). In contrast, in Trypanosoma brucei and influenza virus models, monocyte recruitment impairs pathogen clearance and exacerbates immunomediated pathology (14, 25, 26).
Upon recruitment to the liver, monocytes are referred to as inflammatory monocytes and identified as F4/80low Ly6C+ CD11b+ cells (27–29). Similar to their systemic function, opposing roles during sterile toxin-induced liver inflammation have been identified. For example, in acetaminophen-induced hepatitis, hepatic inflammatory monocytes are endocytic and display an immunoregulatory phenotype (27), while in concanavalin A and CCl4 hepatitis, they promote Th1 cell proliferation and produce proinflammatory cytokines such as TNF and interleukin-6 (IL-6) (28–30). Due to the shortage of specific animal models, the role of these innate immune cells during virus-induced liver disease is less explored. Furthermore, before the recruitment of monocytes, Kupffer cells, together with dendritic cells, liver sinusoidal endothelial cells, and stellate cells, are the first to encounter pathogens upon their passage through the liver sinusoids. Both Kupffer cells and inflammatory monocytes likely play a role in shaping the immune response and thereby affect the outcome of a viral infection in the liver. We and others have previously shown that murine Kupffer cells can be unequivocally identified by the expression of F4/80, CD11b, and CD68 and are predominantly phagocytic in a steady-state condition (31, 32).
In the present study, we set out to characterize in depth the phenotype, function, and gene expression profiles of liver monocytes and Kupffer cells during the early phases of chronic LCMV infection. We demonstrate a strong influx of inflammatory monocytes in the liver within 24 h after LCMV inoculation. Using NanoString gene expression analysis (NanoString Technologies, Seattle, WA) on highly purified Kupffer cells and inflammatory monocytes isolated from mouse liver, we demonstrated that under steady-state conditions these cells are transcriptionally and functionally distinct. As a consequence of LCMV infection, the expression profiles of both Kupffer cells and inflammatory monocytes are strongly altered. Strikingly, 24 h after LCMV infection, differences in gene expression levels of both cell types largely but not completely disappear, resulting in the strong resemblance of Kupffer cells and inflammatory monocytes. These findings provide insight into the function of the cells involved in the early stages of virus-induced liver disease and present potential target cell types for the early control of viral hepatitis.
MATERIALS AND METHODS
Mice, virus, and antibodies.
LCMV Cl13 was propagated in BHK21 cells, and the titer was determined by plaque assay as previously described (33, 34). C57BL/6 mice aged 8 to 12 weeks (Charles River Laboratories, France) received LCMV Cl13 (2 × 106 PFU) intravenously, 5 μg of lipopolysaccharide (LPS; TLR4 ligand, S. Minnesota ultrapure, Invivogen) intraperitoneally, or 200 μl of phosphate-buffered saline (PBS) intraperitoneally. Animals were maintained in a biosafety level III (BSL-III) isolator according to Dutch national biosafety guidelines. Infection was confirmed by plaque assay on liver homogenate. All animal work was conducted according to relevant Dutch national guidelines. The study protocol was approved by the animal ethics committee of the Erasmus University Rotterdam. The antibodies used in flow cytometry included CD45 eFluor450 (30-F11), F4/80-allophycocyanin (APC) or F4/80-fluorescein isothiocyanate (FITC; BM8), CD11b-PECy7 (M1/70), and TNF-PerCPCy5.5 (MP6-XT22) from eBioscience, Ly6C APCCy7 (HK1.4) from Biolegend, MARCO (macrophage receptor with collagenous structure) FITC (ED31) from AbdSerotec, and Aqua Dead Cell Stain from Invitrogen. The VL4 cells, producing LCMV-nucleoprotein (LCMV-NP) monoclonal antibody, were kindly provided by M. Groettrup, University of Constance, Constance, Germany.
Isolation of total liver nonparenchymal cells.
Liver was removed without perfusion, cut into small pieces, incubated in RPMI 1640 containing 30 μg of Liberase TM (Roche)/ml and 20 μg of DNase type I (Sigma)/ml for 20 min, and passed through a 100-μm-pore-size cell strainer. After centrifugation, the cells were resuspended in PBS containing 1% fetal calf serum (FCS) and 2.5 mM EDTA. Parenchymal cells were removed by low-speed centrifugation at 50 × g for 3 min, and erythrocytes were lysed with 0.8% NH4Cl. The remaining nonparenchymal cells were resuspended in culture medium (RPMI 1640, 10% FCS, 10 mM HEPES, 2 mM l-glutamine, penicillin-streptomycin [100 U/ml and 100 μg/ml], 50 μM β-mercaptoethanol) and used for further analysis.
Flow cytometry.
Total liver nonparenchymal cells were stained with Aqua Dead Cell Stain, CD45-eFluor450, F4/80-APC, CD11b-PECy7, and Ly6C-APCCy7 (unless otherwise indicated) and fixed with 2% formaldehyde for 1 h. F4/80high-Kupffer cells were identified as CD45+ F4/80high CD11b+ and inflammatory monocytes were identified as CD45+ F4/80low CD11bhigh Ly6Chigh by using a FACSCanto-II flow cytometer and FACSDiva software (BD Biosciences).
RNA isolation of liver homogenates, generation of cDNA, and real-time PCR.
Liver was homogenized in RNAlater (Qiagen). RNA was extracted using TRIzol (Life Technologies) and a NucleoSpin RNA II kit (Bioké). cDNA was generated by using an iScript cDNA synthesis kit (Bio-Rad Laboratories) according to the manufacturer's protocol. Quantitative PCR (qPCR) were performed using SYBR-green and MyIQ5 detection system (Bio-Rad Laboratories). The sequences of the primers are listed in Table 1. The expression of target genes was normalized to the expression of 18S or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) using the formula 2−ΔCT, ΔCT = CT TLR − CT 18S or ΔCT = CT RNAX − CT GADPH.
TABLE 1.
Gene-specific primers used in qPCR analysis
| Gene | NCBI accession no. | Primer (5′–3′) |
|
|---|---|---|---|
| Orientation | Primer sequence | ||
| GAPDH | NM_008084.2 | Forward | CGTCCCGTAGACAAAATGGT |
| Reverse | TCTCCATGGTGGTGAAGACA | ||
| 18S | NR_003278.3 | Forward | GTAACCCGTTGAACCCCATT |
| Reverse | CCATCCAATCGGTAGTAGCG | ||
| TNF | NM_013693.2 | Forward | CAGGCGGTGCCTATGTCTC |
| Reverse | CGATGACCCCGAAGTTCAGTAG | ||
| IL-10 | NM_010548.2 | Forward | CACAGGGGAGAAATCGATGACA |
| Reverse | ATTTGAATTCCCTGGGTGAGAAG | ||
| IL-6 | NM_031168.1 | Forward | TGGTGACAACCACGGCCTTCC |
| Reverse | AGCCTCCTGACTTGTGAAGTGGT | ||
| MCP-1 | NM_011333.3 | Forward | CAGGTCCCTGTCATGCTTCT |
| Reverse | TCTGGACCCATTCCTTCTTG | ||
| RANTES | NM_013653.3 | Forward | GCGGGTACCATGAAGATCTCTG |
| Reverse | CACTTCTTCTCTGGGTTGGCAC | ||
| IFN-α | NM_010504.2 | Forward | AGGATTTTGGATTCCCCTTG |
| Reverse | TATGTCCTCACAGCCAGCAG | ||
| IFN-β | NM_010510.1 | Forward | ATGAACAACAGGTGGATCCTCC |
| Reverse | AGGAGCTCCTGACATTTCCGAA | ||
| CXCL10 | NM_021274.2 | Forward | CCCCGGTGCTGCGATGGATG |
| Reverse | AGCTGATGTGACCACGGCTGG | ||
RNA isolation of sorted cells and NanoString.
Kupffer cells and inflammatory cells were purified by cell sorting after initial enrichment using CD45-phyoerythrin (PE), followed by anti-PE microbead (Miltenyi Biotec) selection. To obtain sufficient cells, the organs of six mice were pooled. After staining to identify Kupffer cells and inflammatory monocytes as described above, cells were fixed with 2% formaldehyde for 1 h and sorted on a FACSAria SORP flow cytometer (BD Biosciences). Total RNA was isolated from these formaldehyde-fixed cells by using reagents from an RNeasy FFPE kit (Qiagen) according to the manufacturer's protocols, starting with the addition of 150 μl of buffer PKD. An nCounter GX mouse immunology kit (NanoString Technologies) was used to measure the expression of 561 genes in our RNA samples. After hybridization, transcripts were quantitated using the nCounter digital analyzer. Samples were run by the Johns Hopkins deep sequencing and microarray core facility (35). To correct for background levels, the highest negative-control value for each sample was subtracted from each count value of that sample. After background subtraction, any negative count values were considered as 0. Values were normalized by the geometric mean of 13 housekeeping genes provided by the company panel.
Immunohistochemistry for F4/80.
Livers were fixed in 4% formaldehyde, embedded in paraffin, and cut into 5-μm sections. F4/80 antigen was retrieved using proteinase K. Endogenous peroxidase was inactivated using 3% hydrogen peroxide. Liver sections were incubated with rat anti-F4/80 antibody (eBioscience) and rabbit anti-rat horseradish peroxidase (Dako). Upon addition of DAB, liver sections were counterstained with hematoxylin.
Intracellular detection of LMCV-NP.
Intracellular LCMV-NP detection was performed as previously described (36). Total liver nonparenchymal cells were incubated with Aqua Dead Cell Stain, fixed with 2% formaldehyde for 1 h, and permeabilized by using 1% Triton X-100 (Sigma) for 20 min. The cells were further incubated with rat anti-LCMV-NP antibody (VL4), goat anti-rat Alexa 594 (Invitrogen), and blocking buffer (5% bovine serum albumin and 10% rat serum). F4/80-FITC was used in the antibody cocktail. LCMV-NP-positive F4/80high-Kupffer cells were identified.
Cytokine production by total liver nonparenchymal cells.
Total liver nonparenchymal cells were cultured for 5 h at 106 cells/well in a 24-well plate (Costar) in 1 ml of culture medium alone or in combination with phorbol myristate acetate (PMA) and ionomycin (50 and 500 ng/ml, respectively). Brefeldin A (10 μg/ml; Sigma) was added after 1 h. Next, the cells were incubated with Aqua Dead Cell Stain, fixed with 2% formaldehyde for 1 h, permeabilized with 0.5% saponin (Rectapur), and further stained with TNF-PerCPCy5.5. The TNF-positive F4/80high-Kupffer cells and inflammatory monocytes were determined.
In vitro receptor-mediated endocytosis assay.
One million total liver nonparenchymal cells were incubated for 45 min with dextran-FITC (10 μg/ml, 40,000 molecular weight; Invitrogen) at 37°C or on ice. Dextran-positive F4/80high-Kupffer cells and inflammatory monocytes were detected by flow cytometry.
ALT measurement.
Serum alanine aminotransferase (ALT) levels were measured by using an enzyme-linked immunosorbent assay kit for ALT (Biotang USA) according to the manufacturer's protocol.
Data analysis and statistics.
Differences between groups were calculated using one-way analysis of variance (ANOVA; Kruskal-Wallis test, with Dunn's multiple-comparison post-test) or two-way ANOVA with a Bonferroni post-test (GraphPad Prism version 5.01; GraphPad Software). Differences were considered significant when P < 0.05. The results are presented as the means ± the standard errors of the mean (SEM), unless otherwise indicated.
RESULTS
LCMV infection induces a rapid recruitment of inflammatory monocytes to the liver.
Previous studies showed an accumulation of LCMV particles in the liver within minutes after an intravenous challenge (37). In line with this, we observed a reproducible LCMV replication in the liver of mice inoculated 24 h earlier, reaching average titers of 2.27 × 105 PFU/g of liver. To examine the impact of viral replication versus sterile TLR ligand-induced challenge on the innate immune cell repertoire in the liver, we performed a flow cytometric analysis on total liver nonparenchymal cells obtained from LCMV-infected and LPS-treated mice. LPS was used as a model mimicking bacterial infection. F4/80high-Kupffer cells and inflammatory monocytes were identified as CD45+ F4/80high CD11b+ and CD45+ F4/80low CD11bhigh Ly6Chigh cells, respectively (28, 31) (Fig. 1A). After 24 h, LCMV induced an almost 5-fold increase in the fraction of inflammatory monocytes similar to a single LPS injection (from 5.8% in healthy liver to 25.6 and 24.5% of total intrahepatic CD45+ cells in LPS- and LCMV-challenged livers, respectively; Fig. 1B). This increase was not yet observed 4 h after LCMV infection or LPS administration. Interestingly, monocyte recruitment was accompanied by a decrease in the frequency of F4/80high-Kupffer cells in the LPS-treated and LCMV-infected livers (from 10.4% in healthy cells to 5.7 and 4.4% of total intrahepatic CD45+ cells in LCMV-infected and LPS-treated livers, respectively).
FIG 1.

Infiltration of inflammatory monocytes characterizes the activation of the intrahepatic immune response upon challenge by LCMV, as well as LPS. (A) Representative dot plots showing flow cytometric identification of Kupffer cells and inflammatory monocytes in mouse liver. The total liver cells were determined using viability, CD45, and size and granularity gates. Inflammatory monocytes and F4/80high-Kupffer cells were identified as CD45+ F4/80low CD11bhigh Ly6Chigh and CD45+ F4/80high CD11b+ cells, respectively. (B) The frequencies of inflammatory monocytes and F4/80high-Kupffer cells in liver were determined as a percentage of total CD45+ cells at 4 and 24 h after LCMV infection or LPS challenge or in a PBS control. The data were obtained from four separate experiments. Statistical analysis: one-way ANOVA (Kruskal-Wallis test with Dunn's multiple-comparison post-test). ***, P < 0.001. The data show the averages ± the SEM. (C) Liver tissues from PBS-challenged mice (n = 6) and infected mice (24 h postinfection, n = 6) were subjected to F4/80 staining and evaluated by immunohistochemistry. In healthy liver, F4/80-positive cells appear as elongated cells with intense brown F4/80 staining, whereas after 24 h of LCMV infection, F4/80+ cells appear slightly larger in size, with diffuse brown F4/80 staining, compared to the F4/80+ cells in healthy liver. (D) Representative dot plots (from n = 8) of the frequency of LCMV-infected F4/80high-Kupffer cells and inflammatory monocytes, as evidenced by intracellular LCMV-NP staining 24 h postinfection. Control staining without the addition of LCMV-NP antibody or anti-rat CD45 was used to set the threshold gate.
To examine whether the reduction of intrahepatic CD45+ F4/80high CD11b+ cells upon LCMV challenge corresponded to a genuine reduction in the frequency of Kupffer cells or a diminished surface expression of these markers, we performed an immunohistochemical analysis on 24-h-LCMV-infected and healthy livers (Fig. 1C). Interestingly, in contrast to the flow cytometric quantification, there was no clear depletion of Kupffer cells upon LCMV infection. Instead, the morphology of F4/80+ cells, which predominantly consist of Kupffer cells, changed toward larger or swollen cells, with a less intense F4/80 staining (Fig. 1C), suggesting a downregulation of F4/80 expression in comparison to healthy liver. Importantly, no cleaved caspase-3+ cells could be identified in both healthy and 24-h-LCMV-infected livers, thereby excluding the presence of apoptotic parenchymal or nonparenchymal cells (data not shown). Kupffer cells, which retained their F4/80 expression and were still identifiable by flow cytometry, were further termed F4/80high-Kupffer cells.
Corroborating earlier data on infection of Kupffer cells (37), we observed that 29% of F4/80high-Kupffer cells were LCMV-NP+ within 24 h after infection (Fig. 1D). In addition, at this stage F4/80high-Kupffer cells were the predominant LCMV-NP+ cells in the liver (90%), while the remainder LCMV-NP+ cells were inflammatory monocytes (8%) and granulocytes (2%) (data not shown).
During early LCMV infection, F4/80high-Kupffer cells remain endocytic.
Previous data from our group and others have shown that Kupffer cells from healthy mice are specialized phagocytes with only marginal cytokine production ex vivo under steady-state conditions (31) and that Kupffer cells are among the first to be infected with LCMV, thereby limiting viral spread (37–39). We studied here in detail the endocytic ability of F4/80high-Kupffer cells and inflammatory monocytes during early LCMV infection. As shown in Fig. 2A, the endocytic ability of F4/80high-Kupffer cells, as expressed by the fraction of dextran+ F4/80high-Kupffer cells, was reduced in LCMV-infected compared to PBS-injected mice, but the majority of cells were able to perform endocytosis, which was similar to F4/80high-Kupffer cells from LPS-challenged mice. The data at 4 and 24 h were comparable. Inflammatory monocytes exhibited a weaker endocytic ability in control mice, which was completely lost 24 h after LCMV infection, whereas LPS treatment increased it at this time point (14.8% versus 29.3% versus 1.5% dextran+ inflammatory monocytes, in healthy, LPS-treated, and LCMV-infected livers, respectively; Fig. 2A).
FIG 2.
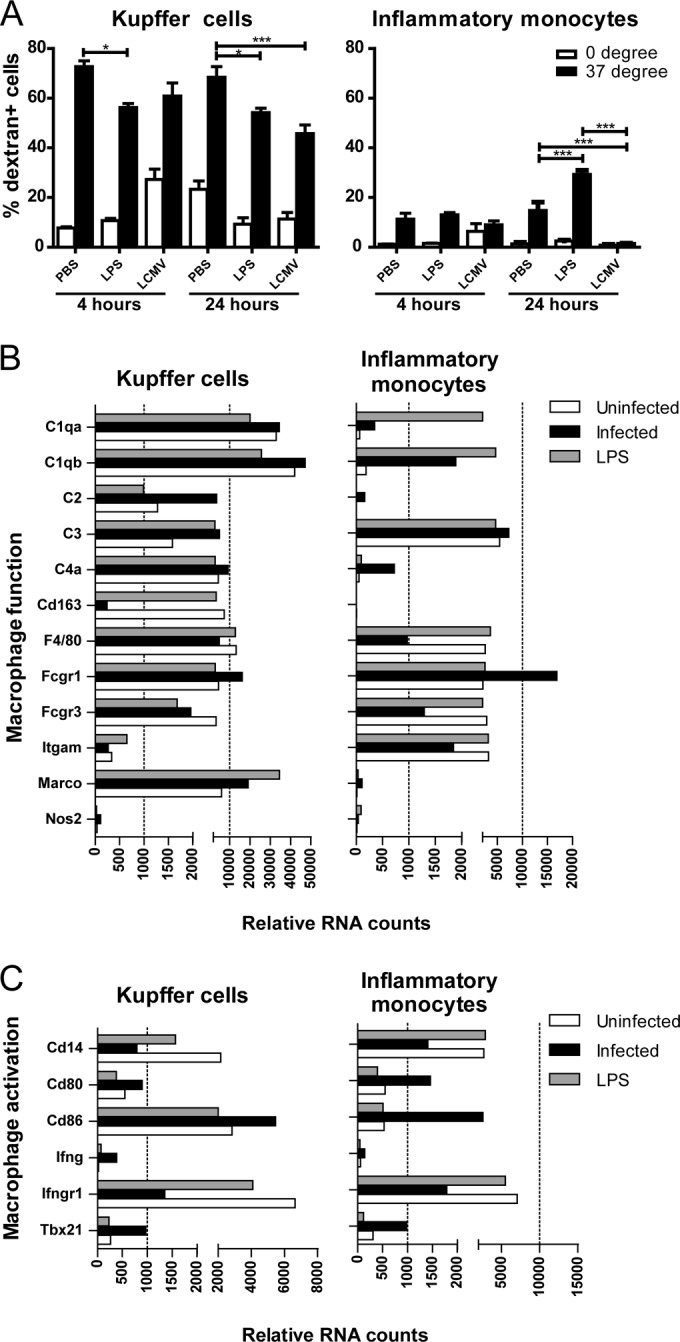
F4/80high-Kupffer cells retain a gene expression profile characteristic of phagocytic ability upon LCMV and LPS challenge, while this is only partially induced in inflammatory monocytes. Mice were infected with LCMV or challenged with LPS or PBS for 4 or 24 h. (A) Endocytosis was determined by flow cytometry by incubating total liver nonparenchymal cells with FITC-conjugated dextran at 0 or 37°C for 45 min. Inflammatory monocytes and F4/80high-Kupffer cells were identified as CD45+ F4/80low CD11bhigh Ly6Chigh and CD45+ F4/80high CD11b+ cells, respectively. Control staining without the addition of dextran was used to set the threshold gate. The assays were performed two times. Statistical analysis was performed using two-way ANOVA with the Bonferroni post-test. ***, P < 0.001. The data show the averages ± the SEM. (B and C) Gene expression was determined by using NanoString technology on total RNA isolated from formaldehyde-fixed, fluorescence-activated cell sorter (FACS)-sorted inflammatory monocytes and F4/80high-Kupffer cells from mice at 24 h after LCMV infection or LPS challenge.
Next, we evaluated whether the endocytic function was reflected by the gene expression profile of the cells. F4/80high-Kupffer cells purified from unchallenged mice show higher expression levels of various complement genes (C1qa, C1qb, C2, C4a, and C6) compared to inflammatory monocytes, both isolated from unchallenged mice (Fig. 2B). Also, gene expression levels of F4/80, genes encoding various Fcγ receptors and Marco were more expressed by F4/80high-Kupffer cells than inflammatory monocytes, indicating a more pronounced activity of the “classical scavenger functions” of F4/80high-Kupffer cells under steady-state conditions. As shown in Fig. 2B, infection with LCMV or challenge with LPS upregulated the expression levels of Marco on F4/80high-Kupffer cells, while the expression of various genes encoding complement remained high, with the exception of C2 and C3, which were further upregulated. In LPS- or LCMV-challenged inflammatory monocytes the expression levels of the complement genes remained lower than those observed in F4/80high-Kupffer cells: i.e., there was minimal upregulation of Marco and the complement genes, with the exception of C1qa and C1qb.
We then examined whether the observed differences in the endocytic response of F4/80high-Kupffer cells and inflammatory monocytes to LPS or LCMV were related to changes in the expression of genes associated with cell activation at 24 h after challenge. As presented in Fig. 2C, induction of mRNA expression of the activation markers CD80 and CD86 was observed for both F4/80high-Kupffer cells and inflammatory monocytes purified from the livers of LCMV-infected mice, whereas CD14 mRNA expression was downregulated. The effects of in vivo exposure to LPS on the expression levels of these markers were minimal at 24 h after challenge. In line with published data on Listeria monocytogenes, as well as in vitro exposure of macrophages to alpha interferon (IFN-α) (40, 41), we now show that both F4/80high-Kupffer cells and inflammatory monocytes isolated from LCMV-infected mice exhibited downregulation of the expression of IFNγR1, despite enhanced expression of IFNγ and Tbx21 mRNA.
Both sorted Kupffer cells and inflammatory monocytes exhibit an active cytokine and chemokine transcriptional profile early after LCMV infection.
Next, we determined the ability of F4/80high-Kupffer cells and inflammatory monocytes to produce cytokines upon stimulation with PMA and ionomycin. As we indicated in a previously published study (31), Kupffer cells produce low or undetectable levels of cytokines (such as TNF and IL-12p40) following in vitro stimulation. We now show that F4/80high-Kupffer cells are weak producers even when isolated from mice challenged with LPS or LCMV (in all conditions on average less than 5% TNF+ cells). In contrast, we demonstrate here that inflammatory monocytes isolated from LCMV-infected mice produced higher levels of TNF compared to control mice after in vitro exposure to PMA and ionomycin. An increased ability to produce TNF by these cells was not seen after LPS treatment (Fig. 3A, 13.4% versus 13.3% versus 27.5% TNF+ inflammatory monocytes in PBS-treated, LPS-treated, and LCMV-infected livers, respectively, upon in vitro restimulation with PMA and ionomycin).
FIG 3.

Both sorted Kupffer cells and inflammatory monocytes exhibit an active cytokine and chemokine transcriptional profile early after LCMV. Mice were infected with LCMV Cl13 or challenged with LPS or PBS (n = 6 to 12 per group) and sacrificed 24 h later. (A) Total liver nonparenchymal cells were stimulated with medium or PMA/ionomycin for 5 h. Flow cytometric analysis and graphic representation of the frequency of TNF-producing inflammatory monocytes (CD45+ F4/80low CD11bhigh Ly6Chigh cells) determined by intracellular cytokine staining. Control staining without the addition of TNF detecting antibody was used to set the threshold gate. Statistical analysis was performed using a two-tailed Student t test. *, P < 0.05; **, P < 0.01. (B) Gene expression was determined in whole liver (n = 4 to 6) by qPCR and in RNA isolated from formaldehyde-fixed, FACS-sorted inflammatory monocytes and F4/80high-Kupffer cells from infected or challenged mice by using NanoString technology. The data shown are obtained from two independent experiments using six pooled livers. The data show the averages ± the SEM.
Next, we examined the hepatic gene expression levels in whole liver of pro- and anti-inflammatory cytokines, chemokines, interferons and interferon-inducible antiviral mediators in both experimental challenge conditions. At 24 h after LCMV inoculation, significant increases of intrahepatic mRNA levels of TNF, IL-6, IL-10, MCP-1, CXCL10, IFNα, IFNβ, and RANTES were noted by evaluation of the whole liver (Fig. 3B). Distinct to the LCMV infection, LPS treatment primarily increased the mRNA levels of innate pro- and anti-inflammatory cytokines and chemokines at an earlier time point (i.e., 4 h after injection [data not shown]), with the exception of RANTES, which remained upregulated 24 h posttreatment. It is important to mention that this early LCMV-induced liver inflammation was not accompanied by a rise in serum transaminases or histological signs of liver damage (data not shown). To determine the possible contribution of F4/80high-Kupffer cells and inflammatory monocytes as the source of the observed gene expression levels, we again determined the gene expression profiles of cells isolated from mice challenged with LPS or LCMV. As shown in Fig. 3B, overall, both F4/80high-Kupffer cells and inflammatory monocytes express mRNA for TNF, IL-6, IL-10, MCP-1, CXCL10, and RANTES 24 h after LCMV infection. In line with the findings in whole liver, challenge with LPS did not exhibit enhanced gene expression levels at 24 h after challenge, except for RANTES mRNA in F4/80high-Kupffer cells. Interestingly, IFNβ mRNA was strongly induced in F4/80high-Kupffer cells from LCMV-infected mice, but not in inflammatory monocytes, whereas IFNα was only weakly induced in both cell types.
Purified F4/80high-Kupffer cells, as well as inflammatory monocytes, induce an interferon response after early LCMV infection.
The induction of IFN-β by F4/80high-Kupffer cells, but not inflammatory monocytes, may indicate that these cell types possess distinct functions in the early events following LCMV infection. We therefore examined the expression of IFN-inducible genes in both cell types to determine to what extent inflammatory monocytes are triggered by IFN production derived from F4/80high-Kupffer cell-derived and other liver cells. As shown in Fig. 4, both F4/80high-Kupffer cells and inflammatory monocytes strongly increase the expression of MxA mRNA early after LCMV infection. Similar findings were observed for Ifi35, Ifih1, Ifit2, and various chemokines, as well as for Irf1 and Irf7. In line with the expected induction of an IFN-dominated response, also the expression of Stat1, Stat2, and Stat3 mRNA was induced in both F4/80high-Kupffer cells and inflammatory monocytes, but not that of Stat4, Stat5, and Stat6 mRNA.
FIG 4.
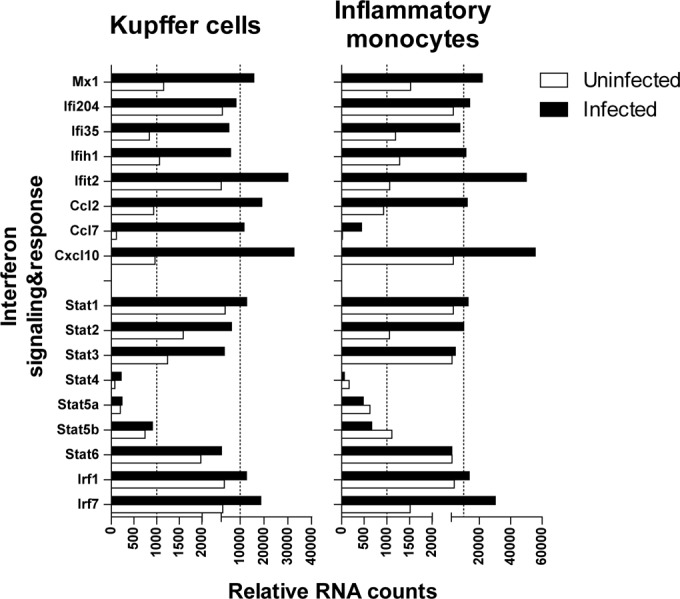
Both purified Kupffer cells and inflammatory monocytes are activated and induce an IFN response following early LCMV infection. Gene expression was determined in RNA isolated from formaldehyde-fixed, FACS-sorted inflammatory monocytes and F4/80high-Kupffer cells from LCMV-infected mice by using NanoString technology. The data shown are obtained from two independent experiments using six pooled livers.
DISCUSSION
Due to a lack of suitable animal models, our current understanding of early virus-host interactions in virus-induced liver disease is limited. In the present study, we make use of a short-term LCMV Cl13 infection in mice to examine phenotypic and functional changes in inflammatory monocytes and F4/80high-Kupffer cells, which are the first innate immune cells to encounter a viral pathogen in the liver. We show here that LCMV induces a marked recruitment of infiltrating monocytes within 24 h after infection. However, we observed that the major LCMV-NP+ population comprises F4/80high-Kupffer cells, which maintain their endocytic activity and present increased expression of several pro- and anti-inflammatory cytokines and chemokines after LCMV infection. On the other hand, inflammatory monocytes obtained from the livers of LCMV-infected mice exhibited weak endocytic activity, while the expression pattern of these inflammatory monocytes strongly resembled those of F4/80high-Kupffer cells early after LCMV infection. The observation that during early LCMV infection inflammatory monocytes outnumber the F4/80high-Kupffer cells indicates their important contribution to the early phase of LCMV-induced liver inflammation.
During early LCMV infection, we show that F4/80high-Kupffer cells become activated, take up LCMV-NP, and remain endocytic. The endocytic ability is in line with the findings of Lang et al. (37), who demonstrated that active uptake of LCMV by Kupffer cells limits viral spread and immunopathology. Similar to the LCMV model, Kupffer cells represent the predominant cell population to eliminate adenovirus type 5 from the liver (42). Previously, activated macrophages have been shown to reduce their F4/80 expression (43, 44). The immunohistological finding corroborates this flow cytometric downregulation of F4/80 surface expression. Similar phenotypic changes in Kupffer cells have been observed after an adenovirus type 5 infection in which cytoplasmic LysM expression was diminished as early as 20 min after infection, whereas membrane-associated major histocompatibility complex II expression was not affected (42).
Despite the observation that ex vivo stimulations show minimal TNF production by F4/80high-Kupffer cells upon polyclonal stimulation, the gene expression profile of sorted F4/80high-Kupffer cells from LCMV-infected mice clearly show induction of pro- and anti-inflammatory cytokines and chemokines, including TNF, IL-6, IL-10, and others. The discrepancy between the results obtained from the in vitro and in vivo data need to be studied in detail but may be due to the nature of the trigger used for restimulation of the cells in vitro. Detailed comparison of the gene expression levels of the cytokines and chemokines in whole liver versus the purified population strongly suggest that both F4/80high-Kupffer cells and inflammatory monocytes contribute, to a large extent, to the production of cytokines early during LCMV infection. Importantly, the mRNA expression levels of TNF, IL-6, MCP, CXCL10, and RANTES are highly similar between both cell types, whereas IL-10 mRNA remains expressed at higher levels by F4/80high-Kupffer cells compared to inflammatory monocytes. Despite the relatively high expression of IFNα mRNA in whole liver 24 h after LCMV infection, low or undetectable levels are observed in F4/80high-Kupffer cells and inflammatory monocytes, indicating that other intrahepatic cells, such as plasmacytoid DC and hepatocytes are the source of this IFN. Another option is that both sorted cell types produce other IFNα subtypes then the IFNα1 and IFNα2 detected by the NanoString probes (NCBI accession numbers NM_010502.2 and NM_010503.2, respectively). Furthermore, due to the known RNA fragmentation after formalin fixation, all tested qPCR primer sets yielded inconsistent results when tested on RNA from sorted F4/80high-Kupffer cells and inflammatory monocytes cells (data not shown) (35). IFNβ, on the other hand, was expressed by F4/80high-Kupffer cells, but not by inflammatory monocytes, which may be the direct result of triggering of IFN-β production upon LCMV infection and replication in F4/80high-Kupffer cells. Importantly, despite distinct and selective expression of type I IFN, both cell types have transcript levels for numerous IFN-stimulated genes (ISGs), encoding both chemokines and proteins that function to suppress viral replication, such as MxA. This might imply that Kupffer cell IFN-β release is instrumental in activating the recruited inflammatory monocytes, which could therefore be an important therapeutic target.
Previous studies have demonstrated the complexity of the functions of inflammatory monocytes found in different organs during a bacterial, fungal, protozoal, or viral infection (14–20). Although LCMV infection and LPS treatment induce the recruitment of inflammatory monocytes to similar extents, we show that the activities of LCMV-induced and LPS-induced inflammatory monocytes are functionally distinct. Similar to their role in colonic inflammation, LCMV-induced inflammatory monocytes are skewed toward a secretory function, as demonstrated by the increase in TNF production ability in comparison to liver monocytic cells from healthy mice and their gene expression profile, and completely lose their steady-state endocytic ability (16). On the other hand, LPS-induced inflammatory monocytes do not increase their ability to produce or express cytokines or chemokines but enhance their endocytosis potential. In mouse studies, TNF production by intrahepatic inflammatory monocytes has been associated with liver damage (29, 30). Similarly, in patients with chronic inflammatory and fibrotic liver diseases, inflammatory monocyte-derived TNF, among other proinflammatory cytokines and chemokines, has been shown to be profibrogenic (45). However, we did not find evidence of liver damage at this early stage after infection. In line with this, Karlmark et al. have shown that the newly recruited inflammatory monocytes are not responsible for the early onset of CCl4-induced hepatitis (28). Alternatively, inflammatory monocyte-derived TNF might act as a positive feedback for further recruitment of inflammatory monocytes to the liver. Recently, the formation of intrahepatic myeloid cell aggregates that can stimulate local T cell proliferation (iMATE) has been attributed to monocyte-derived TNF (46). Our findings support the proinflammatory role of recruited monocytes in the setting of early virus-induced liver disease.
Our study demonstrates that early after LCMV infection a functional dichotomy is observed for inflammatory monocytes and F4/80high-Kupffer cells with respect to endocytosis, but their activation and cytokine gene expression profiles exhibit a strong resemblance. In particular, inflammatory monocytes show a huge capacity for recruitment to the liver and plasticity depending on the nature of the inflammatory signal, i.e., whether it is viral or sterile. While the experiments were performed with a chronic strain of LCMV, recent data show that innate immune responses are independent of the strain used during the first 72 h after inoculation (47). Although the relative contribution of inflammatory monocytes or F4/80high-Kupffer cells to the outcome of the infection needs to be investigated, our study points toward a crucial role for inflammatory monocytes in shaping the inflammatory environment in the liver early after infection.
ACKNOWLEDGMENTS
We thank Vincent Vaes, Judith van Agthoven, and Dennis de Meulder from the Erasmus MC animal care facility for their assistance in performing biotechnical manipulations.
This study was supported by the Virgo consortium, funded by the Dutch government project number FES0908, and by the research collaboration with Janssen Infectious Diseases–Diagnostics BVBA project ICD 551306. T.V. is supported by an Erasmus MC Fellowship 2011.
REFERENCES
- 1.Ganem D, Prince AM. 2004. Hepatitis B virus infection–natural history and clinical consequences. N Engl J Med 350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 2.Lauer GM, Walker BD. 2001. Hepatitis C virus infection. N Engl J Med 345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 3.Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. 2001. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med 194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day CL, Lauer GM, Robbins GK, McGovern B, Wurcel AG, Gandhi RT, Chung RT, Walker BD. 2002. Broad specificity of virus-specific CD4+ T-helper-cell responses in resolved hepatitis C virus infection. J Virol 76:12584–12595. doi: 10.1128/JVI.76.24.12584-12595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lauer GM, Ouchi K, Chung RT, Nguyen TN, Day CL, Purkis DR, Reiser M, Kim AY, Lucas M, Klenerman P, Walker BD. 2002. Comprehensive analysis of CD8+ T-cell responses against hepatitis C virus reveals multiple unpredicted specificities. J Virol 76:6104–6113. doi: 10.1128/JVI.76.12.6104-6113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rehermann B, Nascimbeni M. 2005. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol 5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 7.Claassen MA, Janssen HL, Boonstra A. 2013. Role of T cell immunity in hepatitis C virus infections. Curr Opin Virol 3:461–467. doi: 10.1016/j.coviro.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Chayama K, Hayes CN, Hiraga N, Abe H, Tsuge M, Imamura M. 2011. Animal model for study of human hepatitis viruses. J Gastroenterol Hepatol 26:13–18. doi: 10.1111/j.1440-1746.2010.06470.x. [DOI] [PubMed] [Google Scholar]
- 9.Boonstra A, van der Laan LJ, Vanwolleghem T, Janssen HL. 2009. Experimental models for hepatitis C viral infection. Hepatology 50:1646–1655. doi: 10.1002/hep.23138. [DOI] [PubMed] [Google Scholar]
- 10.Zhou X, Ramachandran S, Mann M, Popkin DL. 2012. Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: past, present and future. Viruses 4:2650–2669. doi: 10.3390/v4112650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. 2008. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe 4:374–386. doi: 10.1016/j.chom.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee LN, Burke S, Montoya M, Borrow P. 2009. Multiple mechanisms contribute to impairment of type 1 interferon production during chronic lymphocytic choriomeningitis virus infection of mice. J Immunol 182:7178–7189. doi: 10.4049/jimmunol.0802526. [DOI] [PubMed] [Google Scholar]
- 13.Shi C, Pamer EG. 2011. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosschaerts T, Guilliams M, Stijlemans B, Morias Y, Engel D, Tacke F, Herin M, De Baetselier P, Beschin A. 2010. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-γ, and MyD88 signaling. PLoS Pathog 6:e1001045. doi: 10.1371/journal.ppat.1001045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. 2003. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 19:59–70. doi: 10.1016/S1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 16.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. 2012. Inflammation switches the differentiation program of Ly6Chi monocytes from anti-inflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med 209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ersland K, Wuthrich M, Klein BS. 2010. Dynamic interplay among monocyte-derived, dermal, and resident lymph node dendritic cells during the generation of vaccine immunity to fungi. Cell Host Microbe 7:474–487. doi: 10.1016/j.chom.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, Jeffrey KL, Anthony RM, Kluger C, Nchinda G, Koh H, Rodriguez A, Idoyaga J, Pack M, Velinzon K, Park CG, Steinman RM. 2010. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209+ dendritic cells for immune T cell areas. Cell 143:416–429. doi: 10.1016/j.cell.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, Mack M, Shpigel N, Boneca IG, Murphy KM, Shakhar G, Halpern Z, Jung S. 2012. Ly6Chi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 37:1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 20.Farache J, Koren I, Milo I, Gurevich I, Kim KW, Zigmond E, Furtado GC, Lira SA, Shakhar G. 2013. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity 38:581–595. doi: 10.1016/j.immuni.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurmaeva E, Bhattacharya D, Goodman W, Omenetti S, Merendino A, Berney S, Pizarro T, Ostanin DV. 2014. Immunosuppressive monocytes: possible homeostatic mechanism to restrain chronic intestinal inflammation. J Leukoc Biol 96:377–389. doi: 10.1189/jlb.3HI0613-340RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurihara T, Warr G, Loy J, Bravo R. 1997. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med 186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skold M, Behar SM. 2008. Tuberculosis triggers a tissue-dependent program of differentiation and acquisition of effector functions by circulating monocytes. J Immunol 181:6349–6360. doi: 10.4049/jimmunol.181.9.6349. [DOI] [PubMed] [Google Scholar]
- 24.Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, Sibley LD. 2008. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. 2000. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol 156:1951–1959. doi: 10.1016/S0002-9440(10)65068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aldridge JR Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, Brown SA, Doherty PC, Webster RG, Thomas PG. 2009. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci U S A 106:5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holt MP, Cheng L, Ju C. 2008. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol 84:1410–1421. doi: 10.1189/jlb.0308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, Merad M, Luedde T, Trautwein C, Tacke F. 2009. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 50:261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 29.Nakamoto N, Ebinuma H, Kanai T, Chu PS, Ono Y, Mikami Y, Ojiro K, Lipp M, Love PE, Saito H, Hibi T. 2012. CCR9+ macrophages are required for acute liver inflammation in mouse models of hepatitis. Gastroenterology 142:366–376. doi: 10.1053/j.gastro.2011.10.039. [DOI] [PubMed] [Google Scholar]
- 30.Helk E, Bernin H, Ernst T, Ittrich H, Jacobs T, Heeren J, Tacke F, Tannich E, Lotter H. 2013. TNFα-mediated liver destruction by Kupffer cells and Ly6Chi monocytes during Entamoeba histolytica infection. PLoS Pathog 9:e1003096. doi: 10.1371/journal.ppat.1003096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Movita D, Kreefft K, Biesta P, van Oudenaren A, Leenen PJ, Janssen HL, Boonstra A. 2012. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J Leukoc Biol 92:723–733. doi: 10.1189/jlb.1111566. [DOI] [PubMed] [Google Scholar]
- 32.Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, Habu Y, Miyazaki H, Hiroi S, Seki S. 2010. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol 53:903–910. doi: 10.1016/j.jhep.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 33.Salvato M, Borrow P, Shimomaye E, Oldstone MB. 1991. Molecular basis of viral persistence: a single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J Virol 65:1863–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med 160:521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russell JN, Clements JE, Gama L. 2013. Quantitation of gene expression in formaldehyde-fixed and fluorescence-activated sorted cells. PLoS One 8:e73849. doi: 10.1371/journal.pone.0073849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basta S, Stoessel R, Basler M, van den Broek M, Groettrup M. 2005. Cross-presentation of the long-lived lymphocytic choriomeningitis virus nucleoprotein does not require neosynthesis and is enhanced via heat shock proteins. J Immunol 175:796–805. doi: 10.4049/jimmunol.175.2.796. [DOI] [PubMed] [Google Scholar]
- 37.Lang PA, Recher M, Honke N, Scheu S, Borkens S, Gailus N, Krings C, Meryk A, Kulawik A, Cervantes-Barragan L, Van Rooijen N, Kalinke U, Ludewig B, Hengartner H, Harris N, Haussinger D, Ohashi PS, Zinkernagel RM, Lang KS. 2010. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology 52:25–32. doi: 10.1016/j.jhep.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Lohler J, Gossmann J, Kratzberg T, Lehmann-Grube F. 1994. Murine hepatitis caused by lymphocytic choriomeningitis virus. I. The hepatic lesions. Lab Invest 70:263–278. [PubMed] [Google Scholar]
- 39.Matloubian M, Kolhekar SR, Somasundaram T, Ahmed R. 1993. Molecular determinants of macrophage tropism and viral persistence: importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J Virol 67:7340–7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. 2010. Induction of IFN-α/β enables Listeria monocytogenes to suppress macrophage activation by IFN-γ. J Exp Med 207:327–337. doi: 10.1084/jem.20091746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu BS, Janssen HL, Boonstra A. 2011. IL-29 and IFNα differ in their ability to modulate IL-12 production by TLR-activated human macrophages and exhibit differential regulation of the IFNγ receptor expression. Blood 117:2385–2395. doi: 10.1182/blood-2010-07-298976. [DOI] [PubMed] [Google Scholar]
- 42.Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li JY, Hoshida Y, Llovet JM, Powers S. 2013. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology 145:1424–1435. doi: 10.1053/j.gastro.2013.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ezekowitz RA, Austyn J, Stahl PD, Gordon S. 1981. Surface properties of bacillus Calmette-Guerin-activated mouse macrophages: reduced expression of mannose-specific endocytosis, Fc receptors, and antigen F4/80 accompanies induction of Ia. J Exp Med 154:60–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ezekowitz RA, Gordon S. 1982. Downregulation of mannosyl receptor-mediated endocytosis and antigen F4/80 in bacillus Calmette-Guerin-activated mouse macrophages: role of T lymphocytes and lymphokines. J Exp Med 155:1623–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liaskou E, Zimmermann HW, Li KK, Oo YH, Suresh S, Stamataki Z, Qureshi O, Lalor PF, Shaw J, Syn WK, Curbishley SM, Adams DH. 2013. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 57:385–398. doi: 10.1002/hep.26016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang LR, Wohlleber D, Reisinger F, Jenne CN, Cheng RL, Abdullah Z, Schildberg FA, Odenthal M, Dienes HP, van Rooijen N, Schmitt E, Garbi N, Croft M, Kurts C, Kubes P, Protzer U, Heikenwalder M, Knolle PA. 2013. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8+ T cells and successful immunotherapy against chronic viral liver infection. Nat Immunol 14:574–583. doi: 10.1038/ni.2573. [DOI] [PubMed] [Google Scholar]
- 47.Norris BA, Uebelhoer LS, Nakaya HI, Price AA, Grakoui A, Pulendran B. 2013. Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit virus-specific T cell immunity. Immunity 38:309–321. doi: 10.1016/j.immuni.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]