

Abstract
Major attention has been focused on the development of gene therapy approaches for the treatment of vascular diseases. In this review, we focus on an alternative use of gene therapy: the use of genetic means to study vascular cell biology and physiology. Both viral and nonviral gene transfer strategies have limitations, but because of the overwhelming inflammatory responses associated with the use of viral vectors, nonviral gene transfer methods are likely to be used more abundantly for future applications in the vasculature. Researchers have made great strides in the advancement of gene delivery to the vasculature in vivo. However, the efficiency of gene transfer seen with most nonviral approaches has been exceedingly low. We discuss how to circumvent and take advantage of a number of the barriers that limit efficient gene delivery to the vasculature to achieve high-level gene expression in appropriate cell types within the vessel wall. With such levels of expression, gene transfer offers the ability to alter pathways at the molecular level by genetically modulating the activity of a gene product, thus obviating the need to rely on pharmacological agents and their foreseen and unforeseen side effects. This genetic ability to alter distinct gene products within a signaling or biosynthetic pathway or to alter structural interactions within and between cells is extremely useful and technologically possible today. Hopefully, with the availability of these tools, new advances in cardiovascular physiology will emerge.
In recent years, the potential to replace defective genes or to use altered genes to combat disease in humans has been realized (2,47). At present, gene therapy protocols are being developed and used in clinical trials to treat or prevent genetic disorders, cancers, and infectious diseases (26). Major attention is also being focused on development of gene therapy approaches to treat a variety of vascular disorders, including atherosclerosis, restenosis, and thrombosis, among others, as well as to promote angiogenesis. Several excellent recent reviews describing the current state of vascular gene therapy and the genetic approaches being tested for various diseases are available (7,78). As such, this review does not extensively summarize these studies. Rather, our goal is to focus on an alternative use of gene therapy: the use of genetic means to study the cell biology and physiology of the vasculature. Moreover, we focus entirely on nonviral methods for gene delivery to the vasculature, with discussions of potential methodologies for studying the vessel wall.
VIRAL VERSUS NONVIRAL APPROACHES
Numerous approaches have been proposed and developed for transferring genes to cells, but all have serious limitations (29). Inefficiency of gene transfer, immunological responses, and nonspecificity of cell targeting are just a few of the problems associated with the current vectors. For example, although adenovirus has been used extensively for vascular gene therapy and has gene transfer efficiencies of almost 95% in vitro, the values are usually much less in vivo and are highly variable: expression of the reporter β-galactosidase gene in smooth muscle and endothelial cells in rat carotid and pulmonary artery models can vary from 0 to 30% of cells transduced, depending on the cell type and the study (33,58). Furthermore, adenovirus cannot traverse cells or tight junctions to infect more than one cell layer. Thus, in uninjured vessels, catheter delivery of virus results in infection of only endothelial cells; to infect the smooth muscle, the endothelium must be damaged and removed. The use of adenoviral vectors also requires administration of upward of 108–109 plaque-forming units (pfu). Because not all viruses within a preparation are viable, this means that typically, 1010–1012 virus particles are used per administration. Consequently, adenovirus-induced cell damage and inflammation are common, making its clinical use doubtful (20,59). Retroviral vectors also give relatively good gene transfer in many systems, but they can only transduce actively dividing cells because their genome, once reverse-transcribed into DNA, cannot enter the nucleus in the absence of cell division and nuclear envelope breakdown (34,45). Thus, retroviral vectors cannot be used on postmitotic or quiescent cells, including terminally differentiated muscle cells and have had little success in vivo in the vasculature (19). From a technical point of view, viral vectors also pose difficulties in production. It remains difficult to produce high titers of many recombinant viruses, and the threat of wild-type virus contamination is always present. Other viruses that have been tested in the vasculature include adeno-associated virus and sendai virus, although many of the same problems are found with these viruses as with adenovirus.
In contrast to the viral vectors, non-viral vectors show great potential for gene therapy in a variety of tissues, especially the vasculature. Unlike their viral counterparts, essentially no immune response is generated against DNA, either as naked plasmid or when complexed with liposomes or other polymers such as polyethylenimine. Furthermore, there is very little inflammation or pathology associated with these non-viral vectors (31,51). Thus, multiple administrations of vector can be given with no decrease in activity. Perhaps most importantly, unlike all of the viral vectors described to date, plasmid production is extremely simple and yields high levels of vector. Plasmids also can be easily purified to avoid contaminants such as wild-type or defective virus particles, as is seen with first-, second-, and third-generation adenovirus vectors, adeno-associated virus vectors, retroviral, and even lentiviral vectors. Thus, there are fewer safety concerns. The advent of commercial kits to purify plasmids based on DNA binding to silica gels (e.g., Qiagen kits) has made the use of plasmids even more attractive. For example, a milligram of plasmid at purities sufficient for animal administration costs less than around $10 and in 1 day a researcher can easily prepare 50 mg or more. Unfortunately, the efficiency of gene transfer to the vasculature in vivo using nonviral approaches has been exceedingly low, thus greatly limiting its successful use. Because of this, there has been a tradeoff for gene transfer technologies: although viruses yield high expression with inflammatory and safety side effects, plasmids are much safer, yet yield little expression. However, there is wide agreement that with the development of new techniques to increase nonviral expression levels, plasmids will be the vector of choice for human applications.
BARRIERS TO GENE TRANSFER
Under physiologically relevant conditions, the levels of gene transfer are low at best. The reason for these low levels of gene transfer is that many barriers exist for the efficient transfer of genes to cells (Fig. 1). First, vectors must be targeted to specific cell types while avoiding many others. Second, before the vector can reach any cell, it must make its way through the extracellular matrix before any cell-specific interactions can occur. Third, the vector must enter the cell by breaking through the plasma membrane. This usually involves the vector being endocytosed into endosomes. The DNA must then escape the endosome before lysosomal fusion to enter the cytoplasm. Fourth, once in the cytoplasm, the DNA is confronted by the nuclear membrane that it must traverse to enter the nucleus. Fifth, the DNA must be maintained within the nucleus and not discarded by random segregation during subsequent mitoses. Finally, the vector must be transcribed appropriately. In addition, if the vectors are to be used in vivo, they also face the task of escaping destruction by serum factors and sequestration by first-pass organs.
Figure 1.

Barriers to gene transfer. Cartoon depicting the various barriers encountered by DNA between administration and protein production.
Viruses have the distinct advantage of having evolved and developed mechanisms to help bypass each of these barriers (25,74). Viral capsids help protect the genome from attack, and capsid proteins mediate binding of the viral particles to the cell surface. Viruses, such as influenza, have developed fusion peptides in envelope proteins to aid in endosomal escape. Many viral capsids or core particles also have the ability to target the genomes to the nucleus. For example, herpesvirus capsids bind to the nuclear pore complex and release the DNA into the nucleus, whereas the HIV genome is complexed into preintegration complexes that enter the nucleus because of the presence of a set of viral proteins bound to the reverse-transcribed DNA that harbor signals for nuclear import. Finally, many of the strongest promoters identified to date are from viruses. By contrast, nonviral vectors rely solely on developments in the laboratory and scientist-driven evolution over just the past 10–20 years.
GENE TRANSFER APPROACHES
The most widely used method for nonviral gene transfer to the vasculature, or any tissue for that matter, has used plasmid-liposome complexes. A variety of cationic and neutral lipids have been developed over the past 10 years and have been shown to complex with plasmids, oligonucleotides, or RNAs, and facilitate cell association, internalization, and gene expression. Because DNA is highly negatively charged, positively charged cationic lipids will interact with the DNA to form complexes. Depending on the lipids used and the methods of observation, the structure of the complexes may vary, but in all cases, the lipids are thought to form classical bilayers with their positively charged head groups interacting with the nucleic acids (53). Many studies have found what appear to be meatballs wrapped with spaghetti; the plasmids are not always completely encased within a perfectly spherical liposome, and portions of the DNA may remain on the surface of the liposome. The resulting complex then displays an outward positive charge that can interact with the negatively charged cell membrane for fusion and endocytosis. Once endocytosed, the complex escapes into the cytoplasm and falls apart to release free DNA that can now enter the nucleus for gene expression.
Although cationic lipids work very well to transfer genes to cells in culture, when used in animals, the results have been much less satisfactory (3). Typical in vitro transfection efficiencies with cationic liposomes can range from 20 to 90% of cells transfected, whereas most in vivo studies report between 1 and 5% of cells transfected (3,64). Systemic delivery of DNA-liposome complexes results in gene transfer mainly to the lung and liver, both first-pass organs with large vascular surface areas (5,36,65,70). Because the complexes are diluted into the large volume of the circulation, limited gene transfer is observed. Indeed, >95% of DNA-liposome complexes are cleared from the circulation within 5 minutes, mainly because of retention within these vascular beds (5). Furthermore, almost all gene expression observed is in cells accessible to the complexes, namely, endothelial cells. Thus, for transfer to the intima or media, vessel damage is a prerequisite (3). However, the constant development of new lipid and liposome formulations as well as techniques to include cell-targeting proteins or ligands will surely increase efficiencies. Indeed, the incorporation of viral fusion proteins into the liposome-DNA complexes has been reported to increase the gene transfer efficiency up to between 10 and 30% (15,73). However, for nonviral vectors to be of clinical use in the vasculature, their ability to transfect cells in vivo must be increased.
Several other cationic polymers have been tested for their ability to mediate gene transfer to the vasculature. Branched chain polycations, such as polyethylenimine (PEI) and dendrimers, have shown high-level gene transfer both in cultured cells and in animals. PEI contains multiple primary, secondary, and tertiary amines, about 20% of which are protonated under physiological conditions. Because of this, PEI has a large proton buffering capacity that may promote endosomal escape and account in part for the increased levels of gene transfer seen with this agent (66). Similar to cationic lipids, PEI complexes with DNA due to electrostatic interactions. Typically, PEI-DNA complexes have been administered systemically, intraluminally using balloon catheters, or extravascularly using a collar placed around a section of vessel, similarly to liposomes (69). As with liposome complexes, the DNA is mainly transferred to lung and liver in the case of systemic delivery (27,48), and in local delivery, only the cells in contact with the complexes become transfected (69). To reduce the amount of nonspecific gene transfer to first pass organs seen with systemic administration of PEI, and to increase the serum stability, several reports have shown that modifying the PEI with either poly(ethylene glycol) or transferrin can reduce serum-induced aggregation and degradation, as well as much of the nonspecific gene transfer (27,48). Dendrimers are also cationic and behave similarly to PEI in terms of DNA condensation, cell association, and transfer efficiency (69,75).
One problem with nonviral gene transfer strategies is that even when the DNA is efficiently transferred to target cells, gene expression is usually transient. In most cases, robust gene expression is detected for only 1 week, after which the levels of gene expression drop precipitously. Although most of the current work to increase the duration of nonviral gene expression is focusing on the use of promoters and other DNA elements, several reports have described the development of controlled-release methods for prolonged gene transfer (30). Thus, even if a plasmid only expresses for 1 week, if the plasmid can be delivered continuously to a site over the course of a month, the total duration of expression will last for 5 weeks from the date of initial treatment. Such methods are being used to coat sutures, stents, and angioplasty balloons for prolonged exposure of vessels to DNA with limited success (24,30). One current limitation is that most studies have encapsulated naked DNA, which in itself is not highly efficient for gene transfer. However, as better coatings are developed to encapsulate DNA-liposome or DNA-polycation complexes, such approaches will become much more valuable.
Another recent development is the use of pressure to deliver DNA to the vasculature. Mann and colleagues cannulated explanted human saphenous veins and applied modest hydrostatic pressure to the vessels that are filled with a solution of either plasmid or oligonucleotide (41). The plasmid or oligonucleotide (between 5 and 100 μM) is delivered through the cannula, and the other end of the vessel is clamped. By using a standard angioplasty insufflator and pressure transducer, pressure is applied to the lumen of the vessel for 10 minutes at 100–200 mm Hg. These conditions resulted in oligonucleotide delivery to 90% of intimal and medial smooth muscle cells, with the occasional adventitial cell being transfected. With use of a similar approach, but with higher pressures, oligonucleotides were delivered to explanted rat hearts that were then used for transplant. When the oligonucleotides were delivered via the coronary circulation and the epicardial and endocardial surfaces at a pressure of 1500 mm Hg for 30 minutes, transfer to 50% of cells in the myocardium was observed. When plasmids were used instead of synthetic oligonucleotides, the use of 100 mm Hg hydrostatic pressure resulted in 10- to 20-fold higher levels of expression than without pressure, although the levels of transgene expression were still rather low (pg/mg protein) (72). However, by using oligonucleotides, this group has shown positive and significant therapeutic outcomes using this method.
Several other groups have used pressure in a different way to transfer DNA to a variety of organs and muscles via the vasculature. By administering a large volume of DNA (approximately 10% of body weight) over a very short period of time (<10 seconds) through the tail vein of rats, significant amounts of gene transfer and expression were obtained in all examined internal organs, with maximal expression being detected in the liver (39,82). It is surprising that little toxicity has been reported for this technique, with only transiently elevated levels of alanine aminotransferase (ALT). In studies in dogs and primates, Wolff and colleagues used a similar hydrodynamics-based method to transfer DNA to skeletal muscle in isolated limbs and achieved high levels of gene expression (81). However, although this method results in high-level gene expression in organs and muscle, essentially no expression was reported in the vasculature itself. Thus, the rapid increase in pressure and volume probably transiently permeabilizes the vasculature to allow the DNA access to target tissues, bypassing the cells of the vessel wall.
Yet another way that has been used successfully to transfer genes to the vasculature is to use a cell-based approach. Smooth muscle or endothelial cells are isolated, grown in culture, and transfected with the gene(s) of interest. Once the cells have been transfected, they are infused into the vasculature of animals where the cells can incorporate into the vessel wall. When fluorescently labeled cells or transfected cells expressing either a reporter (lacZ) or a therapeutic gene (eNOS) were injected into the rat jugular vein, transplantation of the cells into the walls of small pulmonary arteries and arterioles was observed as early as 24 hours after injection (10). Up to 57% of the transferred cells could be identified in the lung within 15 minutes of delivery with 15% remaining for 2 weeks. In another study, SMCs were transduced in vitro with retroviral constructs and then seeded into injured rat carotid arteries (18). By administering the cells to a defined portion of the vessel, the authors were able to show successful cell seeding in the media and intima and thus, gene transfer to a desired area. Because transfection strategies work much better on cells in culture than in animals, this approach may be advantageous if selection strategies are used to isolate and inject only transfected cells. However, because the number of injected cells is much less than the number of cells making up the vascular bed, even in isolated tissues such as the lung, this approach may not be suitable for all desired gene transfer needs.
DELIVERY TO THE VASCULATURE
When delivering cells to the vasculature, several routes are possible. Genes can be systemically delivered, typically by tail vein injection. Alternatively, they can be delivered to defined regions of the vasculature either from the luminal side of the vessel or from the adventitial side. Each of these routes has distinct advantages and disadvantages, and the choice of route should be dictated by the situation.
Most studies aimed at transferring genes to the vasculature have focused on the use of catheter delivery systems to transduce cells. In most cases, gene delivery has used viral particles including adenoviruses, retroviruses, and AAV. Perhaps the greatest use of this approach has been to transfer genes known to inhibit SMC proliferation to vessels injured by balloon angioplasty to develop treatments for restenosis, atherosclerosis, or vein graft stenosis. Genes, including p21 and p27 cyclin-dependent kinase inhibitors (22,76), the retinoblastoma gene product (11), p53 (79), β-interferon (63), the herpesvirus thymidine kinase gene (49), and eNOS (12,73), among others, have been transferred to vessels intraluminally. Although this is an attractive method, especially when genes are delivered immediately after angioplasty by using a second double-balloon catheter, this technique does have limitations. First, catheter-based systems achieve almost no gene transfer to smooth muscle cells within the intima unless the endothelial cell layer is destroyed. During angioplasty, this is not a problem but for procedures to prevent vein graft stenosis or to study the physiology of intact vessels, this represents a serious drawback. Second, even with damage to the endothelium, the elastic lamina of conduit vessels acts as a barrier to prevent any gene transfer to cells in the media (61). The third, and perhaps the major, problem with catheter-based delivery systems is that blood flow is occluded for a period of time, thus causing ischemia to the downstream vascular bed as well as target organs. Although some organs and tissues can tolerate mild ischemia, others cannot. This again leads to a trade-off: localized delivery that may be highly efficient based on vector design or transfer technology versus ischemia, tissue damage, and potential necrosis.
Systemic delivery of DNA complexes in rodents is typically performed via tail vein injection. DNA-liposome, DNA-PEI, DNA-dendrimer, and other complexes administered this way are rapidly disseminated through the circulation and accumulate in first-pass organs. It has been shown that within 5 minutes of injection, almost 95% of DNA-liposome complexes are removed from the circulation (6). Within the first few minutes after injection, most of these DNA-polycation complexes coat the luminal surface of endothelial cells of the microvasculature. Most of the DNA-polycation complexes are retained within the lung and the liver, thus leading to relatively high levels of transfection within small arteries and arterioles. After lung and liver, vessels in the lymph nodes, ovary, and adrenal medulla show the next highest level of gene transfer, followed by those in thymus, uterus, skeletal muscle, heart, and trachea (38). The distribution can be altered when administering a high volume of DNA over a short period of time, because the bolus of fluid under pressure will run contrary to normal circulation. This accounts for the increased levels of gene expression found in liver, spleen, and heart under these conditions (39).
Delivery of DNA to the adventitial surface of blood vessels has the advantage of limiting the distribution of gene expression to that area treated, much like balloon catheter delivery. Unlike catheter delivery, however, adventitial delivery does not occlude blood flow, and thus, causes no ischemia. The major disadvantage to adventitial delivery is obviously that the adventitial surface of the vessel must be exposed and made accessible. One of the greatest features of catheter delivery is that it can be performed with minimal intrusion; adventitial delivery requires that the vessels be exposed through incisions large enough to work through. The main way people have transferred genes to the exterior of the blood vessel is by transiently bathing the vessel in a solution of DNA-polycation complexes, such as DNA-liposomes or PEI-DNA (69). By using such approaches, genes can be transferred, with varying efficiency, to adventitial cells, but cells within the media or intima seldom are transfected.
USE OF ELECTRIC FIELDS TO TRANSFER GENES TO THE VASCULATURE
Electroporation uses electrical fields to create transient pores in the cell membrane that allow the entry of normally impermeable macromolecules into the cytoplasm (62). Although this technique is routinely used to transfer DNA to bacteria, yeast, and mammalian cells in culture (4), it was only recently applied to intact tissues in living animals. The results achieved with electroporation are impressive; in most studies, the increase in gene expression to a tissue injected with DNA is on the order of 100- to 1000-fold compared to DNA in the absence of electroporation (46). Although DNA electroporation protocols have been developed for and work well in solid tissue, such as skeletal muscle in which the DNA can be injected, such an approach is impossible in the vasculature. To circumvent this problem, our group developed a novel electrode design to facilitate DNA transfer to vessels of any size in living animals that does not occlude blood flow or cause ischemia (Fig. 2) (42). The electrode resembles a spoon, with a notch on either side that will allow a vessel to lay within the electrode and bathe in a solution of DNA. The design of the original electrode was such that about a 1-cm-long section of vessel could be placed in the chamber and electroporated. Running parallel to the vessel are two wires that are attached to a square wave electroporator (BTX 830; Genetronics, San Diego, CA). For our initial experiments, we chose to deliver DNA to the rat mesenteric vasculature. The ease of dissection and the ability to use anatomical location to identify treated vessels at a later date allowed up to 10 individual vessels to be treated in a given rat. Briefly, rats are anesthetized and a segment of distal small intestine is exteriorized through a midline incision and fanned out on gauze pads. Portions of the mesentery surrounding a vessel are cut away and the vessel is placed into the electrode. A solution of plasmid DNA (between 0.1 and 2 mg/mL in physiological saline) is pipetted into the electrode so that the solution just covers the vessel segment. A series of 8 square wave pulses of 10-millisecond duration each are applied at 1-second intervals. The vessel is incubated for an additional 1 minute in the chamber, and the solution is removed and saved for the next vessel. The treated vessel is removed from the electrode and the next vessel to be treated is placed in the vessel. The same DNA solution can be reused for multiple vessels, although the volume decreases over time because of the solution being retained on the vessels. To correct for this, additional plasmid solution is added as needed. After all vessels are treated, the intestine is returned to the abdominal cavity, and the incision is closed in layers with suture and metal wound clips. The procedure takes 30–45 minutes once the animal is anesthetized, during which time 6–10 vessels can be treated.
Figure 2.
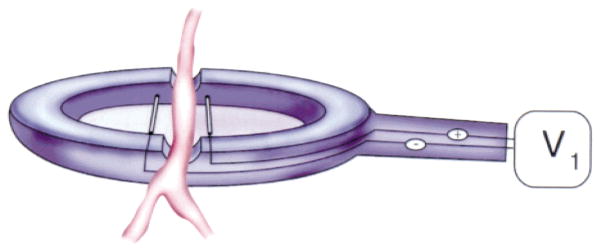
Electrode design for vascular electroporation.
To optimize gene transfer parameters, we chose to use plasmids expressing luciferase and GFP for quantitative and qualitative analysis, respectively. The fact that luciferase can be measured efficiently over a concentration range of 105 makes it an excellent choice for quantitative measurements. The ability to directly observe as few as 300 molecules of GFP within living cells makes this a useful gene product for determining the location of gene expression. Luciferase activity was measured in extracts from vessels that had been electroporated with a CMV promoter-driven luciferase-expressing plasmid. Peak gene transfer was achieved at a field strength of 200 V/cm, a value similar to that found by us and others in other tissues (8). Essentially no gene transfer was observed without an applied electric field, reiterating the relative inability of naked DNA to transfect nonskeletal muscle cells without addition of pressure or polycation. When the field strength was raised to 400 V/cm, gene expression decreased and tissue damage was noted. This has also been reported by others using different tissues. Gene expression was detected as early as 6 hours after electroporation, peaked between days 1 and 3 after transfer and declined to levels just above background by day 5. The level of gene transfer and expression is impressive: we routinely obtain between 0.1 and 1 ng of luciferase per cm of a 200-μm-diameter vessel. By using a GFP-expressing plasmid, gene expression was detected in all cell layers of the treated vessels including endothelial, smooth muscle, and adventitial cells (Fig. 3). High levels of GFP expression were detected over the length of the electroporated segment, and GFP was uniformly distributed throughout the vessel in adjacent sections. In contrast, no GFP expression was detected in vessels that were bathed in DNA but not electroporated or in untreated vessels. These results show that electroporation is capable of producing uniform gene expression along the length of the blood vessel.
Figure 3.
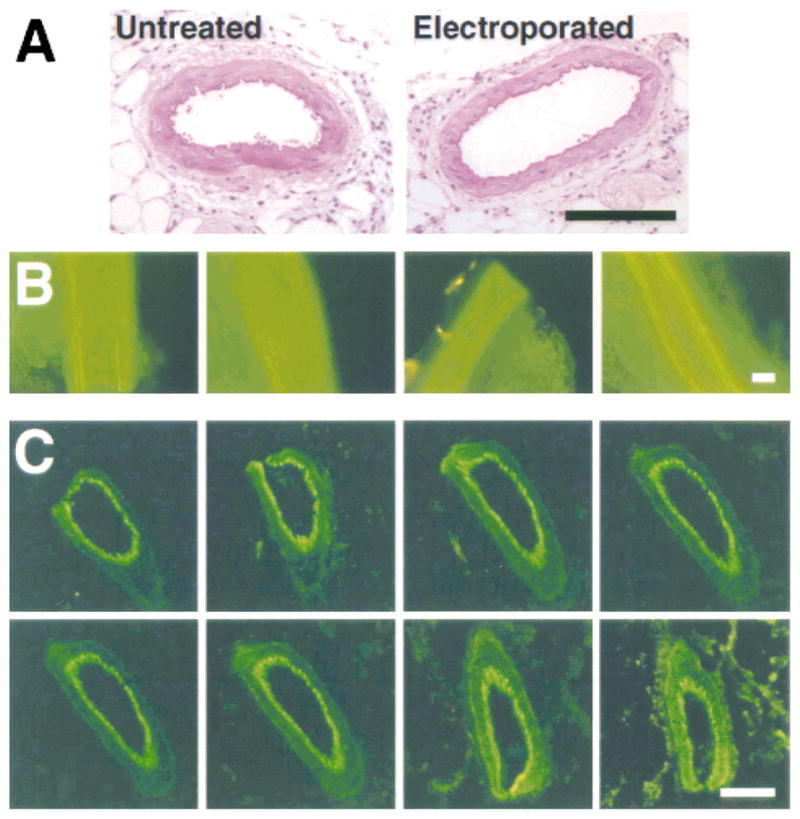
Use of electroporation for vascular gene expression. (A) Electroporation does not damage the vessel. Untreated or electroporated vessels 2 days after gene transfer were fixed, embedded in parafin, sectioned, and stained with hematoxylin and eosin. As can be seen, there are no apparent histological differences between the vessels. (B) High-level gene expression is reproducible. Plasmids expressing green fluorescent protein (GFP) were transferred by electroporation to vessels within multiple animals. Two days after gene transfer, vessels were removed and viewed by fluorescence microscopy for GFP expression. As can be seen, gene transfer is reproducible and is highly efficient. (C) Gene transfer and expression occurs in all cell layers of the vasculature. A set of serial sections from a vessel electroporated with a GFP-expressing plasmid were prepared 2 days after gene transfer. Gene expression can be detected in the adventitial, smooth muscle, and endothelial cell layers.
Although the electroporated vessels appeared healthy on visual inspection at the time of harvest, direct evidence was desired to show that they maintained unaltered vascular function after the procedure. Therefore, the responses of electroporated and control arteries to vasoconstricting stimuli were measured using intravital microscopy. Measurements were made on vessels harvested within the window of gene expression (day 2) or well after transgene expression had ceased (day 40). In both cases, control arteries constricted in a dose-dependent manner to increasing concentrations of phenylephrine. Similarly, the responses of the electroporated vessels were indistinguishable from those of the control vessels. Furthermore, addition of either 0.1 mM adenosine or 50 μM isoproterenol to electroporated or control vessels resulted in nearly identical degrees of vessel dilation. These results show that vascular function, in terms of constriction and dilation, is unaltered by electroporation or the expression of the reporter genes in the vessels, for at least 40 days after the procedure.
More recently, another group showed that electroporation works very well for gene transfer to larger vessels (44). Matsumoto and colleagues delivered plasmids into the lumen of clamped rabbit carotid arteries that were previously rinsed with saline to remove blood. Once rinsed, a solution of plasmid (0.05 to .4 mg/mL) was administered into the lumen of the vessel, and two stainless steel flat plate electrodes (2.5 cm long × 0.5 cm wide) were placed along the DNA-containing vessel, one electrode on either side. A series of 8-, 10-, or 20-millisecond square wave pulses was applied, after which the clamps were removed and the branch used for delivery was ligated, restoring blood flow. Similar to our studies, optimal gene transfer was observed at a field strength of 200 V/cm. A similar time course of expression was also noted: expression peaked at 2 days after treatment and then rapidly declined. By 7 days, the level of expression was <5% than at 2 days and is difficult to distinguish from background. By contrast to our studies, gene expression was detected only in endothelial and medial SMC layers, not in adventitial cells. It makes sense that most expression was detected in cells closer to the DNA solution, and perhaps in larger vessels, DNA can only move through a finite number of cell layers with an applied electric field. Regardless, in contrast to all other techniques developed to date, electroporation promotes gene transfer to multiple cell layers within both large (e.g., carotid) and small (e.g., mesenteric) blood vessels without the need to damage the vessel itself.
LIMITING GENE TRANSFER TO DESIRED CELLS
All of the techniques discussed so far have aimed at transferring genes to the blood vessel as a whole. However, in many instances, it would be desirable to transfer genes only to one cell type within a vessel. For example, transfer of cytostatic genes to smooth muscle cells of the intima appears to be one of the most promising approaches for gene therapy of intimal hyperplasia. Studies in many animal and human explant models have shown that expression of the small cdk inhibitors p21 and p27 may play a predominant role in regulating proliferation and that their relative levels and timing of expression are crucial to the therapy’s efficacy (67,77). These genes are not smooth muscle specific inhibitors, but rather they inhibit progression through the cell cycle of any cells that expresses the gene. If the genes are transferred to endothelial cells, they will not proliferate. Because the endothelium is vitally important for vessel integrity and function, and because endothelial cells play a major role in modulating SMC growth themselves, their unregulated expression of cytostatic genes can abrogate any beneficial effect of the therapy. Furthermore, in a damaged vessel, the endothelium must be allowed to repopulate the luminal surface. Thus, if gene transfer is to be successful for this application, methods to selectively express the gene in one cell type out of many must be developed.
Three different approaches are available for such cell-specific gene expression (Fig. 4). First, as discussed above, is the delivery of genes to certain cell types based on the method of delivery using all techniques except electroporation. If plasmids are delivered via the lumen, the vast majority of gene transfer and expression will be limited to the endothelium. Conversely, if the DNA is added to the adventitial surface, most of the expression will be in these cells. Although this approach can work, it is rather crude and is not absolutely specific.
Figure 4.

Methods for cell-specific gene delivery. (A) Delivery route. Plasmids can be administered via the lumen with or without damage (e.g., balloon angioplasty) to transfer genes to the endothelium or smooth muscle cells, respectively. Alternatively, plasmids can be delivered to the adventitial surface to transfect adventitial cells. (B) Cell-specific promoters. Smooth muscle or endothelial cell specific promoters can be used to drive gene expression in plasmids specifically in smooth muscle or endothelia cells, respectively. (C) Nuclear targeting. Plasmids containing a nuclear localizing DNA sequence from cell type X (e.g., smooth muscle or endothelial cells) can bind to transcription factors present only in cell type X to form a protein-DNA complex that can be imported into the nucleus. By contrast, in all other cell types, these transcription factors are absent; consequently, complexes cannot form, the DNA is not transported into the nucleus, and no gene expression occurs.
The second approach that has been more widely used is to use cell-specific promoters to drive gene expression only in desired cell types. In this case, DNA is delivered to all cells within the vascular wall (or as many as the delivery method allows), and gene expression is limited to those cells in which the promoter is functional. Specific smooth muscle cell promoters that may be used for this approach include those from the smooth muscle myosin heavy chain (40), smooth muscle alpha-actin (50), smooth muscle gamma actin (28), SM22α (35), and calponin (54) genes. Similarly, such endothelial cell specific promoters as those from the genes for von Willebrand Factor (vWF) (17), flk-1/KDR (52), or en-dothelin-1 (32) can be used. The desirable aspect of this approach is that many cell-specific promoters have been identified and can be cloned easily into a desired plasmid. However, not all promoters work in vivo as anticipated from in vitro studies. One of the best examples of this is that of the von Willebrand Factor promoter. Studies characterizing the vWF promoter in cell culture and in transgenic animals found vWF promoter directed gene expression to differ in the two models. In addition, they even observed differences in promoter expression in response to local environments of the cells in vivo (1).
A third way to limit gene expression to certain cell types is to limit nuclear import of the DNA to certain cell types. We and others have shown that the nuclear envelope is one of the major barriers to gene transfer (13,14,60,80). In nondividing cells, the nucleus is surrounded by a double-membraned envelope that is impermeable to large molecules lacking discreet signals for nuclear import. Our laboratory has shown that nuclear import of plasmids in nondividing cells is sequence specific (13,14). Plasmids containing one of these DNA nuclear import sequences are able to enter the nucleus, whereas those lacking such a sequence remain in the cytoplasm until cell division or until they are degraded. The common feature to these import sequences is that they contain binding sites for transcription factors, which in turn harbor protein signals (nuclear localization signals, NLS) that interact with the cell’s machinery for nuclear protein import. Thus, the DNA becomes coated with protein NLSs and is able to enter the nucleus. To date, we have identified several sequences that function as nuclear targeting sequences. One of these is from the SV40 virus and directs DNA nuclear import in all cell types because it binds transcription factors expressed in all cells. This sequence has been shown to function both in cultured cells and in animals (14,37). Another is a portion of the smooth muscle gamma actin promoter (71). Because this promoter binds a collection of transcription factors only expressed in smooth muscle cells, this DNA sequence mediates nuclear import selectivity in smooth muscle cells. In transfection studies, incorporation of this sequence into a plasmid downstream of a gene (i.e., not driving gene expression) increases smooth muscle-specific gene transfer and expression; no expression is seen in non-smooth muscle cells. We are currently testing this sequence for its ability to direct cell-specific gene transfer in vivo, and on the basis of our results with the SV40 sequence, expect this to be a powerful technique for cell-specific gene transfer to the vasculature.
APPLICATIONS OF GENE TRANSFER FOR THE STUDY OF VASCULAR PHYSIOLOGY
Although most of the excitement about gene transfer focuses on applications of gene therapy for human disease, a powerful application is often overlooked. Gene transfer to the vasculature offers the ability to study cell physiology in vivo at the genetic level. For years physiologists have relied on pharmacological agents to inhibit or stimulate pathways under study. However, using such drugs is always a gamble due to foreseen and unforeseen side effects. Furthermore, many drugs inhibit all isotypes of an enzyme or multiple enzymes or cell-signaling proteins that have a common structure or mode of action. Gene transfer offers the ability to alter pathways at the molecular level by modulating the activity of a gene product at the genetic level. Although gene transfer is most often thought of as the transfer and expression of a gene that is either lacking or mutated in an organism, thereby recreating the “normal” condition, gene transfer can also be used to abolish the expression or activity of a gene product. There are several ways to do this, including the use of antisense technology (for a recent review, see Golden et al., 2001, this issue), transgenic animals, dominant negative genes, and DNAzymes.
Transgenic animals are those that have had an exogenous gene recombined randomly into their genome and thus express a gene that they either do not normally express, or do so under different conditions. Examples could be the controlled or uncontrolled expression of a gene by drugs, embryological location, or time, or the constitutive overexpression of a gene. By contrast, animals made by targeted mutagenesis, called “knock-outs,” have had one or both copies of an endogenous gene disrupted, this time by homologous recombination using a targeting construct to the desired gene, so that the resulting animals no longer express a desired gene product. Although these are both very powerful techniques and the generated animals can be used to answer many important questions, they are not without problems. The major problems are those of time and expense. It is not uncommon for the production of transgenic mice to take 6 months to a year and to cost between $2,000 and $5,000. Knock-outs can take 2–3 times as long and cost upwards of $10,000 to $20,000 per line. Moreover, it is not uncommon to find that production of a knock-out animal is impossible due to the essential nature of a gene; if the gene is essential, the knock-out animal will be non-viable. Thus, although these animals have their place in experimentation, they are not always the most appropriate for an individual’s needs.
A more affordable alternative to the production of transgenic animals is the use of dominant negative mutant genes transferred to defined regions of tissue. This is especially attractive for studies in the vasculature, where the effects of the gene can be studied in defined regions of blood vessels. Uniform expression throughout the animal is not required, thus avoiding potential lethality issues raised when an essential gene is deleted from the genome. By essentially “knocking-out” the activity of a gene through the use of a dominant negative mutant limited to a small region of the vasculature, any negative effects of the lack of the gene will be restricted to the region where the dominant negative mutant is expressed, limiting any global effects of disruption. Dominant negative mutants of Rho kinase have been used successfully to inhibit neointima formation in injured vessels after balloon injury (16). Because Rho kinase plays an important role in various cellular functions, including smooth muscle contraction, cytokinesis, and DNA synthesis in smooth muscle cells, its localized expression was necessary to abrogate any global effects. By expressing a dominant negative Rho kinase mutant and observing protection from injury, the authors were able to show that the wild-type enzyme plays a role in the pathogenesis of neointima formation, a finding that could only be obtained from this type of approach.
Dominant negative alleles of genes work by one of several mechanisms to inhibit the activity of the wild type copy of the gene product (Fig. 5). The easiest way to think about dominant negative mutants is in the case of gene products that must dimerize for function. If the dominant negative mutant has no active site but is still capable of binding to a wild type, normal product, the hybrid will have only one half of an active site and will consequently be dead. If the level of expression of the dominant negative mutant is low, a significant number of normal–normal dimers will still exist, causing activity. However, if high levels of the dominant negative mutant are expressed, essentially all of the wild-type monomers will dimerize with the inactive mutants and all activity will be lost. Other mechanisms of dominant negative action could include the mislocalization of the active protein (e.g., a nuclear transcription factor being mislocalized to the cytoplasm where it cannot activate transcription), or the titration of a necessary cofactor or subsequent enzyme in the signaling pathway (e.g., binding of a cofactor by an enzymatically inactive dominant negative mutant so that the concentration of the cofactor is too low to be used by the wild-type protein). One caveat to the use of these mutants is that the level of inhibition of the wild-type activity is highly dependent on the efficiency of gene transfer and expression of the mutant. If very low levels of the mutant gene are transferred to the portion of the vessel under study, very little gene product will be expressed, and the effects of the inhibition will be minimal. Such an effect is often seen even in cell culture where transfection efficiencies can be high and must be appreciated (23). Thus, for dominant negative mutants to be of use, high-level gene transfer and expression must be obtained in the region of interest.
Figure 5.
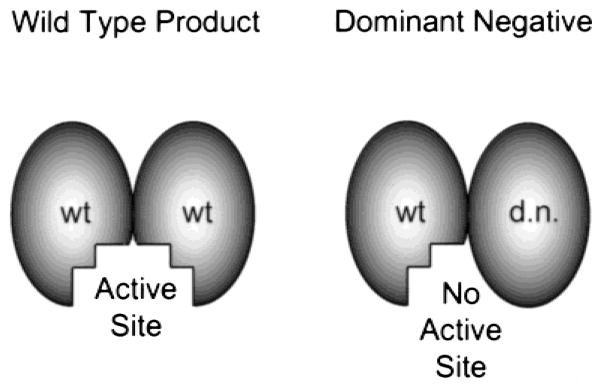
Dominant negative mutants. In the case of a protein that must dimerize for formation of its active site, if high levels of a dominant negative mutant are expressed, the mutant will compete for binding to the wild type protein, producing inactive complexes.
To show the efficacy of electroporation for expression of such gene products, Benoit and colleagues transferred a dominant-negative PKCε mutant gene to portions of the vasculature to study vasoconstriction (Shirasawa and Benoit, manuscript in preparation). Studies have suggested that PKCε may play a role in regulating agonist-induced vascular smooth muscle contraction (9,43). To examine the role of PKCε in adrenoreceptor-mediated contraction of mesenteric arteries, a dominant-negative PKCε (PKCε-KN) (21) was transferred to vessels by electroporation as described (42). Two days after transfer, vessels were assayed for phenylephrine-induced vasoconstriction. PCKε-KN significantly attenuated phenylephrine-induced responses (EC50 = 4.99 ± 1.07 μM) compared to control, non-electroporated vessels (2.81 ± 0.17 μM). Vasoconstriction to KCl did not differ between the groups. Inhibition of all PKC isoforms by the isoform-nonspecific PKC inhibitor, chelerythrine (2.5 μM) attenuated vasoconstrictor function in normal vessels from 2.80 ± 0.31 μM to 4.76 ± 0.75 μM, a decrease in response similar to that found with the dominant negative mutant. These results clearly show that delivery of plasmids using electroporation is a viable method for achieving high level, functional gene expression and that dominant negative mutants can be used to manipulate individual pathways to study vascular physiology.
Another more recent technique that may be used to modulate gene expression levels in vivo is the use of RNA-cleaving catalytic DNAs. Like antisense RNA, these “DNAzymes” can be designed to bind to target sequences on mRNA and mediate the cleavage of the mRNA, causing subsequent degradation and rendering it inactive for translation (Fig. 6). However, un-like antisense oligonucleotides that cleave mRNA in a trimolecular reaction (oligo, mRNA, and RNAse H), DNAzymes do not require RNAse H to degrade the mRNA and thus catalyze the cleavage in a bimolecular reaction (DNAzyme and mRNA) (56,57). DNAzymes require three domains for activity. At the 5-prime and 3-prime ends, 7–9 nucleotide sequences that are complementary to the mRNA target provide the specificity of mRNA binding. Between these two complementary sequences is a 15 nucleotide sequence, termed the “10–23 sequence,” which catalyzes the cleavage of mRNA phosphodiester backbone (56). Another property of DNAzymes that make them well suited for in vivo studies is that they are catalytic (56,57). This means that one molecule of DNAzyme can bind to one mRNA, cleave it, dissociate, and then bind to a second mRNA to repeat the cycle again and again, limiting the need for extremely high level delivery to tissue. Furthermore, because they cleave mRNA between an A (hybridized to the 3-prime portion of the DNAzyme) and an unhybridized U, they can be easily designed to cleave most mRNAs because of the fortuitous presence of such an AU sequence at every start codon (AUG). The only apparent requirement for their successful use is that the complementary sequences are specific for the desired target mRNA.
Figure 6.

DNAzymes. The 15 nucleotide 10–23 catalytic domain is sandwiched between two 7–9 nucleotide arms that are designed to be complementary to the desired target mRNA. Once delivered to cells, the DNAzyme hybridizes to its target mRNA and then cleaves the target, releasing two pieces of the RNA. Because the mRNA has been cleaved, it is no longer capable of directing protein translation; consequently, the levels of product decline on the basis of the half-life of the target protein.
DNAzymes have been used in vitro to degrade target mRNAs as well as in cultured cells to inhibit influenza virus replication (68) and HIV infection (83). They have also been used in the vasculature to inhibit smooth muscle hyperplasia (55). Santiago and colleagues targeted the Egr-1 transcript and showed that transfection of cells with 0.1 μM DNAzyme using dendrimers resulted in a 50% reduction in Egr-1 message levels within 24 hours and inhibited smooth muscle proliferation by 70% over 72 hours. Administration of dendrimer-DNAzyme complexes via a balloon catheter into rat carotids damaged by angioplasty also had a therapeutic effect on intimal hyperplasia. Thus, these oligonucleotides may prove to be useful tools to create transient, localized (or systemic) knock-outs in the vascular wall.
CONCLUSIONS AND FUTURE DIRECTIONS
The ability to transfer genes to the vasculature has already altered the way we think about the prevention and treatment of many diseases. It is our hope that researchers will grasp the immense power of such techniques not to treat disease, but rather to study the physiology of the healthy body. The ability to alter distinct gene products within a signaling or biosynthetic pathway or to alter structural interactions within and between cells is extremely useful and is technologically possible today. The benefits of using nonviral approaches for such delivery are evident: reduced inflammation and immune response, ease and low cost of production, and increasing levels of gene transfer and expression, nearing and perhaps surpassing (especially in the case of electroporation) that of viral vectors. Hopefully, with the availability of these tools, a new era of cardiovascular physiology will emerge.
Acknowledgments
We thank Dr. Joey Benoit for stimulating discussions and for excellent advice and guidance while initiating our studies on in vivo gene transfer.
Supported in part by National Institutes of Health grants HL59956.
References
- 1.Aird WC, Edelberg JM, Weiler-Guettler H, Simmons WW, Smith TW, Rosenberg RD. Vascular bed-specific expression of an endothelial cell gene is programmed by the tissue microenvironment. J Cell Biol. 1997;138:1117–1124. doi: 10.1083/jcb.138.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson WF. Human gene therapy. Science. 1992;256:808–813. doi: 10.1126/science.1589762. [DOI] [PubMed] [Google Scholar]
- 3.Armeanu S, Pelisek J, Krausz E, Fuchs A, Groth D, Curth R, Keil O, Quilici J, Rolland PH, Reszka R, Nikol S. Optimization of nonviral gene transfer of vascular smooth muscle cells in vitro and in vivo. Mol Ther. 2000;1:366–375. doi: 10.1006/mthe.2000.0053. [DOI] [PubMed] [Google Scholar]
- 4.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols Mol Biol. New York: John Wiley & Sons; 1994. [Google Scholar]
- 5.Barron LG, Uyechi LS, Szoka FC., Jr Cationic lipids are essential for gene delivery mediated by intravenous administration of lipoplexes. Gene Ther. 1999;6:1179–1183. doi: 10.1038/sj.gt.3300929. [DOI] [PubMed] [Google Scholar]
- 6.Barron LG, Meyer KB, Szoka FC., Jr Effects of complement depletion on the pharmacokinetics and gene delivery mediated by cationic lipid-DNA complexes. Hum Gene Ther. 1998;9:315–323. doi: 10.1089/hum.1998.9.3-315. [DOI] [PubMed] [Google Scholar]
- 7.Baumgartner I, Isner J. Somatic gene therapy in the cardiovascular system. Annu Rev Physiol. 2001;63:427–450. doi: 10.1146/annurev.physiol.63.1.427. [DOI] [PubMed] [Google Scholar]
- 8.Blair-Parks K, Weston BC, Dean DA. Gene delivery to the cornea by plasmid injection and electroporation. J Gene Med. 2002 doi: 10.1002/jgm.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buus CL, Aalkjaer C, Nilsson H, Juul B, Moller JV, Mulvany MJ. Mechanisms of Ca2+ sensitization of force production by noradrenaline in rat mesenteric small arteries. J Physiol. 1998;510:577–590. doi: 10.1111/j.1469-7793.1998.577bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell AI, Kuliszewski MA, Stewart DJ. Cell-based gene transfer to the pulmonary vasculature: endothelial nitric oxide synthase overexpression inhibits monocrotaline-induced pulmonary hypertension. Am J Respir Cell Mol Biol. 1999;21:567–575. doi: 10.1165/ajrcmb.21.5.3640. [DOI] [PubMed] [Google Scholar]
- 11.Chang MW, Barr E, Seltzer J, Jiang Y, Nabel GJ, Nabel EG, Parmacek MS, Leiden JM. Cytostatic gene therapy for vascular proliferation disorders with a constitutively active form of the retinoblastoma gene product. Science. 1995;267:518–522. doi: 10.1126/science.7824950. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, Daum G, Forough R, Clowes M, Walter U, Clowes AW. Overexpression of human endothelial nitric oxide synthase in rat vascular smooth muscle cells and in balloon-injured carotid artery. Circ Res. 1998;82:862–870. doi: 10.1161/01.res.82.8.862. [DOI] [PubMed] [Google Scholar]
- 13.Dean DA. Import of plasmid DNA into the nucleus is sequence specific. Exp Cell Res. 1997;230:293–302. doi: 10.1006/excr.1996.3427. [DOI] [PubMed] [Google Scholar]
- 14.Dean DA, Dean BS, Muller S, Smith LC. Sequence requirements for plasmid nuclear entry. Exp Cell Res. 1999;253:713–722. doi: 10.1006/excr.1999.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dzau VJ, Mann MJ, Morishita R, Kaneda Y. Fusigenic viral liposome for gene therapy in cardiovascular disease. Proc Natl Acad Sci USA. 1996;93:11421–11425. doi: 10.1073/pnas.93.21.11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eto Y, Shimokawa H, Hiroki J, Morishige K, Kandabashi T, Matsumoto Y, Amano M, Hoshijima M, Kaibuchi K, Takeshita A. Gene transfer of dominant negative Rho kinase suppresses neointimal formation after balloon injury in pigs. Am J Physiol Heart Circ Physiol. 2000;278:H1744–H1750. doi: 10.1152/ajpheart.2000.278.6.H1744. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira V, Assouline Z, Schwachtgen JL, Bahnak BR, Meyer D, Kerbiriou-Nabias D. The role of the 5′-flanking region in the cell-specific transcription of the human von Willebrand factor gene. Biochem J. 1993;293:641–648. doi: 10.1042/bj2930641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher KD, Ulbrich K, Subr V, Ward CM, Mautner V, Blakey D, Seymour LW. A versatile system for receptor-mediated gene delivery permits increased entry of DNA into target cells, enhanced delivery to the nucleus and elevated rates of transgene expression. Gene Ther. 2000;7:1337–1343. doi: 10.1038/sj.gt.3301230. [DOI] [PubMed] [Google Scholar]
- 19.Flugelman MY, Jaklitsch MT, Newman KD, Casscells W, Bratthauer GL, Dichek DA. Low level in vivo gene transfer into the arterial wall through a perforated balloon catheter. Circulation. 1992;85:1110–1117. doi: 10.1161/01.cir.85.3.1110. [DOI] [PubMed] [Google Scholar]
- 20.French BA, Mazur W, Ali NM, Geske RS, Finnigan JP, Rodgers GP, Roberts R, Raizner AE. Percutaneous transluminal in vivo gene transfer by recombinant adenovirus in normal porcine coronary arteries, atherosclerotic arteries, and two models of coronary restenosis. Circulation. 1994;90:2402–2413. doi: 10.1161/01.cir.90.5.2402. [DOI] [PubMed] [Google Scholar]
- 21.Genot EM, Parker PJ, Cantrell DA. Analysis of the role of protein kinase C-alpha, -epsilon, and -zeta in T cell activation. J Biol Chem. 1995;270:9833–9839. doi: 10.1074/jbc.270.17.9833. [DOI] [PubMed] [Google Scholar]
- 22.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 23.Iiri T, Bell SM, Baranski TJ, Fujita T, Bourne HR. A Gsα mutant designed to inhibit receptor signalling through Gs. Proc Natl Acad Sci USA. 1999;96:499–504. doi: 10.1073/pnas.96.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isner JM. Arterial gene transfer of naked DNA for therapeutic angiogenesis: early clinical results. Adv Drug Deliv Rev. 1998;30:185–197. doi: 10.1016/s0169-409x(97)00115-4. [DOI] [PubMed] [Google Scholar]
- 25.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 26.Kerr WG, Mulé JJ. Gene therapy: current status and future prospects. J Leukoc Biol. 1994;56:210–214. doi: 10.1002/jlb.56.2.210. [DOI] [PubMed] [Google Scholar]
- 27.Kircheis R, Wightman L, Schreiber A, Robitza B, Rossler V, Kursa M, Wagner E. Polyethylenimine/DNA complexes shielded by transferrin target gene expression to tumors after systemic application. Gene Ther. 2001;8:28–40. doi: 10.1038/sj.gt.3301351. [DOI] [PubMed] [Google Scholar]
- 28.Kovacs AM, Zimmer WE. Molecular cloning and expression of the chicken smooth muscle γ-actin mRNA. Cell Motil Cytoskeleton. 1993;24:67–81. doi: 10.1002/cm.970240108. [DOI] [PubMed] [Google Scholar]
- 29.Kullo IJ, Simari RD, Schwartz RS. Vascular gene transfer: from bench to bedside. Arterioscler Thromb Vasc Biol. 1999;19:196–207. doi: 10.1161/01.atv.19.2.196. [DOI] [PubMed] [Google Scholar]
- 30.Labhesetwar V, Bonadio J, Goldstein S, Chen W, Levy RJ. A DNA controlled-release coating for gene transfer: transfection in skeletal and cardiac muscle. J Pharm Sci. 1998;87:1347–1350. doi: 10.1021/js980077+. [DOI] [PubMed] [Google Scholar]
- 31.Laitinen M, Hartikainen J, Hiltunen MO, Eranen J, Kiviniemi M, Narvanen O, Makinen K, Manninen H, Syvanne M, Martin JF, Laakso M, Yla-Herttuala S. Catheter-mediated vascular endothelial growth factor gene transfer to human coronary arteries after angioplasty. Hum Gene Ther. 2000;11:263–270. doi: 10.1089/10430340050016003. [DOI] [PubMed] [Google Scholar]
- 32.Lee ME, Block KD, Clifford JA, Quertermous T. Functional analysis of the endothelin-1 gene promoter: evidence for an endothelial cell-specific cis-acting sequence. J Biol Chem. 1990;265:10446–10450. [PubMed] [Google Scholar]
- 33.Lee SW, Trapnell BC, Rade JJ, Virmani R, Dichek DA. In vivo adenoviral vector mediated gene transfer into balloon-injured rat carotid arteries. Circ Res. 1993;73:797–807. doi: 10.1161/01.res.73.5.797. [DOI] [PubMed] [Google Scholar]
- 34.Lewis PF, Emerman M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J Virol. 1994;68:510–516. doi: 10.1128/jvi.68.1.510-516.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Liu Z, Mercer B, Overbeek P, Olson EN. Evidence for serum response factor-mediated regulatory networks governing SM22alpha transcription in smooth, skeletal, and cardiac muscle cells. Dev Biol. 1997;187:311–321. doi: 10.1006/dbio.1997.8621. [DOI] [PubMed] [Google Scholar]
- 36.Li S, Huang L. In vivo gene transfer via intravenous administration of cationic lipid-protamine-DNA (LPD) complexes. Gene Ther. 1997;4:891–900. doi: 10.1038/sj.gt.3300482. [DOI] [PubMed] [Google Scholar]
- 37.Li S, MacLaughlin FC, Fewell JG, Gondo M, Wang J, Nicol F, Dean DA, Smith LC. Muscle-specific enhancement of gene expression by incorporation of the SV40 enhancer in the expression plasmid. Gene Ther. 2001;8:494–497. doi: 10.1038/sj.gt.3301419. [DOI] [PubMed] [Google Scholar]
- 38.Liu D, Knapp JE, Song YK. Mechanisms of cationic liposome-mediated transfection of the lung endothelium. In: Huang L, Hung M-C, Wagner E, editors. Nonviral Vectors for Gene Therapy. San Diego: Academic Press; 1999. pp. 314–337. [Google Scholar]
- 39.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 40.Manabe I, Owens GK. CArG elements control smooth muscle subtype-specific expression of smooth muscle myosin in vivo. J Clin Invest. 2001;107:823–834. doi: 10.1172/JCI11385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mann MJ, Whittemore AD, Donaldson MC, Belkin M, Conte MS, Polak JF, Orav EJ, Ehsan A, Dell’Acqua G, Dzau VJ. Ex vivo gene therapy of human vascular bypass grafts with E2F decoy: the PREVENT single-centre, randomised, controlled trial. Lancet. 1999;354:1493–1498. doi: 10.1016/S0140-6736(99)09405-2. [DOI] [PubMed] [Google Scholar]
- 42.Martin JB, Young JL, Benoit JN, Dean DA. Gene transfer to intact mesenteric arteries by electroporation. J Vasc Res. 2000;37:372–380. doi: 10.1159/000025753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masuo M, Reardon S, Ikebe M, Kitazawa T. A novel mechanism for the Ca(2+)-sensitizing effect of protein kinase C on vascular smooth muscle: inhibition of myosin light chain phosphatase. J Gen Physiol. 1994;104:265–286. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsumoto T, Komori K, Shoji T, Kuma S, Kume M, Yamaoka T, Mori E, Furuyama T, Yonemitsu Y, Sugimachi K. Successful and optimized in vivo gene transfer to rabbit carotid artery mediated by electronic pulse. Gene Ther. 2001;8:1174–1179. doi: 10.1038/sj.gt.3301502. [DOI] [PubMed] [Google Scholar]
- 45.Miller DG, Adam MA, Miller AD. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mir LM, Bureau MF, Gehl J, Rangara R, Rouy D, Caillaud J-M, Delaere P, Branellec D, Schwartz B, Scherman D. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc Natl Acad Sci USA. 1999;96:4262–4267. doi: 10.1073/pnas.96.8.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mulligan RC. The basic science of gene therapy. Science. 1993;260:926–932. doi: 10.1126/science.8493530. [DOI] [PubMed] [Google Scholar]
- 48.Ogris M, Brunner S, Schuller S, Kircheis R, Wagner E. PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery [In Process Citation] Gene Ther. 1999;6:595–605. doi: 10.1038/sj.gt.3300900. [DOI] [PubMed] [Google Scholar]
- 49.Ohno T, Gordon D, San H, Pompili VJ, Imperiale MJ, Nabel GJ, Nabel EG. Gene therapy for vascular smooth muscle cell proliferation after arterial injury. Science. 1994;265:781–784. doi: 10.1126/science.8047883. [DOI] [PubMed] [Google Scholar]
- 50.Owens GK, Loeb A, Gordon D, Thompson MM. Expression of smooth muscle-specific α-iso-actin in cultured vascular smooth muscle cells: relationship between growth and cytodifferentiation. J Cell Biol. 1986;102:343–352. doi: 10.1083/jcb.102.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parker SE, Vahlsing HL, Serfilippi LM, Franklin CL, Doh SG, Gromkowski SH, Lew D, Manthorpe M, Norman J. Cancer gene therapy using plasmid DNA: safety evaluation in rodents and non-human primates [see comments] Hum Gene Ther. 1995;6:575–590. doi: 10.1089/hum.1995.6.5-575. [DOI] [PubMed] [Google Scholar]
- 52.Patterson C, Perrella MA, Hsieh CM, Yoshizumi M, Lee ME, Haber E. Cloning and functional analysis of the promoter for KDR/flk-1, a receptor for vascular endothelial growth factor. J Biol Chem. 1995;270:23111–23118. doi: 10.1074/jbc.270.39.23111. [DOI] [PubMed] [Google Scholar]
- 53.Safinya C, Koltover I. Self-assembled structures of lipid DNA nonviral gene delivery systems from synchontron X-ray diffraction. In: Huang L, Hung M-C, Wagner E, editors. Nonviral Vectors for Gene Therapy. San Diego: Academic Press; 1999. pp. 92–119. [Google Scholar]
- 54.Samaha F, Ip JHS, Morrisey E, Seltzer J, Tang Z, Solway J, Parmacek M. Developmental pattern of expression and genomic organization of the calponin-hi gene. J Biol Chem. 1996;271:395–403. doi: 10.1074/jbc.271.1.395. [DOI] [PubMed] [Google Scholar]
- 55.Santiago FS, Lowe HC, Kavurma MM, Chesterman CN, Baker A, Atkins DG, Khachigian LM. New DNA enzyme targeting Egr-1 mRNA inhibits vascular smooth muscle proliferation and regrowth after injury. Nat Med. 1999;5:1438. doi: 10.1038/71020. [DOI] [PubMed] [Google Scholar]
- 56.Santoro SW, Joyce GF. A general purpose RNA-cleaving DNA enzyme. Proc Natl Acad Sci USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Santoro SW, Joyce GF. Mechanism and utility of an RNA-cleaving DNA enzyme. Biochemistry. 1998;37:13330–13342. doi: 10.1021/bi9812221. [DOI] [PubMed] [Google Scholar]
- 58.Schachtner SK, Rome JJ, Hoyt RF, Newman KD, Virmani R, Dichek DA. In vivo Adenovirus-mediated gene transfer via the pulmonary artery of rats. Circ Res. 1995;76:701–709. doi: 10.1161/01.res.76.5.701. [DOI] [PubMed] [Google Scholar]
- 59.Schulick AH, Newman KD, Virmani R, Dichek DA. In vivo gene transfer into injured carotid arteries: optimization and evaluation of acute toxicity. Circulation. 1995;91:2407–2414. doi: 10.1161/01.cir.91.9.2407. [DOI] [PubMed] [Google Scholar]
- 60.Sebestyen MG, Wolff JA. Nuclear transport of exogenous DNA. In: Huang L, Hung M-C, Wagner E, editors. Nonviral Vectors for Gene Therapy. San Diego: Academic Press; 1999. pp. 140–169. [Google Scholar]
- 61.Simari RD, San H, Rekhter M, Ohno T, Gordon D, Nabel GJ, Nabel EG. Regulation of cellular proliferation and intimal formation following balloon injury in atherosclerotic rabbit arteries. J Clin Invest. 1996;98:225–235. doi: 10.1172/JCI118770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Somiari S, Glasspool-Malone J, Drabick JJ, Gilbert RA, Heller R, Jaroszeski MJ, Malone RW. Theory and in vivo application of electroporative gene delivery. Mol Ther. 2000;2:178–187. doi: 10.1006/mthe.2000.0124. [DOI] [PubMed] [Google Scholar]
- 63.Stephan D, San H, Yang ZY, Gordon D, Goelz S, Nabel GJ, Nabel EG. Inhibition of vascular smooth muscle cell proliferation and intimal hyperplasia by gene transfer of beta-interferon. Mol Med. 1997;3:593–599. [PMC free article] [PubMed] [Google Scholar]
- 64.Takeshita S, Gal D, Leclerc G, Pickering JG, Riessen R, Weir L, Isner JM. Increased gene expression after liposome-mediated arterial gene transfer associated with intimal smooth muscle cell proliferation. In vitro and in vivo findings in a rabbit model of vascular injury. J Clin Invest. 1994;93:652–661. doi: 10.1172/JCI117017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan Y, Liu F, Li Z, Li S, Huang L. Sequential injection of cationic liposome and plasmid DNA effectively transfects the lung with minimal inflammatory toxicity. Mol Ther. 2001;3:673–682. doi: 10.1006/mthe.2001.0311. [DOI] [PubMed] [Google Scholar]
- 66.Tang MX, Szoka FC. The influence of polymer structure on the interactions of cationic polymers with DNA and morphology of the resulting complexes. Gene Ther. 1997;4:823–832. doi: 10.1038/sj.gt.3300454. [DOI] [PubMed] [Google Scholar]
- 67.Tanner FC, Yang ZY, Duckers E, Gordon D, Nabel GJ, Nabel EG. Expression of cyclin-dependent kinase inhibitors in vascular disease. Circ Res. 1998;82:396–403. doi: 10.1161/01.res.82.3.396. [DOI] [PubMed] [Google Scholar]
- 68.Toyoda T, Imamura Y, Takaku H, Kashiwagi T, Hara K, Iwahashi J, Ohtsu Y, Tsumura N, Kato H, Hamada N. Inhibition of influenza virus replication in cultured cells by RNA-cleaving DNA enzyme. FEBS Lett. 2000;481:113–116. doi: 10.1016/s0014-5793(00)01974-8. [DOI] [PubMed] [Google Scholar]
- 69.Turunen MP, Hiltunen MO, Ruponen M, Virkamäki L, Szoka JFC, Urtti A, Ylä-Herttuala S. Efficient adventitial gene delivery to rabbit carotid artery with cationic polymer–plasmid complexes. Gene Ther. 1999;6:6–11. doi: 10.1038/sj.gt.3300800. [DOI] [PubMed] [Google Scholar]
- 70.Tyler RC, Fagan KA, Unfer RC, Gorman C, McClarrion M, Bullock C, Rodman DM. Vascular inflammation inhibits gene transfer to the pulmonary circulation in vivo. Am J Physiol. 1999;277:L1199–L1204. doi: 10.1152/ajplung.1999.277.6.L1199. [DOI] [PubMed] [Google Scholar]
- 71.Vacik J, Dean BS, Zimmer WE, Dean DA. Cell-specific nuclear import of plasmid DNA. Gene Ther. 1999;6:1006–1014. doi: 10.1038/sj.gt.3300924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.von der Leyen HE, Braun-Dullaeus R, Mann MJ, Zhang L, Niebauer J, Dzau VJ. A pressure-mediated nonviral method for efficient arterial gene and oligonucleotide transfer. Hum Gene Ther. 1999;10:2355–2364. doi: 10.1089/10430349950017004. [DOI] [PubMed] [Google Scholar]
- 73.von der Leyen HE, Gibbons GH, Morishita R, Lewis NP, Zhang L, Nakajima M, Kaneda Y, Cooke JP, Dzau VJ. Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci USA. 1995;92:1137–1141. doi: 10.1073/pnas.92.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Walther W, Stein U. Viral vectors for gene transfer: a review of their use in the treatment of human diseases. Drugs. 2000;60:249–271. doi: 10.2165/00003495-200060020-00002. [DOI] [PubMed] [Google Scholar]
- 75.Wang Y, Boros P, Liu J, Qin L, Bai Y, Bielinska AU, Kukowska-Latallo JF, Baker JR, Jr, Bromberg JS. DNA/dendrimer complexes mediate gene transfer into murine cardiac transplants ex vivo. Mol Ther. 2000;2:602–608. doi: 10.1006/mthe.2000.0201. [DOI] [PubMed] [Google Scholar]
- 76.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 77.Yang ZY, Simari RD, Perkins ND, San H, Gordon D, Nabel GJ, Nabel EG. Role of the p21 cyclin-dependent kinase inhibitor in limiting cell proliferation in response to arterial injury. Proc Natl Acad Sci USA. 1996;93:7905–7910. doi: 10.1073/pnas.93.15.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yla-Herttuala S, Martin JF. Cardiovascular gene therapy. Lancet. 2000;355:213–222. doi: 10.1016/S0140-6736(99)04180-X. [DOI] [PubMed] [Google Scholar]
- 79.Yonemitsu Y, Kaneda Y, Tanaka S, Nakashima Y, Komori K, Sugimachi K, Sueishi K. Transfer of wild-type p53 gene effectively inhibits vascular smooth muscle cell proliferation in vitro and in vivo. Circ Res. 1998;82:147–156. doi: 10.1161/01.res.82.2.147. [DOI] [PubMed] [Google Scholar]
- 80.Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem. 1995;270:18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]
- 81.Zhang G, Budker V, Williams P, Subbotin V, Wolff JA. Efficient expression of naked DNA delivered intraarterially to limb muscles of nonhuman primates. Hum Gene Ther. 2001;12:427–438. doi: 10.1089/10430340150504046. [DOI] [PubMed] [Google Scholar]
- 82.Zhang G, Song YK, Liu D. Long-term expression of human alpha1-antitrypsin gene in mouse liver achieved by intravenous administration of plasmid DNA using a hydrodynamics-based procedure. Gene Ther. 2000;7:1344–1349. doi: 10.1038/sj.gt.3301229. [DOI] [PubMed] [Google Scholar]
- 83.Zhang X, Xu Y, Ling H, Hattori T. Inhibition of infection of incoming HIV-1 virus by RNA-cleaving DNA enzyme. FEBS Lett. 1999;458:151–156. doi: 10.1016/s0014-5793(99)01149-7. [DOI] [PubMed] [Google Scholar]