

Abstract
Irreversible photooxidation based on N–O bond fragmentation is demonstrated for N-methoxyheterocycles in both the singlet and triplet excited state manifolds. The energetic requirements for bond fragmentation are studied in detail. Bond fragmentation in the excited singlet manifold is possible for ππ* singlet states with energies significantly larger than the N–O bond dissociation energy of ca 55 kcal mol−1. For the nπ* triplet states, N–O bond fragmentation does not occur in the excited state for orbital overlap and energetic reasons. Irreversible photooxidation occurs in the singlet states by bond fragmentation followed by electron transfer. Irreversible photooxidation occurs in the triplet states via bimolecular electron transfer to the donor followed by bond fragmentation. Using these two sensitization schemes, donors can be irreversibly oxidized with oxidation potentials ranging from ca 1.6–2.2 V vs SCE. The corresponding N-ethylheterocycles are characterized as conventional reversible photooxidants in their triplet states. The utility of these sensitizers is demonstrated by irreversibly generating the guanosine radical cation in buffered aqueous solution.
INTRODUCTION
Photoinduced one-electron oxidation of a donor D is usually accomplished by electron transfer to an acceptor A, as indicated in Fig. 1 for the case of an excited acceptor (1–3). In all photoinduced electron transfer processes, however, there are two electron transfer reactions, the initial charge separation step, ket, to form the geminate radical-ion pair A•− D•+, and return electron transfer with this pair, k−et, Fig. 1 (3). Return electron transfer wastes the photon energy and also removes D•+ (4). Return electron transfer in the geminate radical-ion pair is often faster than many chemical reactions of D•+ (5–8). Even in polar solvents where separation of the radical-ion pair may occur, ksep, return electron transfer still decreases the yield of separated D•+ (9–13). In more rigid environments, where separation within the geminate radical-ion pair is slower or is impossible, return electron transfer may dominate (14–17). Irreversible bond fragmentation can be a useful strategy to mitigate the effects of return electron transfer (18–22). Two such approaches are illustrated in Fig. 2 using a positively charged electron acceptor X–Y+.
Figure 1.

Conventional scheme for photoinduced oxidation of a donor, D, to form the radical cation, D•+, illustrating the energy wasting return electron transfer process, k−et.
Figure 2.

Two schemes for irreversible photooxidation of a donor, D. a) Fragmentation in the excited state of X–Y+ gives radical cation Y•+ that irreversibly oxidizes D to give D•+. b) Electron transfer from excited D to X–Y+ results in fragmentation of the acceptor, which makes the electron transfer irreversible.
In Fig. 2a, excitation of X–Y+ results in rapid fragmentation of the excited state, kfr*, to form a radical X• and a radical cation Y•+. If the oxidation potential of Y is larger than that of D, exothermic electron transfer occurs from D to Y•+, ket•+. In Fig. 2b, excitation of D results in photoinduced electron transfer to X–Y+ to form D•+ and the radical, X–Y•, which undergoes rapid bond fragmentation, kfr•, to form a radical X• and a stable molecule Y. The overall products of both of these reaction sequences are the same, the radical cation D•+, a radical X• and a stable organic molecule Y. Importantly, there is no radical anion, and thus conventional return electron transfer cannot occur. Although some organic radicals have low-oxidation potentials (23,24), most will not undergo electron transfer to D•+. Using either of the mechanisms in Fig. 2, oxidation of D may be accomplished essentially irreversibly.
Here, we discuss the photochemistry of a series of X–Y+ molecules, N-methoxyheterocycles, 1–8, Fig. 3, which can serve as irreversible photooxidants according to the mechanisms shown in Fig. 2. The corresponding N-ethylheterocycles 1E–8E are also studied as nonfragmenting analogues. N-alkoxyheterocycles are nitrogen onium salts. The photochemistry of onium salts, in particular iodonium, sulfonium and phosphonium, has been extensively investigated, as several of these find applications in photoimaging applications and as photoacids (25–30). Ammonium salts have been much less investigated from a photochemical perspective (31–35).
Figure 3.

N-methoxyheterocycles studied as irreversible photooxidants, used as the tetrafluoroborate salts unless otherwise specified. Also studied are the corresponding N-ethylheterocycles, 1E–8E, as tetrafluoroborate or hexafluorophosphate salts.
The useful processes in onium salt photochemistry involve fragmentation of one of the sigma bonds to the hypervalent atom, which can be achieved by photochemical excitation, or by photoinduced one-electron reduction. The one-electron reduction route represents the central step in Fig. 2b. Fragmentation of N-alkoxyheterocycles according to Fig. 2b is the basic chemical process behind an important light-initiated radical polymerization technology (36,37), and has been thoroughly described both from an experimental and theoretical perspective (38–41). Here, we focus on the corresponding photochemical fragmentation scheme of Fig. 2a.
Photochemical activation of N-alkoxyheterocycles has been described previously. In 1970, Mee et al. showed that irradiation of N-methoxyphenanthridinium (3) resulted in products consistent with N–O bond cleavage (42). The substitution pattern resulting from trapping with substituted benzenes was taken as evidence for formation of a methoxy radical. In 1992, Schnabel showed that irradiation of N-methoxyisoquinolinium (2) resulted in the formation of protons (43). The radical cation intermediate was subsequently identified in a transient absorption experiment (44). Huntley et al. also showed that excitation of 2 could be used to oxidize the thymidine dimer via the mechanism shown in Fig. 2a (45). In studies of benzothiazoline radical cations, Shukla et al. used excited 3 to generate the phenanthridine radical cation, which was identified spectroscopically and used as a one-electron oxidant according to Fig. 2a (46). The same approach, using 3, has subsequently been used in studies of triphenylmethyl sulfide and other radical cations (47,48).
This previous work clearly demonstrates the utility of the N-methoxyheterocycles as irreversible oxidants. Here, we describe more detailed photochemical studies that are designed to examine the efficiency of sensitization and expand the scope and utility of these structures. The nature of the excited states responsible for fragmentation are examined, measurements have been made of the kinetics and efficiencies of generation of the oxidizing species, and different approaches depending upon the spin state of the excited state are discussed. Finally, an application of these structures as irreversible photooxidants of a DNA purine base is described.
MATERIALS AND METHODS
General materials
Phenanthridine, 1-isoquinolinyl phenyl ketone, 6-methoxyquinoline, trans-stilbene, diphenyl methanol, deoxyguanosine mononucleotide were obtained from Sigma Aldrich. 8-methoxyquinoline was synthesized according to a literature procedure (49). 2-(4′-methoxystyryl) pyridine was a gift from the Eastman Kodak Company. Compounds 1/1E, 2/2E, 3/3E, 4/4E and 6/6E were available from previous studies (41). The N-methoxy structures 1–8 were all prepared as tetrafluoroborate salts, the corresponding N-ethyl structures (1E–8E) were prepared as either tetrafluoroborate or hexafluorophosphate salts.
General synthetic procedures
The other N-ethyl- and N-methoxyheterocycles (5/5E, 7/7E, 8/8E) were prepared by alkylation of the corresponding heterocycles and their N-oxides. The general procedures for the alkylations and preparation of the N-oxides have been described previously (41). The synthesized products were characterized by proton and carbon NMR spectroscopy using the Varian Gemini 300, Varian Inova 400 and Varian Inova 500 spectrometers. The samples were dissolved in CDCl3, DMSO-d6, CD3CN, or CD3OD (Cambridge Isotopes). Mass verification was accomplished using a Vestec MALDITOF mass spectrometer with 337 nm excitation; the compounds were sometimes excited without a matrix (LD-TOF).
N-methoxy-2-(4′-methoxystyryl) pyridinium tetrafluoroborate (5)
Following the literature procedure (41), 2-(4′-methoxystyryl) pyridine N-oxide (1.40 g, 6.16 mmol, synthesized according to (41)) and trimethyloxonium tetrafluoroborate (1.05 g, 7.08 mmol) were stirred in 50 mL dichloromethane. The precipitate was filtered off and yielded 1.49 g (4.53 mmol, 73%) of 5. 1H-NMR (300 MHz, CD3CN) δ (ppm) = 3.74 (s, 3 H), 4.19 (s, 3 H), 6.88–6.93 (m, 3 H), 7.23 (d, 1H, J = 16.5 Hz), 7.59–7.66 (m, 3 H), 7.81 (d, 1 H, J = 16.5 Hz), 8.17–8.25 (m, 1 H), 8.62 (d, 1 H, J = 7.2 Hz). 13C-NMR (75 MHz, CD3CN) δ (ppm) = 56.2, 70.0, 110.8, 115.6, 126.1, 126.5, 131.3, 131.7, 141.0, 144.3, 146.2, 151.6, 163.5. MS m/z 242.4 (M+).
N-ethyl-2-(4′-methoxystyryl)pyridinium hexafluorophosphate (5E)
Following the general procedure (41), 2-(4′-methoxystyryl)pyridine (1.80 g, 8.52 mmol) and triethyloxonium hexafluorophosphate (2.56 g, 10.3 mmol) were stirred in 40 mL of dichloromethane. Recrystallization from methanol/dichloromethane afforded 2.49 g (6.46 mmol, 75%) of 5E. 1H-NMR (300 MHz, CD3CN) δ (ppm) = 1.53 (t, 3 H, J = 7.2 Hz), 3.85 (s, 3 H), 4.63 (q, 2 H, J = 7.2 Hz), 7.00–7.04 (m, 2 H), 7.22 (d, 1 H, J = 15.9 Hz), 7.72 (d, 1 H, J = 15.9 Hz), 7.73–7.76 (m, 3 H), 8.22–8.34 (m, 2 H), 8.50 (d, 1H, J = 6.0 Hz).
N-methoxy-1-isoquinolinyl phenyl ketone tetrafluoroborate (7)
Following the general procedures (41), trimethyloxonium tetrafluoroborate (0.27 g, 1.83 mmol) and 1-isoquinolinyl phenyl ketone N-oxide (0.40 g, 1.58 mmol, synthesized according to the general procedures (41)) were stirred in minimum amount of dichloromethane to dissolve the reactants, and allowed to react for 4 h. Recrystallization from a methanol/ethyl acetate mixture afforded .49 g (1.39 mmol, 88%) of 7. 1H NMR (499.92 MHz, CD3CN) δ (ppm) = 4.50 (s, 3H), 7.59 (t, 2H, J = 7.9 Hz), 7.81 (t, 1H, J = 8.0 Hz), 8.00 (d, 2H, J = 7.9 Hz), 8.04–8.11 (m, 2H), 8.31 (t, 1H, J = 8.0 Hz), 8.53 (d, 1H, J = 8.0 Hz), 8.91 (d, 1H, J = 7.5 Hz), 9.29 (d, 1H, J = 7.0 Hz). 13C NMR (125.718 MHz, CD3CN) δ (ppm) = 42.63, 98.31, 100.11, 101.18, 102.07, 102.25, 102.85, 104.35, 105.49, 106.66, 109.36, 110.15, 110.80, 185.6. MS m/z 264.04 (M+).
N-ethyl-1-isoquinolinyl phenyl ketone tetrafluoroborate (7E)
Following the general procedure (41), triethyloxonium tetrafluoroborate (3.0 g, 15.8 mmol) and 1-isoquinolinyl phenyl ketone (3.2 g, 13.7 mmol) were stirred in minimum amount of dichloromethane to dissolve the reactants, and allowed to react for 4 h. Recrystallization from a methanol/ethyl acetate afforded 3.6 g (10.4 mmol, 76%) of 7E. 1H NMR (499.92 MHz, CD3CN) δ (ppm) = 1.60 (t, 3H, J = 7.0 Hz), 4.69 (m, 1H, J = 6.9 Hz), 4.85 (m, 1H, J = 6.9 Hz), 7.60 (t, 2H, J = 7.8 Hz), 7.83 (tt, 1H, J = 1.1, 7.5 Hz), 7.96–8.02 (m, 4H), 8.27 (m, 1H), 8.45 (d, 1H, J = 8.0 Hz), 8.79 (d, 1H, J = 7.0 Hz), 8.99 (d, 1H, J = 6.5 Hz),. 13C NMR (125.718 MHz, CD3CN) δ (ppm) = −11.7, 28.0, 98.3, 100.3, 100.4, 100.8, 102.4, 103.1, 104.8, 106.8, 108.4, 109.6, 109.9, 111.0, 185.6. MS m/z 262.10 (M+).
N-methoxy-6-benzoylphenanthridine tetrafluoroborate (8)
8 was prepared from 6-benzyl-5,6-dihyrophenanthridine (8a), which was prepared according to a literature procedure (50). 8a (0.813 g, 3.0 mmol) was dissolved in warm glacial acetic acid and excess Na2Cr2O7 was added over 30 min. The solution was refluxed for 90 min, poured into water and cooled over night (51). Recrystalization from ethanol afforded 0.73 g (2.58 mmol, 86%) of 6-benzoylphenanthridine (8b). Compound 8b, 0.365 g (1.29 mmol) was oxidized to the N-oxide using perbenzoic acid (2.972 mmol) (52). The resulting emulsion was extracted with CHCl3 to afford 0.33 g (1.11 mmol, 86%) 6-benzoylphenanthridine N-oxide (8c). Alkylation was carried out according to the general procedures to yield 0.4 g (.99 mmol, 89%) of 8. 1H NMR (499.92 MHz, CD3CN) δ (ppm) = 4.46 (s, 3H), 7.68 (t, 2H, J = 7.82 Hz), 7.92 (t, 1H, J = 7.57 Hz), 8.03–8.08 (m, 3H), 8.19 (d, 1H, J = 8.30 Hz), 8.29–8.33 (m, 2H), 8.49 (t, 1H, J = 7.82 Hz), 8.57 (d, 1H, J = 8.66 Hz), 9.17 (t, 2H, J = 9.5 Hz). 13C NMR (125.718 MHz, CD3CN) δ (ppm) = 71.96, 118.75, 122.83, 125.13, 126.02, 129.14, 130.84, 131.15, 131.51, 132.60, 132.62, 133.27, 134.55, 134.75, 136.78, 138.33, 140.19, 154.77, 185.53. MS m/z 314.1 (M+)
N-ethyl-6-benzoylphenanthridine tetrafluoroborate (8E)
Preparation of 8b was performed according to the procedure outlined above. Alkylation of 6-benzoylphenanthridine, 8b, (0.365 g, 1.29 mmol) was performed according to the general procedures, affording 0.36 g (0.89 mmol, 69%) of 8E. 1H NMR (399.86 MHz, CD3CN) δ (ppm) = 7.64 (t, 2H, J = 7.3 Hz), 7.87 (t, 1H, J = 7.5 Hz), 7.87 (t, 1H, J = 7.5 Hz), 7.98 (d, 3H, J = 8.4 Hz), 8.14 (m, 2H), 8.27 (m, 2H), 8.39 (t, 1H, J = 7.8 Hz), 9.03 (m, 1H), 9.09 (d, 1H, J = 8.5 Hz). 13C NMR (125.718 MHz, CD3CN) δ (ppm) = 123.02, 123.60, 124.80, 124.90, 126.86, 130.53, 131.21, 131.58, 131.80, 132.36, 133.31, 135.56, 137.49, 137.59, 139.09. MS m/z 314.08 (M+)
Measurement methods and instrumentation
Nanosecond and picosecond transient absorption spectroscopy was performed as described previously (41). Emission spectroscopy was performed using a Spex Fluorolog 1-1-2 spectrometer. Spectra at room temperature were measured in 1 cm pathlength cuvettes with arms that could be sealed with stopcocks, if necessary, after deoxygenation by purging with argon. Experiments at liquid nitrogen temperature were performed by immersion of the samples, contained in NMR tubes that had been sealed after purging with argon, into a specially designed Dewar that fitted the standard sample compartment of the fluorimeter. Samples for study at liquid nitrogen temperature were prepared in either an EPA solution (a 2:5:5 mixture of ethanol:isopentane:diethyl ether), or a 50:50 mixture of ethanol and methanol with 1.0 M methyl iodide, as indicated in the figure captions.
The reduction potentials for 7E and 8E were estimated using cyclic voltammetry (CV) in acetonitrile at room temperature, in the presence of 0.1 M LiClO4 as electrolyte and with 1 mM of the analyte, using a CHI 900C potentiostat. Solutions were purged with N2 for 10 min. Ag/AgCl was used as a reference electrode and Pt wire was used as a counter electrode. The working electrode was a glassy carbon disk of surface area 0.07065 cm2. CV experiments were done at a scan rate of 5 V/s. Highly reversible reduction was observed for 7E, but 8E was not reversible. The reduction potential for 8E was thus estimated by adding 0.17 V to the literature potential for 3E, as this is the observed potential difference between 1E and 7E. As described in the text, the reduction potentials for all of the N-methoxy structures were then estimated from those for the corresponding N-ethyl structures by adding 0.14 V (37).
Product Analysis
25 mg of N-methoxyquinolinium tetrafluoroborate (1) was dissolved in 25 mL of acetonitrile and purged with nitrogen. Irradiation was performed in a Luzchem photoreactor with UV-B lamps until no starting material could be detected (by TLC). Aqueous sodium bicarbonate solution was added to the irradiated solution, which was then extracted with methylene chloride. The organic layer was dried over magnesium sulfate and filtered.
GC-MS analysis was performed using a Shimadzu GCMS QP5000 with a Restek RTX-XLB (Cat #12823) column. A typical temperature program was 11°C/min from 125 to 250°C, with an initial hold time of 8 min and a final hold time of 30 min. The retention times were 6.5 min for quinoline and between 13.1 and 13.6 min for methoxylated quinolines. The mass spectra were matched with the NIST/EPA/NIH Mass Spectral Library 2002.
Proton Yield Measurements
N-methoxyheterocycle tetrafluoroborate (~25 mg, 0.1 mmol) was irradiated in 25 mL of nitrogen-purged solvent as described above until complete consumption of the starting material was observed by TLC. The pH of the resulting solution was measured using three methods. Method A: Appropriately diluted aliquots of the solution were mixed with a phosphate buffer solution (3 mM, pH ~ 7.6) and the pH was then measured. The change in pH relative to a reference sample yielded the protons produced in the irradiation sample. The same procedure was performed with a TRIS buffer (3 mM, pH ~ 8.3). Method B: Spectrophotometric analysis of methanolic p-nitrophenolate solutions (λmax = 400 nm) (53) with and without exposure to aliquots of the irradiation solution was used to calculate the concentration of the photogenerated protons. Method C: The N-methoxyheterocycle was dissolved in a buffer solution (phosphate buffer ~3 mM, pH ~ 7.6, or TRIS buffer ~3 mM, pH ~ 8.3), and the pH was constantly monitored during the irradiation using a pH meter. The change in pH allowed for calculation of the protons produced during the irradiation.
Electronic Structure Calculations
All calculations were performed using Gaussian 09, Revision. A.02 (54) using B3LYP with the aug-cc-pVDZ basis set. All species were fully optimized in the gas phase and optimizations were verified to be true minima.
RESULTS AND DISCUSSION
Singlet state sensitizers
Nanosecond transient absorption spectroscopy
Nanosecond transient absorption spectroscopy of compounds 1–5 was performed in acetonitrile at room temperature. A typical result is illustrated in Fig. 4. The main absorptions in the visible regions are due to the relevant radical cations formed by fragmentation of the N–O bonds. These assignments are based on previous work on some of the same systems (44,46), the similarity of the absorptions to those previously reported (55), and on the insensitivity of their lifetimes to dissolved oxygen. The transients also reacted with low-oxidation potential donors such as amines. Shown as an inset in Fig. 4 is a plot of the observed pseudo-first-order rate constant for decay of the quinoline radical cation from 1, as a function of concentration of added N, N-dimethylaniline. The slope of the plot yields a bimolecular rate constant of 2.1 × 1010 M−1 s−1, consistent with diffusion-controlled electron transfer (1). Similar results were obtained for the other radical cations using stilbene as the donor, Table 1. The radical cations were formed within the nanosecond laser pulse, which is consistent with rapid cleavage from presumably the first excited singlet state. Other absorptions were also observed after decay of the radical cations (for example, see Fig. 4). These absorptions decayed on the millisecond timescale and are assigned to radical/radical cation coupling products as discussed further below.
Figure 4.

Transient absorption spectra for nanosecond pulsed laser photolyis of N-methoxyquinolinium tetrafluoroborate (1) in acetonitrile at room temperature, showing (closed circles) absorption by the radical cation with an absorption maximum at ca 600 nm immediately after the pulse, and (open circles) absorptions assigned to radical/radical cation addition products, see text, taken 50 ms after the pulse. The inset shows the pseudo-first-order rate constant for decay of the radical cation at 600 nm as a function of added dimethylaniline (DMA): the slope gives a bimolecular rate constant for reaction of 2.1 × 1010 M−1 s−1.
Table 1.
Photophysical and electrochemical parameters related to singlet state photoinduced cleavage and sensitization ability of tetrafluoroborate salts of N-alkoxyheterocycles in acetonitrile at room temperature.
| Structure | λmax* (nm) | 1E*0,0† (kcal mol−1) | λ+max‡ (nm) | ε+max§ (M−1 cm−1) | k+e|| (stilbene) (M−1 s−1) | Φions¶ | Φradical# | Eox** (V vs. SCE) | t††(μs) | |
|---|---|---|---|---|---|---|---|---|---|---|

|
1 | 315 | 80.1 | 610 | 1650 | 2.0 × 1010 | 0.48 ± 0.08 | 0.43 ± 0.06 | 2.1 | 3 |

|
2 | 334 | 80.4 | 670 | 2135 | 1.9 × 1010 | 0.50 ± 0.10 | 0.39 ± 0.07 | 2.1 | 3 |

|
3 | 322 | 75.0 | 695 | 15 100 | 1.9 × 1010 | 0.53 ± 0.09 | 0.43 ± 0.12 | 2.0 | 2.5 |

|
4 | 345 | 78.8 | 475 | 36 000 | 1.4 × 1010 | 0.08 ± 0.04 | – | 1.8 | 44 |

|
5 | 379 | 69.2 | 460 | 15 000 | –‡‡ | 0.07 ± 0.04 | – | 1.4 | – |
Absorption maximum of the first absorption band of the N-methoxyheterocycle;
Energy of the excited singlet state of the corresponding N-ethyl structures (1E–5E), determined as the midenergy point of the absorption and fluorescence spectrum, see text;
Absorption maximum of the radical cation;
Extinction coefficient at the absorption maximum of the radical cation. Estimated errors are ±10% for 1–3, and ± 20% for 4 and 5;
Bimolecular rate constant for oxidation of stilbene by the radical cations. The experimental errors are ca 10%;
Quantum yield of formation of separated radical cations, determined as described in the text using trapping with stilbene, errors derived from multiple measurements;
Quantum yield of formation of separated methoxy radicals, determined as described in the text using trapping with diphenylmethanol, errors derived from multiple measurements;
Estimated oxidation potential of the neutral heterocycle, determined by adding 0.25 V to the oxidation potential of the corresponding hydrocarbon (46), using 1.80 V for naphthalene (55), 1.73 V for phenanthrene (56), 1.57 for trans-stilbene (56) and 1.15 V for trans-4-methoxystilbene (57), all versus SCE;
Approximate lifetimes of the radical cations under the experimental conditions in acetonitrile at room temperature;
Oxidation of stilbene is too slow in this case.
In contrast, no evidence for a radical cation could be detected in transient absorption for the 4-benzoylmethoxypyridinium 6. A transient was observed that was quenched by dissolved oxygen and was assigned to the excited triplet state (see further below). N–O bond fragmentation evidently does not compete with intersystem crossing in the excited singlet state for 6. The excited states of the N-methoxyheterocycles are therefore not necessarily dissociative with respect to the N–O bond. This is expected, as the excited states involve π* excitation and the configurations required for bond cleavage require σ* occupation. The reaction thus requires mixing of π* and σ* configurations which usually results in a barrier (56). This same situation applies to N–O bond cleavage in their corresponding radicals formed upon one-electron reduction (40,41).
Fragmentation in the excited state generates a radical cation and a methoxy radical. Absolute quantum yields were measured for formation of both the radical cations and the corresponding methoxy radicals for 1–3. The heterocycle radical cations can be trapped by reduction using stilbene. At sufficiently high concentrations of the trap, essentially all of the heterocycle radical cation is converted into the stilbene radical cation. The stilbene radical cation has a well-characterized absorption spectrum and a large extinction coefficient (57). Quantum yields are obtained by comparing the stilbene radical cation signals with the corresponding signals for trapping in other systems that have known quantum yields for formation of separated radical cations (58). The quantum yields for formation of separated radical cations obtained in this way are summarized in Table 1. For 5, dimethoxystilbene was used in place of stilbene due to the lower oxidizing ability of the radical cation of 5.
An analogous experiment can be performed to trap the separated methoxy radicals. Diphenylmethanol reacts with oxygen centered radicals as shown in Eq. (1) to
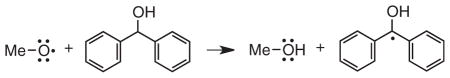
form the diphenylketyl radical, which has an absorbance maximum at ca 540 nm (59). Pulsed laser excitation of 1–3 in the presence of 2.5 M diphenylmethanol results in time-resolved formation of the diphenylketyl radical at 540 nm with a time constant of ca, 75 ns. This is consistent with the rate constant for hydrogen atom abstraction from diphenylmethanol by the methoxy radical of 5.3 × 106 M−1 s−1, which is similar to the rate constant reported previously for the analogous reaction of the t-butoxy radical with diphenylmethanol (59). In this case the diphenylketyl radical signals can be compared to those from reaction of benzophenone triplet with diphenylmethanol, which is known to yield diphenylketyl radicals with a quantum yield of 2.0 (60). The quantum yields for formation of methoxy radicals obtained in this way are also summarized in Table 1.
The yield experiments were performed multiple times for both the radicals and the radical cations, and although the averaged radical yields are somewhat lower than those for the radical cations, they are the same within the realistic experimental uncertainty.
The yields of radical cations from the N-methoxystyrylpyridiniums 4 and 5 are considerably lower than from 1–3, Table 1. The excited singlet states of N-alkylstyrylpyridiniums are known to undergo cis-trans isomerization in their excited states (61,62), which undoubtedly represents a major route for energy dissipation in 4 and 5, lowering the yields of both the radical cations and the methoxy radicals. The excited state singlet energies for 1–5 can in principle be measured from the midpoint of the maxima of the absorption and fluorescence spectra plotted on a linear energy scale. Fluorescence is difficult to detect from 1–5, however, not only because it is weak due to excited state bond cleavage but also due to interfering emission from the highly fluorescent protonated forms of the parent heterocycles, which are formed by photolysis of 1–5 in the spectrometer. However, fluorescence is readily observed for the corresponding N-ethyl substituted structures 1E–5E. The excited singlet energies of the N-ethyl 1E–5E were thus determined from their absorption and fluorescence spectra, and it was assumed that these give good estimates of the corresponding values for 1–5. These values are summarized in Table 1. Although the radical cation yields for both 4 and 5 are low, they are similar (Table 1), even though the excited singlet energy for 5 is significantly lower than that for 4. The singlet excited state energy of 69 kcal mol−1 for 5 is evidently sufficient to break the N–O bond, which is consistent with the estimated N–O bond dissociation energy, see below.
Steady-state photolysis
Steady-state photolysis of compounds 1–3 was performed in deoxygenated acetonitrile. In each case NMR analysis of the products revealed the formation of methanol, the corresponding neutral parent heterocycle, and multiple radical/radical cation coupling products and other methoxylated structures. A detailed investigation was made for 1.
Irradiation of a 10 mM deoxygenated solution of the tetrafluoroborate salt of 1 in deuterated acetonitrile in an NMR tube resulted in steady decrease in the concentration of the starting material (observed via the characteristic N-methoxy peak at 4.50 ppm), and formation of methanol (at 3.27 ppm), as well as several methoxylated structures that had absorptions in the 4.58–3.33 ppm region. Of the methoxylated products, three were formed in significantly greater yield than the others. The disappearance of the starting material correlated directly with the formation of the major methoxylated products and also with methanol, Fig. 5, which suggests that they are all primary photo-products. The chemical yield of methanol observed in the NMR experiment is lower than expected based on the yield of separated methoxy radicals, which indicates that most of the methoxy radicals do not form methanol directly in the absence of a work-up procedure in the NMR tube. The number of methoxylated peaks observed in the raw NMR spectrum was too large to allow identification of any other products. GC-MS analysis of the irradiated solution after basic extraction confirmed formation of quinoline as a product (m/z = 129), and gave four major additional peaks with longer retention times, presumably corresponding to some of the major product peaks observed in the NMR experiment. A parent ion of m/z = 159 was observed for each peak, consistent with formation of isomers of methoxyquinoline by coupling of the methoxy radical and the quinoline radical cation. Matching the mass spectra of those peaks to the NIST/EPA/NIH Mass Spectral Library 2002 suggested that they were the 3-, 5-, 6- and 8-methoxyquinolines. Confirmation for the formation of 6-methoxyquinoline was obtained by matching the GC-MS peak with a commercially available sample. An attempt to separate the irradiation mixture by column chromatography yielded only 8-methoxyquinoline, which was confirmed by independent synthesis (49).
Figure 5.

Time dependence of 1H-NMR integration signals as a function of photolysis time in acetonitrile at room temperature for, (filled circles, left vertical axis) N-methoxyquinolinium tetrafluoroborate, and (open squares, right vertical axis) three of the major methoxyquinoline products, and (open circles, right axis) the methanol product. The vertical axes give relative magnitudes for the signal intensities, but have arbitrary absolute value. The curves through the data points are for guidance only.
Addition of triethylamine, benzyl trimethylsilane or hexamethylbenzene to the reaction solution at concentrations up to 10−3 M, which should trap any freely diffusing radical cations, did not significantly change the number of peaks in the methoxy region of the NMR spectrum. This suggests that these products are formed only as a result of in-cage coupling in a primary geminate radical/radical cation pair. A proposed overall reaction scheme for photolysis of 1 (that presumably also applies to 2–5) is shown in Fig. 6. The quinoline radical cation and methoxy radical that separate from the singlet geminate radical cation/radical pair, ksep, do not couple, but instead are reduced to form quinoline (k+e) and gain a hydrogen atom to form methanol (k•H), respectively. Presumably these reactions occur with the solvent and/or impurities under the experimental or workup conditions. That the radical cation is reduced by solvent impurities is supported by the observation that all of the radical cations from 1–5 decay via pseudo-first-order kinetics, rather than the second-order kinetics as expected for radical coupling (Table 1). The radical cation lifetimes generally increase with decreasing oxidizing ability of the radical cation due to decreasing susceptibility to impurity reduction, Table 1.
Figure 6.

Reaction scheme for photolysis of N-methoxyquinolinium tetrafluoroborate (1). The first excited singlet state undergoes N–O bond cleavage, k*fr, to form a geminate singlet radical cation/radical pair, which undergoes recombination, krec, to form coupling products (for clarity, only one representative primary coupling at the 6-position is shown), which deprotonate to form isomeric methoxyquinolines (of which four are major) and a proton. Separation within the geminate pair, ksep, generates the radical cation that is detected in the nanosecond transient absorption experiment and a methoxy radical, which are trapped by an electron (k+e) and a hydrogen atom (k• H), respectively, to form quinoline and methanol.
Formation of the methoxyquinoline products from the singlet radical cation/radical pair via recombination, krec, requires elimination of a proton to rearomatize the ring systems, Fig. 6. This proton generation provides an alternate method to measure the extent of the coupling reaction. Schnabel et. al previously reported a quantum yield for proton formation of 0.48 for irradiation of 1, by titration with p-nitrophenolate (43). We measured the quantum yield for proton formation indirectly by irradiating solutions of 1–3 in aqueous buffer systems (phosphate buffer at pH ~ 7.5 and Tris buffer at pH ~ 8.3) to complete conversion, and measured the proton concentration with a pH meter. The results were somewhat irreproducible, but average chemical yields (equivalent to quantum yields) of 58% ± 4, 59% ± 16 and 48% ± 16 were obtained for 1, 2 and 3, respectively. Solutions of 1–3 were also irradiated to completion in acetonitrile, and the proton concentration estimated by titration with the p-nitrophenolate anion (53). The chemical yields of protons determined this way were again somewhat irreproducible, but from repeated measurements we obtained values of 49% ± 9, 53% ± 7 and 59% ± 9 for 1, 2 and 3, respectively. The results are generally consistent with the quantum yield for 1 reported previously by Schnabel (43). Taking into consideration the accuracy with which we are able to measure the yields of the protons, radical cations and methoxy radicals, we conclude that their sum effectively accounts for the fate of all of the excited states in the photochemical reactions of 1–3. In turn this suggests that the primary geminate pairs mainly form separated radicals or recombine to liberate a proton. Recombination in the geminate pair to reform starting material appears to be inefficient. This is presumably because the radical cations for these heterocycles are π-radical cations rather than nitrogen-centered radical cations, which will favor bond formation with the ring carbons rather than with the nitrogen.
Finally, the primary coupling products in Fig. 6, krec, are Wheland intermediates, which are reported to have broad absorptions in the UV–visible range (63). For the structures studied here, the large number of possible intermediates would be expected to produce a rather broad and featureless composite absorption, and indeed, this is what is observed for 1–3 in nanosecond transient absorption spectroscopy, see Fig. 4 for example. No attempt has been made to analyze the kinetics of these absorptions due to their obviously complex nature.
Picosecond transient absorption spectroscopy
Picosecond transient absorption spectroscopy of compounds 1–3 was performed in acetonitrile at room temperature. For 1 and 2, absorptions due to the corresponding radical cations are observed within roughly 30 ps, Fig. 7. Decay in these absorptions as a function of time was observed, to a level that was constant on the picosecond timescale. This decay is most reasonably assigned to geminate recombination within the methoxy radical/radical cation pair, krec, in competition with separation, ksep. The percentage of the observed signal decay, ca 15%–20%, is smaller than the extent of cage recombination estimated from the studies of proton formation and separated radicals. However, the absorbance signal after the decay must also contain contributions from the coupling products. These will add to the constant absorbance signal after the decay, making the constant signal larger and thus make the decaying part of the absorbance signal appear smaller. Also, if k*fr is not significantly larger than (krec + ksep), i.e., if the geminate pair is not formed instantaneously, then the maximum observed absorbance due to the pair is reduced if recombination and fragmentation are even slightly competitive. Although the excited singlet state could not be directly detected for 1 and 2, the singlet state was observed for 3, see below. For 1, a first-order kinetic fit to the observed decay data yields (ksep + krec) = 6 × 109 s−1. Taking the percent geminate recombination to be ca 50% from the proton production experiment described above, then ksep ~ 3 × 109 s−1 and krec ~ 3 × 109 s−1. The rate constant for separation is in reasonable agreement with previously reported values for other geminate pairs in polar aprotic solvents (13).
Figure 7.

Normalized absorbance as a function of time for the singlet geminate quinoline radical cation/methoxy radical pair, observed at 600 nm, for picosecond excitation of N-methoxyquinolinium tetrafluoroborate, 1, in acetonitrile at room temperature. The gray curve represents an exponential fit to the data with a rate constant of 6 × 109 s−1, which is assigned to the sum (ksep + krec). The inset shows the corresponding transient absorption spectra taken ca 30 ps after the pulse for (gray curve) the quinoline radical cation geminate pair from excitation of 1, and (black curve) the isoquinoline radical cation geminate pair from excitation of 2.
In the case of 3, the radical cation signal at ca 690 nm was not observed immediately after the pulse, instead a different absorbance with maximum at ca 630 nm was observed, Fig. 8. This signal decayed with time and was replaced with a radical cation signal, Fig. 8. The 630 nm transient is the precursor to the radical cation, presumably the first excited singlet state. The 630 nm transient decayed with a rate constant of 1.5 × 109 s−1, which is thus assigned to k*fr. No geminate recombination at all can be observed for 3 because in this case the pair is formed more slowly than it reacts. No direct evidence for an excited state is obtained for 1 and 2, however, these have higher singlet excited state energies, Table 1, which should increase the energy of the π* configuration compared to that of the σ*, resulting in a lower energy curve crossing and faster bond fragmentation rate (56).
Figure 8.

Normalized absorbance spectra observed upon excitation of N-methoxyphenanthridinium tetrafluoroborate 3 in acetonitrile at room temperature (black curve) 25 ps after the pulse, assigned to the excited singlet state, see text, and (gray curve) 2 ns after the pulse, assigned to the phenanthridine radical cation, see text. The inset shows the time dependence of the absorbance at 630 nm as a function of time fitted to an exponential function with time constant 1.5 × 109 s−1, assigned to the rate constant for cleavage of the N–O bond.
Utility as irreversible photooxidants
The utility of the N-methoxyheterocycles as irreversible photooxidants depends upon several factors: the ability to excite the sensitizer in the presence of a donor (determined by its absorption spectrum), the ability of the excited state to cleave the N–O bond (determined by the singlet excitation energy, lifetime and competing processes), the efficiency of formation of the oxidizing radical cations, and the oxidizing power and lifetimes of the radical cations. Relevant data are collected in Table 1.
The enthalpy of fragmentation of the N–O bond in N-methoxy-4-phenylpyridinium has previously been reported to be 55 kcal mol−1, from DFT calculations (37). As discussed in detail below, we have also obtained values close to this for the other N-methoxyheterocycles. Thus, N–O bond cleavage for all of the excited singlet states of Table 1 is exothermic. The barrier to cleavage even for 5 which has the lowest excited state energy does not prevent fragmentation, but bond cleavage appears to be much less efficient than isomerization of the central C=C double bond. Neither 4 nor 5 are particularly efficient sensitizers, although they suggest that excited states with energies as low as 69 kcal mol−1 could, in principle, be useful, which corresponds to absorption just within the visible region.
The oxidizing abilities of the photogenerated radical cations are given simply by the oxidation potentials of the corresponding neutral heterocycles. In our hands, electrochemical oxidation of all of the heterocycles is highly irreversible in CV. The oxidation potentials in Table 1 are thus estimated values, obtained as described previously by adding 0.25 V to the oxidation potential of the corresponding hydrocarbon (37), i.e. naphthalene for quinoline and isoquinoline etc. Structures 1–3 generate oxidizing radical cations with reasonable efficiency, and with the ability to oxidize donors with potentials as high as ca 2 V vs SCE.
Triplet state sensitizers
As mentioned above, the excited triplet state of the benzoylpyridinium 6 does not undergo N–O cleavage. Nevertheless, the triplet states of the N-alkoxyheterocycles can still be used as irreversible photooxidants according to Fig. 9a, which is related to the mechanism given in Fig. 2b. Electron-transfer quenching of the excited triplet states using a one-electron donor generates a neutral radical. These radicals have previously been shown to undergo rapid N–O bond cleavage (38,39). This triplet oxidation mechanism is investigated in some detail in this section. One motivation for exploring the triplet-sensitized routes was that covalent bond formation via recombination in the primary geminate radical cation/radical pairs, Fig. 6, is spin forbidden in the triplet manifold. These recombinations can result in the formation of interfering transient absorptions from the Wheland intermediates, discussed above. Elimination of this recombination would result in fewer complicating transient absorptions. Based on the transient absorptions observed already with the benzoylpyridinium 6, we anticipated that structures 7 and 8 (Fig. 3) might also undergo efficient intersystem crossing to form excited triplet states upon excitation.
Figure 9.
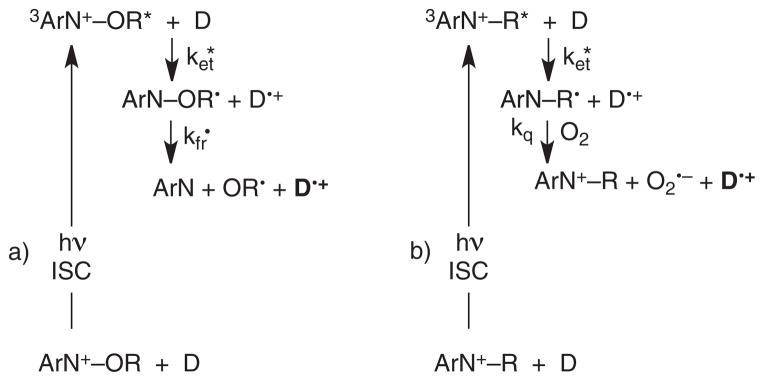
Two schemes for triplet sensitization. a) Excitation of an N-methoxyheterocycle followed by intersystem crossing forms the excited triplet state that undergoes electron transfer followed by rapid bond fragmentation. a) Mechanism is related to the mechanism given in Fig. 2b), but occurs in the triplet manifold, and results in irreversible photooxidation. b) Excitation of an N-alkylheterocycle results in conventional triplet state photoinduced electron transfer, which is not irreversible, but regenerates the triplet sensitizer. The mechanisms are different because cleavage can occur in the weaker N–O bond in a) but not in the stronger N–C bond in b).
Nanosecond transient absorption spectroscopy
Pulsed laser excitation of 7,8, 6E–8E and also 3E in acetonitrile at room temperature gave transient absorptions that behaved similarly to that observed upon excitation of 6. These transients decayed on the nanosecond to microsecond timescale. Representative spectra are shown in Fig. 10 and the absorbance maxima for the transients in the visible region are summarized in Table 2. The spectra for the N-methoxy and their corresponding N-ethyl derivatives are essentially the same for the structures 6/6E, 7/7E and 8/8E. This implies that the same species is formed upon excitation of both. The measured lifetimes are too long to be excited singlet states. If the transients are triplet states and not radical cations, then N–O bond cleavage does not occur efficiently in the excited singlet states because intersystem crossing dominates. Furthermore, if no radical cations are observed, N–O bond cleavage does not occur in the triplet states either.
Figure 10.

Normalized transient absorption spectra of the N-ethyl triplet states (open circles) and the N-methoxy triplet states (closed circles) upon pulsed laser excitation in acetonitrile at room temperature for the structures indicated, ca 100 ns after the laser pulse. The absolute vertical scale is arbitrary, only the relative shapes of the spectra can be compared.
Table 2.
Spectral, kinetic and energetic properties of N-methoxy and N-ethylheterocycle excited triplet states in acetonitrile at room temperature.
| Structure* | 3λ*max† (nm) | 3kq*(O2)‡ (M−1 s−1) | 3E*0,0§ (kcal mol−1) | Ered|| (V vs. SCE) | 3Ered*¶(V vs. SCE) | 3t*# (ns) | |
|---|---|---|---|---|---|---|---|

|
6 | 700 | 6.2 × 108 | 67.2 | −0.53 | 2.4 | 80 |

|
6E | 700 | 7.8 × 108 | 67.2 | −0.67** | 2.2 | 50 |

|
7 | 550 | 1.1 × 109 | 56.5 | −0.65 | 1.8 | 600 |

|
7E | 550 | 1.0 × 109 | 56.5 | −0.79 | 1.7 | 500 |
|
8 | 525 | 1.8 × 109 | 57.2 | −0.63 | 1.9 | 400 |

|
8E | 525 | 1.2 × 109 | 58.1 | −0.77 | 1.7 | 400 |

|
3E | 500 | 4.5 × 108 | 60.3 | −0.96†† | 1.7 | 1000 |
|
1 | 440 (500)‡‡ | 5.3 × 108 | – | −0.84 | – | 2700 |
|
1E§§ | (440) 500‡‡ | 6.0 × 108 | 60.4§§ | −0.98|||| | 1.6 | 1900 |
Experiments performed using tetrafluoroborate or hexafluorophosphate salts except where noted;
Absorption maximum of the excited triplet state;
Bimolecular rate constant for quenching of the excited triplet by oxygen;
Energy of the 0,0 phosphorescence transition measured in EPA or ethanol/methanol/methyl iodide glass at 77 K;
Estimated reduction potential of the N-methoxy or N-ethylheterocycle versus SCE, from measurements as part of this work or as indicated. The reduction potentials of the N-methoxy structures were not measured but are obtained by adding 0.15 V to those for the corresponding N-ethyl structure, as described in Ref. (37);
Reduction potential of the excited triplet state estimated as Ered* = Ered + (3E*0,0/23.06);
Measured lifetime of the excited triplet state;
from Ref. (64);
from Ref. (65);
Two maxima observed, the weaker absorbance is in parenthesis;
Iodide salt;
From Ref. (66).
Sensitivity to dissolved oxygen confirmed that the transients were triplet states: the relevant rate constants, 3kq*(O2), are summarized in Table 2. The Wigner spin rules state that oxygen should react with triplet excited states to give singlet oxygen, with a rate constant that is ca one ninth of the diffusion-controlled limit (67). Actual rate constants for oxygen quenching often vary slightly from this value due to differences in the Franck-Condon factors for energy transfer, determined mainly by reaction exothermicity (67,68). Thus, the fact that all of the rate constants are ca 109 M−1 s−1 is good evidence in favor of reaction of oxygen with an excited triplet state in each case.
Corresponding excitation of 1 and 3 gave no identifiable triplet states, as they yield radical cations, as discussed above. For N-ethylquinolinium, 1E, a very weak transient absorption with maximum at ca 500 nm was observed, the intensity of which increased slightly upon the addition of methyl iodide, presumably as a result of an external heavy atom effect (69). Efficient formation of both the triplet states of 1 and 1E was, however, accomplished by using triplet sensitization.
Excitation of benzophenone in the presence of ca 10−2 M 1 or 1E resulted in rapid quenching of the benzophenone triplet, observed at 525 nm, and formation of new transient absorptions with spectra shown in Fig. 11. These spectra have absorption maxima at the same wavelengths and are similar in shape, only the relative intensities of the vibronic bands are different. Both of these can be assigned to triplet states based on the fact that they react with oxygen with rate constants close to 109 M−1 s−1, Table 2.
Figure 11.

Triplet–triplet absorption spectra for 1 (closed circles) and 1E (open circles) obtained by sensitization using benzophenone, in acetonitrile solution at room temperature.
The lifetimes of the triplet states of both 1 and 1E are similar, which suggests that they are determined by factors other than bond cleavage. In fact, the measured lifetimes in acetonitrile are quite short for triplet states, Table 2, which is almost certainly a consequence of solution phase quenching. This is supported by experiments described below in which it is found that the lifetimes are much longer in rigid solution. The excited states of these cations are expected to be very strong one-electron oxidants. Similar to the behavior of the radical cations discussed above, the lifetimes of the triplet correlate roughly with their oxidizing ability. The solution phase reactions that shorten the lifetimes of these triplet states are reasonably assigned to one-electron reduction via reaction with solvent impurities.
Emission spectroscopy
As mentioned above, 1E and 3E fluoresce quite strongly at room temperature. The excited singlet states are ππ* in these cases, and weak fluorescence can even be observed for 3, which undergoes bond cleavage. No significant fluorescence or phosphorescence above background is observed for the benzoyl-substituted compounds 6/6E and 7/7E in fluid solution at room temperature. For 8 and 8E, weak fluorescence can be observed, suggesting some ππ* character in the excited states as a result of decreasing ππ* energy as the conjugation in the aromatic ring increases (70,71).
In a glass at liquid nitrogen temperature (see methods and materials for details), phosphorescence is observed for all of the benzoyl-substituted structures 6–8 and 6E–8E. The fluorescence from 8 and 8E also becomes better resolved. Fluorescence can still not be distinguished above the scattered light background for 6/6E and 7/7E, suggesting very rapid intersystem crossing in these cases. Representative spectra are shown in Figs. 12 and 13. The emission energies corresponding to the zero–zero transitions for phosphorescence, 3E*0,0, are summarized in Table 2.
Figure 12.

Phosphorescence spectra of (thin black curve) N-methoxy-4-benzoylpyridinium, 6, and (heavier gray curve) N-ethyl-4-benzoylpyridinium, 6E, as tetrafluoroborate salts, in an EPA glass at 77 K. The vertical scale is arbitrary for both spectra.
Figure 13.

Phosphorescence and fluorescence spectra of sensitizers, for the structures indicated, measured at 77 K in (thin black curves) an EPA glass, and (heavier gray curves) a 1:1 ethanol/methanol glass with ca 1 M methyl iodide.
The phosphorescence spectra for 6/6E and 7/7E exhibit classical nπ* behavior, with vibronic progressions corresponding to the carbonyl stretching frequency (60,70,71). Indeed, the spectra for 6 and 6E are similar to that of benzophenone in a low-temperature glass (60,70), Fig. 12. For 8 and 8E, the phosphorescence spectra are a little broader, and for 8, the phosphorescence is weaker than fluorescence when measured in EPA glass. Phosphorescence in 8 was enhanced by the external heavy atom effect in a 1:1 ethanol/methanol glass in presence of methyl iodide, Fig. 13. This behavior indicates that the nature of the excited triplet states for 8 and 8E is changing from being highly nπ* to having substantial ππ* character as the energies of the ππ* configurations decrease with increasing size of the conjugated π system (70,71). That the lowest excited triplet state for 8 and 8E is highly ππ* in character and is phenanthridinium like is further supported by the observation that both the phosphorescence and triplet–triplet absorption spectra for 8 and 8E are very similar to those of 3E, Figs. 10 and 13.
For the quinoliniums 1 and 1E, no significant phosphorescence could be detected even at low temperature for the tetrafluoroborate salts. For 1E, however, phosphorescence could be measured for the iodide salt. Presumably the salt is ion paired in EPA, and the external heavy atom effect of the iodide enhances phosphorescence in this case.
The strong phosphorescence for the benzoyl-substituted structures 6–8 in EPA at 77 K confirms that N–O bond cleavage does not occur in the triplet states (at least at this temperature), and that the short lifetimes in room temperature are almost certainly a result of bimolecular quenching, which is precluded in the rigid glass medium.
N-Ethylheterocycles as sensitizers
The N-ethyl structures can also act as triplet photooxidation sensitizers that operate via the mechanism shown in Fig. 9b. In this case, electron transfer quenching of the excited triplet state occurs to generate a neutral radical, which does not undergo cleavage but that can be trapped by oxygen to generate the superoxide anion. Quenching by oxygen can thus remove transient absorptions due to the heterocycle radicals. Obviously, electron transfer in this scheme is reversible, since a radical anion is still generated, but this route avoids generating alkoxy radicals in cases where their own chemistry could be complicating. The mechanism of Fig. 9b also has the advantage that the sensitizer is not consumed. This is useful in cases where the sensitizer must be recycled.
Spectroscopic characterization of the N-ethylheterocycle radicals that are formed upon one-electron reduction in the sensitizers was accomplished by reducing their triplet excited states with benzyltrimethylsilane as a one-electron donor, which does not form any interfering transient absorptions in the region of interest. The radical cation of benzyltrimethylsilane undergoes very rapid C–Si bond fragmentation in acetonitrile (72). The benzyl radical product of this fragmentation is transparent in the visible region (73). The absorption maxima of the radicals measured using this method are summarized in Table 3. These radicals could be quenched by molecular oxygen, and the rate constants for these reactions, kq•(O2), are given in Table 3. The mechanism of oxygen quenching is presumably electron transfer to form the superoxide anion. The reduction potential of oxygen in acetonitrile is ca −0.8 V vs SCE (74). The oxidation potentials of the N-ethyl radicals are the same as the reduction potentials of the corresponding parent cations. These were obtained from literature values, and where these do not exist, from measurements as part of this work, Table 2. The reductions are all quasi-reversible, and consequently the errors in the cation reduction potentials are not known. Based on the values in Table 2, electron transfer to oxygen from the radical from 6E should be slightly endothermic, and the corresponding reactions for the radicals from 7E and 3E should be close to thermoneutral or slightly exothermic. This is consistent with the observed rate constants for reaction with oxygen which are all just below the diffusion-controlled limit (1,2).
Table 3.
Absorption and kinetic properties of N-ethylheterocycle radicals in acetonitrile at room temperature.
| Radical Structure* | Radical Precursor† | λmax‡ (nm) | kq•(O2)§ (M−1 s−1) |
|---|---|---|---|

|
6E | 510 | 3.3 × 107 |

|
7E | 700 | 7.3 × 108 |

|
3E | 575 | 4.3 × 108 |
Several other resonance contributors can be drawn;
Studied as either the tetrafluoroborate or hexafluorophosphate salts;
Approximate absorption maximum of the radical;
Rate constant for radical reaction with molecular oxygen.
Oxidizing power of the triplet sensitizers
Table 2 summarizes the oxidizing abilities of the sensitizer triplet states. These are the reduction potentials of the triplet excited states (3Ered*, vs SCE), which are estimated according to Eq. (2). Here, Ered is the reduction potential of the ground state of the sensitizer, and 3E*0,0 is the sensitizer excited state energy in kcal mol−1. The reduction potentials for the N-methoxyheterocycle cannot be determined
| (2) |
electrochemically, as they undergo rapid N–O bond cleavage upon reduction (37,40,41). For these we used an approximation described previously, i.e. that the N-methoxy structures would have potentials that are less negative by 0.14 V than those for the N-ethyl structures (37), Table 2. The reduction potentials represent the largest source of error for the estimated excited state reduction potentials summarized in Table 2. For all donors with oxidation potentials that are less positive than 3Ered*, electron transfer would be exothermic. The oxidizing powers of the triplet states are in the same range as those of the radical cations derived from the singlet sensitizers summarized in Table 1.
N–O vs N–C bond fragmentation
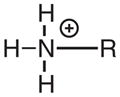
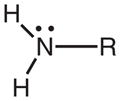
A minimum requirement for N–O bond dissociation is that the energy of the excited state be larger than the bond dissociation energy (BDE). The bond dissociation enthalpy for the N–O bond of N-methoxy-4-phenylpyridinium has previously been reported to be 55 kcal mol−1, using the B3LYP/6-311G*//B3LYP/6-31G* DFT method (37). We have performed similar calculations in the gas phase using B3LYP/aug-cc-pVDZ for the N–O bonds in N-methoxyquinolinium (1), N-methoxy-4-benzoylpyridinium (6) and N-methoxy-4-phenylpyridinium, as well as their corresponding N–ethyl structures, Table 4. The results are very consistent with the previously published value from Ref. (37) and readily explain why bond cleavage occurs in the singlet excited states of the N-methoxy (N–O bonded) structures and not the N-ethyl (N–C structures). Simply, the N–C BDE is larger than any of the singlet excited state energies, whereas the N–O BDE is smaller. There are several competing factors that determine the magnitude of the dissociation energy for covalent bonds such as these (see Ref. (76), for example), including the electrostatic energy (that includes interatom electron repulsion), orbital overlap and exchange energies. In turn, these depend upon atomic size and electronegativity, so that prediction of bond dissociation energies is difficult. The N–O bond in the N-methoxy structures could be weaker than the N–C bond in the N-ethyl structures due to electron repulsion between the nonbonding electrons on oxygen and the π-electrons in the ring. The fact that the N–O BDE in methoxyamine (MeO–NH2, with nonbonding electrons on both fragmenting atoms) is smaller than that for the N–C BDE in methylamine (Me–NH2, with nonbonding electrons on only one fragmenting atoms), Table 4, would support this hypothesis. However, DFT calculations indicate that the N–O bond is also significantly weaker than the N–C bond in the protonated forms of these structures where there are no nonbonding electrons on the nitrogens, Table 4. The data in Table 4 show that the BDEs in the protonated structures are stronger than the neutral counterparts, consistent with a previous study (77), which suggests that electron repulsion can only be a partial explanation at best, and that electronegativity and atomic size effects are probably more important. The strengths of bonds to carbon tend to increase with increasing electronegativity of the other atom (78). However, bond strengths between two increasingly electronegative elements of constant electronegativity difference tend to decrease (77). This latter effect has been assigned to a reduced electrostatic contribution to the strength of a bond between two very electronegative elements due to polarization of the shared electrons away from the internuclear space toward the two nuclei (76,78). The electronegativity difference between carbon and nitrogen is similar to that between nitrogen and oxygen. However, the formal positive charge on the nitrogen in the N-methoxy and N-ethylheterocycles effectively increases the electronegativity of the nitrogen. Thus, the N–C bond in the N-ethylheterocycles is stronger, as expected for a bond from carbon to a very electronegative element. However, the N–O bond is weaker, as expected for a bond between two very electronegative elements. We assume that this accounts for the very different photochemistry for the N-methoxy and the N-ethylheterocycles.
Table 4.
N–O and N–C bond dissociation enthalpies in the gas phase, calculated using B3LYP/aug-cc-pVDZ DFT.


Experimental values from Ref. (75).
The lack of N–O bond cleavage in the excited triplet states then requires an explanation, since the excited state energies of all of the N-methoxy triplet states are close to or greater than the N–O BDE of ca 55 kcal mol−1, Table 2. In particular, the 4-benzoyl structure 6 has a triplet energy of ca 67 kcal mol−1. Whereas all of the excited singlet states that undergo cleavage summarized in Table 1 are ππ*, the triplet states have significant nπ* character, and in the case of 6 the triplet state is essentially exclusively nπ*. As mentioned above, bond fragmentation requires mixing of the ππ* or nπ* (as appropriate) configurations with the relevant σσ* configuration (56). This mixing allows the reaction to occur, but creates an energetic barrier. A detailed state or orbital analysis of the fragmentation reaction is beyond the scope of this work, but it is readily apparent that one of the contributing factors is different for the different excited states. The σσ*/ππ* or σσ*/nπ* mixing must effectively transfer a “hole” to the sigma bond. This mixing or “hole transfer” is readily accomplished as a consequence of the orbital overlap that occurs by bending the N–O bond out of the plane of the aromatic system for a ππ* excited state (41,56). However, there is no equivalent geometric distortion that will allow appropriate orbital overlap to transfer a hole from an nπ* state, where the hole is localized on the remote carbonyl oxygen atom. On this basis it is reasonable to expect much weaker configuration mixing and thus a slower cleavage reaction for the nπ* excited states, as observed. The triplet states develop more ππ* character with increasing conjugation, but the excited triplet energies also decrease, which will increase the energy gap between the π* and σ* configurations, increasing the reaction barrier and decreasing reactivity. Comparing the excited singlet and triplet state reactions is complicated by the fact that the 1σσ* and 3σσ* configurations are different, but we note that 69 kcal mol−1 is sufficiently high in an excited ππ* singlet state to allow reaction (structure 5), whereas ca 60 kcal mol−1 is not high enough in an excited ππ* triplet state to allow reaction (assuming the triplet energy for structure 1 is similar to that for 1E).
Irreversible photooxidation
The oxidizing abilities of the sensitizers discussed here are quite large, up to ca 2.0 vs SCE. The sensitizers are also soluble in both organic solvents and water. Their utility is illustrated in the oxidation of guanosine as a donor, the radical cation of which is not easy to detect due to its low extinction coefficient. The best transient absorption spectrum of the guanosine radical cation was reported in 1989 by Candeias and Steenken using pulse radiolysis (79). Pulse radioloysis is an ideal time-resolved technique for detection of low extinction species due to the large sample sizes, path lengths and irradiation energies (80). Direct observation of the guanosine radical cation in solution at room temperature using photosensitization is considerably more difficult as formation via conventional photoinduced electron transfer generates a reduced acceptor, the absorbance of which can easily mask the guanosine radical cation absorption. However, the transient absorption spectrum of the guanosine radical cation is readily generated using the sensitizers described here.
In aqueous solution buffered at pH 2.0, guanosine is found to react with the quinoline radical cation formed by excitation of 2, with the excited triplet state of 6, and with the excited triplet state of 3E with bimolecular rate constants of between 2 and 8 × 109 M−1 s−1. Using 2, 6 and 3E, formation of the guanosine radical cation occurs via the mechanisms shown in Figs. 2a, 9a and b, respectively. Using 50 mM guanosine, the radical cation spectrum is formed within ca 20 ns of the laser pulse using 2 and 6, and within ca 2 μs of the pulse in oxygen saturated solution using 3E, to allow time for complete reoxidation of the reduced 3E by oxygen. The transient absorption spectra that were obtained using these three sensitization mechanisms are identical to that obtained by pulse radiolysis, Fig. 14.
Figure 14.

Normalized transient absorption spectra of the radical cation of deoxyguanosine mononucleotide in buffered pH 2.0 aqueous solution at room temperature, formed by photooxidation using (red curve) N-methoxyisoquinolinium tetrafluoroborate (2), (blue curve) N-ethylphenanthridinium tetrafluoroborate (3E) and (green curve) N-methoxy-4-benzoylpyridinium tetrafluoroborate (6) sensitizers, compared to (black squares) the corresponding spectrum obtained by Candeias and Steenken (79) using pulse radiolysis.
CONCLUSION
The excited states of N-methoxyheterocycles can be used as irreversible photooxidants in either the singlet or the triplet spin manifold. The excited state energy must significantly exceed the N–O bond dissociation energy of ca 55 kcal mol−1 to allow reactions to compete with other excited state processes. The ππ* excited singlet states have energies of at least 69 kcal mol−1, and all undergo N–O bond fragmentation with varying efficiencies, depending upon competing excited state processes. The most efficient singlet states generate separated radical cations with a quantum yield of ca 0.5. These can act as irreversible oxidants for donors oxidation potentials less than ca 2 V vs SCE in a fragmentation followed by electron transfer mechanism. The N-methoxyheterocycle triplet states do not undergo N–O bond fragmentation, but can still act as irreversible oxidants for donors with oxidation potentials in the range ca 1.6–2.2 V vs SCE. For the triplet states oxidation takes place in an electron transfer followed by fragmentation mechanism. The corresponding N-ethyl structures have a similar oxidizing ability, but do not fragment and thus act as conventional reversible photooxidation sensitizers.
Acknowledgments
The authors at Arizona State University acknowledge financial support from the National Science Foundation (CHE-0213445). The authors at Millsaps College acknowledge financial support by the Mississippi INBRE, funded by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103476. The authors also thank one of the reviewers for a useful suggestion.
Footnotes
This paper is part of the Special Issue honoring the memory of Nicholas J. Turro
References
- 1.Kavarnos GJ, Turro NJ. Photosensitization by reversible electron transfer: Theories, experimental evidence, and examples. Chem Rev. 1986;86:401–449. [Google Scholar]
- 2.Beens H, Weller A. Excited molecular π-complexes in solution. In: Birks JB, editor. Organic Molecular Photophysics. Vol. 2. Wiley; London: 1975. pp. 159–213. Chapter 4. [Google Scholar]
- 3.Gould IR, Young RH, Farid S. Dynamics of photoinduced electron transfer in solution. In: Honda K, editor. Photochemical Processes in Organized Molecular Systems. Elsevier; New York, NY: 1991. pp. 19–40. [Google Scholar]
- 4.Gould IR, Farid S. Dynamics of Bimolecular Photoinduced Electron-Transfer Reactions. Acc Chem Res. 1996;29:522–528. [Google Scholar]
- 5.Mattes SL, Farid S. Photochemical electron-transfer reactions of olefins and related compounds. In: Padwa A, editor. Organic Photochemistry. Marcel Dekker; New York, NY: 1983. pp. 233–326. [Google Scholar]
- 6.Zou C, Miers JB, Ballew RM, Dlott DD, Schuster GB. Electron transfer and back electron transfer in photoexcited ion pairs: Forward and back directions have different maximum rates. J Am Chem Soc. 1991;113:7823–7825. [Google Scholar]
- 7.Lucia LA, Wang Y, Nafisi K, Netzel TL, Schanze KS. Direct Observation of Ultrafast C-C Bond Fragmentation in a Diamine Radical Cation. J Phys Chem. 1995;99:11801–11804. [Google Scholar]
- 8.Bockman TM, Hubig SM, Kochi JK. Direct Observation of Ultrafast Decarboxylation of Acyloxy Radicals via Photoinduced Electron Transfer in Carboxylate Ion Pairs. J Org Chem. 1997;62:2210–2221. doi: 10.1021/jo9617833. [DOI] [PubMed] [Google Scholar]
- 9.Weller A. Mechanism and Spindynamics of Photoinduced Electron Transfer Reactions. Z Phys Chem (Munich) 1982;130:129–138. [Google Scholar]
- 10.Goodman JL, Peters KS. Picosecond decay dynamics of the trans-stilbene-olefin contact ion pair: Electron-transfer vs. ion-pair separation. J Am Chem Soc. 1985;107:6459–6463. [Google Scholar]
- 11.Mataga N. Photoinduced charge separation and charge recombination of transient ion-pair states. Ultrafast laser photolysis. In: Bolton JR, Mataga N, McLendon G, editors. Electron Transfer in Inorganic, Organic and Biological Systems, Advances in Chemistry Series. Vol. 228. American Chemical Society; Washington, D.C: 1991. pp. 91–115. [Google Scholar]
- 12.Arnold BR, Noukakis D, Farid S, Goodman JL, Gould IR. Dynamics of Interconversion of Contact and Solvent-Separated Radical-Ion Pairs. J Am Chem Soc. 1995;117:4399–4400. [Google Scholar]
- 13.Arnold BR, Farid S, Goodman JL, Gould IR. Absolute Energies of Interconverting Contact and Solvent-Separated Radical-Ion Pairs. J Am Chem Soc. 1996;118:5482–5483. [Google Scholar]
- 14.Chen P, Meyer TJ. Electron Transfer in Frozen Media. Inorg Chem. 1996;35:5520–5524. doi: 10.1021/ic9512845. [DOI] [PubMed] [Google Scholar]
- 15.Baessler H. Electronic Transport in Disordered Organic Solids. Matr Sci. 1981;7:5–12. [Google Scholar]
- 16.Wasielewski MR, O’Neil MP, Gosztola D, Niemczyk MP, Svec WA. Ultrafast photoinduced electron transfer reactions in supramolecular arrays: From charge separation and storage to molecular switches. Pure Appl Chem. 1992;64:1319–1326. [Google Scholar]
- 17.Rhodes TA, Farid S, Goodman JL, Gould IR, Young RH. Charge-Transfer Complexes and Energetic Disorder in Amorphous Organic Solids. J Am Chem Soc. 1999;121:5340–5341. [Google Scholar]
- 18.Chen L, Farahat MS, Gaillard ER, Farid S, Whitten DG. Photoinduced electron transfer double fragmentation: An oxygen-mediated radical chain process in the co-fragmentation of substituted pinacol donors with carbon tetrachloride. J Photochem Photobiol, A. 1996;95:21–25. [Google Scholar]
- 19.Karki SB, Dinnocenzo JP, Farid S, Goodman JL, Gould IR, Zona TA. Bond-Coupled Electron Transfer Processes: A New Strategy for High-Efficiency Photoinduced Electron Transfer Reactions. J Am Chem Soc. 1997;1997(119):431–432. [Google Scholar]
- 20.Al-Kaysi RO, Goodman JL. Bond-Coupled Electron Transfer Processes: Cleavage of Si–Si Bonds in Disciplines. J Am Chem Soc. 2005;127:1620–1621. doi: 10.1021/ja0432564. [DOI] [PubMed] [Google Scholar]
- 21.Beckman TM, Hubig SM, Kochi JK. Direct Measurement of Ultrafast Carbon–Carbon Cleavage Rates via the Subpicosecond Charge-Transfer Activation of Pinacols. J Am Chem Soc. 1998;120:6542–6547. [Google Scholar]
- 22.Fox MA, Dulay MT, Krosley K. Comparison of Oxidative and Excited State Cyclizations of N-Benzyldiphenylamines to N-Benzylcarbazoles. J Am Chem Soc. 1994;116:10992–10999. [Google Scholar]
- 23.Wayner DDM, McPhee DJ, Griller DJ. Oxidation and reduction potentials of transient free radicals. J Am Chem Soc. 1988;110:132–137. [Google Scholar]
- 24.Wayner DDM, Parker VD. Bond energies in solution from electrode potentials and thermochemical cycles. A simplified and general approach. Acc Chem Res. 1993;26:287–294. [Google Scholar]
- 25.Tasdelen MA, Kumbaraci V, Jockusch S, Turro NJ, Talinli N, Yagci Y. Photoacid Generation by Stepwise Two-Photon Absorption: Photoinitiated Cationic Polymerization of Cyclohexene Oxide by Using Benzodioxinone in the Presence of Iodonium Salt. Macromolecules. 2008;41:295–297. [Google Scholar]
- 26.Yagci Y, Jockusch S, Turro NJ. Mechanism of Photo-induced Step Polymerization of Thiophene by Onium Salts: Reactions of Phenyliodinium and Diphenylsulfinium Radical Cations with Thiophene. Macromolecules. 2007;4:4481–4485. [Google Scholar]
- 27.Saeva FD. Intramolecular photochemical electron transfer (PET)-induced bond cleavage reactions in some sulfonium salt derivatives. Adv Electron Transfer Chem. 1994;4:1–25. [Google Scholar]
- 28.Crivello JV, Liu S. Free Radical Induced Acceleration of Cationic Photopolymerization. Chem Matr. 1998;10:3724–3731. [Google Scholar]
- 29.Dektar JL, Hacker NP. Photochemistry of diaryliodonium salts. J Org Chem. 1990;55:639–647. [Google Scholar]
- 30.DeVoe RJ, Olofson PM, Sahyun MRV. Photochemistry and Photophysics of ‘Onium Salts. Adv Photochem. 1992;17:313–355. [Google Scholar]
- 31.Hostetler KJ, Crabtree KN, Poole JS. The Photochemistry of 4-Azidopyridine-1-oxide. J Org Chem. 2006;71:9023–9029. doi: 10.1021/jo061259o. [DOI] [PubMed] [Google Scholar]
- 32.Peng L, Wirz J, Goeldner M. 2-Nitrobenzyl Quaternary Ammonium Derivatives Photoreleasing Nor-butyrylcholine in the Microsecond Time Range. Tetrahedron Lett. 1997;38:2961–2964. [Google Scholar]
- 33.Larson JR, Epiotis ND, McMurchie LE, Shaik SS. The role of spin inversion in the triplet photochemistry of benzyl halides and benzylammonium salts. J Org Chem. 1980;45:1388–1393. [Google Scholar]
- 34.Arnold B, Donald L, Jurgens A, Pincock JA. Homolytic versus heterolytic cleavage for the photochemistry of 1-naphthylmethyl derivatives. Can J Chem. 1985;63:3140–3146. [Google Scholar]
- 35.Foster B, Gaillard B, Mathur N, Pincock AL, Pincock JA, Sehmbey C. Substituent effects on homolytic versus heterolytic photocleavage of (1-naphthylmethyl)trimethylammonium chloride. Can J Chem. 1987;65:1599–1607. [Google Scholar]
- 36.Shukla D, Adiga SP, Ahearn WG, Dinnocenzo JP, Farid S. Chain-Amplified Photochemical Fragmentation of N-Alkoxypyridinium Salts: Proposed Reaction of Alkoxyl Radicals with Pyridine Bases To Give Pyridinyl Radicals. J Org Chem. 2013;78:1955–1964. doi: 10.1021/jo301975j. [DOI] [PubMed] [Google Scholar]
- 37.Gould IR, Shukla D, Giesen D, Farid S. Energetics of Electron-Transfer Reactions of Photoinitiated Polymerization: Dye-Sensitized Fragmentation of N-Alkoxypyridinium Salts. Helv Chim Acta. 2001;84:2796–2812. [Google Scholar]
- 38.Lorance ED, Gould IR. A Quantitative Curve-Crossing Model for Radical Fragmentation. J Phys Chem A. 2005;109:2912–2919. doi: 10.1021/jp0449713. [DOI] [PubMed] [Google Scholar]
- 39.Lorance ED, Hendrickson K, Gould IR. Density Functional Theory Predicts the Barriers for Radical Fragmentation in Solution. J Org Chem. 2005;70:2014–2020. doi: 10.1021/jo040259q. [DOI] [PubMed] [Google Scholar]
- 40.Lorance ED, Kramer WH, Gould IR. Barrierless Electron Transfer Bond Fragmentation Reactions. J Am Chem Soc. 2004;126:14071–14078. doi: 10.1021/ja030438+. [DOI] [PubMed] [Google Scholar]
- 41.Lorance ED, Kramer WH, Gould IR. Kinetics of Reductive N–O Bond Fragmentation: The Role of a Conical Intersection. J Am Chem Soc. 2002;124:15225–15238. doi: 10.1021/ja020768e. [DOI] [PubMed] [Google Scholar]
- 42.Mee JD, Heseltine DW, Taylor EC. Photolysis of N-alkoxy Quaternary Ammonium Salts. A Potential New Method of Aromatic Methoxylation. J Am Chem Soc. 1970;92:5814–5816. [Google Scholar]
- 43.Yagci Y, Kornowski A, Schnabel W. N-Alkoxy Pyridinium and N-Alkoxy Quinolinium Salts as Initiators for Cationic Photopolymerization. J Polym Sci: Part A: Polymer Chem. 1992;30:1987–1991. [Google Scholar]
- 44.Yagci Y, Schnabel W. Flash-Photolysis Experiments with Pyridinium Salts Regarding the Initiation of Cationic Polymerizations. Journal of Macromolecular Science Macro Rep. 1993;A30(Suppls. 3 & 4):175–182. [Google Scholar]
- 45.Huntley JJA, Nieman RA, Rose SD. Development and Investigation of a Novel Oxidative Pyrimidine Dimer Splitting Model. Photochem Photobiol. 1999;69:1–7. [Google Scholar]
- 46.Shukla D, Liu G, Dinnocenzo JP, Farid S. Controlling parameters for radical cation fragmentation reactions: Origin of the intrinsic barrier. Can J Chem. 2003;81:744–757. [Google Scholar]
- 47.Del Giacco T, Lanzalunga O, Mazzonna M, Mencarelli P. Structural and Solvent Effects on the C-S Bond Cleavage in Aryl Triphenylmethyl Sulfide Radical Cations. J Org Chem. 2012;77:1843–1852. doi: 10.1021/jo202418d. [DOI] [PubMed] [Google Scholar]
- 48.Baciocchi E, Bettoni M, Del Giacco T, Lanzalunga O, Mazzonna M, Mencarilli P. Structure and C–S Bond Cleavage in Aryl 1-Methyl-1-arylethyl Sulfide Radical Cations. J Org Chem. 2011;76:573–582. doi: 10.1021/jo102086f. [DOI] [PubMed] [Google Scholar]
- 49.Trecourt F, Mallet M, Mongin F, Queguiner G. Synthesis of substituted 8-methoxyquinolines by regioselective bromine-lithium exchange of 5,7-dihalo-8-methoxyquinolines and 7-bromo-8-methoxyquinoline. Synthesis. 1995;9:1159–1162. [Google Scholar]
- 50.Kant J, Popp FD. Reissert Compound Studies. XLV. The Phenanthridine Reissert Compound. J Heterocyclic Chem. 1984;21:425–427. [Google Scholar]
- 51.Gilman H, Eisch J. The Preparation and Dichromate Oxidation of Certain 6-Substituted Phenanthridines. J Am Chem Soc. 1957;79:4423–4426. [Google Scholar]
- 52.Ogata Y, Sawaki Y. The Formation of Peracids by the Perhydrolysis with Alkaline Hydrogen Peroxide. Tetrahedron. 1967;23:3327–3332. [Google Scholar]
- 53.Browers GB, Jr, McComb RB, Christensen RG, Schaffer R. High-purity 4-nitrophenol: Purification, characterization, and specifications for use as a spectrophotometric reference material. Clin Chem. 1980;26:724–729. [PubMed] [Google Scholar]
- 54.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A02. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 55.Shida T. Electronic Absorption Spectra of Radical Ions. Elsevier; Amsterdam: 1988. [Google Scholar]
- 56.Michl J, Bonacic-Koutecky V. Electronic Aspects of Organic Photochemistry. Wiley-Interscience; New York, NY: 1990. [Google Scholar]
- 57.Lewis FD, Bedell AM, Dykstra RE, Elbert JE, Gould IR, Farid S. Photochemical Generation, Isomerization, and Oxygenation of Stilbene Cation Radicals. J Am Chem Soc. 1990;112:8055–8064. [Google Scholar]
- 58.Gould IR, Ege D, Moser JE, Farid S. Efficiencies of photoinduced electron-transfer reactions: Role of the Marcus inverted region in return electron transfer within geminate radical-ion pairs. J Am Chem Soc. 1990;112:4290–4301. [Google Scholar]
- 59.Small RD, Scaiano JC. Absolute rates of hydrogen abstraction by tert-butoxy radicals. J Am Chem Soc. 1978;100:296–298. [Google Scholar]
- 60.Turro NJ. Modern Molecular Photochemistry. University Science Books; Sausolito, CA: 1991. [Google Scholar]
- 61.Görner H, Schulte-Frohlinde D. Trans-to-Cis Photoisomerization of the Quaternary Iodides of 4-Cyano- and 4-Nitro-4′-Azastilbene in Ethanol Solution: Singlet versus Triplet Mechanism. Chem Phys Lett. 1983;101:79–85. [Google Scholar]
- 62.Görner H. Photoinduced Electron Transfer vs. Trans -Cis Photoisomerization for Quaternary Salts of 4-Nitro-4′-azastilbene and Their Quinolinium Analogues. J Phys Chem. 1985;89:4112–4119. [Google Scholar]
- 63.Hubig SM, Kochi JK. Direct Observation of the Wheland Intermediate in Electrophilic Aromatic Substitution. Reversible Formation of Nitrosoarenium Cations. J Am Chem Soc. 2000;122:8279–8288. [Google Scholar]
- 64.Leventis N, Rawaswdeh AM, Zhang G, Elder IA, Leventis CS. Tuning the Redox Chemistry of 4-Benzoyl-N-methylpyridinium Cations through Para Substitution. Hammett Linear Free Energy Relationships and the Relative Aptitude of the Two-Electron Reduced Forms for H-Bonding. J Org Chem. 2002;67:7501–7510. doi: 10.1021/jo020489+. [DOI] [PubMed] [Google Scholar]
- 65.Fukuzumi S, Nishimne M, Ohkubo K, Tkachenko NV, Lemmetyinen H. Driving Force Dependence of Photoinduced Electron Transfer Dynamics of Intercalated Molecules in DNA. J Phys Chem B. 2003;107:12511–12518. [Google Scholar]
- 66.Fukuzumi S, Kitano T. Mechanisms of reductive methylation of NAD+ analogues by a trans-dimethylcobalt(III) complex. J Chem Soc Perkin Trans. 1991;2:41–45. [Google Scholar]
- 67.Gijzman OLJ, Kaufman F, Porter GJ. Oxygen quenching of aromatic triplet states in solution. Part 1. J Chem Soc (Far II) 1973;69:708–720. [Google Scholar]
- 68.Wirp C, Bendig J, Brauer HD. Singlet and triplet state quenching of aromatic cations by molecular oxygen. Ber Bunsen-Ges. 1997;10:961–967. [Google Scholar]
- 69.Kasha M, McGlynn SP. Molecular Electronic Spectroscopy. Ann Rev Phys Chem. 1956;7:403–424. [Google Scholar]
- 70.Turro NJ, Gould IR, Liu J, Jenks WS, Staabb H, Alt R. Investigations of the influence of molecular geometry on the spectroscopic and photochemical properties of.alpha.-oxo[1.n]para-cyclophanes (cyclophanobenzophenones) J Am Chem Soc. 1989;111:6378–6383. [Google Scholar]
- 71.Lim EC. Vibronic interactions and luminescence in aromatic molecules with non-bonding electrons. In: Lim EC, editor. Excited States. Academic Press; New York: 1977. pp. 305–336. [Google Scholar]
- 72.Dockery KP, Dinnocenzo JP, Farid S, Goodman JL, Gould IR, Todd WP. Nucleophile-Assisted Cleavage of Benzyltrialkylsilane Cation Radicals. J Am Chem Soc. 1997;119:1876–1883. [Google Scholar]
- 73.Turro NJ, Gould IR, Baretz BH. Absolute rate constants for decarbonylation of phenylacetyl and related radicals. J Phys Chem. 1983;87:531–532. [Google Scholar]
- 74.Mann CK, Barnes KK. Electrochemical Reactions in Nonaqueous Systems. Marcel Dekker; New York, NY: 1970. [Google Scholar]
- 75.Luo Y-R. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton, FL: 2007. [Google Scholar]
- 76.Otilia M, Yanez M, Eckert-Maksic M, Maksic ZB, Alkorta I, Elguero J. Periodic Trends in Bond Dissociation Energies. A Theoretical Study. J Phys Chem A. 2005;109:4359–4365. doi: 10.1021/jp050857o. [DOI] [PubMed] [Google Scholar]
- 77.Boyd RJ, Mark Glover JN, Pincock JA. A Theoretical Study of the Change in Homolytic Bond Dissociation Energy on Conversion of A-B to A-B+H. J Am Chem Soc. 1989;111:5152–5155. [Google Scholar]
- 78.Forslund LE, Kaltsoyannis N. Why is the F2 bond so weak? A bond energy decomposition analysis. New J Chem. 2003;27:1108–1114. [Google Scholar]
- 79.Candeias LP, Steenken S. Structure and acid-base properties of one-electron-oxidized deoxyguanosine, guanosine, and 1-methylguanosine. J Am Chem Soc. 1989;111:1094–1099. [Google Scholar]
- 80.Herkstroeter WG, Gould IR. Absorption spectroscopy of transient species. In: Rossiter V, Baetzold R, editors. Physical Methods of Chemistry Series. 2. Vol. 8. Wiley; New York, NY: 1993. pp. 225–319. [Google Scholar]