

Abstract
Diabetic kidney disease (DKD) is a progressive proteinuric renal disorder in patients with type 1 or type 2 diabetes mellitus. It is a common cause of end-stage kidney disease worldwide, particularly in developed countries. Therapeutic targeting of the renin–angiotensin system (RAS) is the most validated clinical strategy for slowing disease progression. DKD is paradoxically a low systematic renin state with an increased intrarenal RAS activity implicated in its pathogenesis. Angiotensin II (AngII), the main peptide of RAS, is not only a vasoactive peptide but functions as a growth factor, activating interstitial fibroblasts and mesangial and tubular cells, while promoting the synthesis of extracellular matrix proteins. AngII also promotes podocyte injury through increased calcium influx and the generation of reactive oxygen species. Blockade of the RAS using either angiotensin converting enzyme inhibitors, or angiotensin receptor blockers can attenuate progressive glomerulosclerosis in animal models, and slows disease progression in humans with DKD. In this review, we summarize the role of intrarenal RAS activation in the pathogenesis and progression of DKD and the rationale for RAS inhibition in this population.
Keywords: renin-angiotensin system, diabetic kidney disease, angiotensin II, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers
Introduction
Worldwide, more than 347 million people are suffering from diabetes mellitus (DM) and its complications which are projected to be the seventh leading cause of death by 2030.1 The incidence has doubled in the last two decades,2–4 and DM is the underlying cause of both micro- and macrovascular disorders such as diabetic kidney disease (DKD), retinopathy, coronary artery disease, and peripheral vascular disease. DKD is a progressive disease that affects patients with long-standing type 1 (insulin dependent) or type 2 (non-insulin-dependent) DM, and is functionally characterized by different degrees of albuminuria and chronic kidney disease.5 Associated histological structural changes include mesangial expansion, glomerular basement membrane thickening, glomerular sclerosis, and in advanced stages, tubulointerstitial fibrosis.5 In the US, it is estimated that 44% of all new end-stage renal disease (ESRD) cases are due to DKD.6 Interestingly, despite the increased incidence rate of DM, recent studies have shown that the incidence of DKD has dramatically decreased with the implementation of tight glycemic control and the use of renin–angiotensin system (RAS) blockers.7 The use of RAS blockers in DKD not only decreases albuminuria, but slows disease progression, which is only partially explained by their hemodynamic effects. The RAS cascade starts with the production of prorenin in the juxtaglomerular cells, which is cleaved into renin.8 Renin then cleaves angiotensinogen (AGT) to form angiotensin I (AngI) which later is converted by angiotensin converting enzyme (ACE) to the octapeptide angiotensin II (AngII). AngII in turn activates both angiotensin type 1 and 2 (AT1, AT2, respectively) receptors (Figure 1). AngII was previously considered to be the only active metabolite of this cascade, but evidence in the last few decades has shed light on the importance and biological role of each product including prorenin, renin, and AngI. In this review, we summarize the role of RAS in diabetic nephropathy and the mechanisms by which RAS inhibition slows disease progression.
Figure 1.
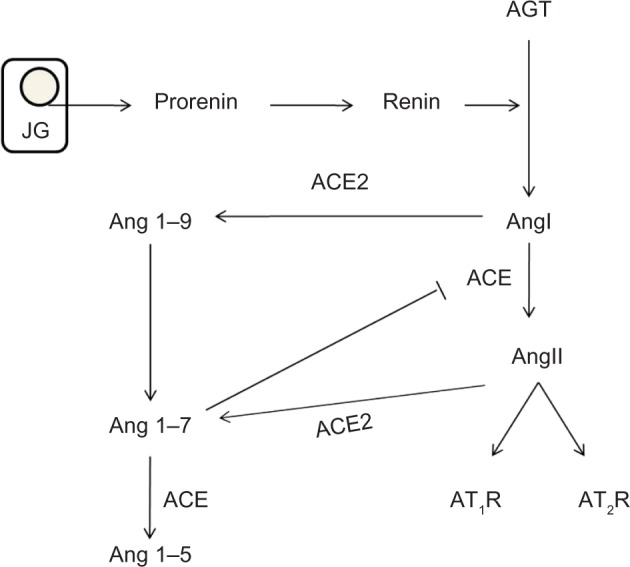
RAS cascade.
Notes: Juxtaglomerular (JG) cells in the kidney activate prorenin and secrete renin into the circulation. Renin hydrolyzes angiotensinogen (AGT) to angiotensin I (AngI) which is subsequently converted by angiotensin converting enzyme (ACE) to angiotensin II (AngII). AngII exerts its action by binding to angiotensin II Types I and II receptors (AT1R and AT2R). ACE2 catalyzes the conversion of AngI to angiotensin 1–9 (Ang 1–9) which is subsequently converted to angiotensin 1–7 (Ang 1–7) by ACE. Ang 1–7 also inhibits ACE and opposes AngII signaling.
Abbreviation: RAS, renin–angiotensin system.
Genetics of RAS
Host genetic variation in RAS components may predict the risk of developing DKD. It is well documented that there is an increased likelihood of developing DKD in family members who have siblings affected with the disease. Genetic studies have sought to determine variants and factors predisposing individuals to DKD.9–11 Some studies examined the role of the ACE gene genotype as a potential genetic risk factor and showed conflicting results. Investigators analyzed the insertion/deletion (I/D) and DD polymorphism in ACE gene in type 2 DM patients and concluded that DD polymorphisms are associated with greater risk of renal function decline, severe proteinuria, and worse outcomes.12–14 These findings were not seen in type 1 DM patients or in a large review of 19 studies that included type 1 and 2 patients.15–17 Male, and not female, patients with type 1 diabetes and the AA haplotype of the AT2 receptor gene on the X-chromosome had a lower glomerular filtration rate than those with the GT haplotype.18
Hemodynamic effects of RAS on the glomerular filter
Renal autoregulation enables the kidneys to maintain a stable intraglomerular pressure and glomerular filtration rate (GFR) as the mean arterial pressure varies from 80–160 mmHg.19 This is achieved through tubular–glomerular feedback (TGF) and myogenic response affecting the afferent arteriole’s tonicity, and angiotensin II (AngII) affecting the tonicity of both the afferent and the efferent arterioles.20 This autoregulatory mechanism is disrupted in different diseases, including hypertension, chronic kidney disease, and diabetes (Figure 2). Early in the course of DKD, an elevation in the GFR is seen, accompanied later by glomerular and renal hypertrophy along with increased intraglomerular pressure. This in turn increases the shear stress on the glomerular capillary wall resulting in progressive and sustained renal damage.21
Figure 2.

Hemodynamic changes in diabetic kidney disease (DKD).
Notes: Early in the course of diabetes mellitus (DM), there is volume expansion and an increase in sodium (Na) reabsorption from the proximal tubule (PT) mediated by increased insulin production, and the augmented sodium–glucose cotransport leading to increased tubuloglomerular feedback (TGF) mediated afferent arteriolar (AA) dilatation and increased flow rate. This is accompanied by an increased production of local angiotensin II (AngII) augmenting the dilatory effects of TGF on AA. Increased local AngII will cause mesangial cell constriction and efferent arteriolar (EA) vasoconstriction, causing along with the increased flow rate an increased intraglomerular pressure (IGP). This will increase sheer stress and chronically worsens kidney damage.
A key mechanistic factor in this hyperfiltration response in DKD is RAS mediated glomerular afferent arteriole dilatation which increases renal blood flow and intraglomerular pressure.22 Other contributors to increased afferent arteriole dilatation include insulin-like growth factor I (IGF-1),23 atrial natriuretic peptide,24 and advanced glycation end products, the latter also influencing the micro- and macrovascular changes seen in DM.25 Additionally in DM, there is an augmented sodium–glucose cotransport leading to volume expansion, along with hyperinsulinemia and local AngII stimulating sodium reabsorption in the proximal tubule.26–28 This increased proximal sodium reabsorption activates the tubuloglomerular feedback mechanism in the macula densa, and can then raise the GFR via dilatation of the afferent arteriole.26
AngII has vasoconstrictive effects on the afferent and the efferent arterioles as well as the interlobular arteries, mediated in part by the local generation of thromboxane A2. The constrictive efficacy is greatest at the efferent arteriole due to its smaller basal diameter. This leads to a net reduced renal blood flow, increased intraglomerular pressure, and an increase in GFR.29 Concurrently, AngII stimulates the release of vasodilator prostaglandins from the glomeruli and constricts glomerular mesangial cells, thereby lowering the surface area available for filtration. AngII also sensitizes the afferent arteriole to the constricting signal of tubuloglomerular feedback.30–33
RAS activation and its importance in DKD
The beneficial effects of RAS blockers in slowing the progression of diabetic nephropathy have been well documented,34–36 despite the low systemic renin state in DKD. This phenomenon is thought to be reflecting the activated intrarenal renin system or increased intrarenal sensitivity to AngII.37 Likely reflecting different stages of kidney disease and different measurement methods (renin activity, protein level, RNA expression, serum potassium and bicarbonate, and immunohistochemistry and fluorescence), human clinical data measuring RAS activation have been conflicting in DKD.38–41 Experimental models however tend to show a consistent increase in RAS activation.42–46 This activation in RAS involves all factors and steps in the cascade and takes place in a paracrine fashion. Table 1 summarizes the deleterious effects of RAS cascade activation in DKD.
Table 1.
The deleterious effects of RAS cascade activation in DKD
| Prorenin |
| Glomerulosclerosis |
| Renal fibrosis |
| Renin |
| Nonenzymatic enhancement of angiotensin II synthesis |
| Mesangial hypertrophy |
| Renal fibrosis |
| Angiotensin II |
| Increased intraglomerular pressure |
| Water and solute retention |
| Mesangial cell proliferation and hypertrophy |
| Renal fibrosis |
| Glomerular and tubolointerstitial microinflammation |
| Insulin resistance |
| Decreased AT2R synthesis |
Abbreviations: AT2R, angiotensin type 2 receptor; RAS, renin–angiotensin system; DKD, diabetic kidney disease.
Prorenin
The importance of prorenin and its biological role beyond the formation of renin was first established in the 1980s when it was found that its levels, despite the low systemic renin levels and activity, are increased in diabetic subjects, and correlate with the presence of retinopathy and nephropathy.39 In DM, prorenin represents 95% of circulating renin, and the mechanism by which prorenin is implicated in the development of DKD and retinopathy in this population is under investigation.47 It binds to the prorenin receptor (PRR) and, independently of AngII, triggers intracellular signaling and the activation of the mitogen-activated protein (MAP) kinases ERK1/2, leading to upregulation of TGFβ1, PAI1, collagens, fibronectin, and cyclooxygenase-2.48–52 This raises the possibility that high prorenin levels play a role in diabetic nephropathy by stimulating PRR and inducing profibrotic protein syntheses.8 Indeed, transgenic rats overexpressing human PRR develop proteinuria, and a slowly progressive glomerulosclerosis and kidney damage independent of AngII.42 Interestingly, inducible overexpression of prorenin does not result in glomerulosclerosis suggesting that prorenin may augment the fibrosis rather than causing it.43,53 Prorenin also participates in the generation of AngI. Prorenin binding to PRR induces a conformational change involving unfolding of the peptide from the enzymatic cleft so the cleft is now accessible to AGT and generates AngI.54,55
ACE
The best evidence for intrarenal renin production and activity in DM comes from spontaneous or streptozotocin (STZ)-induced diabetes animal models.44,56 In spite of the suppressed or normal plasma renin activity, the intrarenal Ren mRNA and protein levels, most significantly in the proximal tubule, are increased in DM.44 The total renal ACE activity is significantly reduced in DM rats, with specific redistribution in diabetic kidneys.56 While proximal tubule ACE activity is reduced, ACE staining intensity is enhanced in diabetic glomeruli and renal vasculature. This suggests a role for glomerular ACE in mediating nephron injury, possibly by increasing local intraglomerular AngII formation.
Renin
Early diabetes causes a significant stimulation of the proximal tubule renin mRNA expression.44 Renin, independent of its enzymatic action to enhance AngII synthesis, directly increases production of transforming growth factor β (TGF-β), the fibrogenic cytokine.48 Renin binds to its specific receptor on the cell surface of mesangial cells,57 leading to hypertrophy,58 and enhanced efficiency of angiotensinogen cleavage by renin, thereby unmasking prorenin catalytic activity.57 The renin receptor was also localized in the sub-endothelium of the renal arteries suggesting that renin has a novel receptor-mediated action that could play a role in renal fibrosis.59 In podocytes, high glucose is shown to cause increased AngII generation through increasing renin mRNA expression with a concomitant increase in PRR and hence augmenting the conversion from AGT to AngI.60
Angiotensinogen
The high extracellular glucose in DM stimulates the synthesis of AGT in a concentration dependent manner and increases the expression of its gene (Agt) in the proximal tubule of the kidneys.56,59 This increased AGT protein level in the proximal tubules is due to many factors, including the effects of high extracellular glucose on Agt expression via reactive oxygen species45 and the direct effect of glucose on its promoter. A glucose response element has been located on the Agt promoter.46
Angiotensin II
As previously mentioned, in diabetic nephropathy there is an increase in the generation of the intrarenal AngII despite the systemic suppression of RAS. The deleterious effects of this rise in AngII go beyond the hemodynamic changes to involve insulin resistance, growth promotion, and tubular damage. One of the most important roles of AngII in DKD is its association with volume expansion through water and Na reabsorption. It activates the Na+–H+ antiporter in the luminal membrane through stimulation of an inhibitory G protein that decreases cyclic AMP (adenosine monophosphate) generation, minimizing the normally suppressive effect of cyclic AMP on Na+–H+ exchange.61 AngII stimulates phosphatidylinositol turnover, resulting in the generation of protein kinase C.61,62 It also increases the secretion of aldosterone from the adrenal cortex enhancing Na+ transport in the cortical collecting tubule.63 AngII inhibits proteinase activity in the proximal tubule and causes mesangial cell expansion via decreasing the activity of plasminogen activator. AngII mediated TGF-β1 upregulation and vascular endothelial growth factor release from the glomerular epithelial and mesangial cells contribute to mesangial matrix expansion.64 Renal fibroblasts express AT1 receptor and respond to AngII stimuli by cell proliferation, matrix expansion, and synthesis of fibronectin, by a TGF-β-dependent mechanism.65 Microinflammation of the glomeruli and tubulointerstitial regions and subsequent extracellular matrix expansion are common pathways for the progression of DKD.66 AngII activates inflammatory cells by direct chemotaxis including osteopontin (OPN), RANTES and the production of other proinflammatory mediators, including MCP-1 and TGFβ. It activates protein kinase C, protein tyrosine kinases (PTK), mitogen-activating protein kinases (MAPK), extracellular signal-regulated kinase (ERK), c-Jun amino terminal kinase (JNK), p38 MAP kinase (p38 MAPK), and the activator protein-1 (AP-1). These factors are implicated and involved in proliferation, differentiation, fibrosis, and inflammation processes.65 AngII has also been implicated in insulin resistance. AngII inhibits insulin-mediated GLUT4 translocation in this skeletal muscle model through a transient activation of ERK1/2 inhibiting insulin receptor substrate 1/2 (IRS-1/2) and through a direct inhibitory nitration of Akt. It induces tyrosine phosphorylation of IRS-1 by Janus kinase 2 associated with AT1 receptor stimulation which attenuates insulin-induced activation of phosphatidylinositol-3-kinase associated with IRS-1, leading to decreased insulin sensitivity.67 Treatment with the angiotensin II receptor blocker (ARB) candesartan increases renal insulin receptor expression in STZ-induced diabetic rat models and the insulin resistant Zucker rats irrespective of insulin levels.68
It appears that high glucose directly stimulates podocytes to increase AngII production independent of ACE activity, and increases AT1 receptor levels promoting the deleterious effects of AngII on podocytes.60 AngII directly promotes podocyte injury via release of calcium from intracellular stores as well as influx from the extracellular space.69 Finally, AngII activates enzyme systems like Nox4 that use NADPH oxidases as substrates for superoxide generation.70 These mechanisms contribute to proteinuria and progressive renal disease.
Aldosterone
Aldosterone secretion from the adrenal cortex is mainly mediated by AngII and plasma potassium concentration. This secretion might result in a vicious cycle of local RAS stimulation causing further RAS activation and aldosterone generation.71 The effects of aldosterone go beyond solute and water retention, vasoconstriction, and subsequently, increased blood pressure. In the diabetic kidney, aldosterone activity contributes to tubulointerstitial fibrosis and inflammation, key features of disease progression. Aldosterone increases extracellular matrix production through enhanced TGF-β1 and type IV collagen gene expression.72 In diabetic rats, aldosterone induces glomerular and tubulointerstitial macrophage infiltration through TGFβ.73 Aldosterone also stimulates ROS production through activation of NADPH oxidase in mesangial cells.74 Systemically, higher circulating levels of aldosterone are associated with insulin resistance.75 There is a need for large prospective trials to determine the efficacy of aldosterone inhibition in the treatment of DKD.
ACE2 and angiotensin 1–7 (Ang 1–7)
Angiotensin converting enzyme 2 (ACE2) is an enzyme related to ACE, expressed in the intrarenal endothelium and the tubular epithelial cell.76 It hydrolyzes AngI to Ang 1–9, which is subsequently cleaved by ACE into Ang 1–7. ACE2 also catalyzes the conversion of AngII to Ang 1–7 (the major pathway of Ang 1–7 production). ACE2 does not produce AngII, or metabolize bradykinin (BK), and is not inhibited by ACE inhibition.76,77 Cardiac ischemia studies suggest that ACE2 and ACE have counterbalancing effects. Animal models with disrupted ACE2 gene showed significant cardiac abnormalities including decreased contractility and increased damage, probably mediated by the upregulation of hypoxia-induced genes. These histological findings were abrogated by concomitant ACE gene ablation.78
Ang 1–7 causes intrarenal vasodilation and counterbalances the vasoconstrictor effects of AngII via stimulation of NO and prostaglandins, and synergistically acts with BK.77 The effects are not limited to hemodynamics. In animal models of DKD, Ang 1–7 treatment ameliorates mesangial expansion and renal fibrosis, and reduces oxidative stress, inflammation, and lipotoxicity through blocking the activation of MAPKs by AngII and selective inhibition of glucose-stimulated protein synthesis, TGF-β1/Smad3- and VEGF-mediated pathways.79,80 There is a dynamic reciprocal interaction between Ang 1–7 and ACE; Ang 1–7 inhibits ACE by 30%–70%, and at the same time is degraded by it. In addition to its vasodilatory and natriuretic actions, Ang (1–7) has an antiproliferative effect on vascular smooth muscle cells.77
Types of RAS blockades and their pleiotropic effects
Direct renin inhibitors (DRI), angiotensin converting enzyme inhibitors (ACEi), and angiotensin II receptor blockers (ARB) are available to treat hypertension and proteinuric kidney diseases. The use of these medications results in either decreased AngII (DRI and ACEi) or blocking its receptor AT1 (ARB). To better understand the differences in effects and actions of these medications, attention should first be paid to AngII receptor distribution. AT1 receptors are distributed throughout the kidney compartments. They are localized in the afferent and efferent arterioles, vascular smooth muscle cells, podocytes, glomerular mesangial and macula densa cells, the proximal tubule cell brush border, basolateral membranes, thick ascending limb epithelia, distal tubules, and the cortical collecting ducts.81 The AT2 receptor is localized in the afferent arteriole, glomerular endothelial and mesangial cells, proximal tubule cells, and interstitial cells.81,82 AT1 receptor stimulation leads to renal arterial vasoconstriction, tubule epithelial sodium reabsorption, augmentation of tubulo–glomerular feedback sensitivity, and inhibition of pressure-natriuresis.77 Conversely, AT2 receptor activation exerts the opposite effects with respect to cardiovascular hemodynamics and cell growth. AT2 receptor activation stimulates BK and NO production, vasodilatation, and modulates the vasoconstrictor action mediated by the AT1 receptor through endothelium-dependent vasodilatation.83
DRI
AngII has a negative feedback on RAS, and during ACEi therapy, there is a rise in plasma renin activity (PRA) and concentration (PRC) as a result of decreased renal parenchymal AngII. This led to the development of DRIs for monotherapy or in combination with ACEi therapy. They have been shown to have a similar antiproteinuric and antihypertensive effects, but without significant cardiovascular benefits.84,85
ACEi
ACEi is probably one of the most studied and used class of antihypertensive medication. Since the late 1970s and the use of captopril, ACEi has become the cornerstone in treating hypertensive patients with proteinuria. It inhibits ACE activity by blocking the conversion from AngI to AngII, and inhibits BK degradation. Bradykinin, via its B2 receptors, stimulates NO, cGMP, prostaglandin E2, and prostacyclin. Therefore, ACEi therapy not only inhibits AngII production but also increases the production of the vasodilatory factors.86 ACEi therapy also inhibits the ACE mediated degradation of Ang 1–7 augmenting the renoprotective effects of Ang 1–7. The pleiotropic effects of ACEi include anti-inflammatory and antioxidant,87 antithrombotic, and profibrinolytic activities.88 ACEi is also important in significantly improving arterial compliance through cytoprotection of vascular endothelium,89 along with better regression of left ventricular hypertrophy compared to beta blockers.90
ARB
The major clinical and physiological differences between ACEi and ARBs are related to activation of AT2 receptors. ARBs selectively block AT1 receptor without directly affecting the synthesis of AngII and subsequent AT2 activation. Some of the beneficial effects of using ARB are thought to be due to selective inhibition of AT1 and the concomitant renoprotective stimulation of AT2 receptors.
It has been proposed that ARBs might have a greater potential in preventing renal interstitial fibrosis compared to ACEi mediated by AT2 receptor proapoptotic properties.91 This is supported by the fact that renal interstitial fibrosis after ureteral obstruction is enhanced in rats with either AT2 deletion, mutation, or pharmacologic blockade.92,93
RAS blockers and renal outcome studies
The effects and roles of RAS blockers in the progression of kidney disease are probably mostly seen due to their hemodynamic effects and blood pressure control. It is well known that blood pressure control is considered a cornerstone in the management and prevention of chronic kidney diseases.94 Thus, different studies have evaluated the hypothesis whether a stricter lower blood pressure control will confer a better renal outcome, and initial societies guidelines95–97 have recommended a lower blood pressure target (<130 mmHg systolic) for patients with CKD and significant proteinuria, based on the findings of post hoc analysis of major trials like MDRD98–100 AASK,101,102 and a patient level meta-analysis of eleven randomized trials.103 These studies mostly included nondiabetic kidney disease patients, and thus it is hard to extrapolate their findings and assume that DKD patients will respond similarly. To address this issue, post hoc analysis of RENAAL104 and Irbesartan Diabetic Nephropathy Trial (IDNT)105 found that higher BP targets (140–149 mmHg for RENAAL, and >145 mmHg for IDNT) resulted in higher risk of composite outcome. These studies also found that a tighter BP control (systolic <120 mmHg) resulted in increased mortality risk. It is important to mention that these studies were post hoc analyses that included a RAS blockers arm and a placebo arm, and were not designed to answer the question whether achieving a lower BP goal (<130 mmHg systolic) by increasing RAS blockers dose will have any benefit in this population. At the same time, these data were later challenged and questioned by the findings of increased risk of adverse outcomes (cardiac events, stroke, and falls) in different populations (especially elderly) and current guidelines have raised the target to 140 mmHg systolic.106–109
Several studies have evaluated the role of ACEi in halting DKD progression. The Captopril-Diabetes study evaluated type 1 diabetic patients with nephrotic range proteinuria and showed a significant reduction in proteinuria along with lower GFR decline rate compared to placebo.110,111 The ADVANCE trial compared perindopril–indapamide combination to placebo in over 11,000 patients with DM, and found a significantly lower microalbuminuria, and lower worsening macroalbuminuria incidence rate in all groups studied, including normotensive patients.112,113 The importance and significance of the effects of proteinuria reduction on renal outcomes was questioned since the treatment groups had better blood pressure control, which might have played a big role in preventing disease progression. Later studies showed that lowering proteinuria in patients with diabetic nephropathy (regardless of the agent used) is associated with improved outcomes and a decreased risk of ESRD.114–117
The IDNT and the RENAAL trial studied ARBs therapy in patients with DKD and found similar beneficiary results. ARB therapy was superior to placebo and amlodipine in decreasing proteinuria, lower rates of doubling creatinine, or the development of ESRD and mortality.34,118
No significant difference was found between ACEi and ARB in patients with early DKD. In the DETAIL trial, both groups (enalapril vs telmisartan) had similar 5-year GFR decline, annual changes in the GFR, blood pressure, serum creatinine, urinary albumin excretion, ESRD, cardiovascular events, and mortality.119
Trials evaluating dual RAS blockades and controversies
It was previously demonstrated that ACEi/ARB combination therapy decreases proteinuria, more than monotherapy with either drug.120–122 These studies found no difference in adverse events in the combination group compared to monotherapy, but bore two major limitations that made their results and conclusion debatable. The sample sizes in these studies were small and patients were followed for a maximum of 16 weeks. Large scale randomized controlled trials conducted in different patient populations over a longer follow-up period failed to show any benefit on hard outcomes, including 50% drop in GFR, ESRD, and mortality.123,124 Patients on dual therapy had significantly higher adverse events, including hyperkalemia and hospitalization for acute kidney injury (AKI), as seen in the VA NEPHRON-D study,123 and even a statistically nonsignificant trend of higher incidence of chronic dialysis, doubling in serum creatinine and death, along with a statistically significant higher incidence rate of AKI requiring dialysis, hypotension, and hyperkalemia (ONTARGET trial).124 The results from studies evaluating the benefit of adding DRI (Aliskiren) to either ACEi or ARB in patients with DKD showed similar trend.125,126 Initially, the AVOID trial studied the combination between DRI and ARB in a group of 599 patients for 6 months and concluded that aliskiren may have renoprotective effects that are independent of its blood pressure-lowering effect in patients with hypertension and DKD, without a significant increase in adverse events.125 Later, the ALTITUDE investigators evaluated 8,561 patients for a median follow-up of 32.9 months and found that the combination therapy increased the risk of composite primary endpoint (ESRD, doubling of serum creatinine, renal death, cardiovascular death, cardiac arrest, heart failure, nonfatal myocardial infarction, or nonfatal stroke), along with hyperkalemia.126
As many as 40% of subjects treated with RAS blockers (ACEi or ARBs) have above normal limits levels of aldosterone (aldosterone breakthrough).127,128 This is thought to be due to different mechanisms that include: stimulation from tissue not inhibited by ACEi/ARBs, AngII generation via pathway not requiring ACE, or other stimuli such as high serum potassium concentrations.129 Adding spironolactone (aldosterone receptor antagonist) to ACEi in DN patients with aldosterone breakthrough can decrease proteinuria.128 This study, though promising and reporting no significant change in serum potassium, was limited to a small number of patients and was of short duration. Similar findings on proteinuria were noted in different small and short duration randomized controlled studies using a combination therapy of spironolactone and either ACEi or ARBs.130–135 Not surprisingly, all studies reported either a trend or a statistically significant higher levels of serum potassium, and greater eGFR decline in the combination therapy group. Similar findings were noted in adding eplerenone (aldosterone antagonist) to ACEi or ARBs in patients with DN and preserved renal function.136
There are no long-term data regarding the beneficial effects of adding aldosterone antagonist, and the current literature suggests possible increased risk of adverse events.
Adverse events
The major adverse events in patients using RAS are hypotension, hyperkalemia, angioedema, and reduction in GFR. ACEi can result in dry cough in up to 20% of cases, while ARB is associated with cough in up to 9.9% of cases.137,138 Recently there is an emerging concern about the development of sprue-like enteropathy in patients using olmesartan.139 Patients with bilateral renal artery stenosis, polycystic kidney disease, and advanced kidney disease may sustain a significant decrease in GFR, and in some cases AKI. The intrarenal perfusion pressure in these cases is already reduced, and patients are highly dependent on AngII to maintain a stable intraglomerular pressure and GFR. Similar but modest intrarenal hemodynamic changes are seen in elderly and DKD patients on RAS, increasing their risk of sustaining AKI in the setting of volume depletion, contrast agents, and major stressors like surgery.140–142
Summary and recommendations
Intrarenal activation of RAS in DKD plays a major role in disease pathophysiology and is implicated in its progression. The intrarenal generation is increased despite the systemic RAS suppression. This activation further downregulates the expression of both AT1 and AT2 receptors contributing to early diabetic nephropathy. This reduces AT2 receptor mediated beneficial counterregulatory actions. The negative impact of increased AngII on the kidneys goes beyond the regulation of intraglomerular pressure. It leads to increased insulin resistance, solute retention, stimulation of TGF-β secretion, increased synthesis, and decreased degradation of matrix proteins, all leading to matrix accumulation, kidney fibrosis, and further kidney damage. The use of ACEi and ARBs has significantly changed the incidence of ESRD and has provided renal protection through their effects on RAS, hemodynamic changes, and other pleiotropic effects that include anti-inflammatory, antioxidant, antithrombotic effects, and profibrinolytic activities. Even though a combination therapy of both ACEi and ARBs has decreased protein excretion in DKD patients, it is associated with worse renal outcomes compared to monotherapy. Studies evaluating other components of the RAS, including ACE2 and Ang 1–7 along with genetic animal models are warranted.
Studies to date have found no difference in renal outcomes between ACEi and ARBs, thus all hypertension patients with proteinuric kidney disease should be started on either ACEi or ARBs to halt disease progression. In our opinion, we believe that the evidence and the risk of adverse events overweigh the benefits of RAS blockers combination therapy. This therapy, though resulting in decreased proteinuria, carries a significant risk of adverse outcomes including hyperkalemia, increased hospitalization events, acute kidney injury, and a faster decline in renal function, and should be avoided. Another important issue we as clinicians face on a daily basis is the increase in serum potassium level in patients on RAS blockers. Little is available in the literature in regards to the use of sorbitol free sodium polystyrene sulfonate (Kayexalate), or other resins, as a secondary prevention method. A small study reports that this approach is effective and well tolerated.143 We believe that all efforts to keep patients on the RAS blockers should be implemented, and other means and therapies aiming at decreasing serum potassium levels should be considered before discontinuing RAS blockers in cases of mild/non-life-threatening hyperkalemia. To date, no studies evaluating the long-term effects of such an approach are available, and the need for further study is warranted. There is also little evidence to guide decision making regarding the use of RAS blockers in predialysis advanced kidney disease patients, eg, eGFR <30 mL/min. We believe that each patient should be evaluated individually, and a risk and benefit evaluation to aid the decision should be made.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Yacoub R, Lee K, He JC. The role of SIRT1 in diabetic kidney disease. Front Endocrinol. 2014;5:166. doi: 10.3389/fendo.2014.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fox CS, Pencina MJ, Meigs JB, Vasan RS, Levitzky YS, D’Agostino RB., Sr Trends in the incidence of type 2 diabetes mellitus from the 1970s to the 1990s: the Framingham Heart Study. Circulation. 2006;113(25):2914–2918. doi: 10.1161/CIRCULATIONAHA.106.613828. [DOI] [PubMed] [Google Scholar]
- 3.Hu D, Fu P, Xie J, et al. Increasing prevalence and low awareness, treatment and control of diabetes mellitus among Chinese adults: the InterASIA study. Diabetes Res Clin Pract. 2008;81(2):250–257. doi: 10.1016/j.diabres.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y, Wang L, He J, et al. Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310(9):948–959. doi: 10.1001/jama.2013.168118. [DOI] [PubMed] [Google Scholar]
- 5.Tervaert TW, Mooyaart AL, Amann K, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21(4):556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 6.Mauer M, Zinman B, Gardiner R, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med. 2009;361(1):40–51. doi: 10.1056/NEJMoa0808400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bojestig M, Arnqvist HJ, Hermansson G, Karlberg BE, Ludvigsson J. Declining incidence of nephropathy in insulin-dependent diabetes mellitus. N Engl J Med. 1994;330(1):15–18. doi: 10.1056/NEJM199401063300103. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen G, Muller DN. The biology of the (pro)renin receptor. J Am Soc Nephrol. 2010;21(1):18–23. doi: 10.1681/ASN.2009030300. [DOI] [PubMed] [Google Scholar]
- 9.Krolewski AS. Genetics of diabetic nephropathy: evidence for major and minor gene effects. Kidney Int. 1999;55(4):1582–1596. doi: 10.1046/j.1523-1755.1999.00371.x. [DOI] [PubMed] [Google Scholar]
- 10.Trevisan R, Viberti G. Genetic factors in the development of diabetic nephropathy. J Lab Clin Med. 1995;126(4):342–349. [PubMed] [Google Scholar]
- 11.Satko SG, Langefeld CD, Daeihagh P, Bowden DW, Rich SS, Freedman BI. Nephropathy in siblings of African Americans with overt type 2 diabetic nephropathy. Am J Kidney Dis. 2002;40(3):489–494. doi: 10.1053/ajkd.2002.34888. [DOI] [PubMed] [Google Scholar]
- 12.Jeffers BW, Estacio RO, Raynolds MV, Schrier RW. Angiotensin-converting enzyme gene polymorphism in non-insulin dependent diabetes mellitus and its relationship with diabetic nephropathy. Kidney Int. 1997;52(2):473–477. doi: 10.1038/ki.1997.355. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida H, Kuriyama S, Atsumi Y, et al. Angiotensin I converting enzyme gene polymorphism in non-insulin dependent diabetes mellitus. Kidney Int. 1996;50(2):657–664. doi: 10.1038/ki.1996.362. [DOI] [PubMed] [Google Scholar]
- 14.Kuramoto N, Iizuka T, Ito H, et al. Effect of ACE gene on diabetic nephropathy in NIDDM patients with insulin resistance. Am J Kidney Dis. 1999;33(2):276–281. doi: 10.1016/s0272-6386(99)70300-8. [DOI] [PubMed] [Google Scholar]
- 15.Boright AP, Paterson AD, Mirea L, et al. Genetic variation at the ACE gene is associated with persistent microalbuminuria and severe nephropathy in type 1 diabetes: the DCCT/EDIC Genetics Study. Diabetes. 2005;54(4):1238–1244. doi: 10.2337/diabetes.54.4.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hadjadj S, Tarnow L, Forsblom C, et al. Association between angiotensin-converting enzyme gene polymorphisms and diabetic nephropathy: case-control, haplotype, and family-based study in three European populations. J Am Soc Nephrol. 2007;18(4):1284–1291. doi: 10.1681/ASN.2006101102. [DOI] [PubMed] [Google Scholar]
- 17.Kunz R, Bork JP, Fritsche L, Ringel J, Sharma AM. Association between the angiotensin-converting enzyme-insertion/deletion polymorphism and diabetic nephropathy: a methodologic appraisal and systematic review. J Am Soc Nephrol. 1998;9(9):1653–1663. doi: 10.1681/ASN.V991653. [DOI] [PubMed] [Google Scholar]
- 18.Pettersson-Fernholm K, Frojdo S, Fagerudd J, et al. The AT2 gene may have a gender-specific effect on kidney function and pulse pressure in type I diabetic patients. Kidney Int. 2006;69(10):1880–1884. doi: 10.1038/sj.ki.5000348. [DOI] [PubMed] [Google Scholar]
- 19.Palmer BF. Renal dysfunction complicating the treatment of hypertension. N Engl J Med. 2002;347(16):1256–1261. doi: 10.1056/NEJMra020676. [DOI] [PubMed] [Google Scholar]
- 20.Badr KF, Ichikawa I. Prerenal failure: a deleterious shift from renal compensation to decompensation. N Engl J Med. 1988;319(10):623–629. doi: 10.1056/NEJM198809083191007. [DOI] [PubMed] [Google Scholar]
- 21.Bakris GL, Slataper R, Vicknair N, Sadler R. ACE inhibitor mediated reductions in renal size and microalbuminuria in normotensive, diabetic subjects. J Diabetes Complications. 1994;8(1):2–6. doi: 10.1016/1056-8727(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 22.Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med. 2007;357(8):797–805. doi: 10.1056/NEJMra064398. [DOI] [PubMed] [Google Scholar]
- 23.Hirschberg R, Brunori G, Kopple JD, Guler HP. Effects of insulin-like growth factor I on renal function in normal men. Kidney Int. 1993;43(2):387–397. doi: 10.1038/ki.1993.57. [DOI] [PubMed] [Google Scholar]
- 24.Anderson S, Vora JP. Current concepts of renal hemodynamics in diabetes. J Diabetes Complications. 1995;9(4):304–307. doi: 10.1016/1056-8727(95)80028-d. [DOI] [PubMed] [Google Scholar]
- 25.Vlassara H. Protein glycation in the kidney: role in diabetes and aging. Kidney Int. 1996;49(6):1795–1804. doi: 10.1038/ki.1996.270. [DOI] [PubMed] [Google Scholar]
- 26.Hannedouche TP, Delgado AG, Gnionsahe DA, Boitard C, Lacour B, Grunfeld JP. Renal hemodynamics and segmental tubular reabsorption in early type 1 diabetes. Kidney Int. 1990;37(4):1126–1133. doi: 10.1038/ki.1990.95. [DOI] [PubMed] [Google Scholar]
- 27.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol. 1999;10(12):2569–2576. doi: 10.1681/ASN.V10122569. [DOI] [PubMed] [Google Scholar]
- 28.Yanagawa N. Potential role for local luminal angiotensin II in proximal tubule sodium transport. Kidney Int Suppl. 1991;32:S33–S36. [PubMed] [Google Scholar]
- 29.Denton KM, Fennessy PA, Alcorn D, Anderson WP. Morphometric analysis of the actions of angiotensin II on renal arterioles and glomeruli. Am J Physiol. 1992;262(3 Pt 2):F367–F372. doi: 10.1152/ajprenal.1992.262.3.F367. [DOI] [PubMed] [Google Scholar]
- 30.Stahl RA, Paravicini M, Schollmeyer P. Angiotensin II stimulation of prostaglandin E2 and 6-keto-F1 alpha formation by isolated human glomeruli. Kidney Int. 1984;26(1):30–34. doi: 10.1038/ki.1984.130. [DOI] [PubMed] [Google Scholar]
- 31.Oliver JA, Pinto J, Sciacca RR, Cannon PJ. Increased renal secretion of norepinephrine and prostaglandin E2 during sodium depletion in the dog. J Clin Invest. 1980;66(4):748–756. doi: 10.1172/JCI109912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vallotton MB, Gerber-Wicht C, Dolci W, Wuthrich RP. Interaction of vasopressin and angiotensin II in stimulation of prostacyclin synthesis in vascular smooth muscle cells. Am J Physiol. 1989;257(5 Pt 1):E617–E624. doi: 10.1152/ajpendo.1989.257.5.E617. [DOI] [PubMed] [Google Scholar]
- 33.Ichikawi I, Harris RC. Angiotensin actions in the kidney: renewed insight into the old hormone. Kidney Int. 1991;40(4):583–596. doi: 10.1038/ki.1991.249. [DOI] [PubMed] [Google Scholar]
- 34.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 35.Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet. 2000;355(9200):253–259. No authors listed. [PubMed] [Google Scholar]
- 36.ACEInhibitors in Diabetic Nephropathy Trialist Group Should all patients with type 1 diabetes mellitus and microalbuminuria receive angiotensin-converting enzyme inhibitors? A meta-analysis of individual patient data. Ann Intern Med. 2001;134(5):370–379. doi: 10.7326/0003-4819-134-5-200103060-00009. [DOI] [PubMed] [Google Scholar]
- 37.Price DA, Porter LE, Gordon M, et al. The paradox of the low-renin state in diabetic nephropathy. J Am Soc Nephrol. 1999;10(11):2382–2391. doi: 10.1681/ASN.V10112382. [DOI] [PubMed] [Google Scholar]
- 38.Gurley SB, Coffman TM. The renin-angiotensin system and diabetic nephropathy. Semin Nephrol. 2007;27(2):144–152. doi: 10.1016/j.semnephrol.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Luetscher JA, Kraemer FB, Wilson DM, Schwartz HC, Bryer-Ash M. Increased plasma inactive renin in diabetes mellitus. A marker of microvascular complications. N Engl J Med. 1985;312(22):1412–1417. doi: 10.1056/NEJM198505303122202. [DOI] [PubMed] [Google Scholar]
- 40.Lai KN, Leung JC, Lai KB, To WY, Yeung VT, Lai FM. Gene expression of the renin-angiotensin system in human kidney. J Hypertens. 1998;16(1):91–102. doi: 10.1097/00004872-199816010-00014. [DOI] [PubMed] [Google Scholar]
- 41.Tuck ML, Sambhi MP, Levin L. Hyporeninemic hypoaldosteronism in diabetes mellitus. Studies of the autonomic nervous system’s control of renin release. Diabetes. 1979;28(3):237–241. doi: 10.2337/diab.28.3.237. [DOI] [PubMed] [Google Scholar]
- 42.Kaneshiro Y, Ichihara A, Sakoda M, et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol. 2007;18(6):1789–1795. doi: 10.1681/ASN.2006091062. [DOI] [PubMed] [Google Scholar]
- 43.Peters B, Grisk O, Becher B, et al. Dose-dependent titration of prorenin and blood pressure in Cyp1a1ren-2 transgenic rats: absence of prorenin-induced glomerulosclerosis. J Hypertens. 2008;26(1):102–109. doi: 10.1097/HJH.0b013e3282f0ab66. [DOI] [PubMed] [Google Scholar]
- 44.Zimpelmann J, Kumar D, Levine DZ, et al. Early diabetes mellitus stimulates proximal tubule renin mRNA expression in the rat. Kidney Int. 2000;58(6):2320–2330. doi: 10.1046/j.1523-1755.2000.00416.x. [DOI] [PubMed] [Google Scholar]
- 45.Hsieh TJ, Zhang SL, Filep JG, Tang SS, Ingelfinger JR, Chan JS. High glucose stimulates angiotensinogen gene expression via reactive oxygen species generation in rat kidney proximal tubular cells. Endocrinology. 2002;143(8):2975–2985. doi: 10.1210/endo.143.8.8931. [DOI] [PubMed] [Google Scholar]
- 46.Choi KC, Kim NH, An MR, Kang DG, Kim SW, Lee J. Alterations of intrarenal renin-angiotensin and nitric oxide systems in streptozotocin-induced diabetic rats. Kidney Int Suppl. 1997;60:S23–S27. [PubMed] [Google Scholar]
- 47.Hollenberg NK. Direct renin inhibition and the kidney. Nat Rev Nephrol. 2010;6(1):49–55. doi: 10.1038/nrneph.2009.201. [DOI] [PubMed] [Google Scholar]
- 48.Huang Y, Wongamorntham S, Kasting J, et al. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. 2006;69(1):105–113. doi: 10.1038/sj.ki.5000011. [DOI] [PubMed] [Google Scholar]
- 49.Huang Y, Noble NA, Zhang J, Xu C, Border WA. Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int. 2007;72(1):45–52. doi: 10.1038/sj.ki.5002243. [DOI] [PubMed] [Google Scholar]
- 50.Sakoda M, Ichihara A, Kaneshiro Y, et al. (Pro)renin receptor-mediated activation of mitogen-activated protein kinases in human vascular smooth muscle cells. Hypertens Res. 2007;30(11):1139–1146. doi: 10.1291/hypres.30.1139. [DOI] [PubMed] [Google Scholar]
- 51.Kaneshiro Y, Ichihara A, Takemitsu T, et al. Increased expression of cyclooxygenase-2 in the renal cortex of human prorenin receptor gene-transgenic rats. Kidney Int. 2006;70(4):641–646. doi: 10.1038/sj.ki.5001627. [DOI] [PubMed] [Google Scholar]
- 52.Feldt S, Batenburg WW, Mazak I, et al. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension. 2008;51(3):682–688. doi: 10.1161/HYPERTENSIONAHA.107.101444. [DOI] [PubMed] [Google Scholar]
- 53.Mercure C, Prescott G, Lacombe MJ, Silversides DW, Reudelhuber TL. Chronic increases in circulating prorenin are not associated with renal or cardiac pathologies. Hypertension. 2009;53(6):1062–1069. doi: 10.1161/HYPERTENSIONAHA.108.115444. [DOI] [PubMed] [Google Scholar]
- 54.Danser AH. (Pro)renin receptor and vacuolar H+-ATPase. Hypertension. 2009;54(2):219–221. doi: 10.1161/HYPERTENSIONAHA.109.135236. [DOI] [PubMed] [Google Scholar]
- 55.Advani A, Kelly DJ, Cox AJ, et al. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension. 2009;54(2):261–269. doi: 10.1161/HYPERTENSIONAHA.109.128645. [DOI] [PubMed] [Google Scholar]
- 56.Anderson S, Jung FF, Ingelfinger JR. Renal renin-angiotensin system in diabetes: functional, immunohistochemical, and molecular biological correlations. Am J Physiol. 1993;265(4 Pt 2):F477–F486. doi: 10.1152/ajprenal.1993.265.4.F477. [DOI] [PubMed] [Google Scholar]
- 57.Huang Y, Border WA, Noble NA. Functional renin receptors in renal mesangial cells. Curr Hypertens Rep. 2007;9(2):133–139. doi: 10.1007/s11906-007-0024-4. [DOI] [PubMed] [Google Scholar]
- 58.Nguyen G, Delarue F, Berrou J, Rondeau E, Sraer JD. Specific receptor binding of renin on human mesangial cells in culture increases plasminogen activator inhibitor-1 antigen. Kidney Int. 1996;50(6):1897–1903. doi: 10.1038/ki.1996.511. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109(11):1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Durvasula RV, Shankland SJ. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol. 2008;294(4):F830–F839. doi: 10.1152/ajprenal.00266.2007. [DOI] [PubMed] [Google Scholar]
- 61.Liu FY, Cogan MG. Angiotensin II stimulates early proximal bicarbonate absorption in the rat by decreasing cyclic adenosine monophosphate. J Clin Invest. 1989;84(1):83–91. doi: 10.1172/JCI114174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu FY, Cogan MG. Role of protein kinase C in proximal bicarbonate absorption and angiotensin signaling. Am J Physiol. 1990;258(4 Pt 2):F927–F933. doi: 10.1152/ajprenal.1990.258.4.F927. [DOI] [PubMed] [Google Scholar]
- 63.Kifor I, Moore TJ, Fallo F, et al. Potassium-stimulated angiotensin release from superfused adrenal capsules and enzymatically dispersed cells of the zona glomerulosa. Endocrinology. 1991;129(2):823–831. doi: 10.1210/endo-129-2-823. [DOI] [PubMed] [Google Scholar]
- 64.Singh R, Alavi N, Singh AK, Leehey DJ. Role of angiotensin II in glucose-induced inhibition of mesangial matrix degradation. Diabetes. 1999;48(10):2066–2073. doi: 10.2337/diabetes.48.10.2066. [DOI] [PubMed] [Google Scholar]
- 65.Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension. 2001;38(3 Pt 2):635–638. doi: 10.1161/hy09t1.094234. [DOI] [PubMed] [Google Scholar]
- 66.Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci. 2013;124(3):139–152. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 67.Velloso LA, Folli F, Perego L, Saad MJ. The multi-faceted cross-talk between the insulin and angiotensin II signaling systems. Diabetes Metab Res Rev. 2006;22(2):98–107. doi: 10.1002/dmrr.611. [DOI] [PubMed] [Google Scholar]
- 68.Tiwari S, Halagappa VK, Riazi S, Hu X, Ecelbarger CA. Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J Am Soc Nephrol. 2007;18(10):2661–2671. doi: 10.1681/ASN.2006121410. [DOI] [PubMed] [Google Scholar]
- 69.Gloy J, Henger A, Fischer KG, et al. Angiotensin II modulates cellular functions of podocytes. Kidney Int Suppl. 1998;67:S168–S170. doi: 10.1046/j.1523-1755.1998.06736.x. [DOI] [PubMed] [Google Scholar]
- 70.Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009;302(2):148–158. doi: 10.1016/j.mce.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harada E, Yoshimura M, Yasue H, et al. Aldosterone induces angiotensin-converting-enzyme gene expression in cultured neonatal rat cardiocytes. Circulation. 2001;104(2):137–139. doi: 10.1161/01.cir.104.2.137. [DOI] [PubMed] [Google Scholar]
- 72.Kang YS, Ko GJ, Lee MH, et al. Effect of eplerenone, enalapril and their combination treatment on diabetic nephropathy in type II diabetic rats. Nephrol Dial Transplant. 2009;24(1):73–84. doi: 10.1093/ndt/gfn448. [DOI] [PubMed] [Google Scholar]
- 73.Han SY, Kim CH, Kim HS, et al. Spironolactone prevents diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. J Am Soc Nephrol. 2006;17(5):1362–1372. doi: 10.1681/ASN.2005111196. [DOI] [PubMed] [Google Scholar]
- 74.Miyata K, Rahman M, Shokoji T, et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol. 2005;16(10):2906–2912. doi: 10.1681/ASN.2005040390. [DOI] [PubMed] [Google Scholar]
- 75.Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150(11):776–783. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87(5):E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 77.Carey RM, Siragy HM. The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrinol Metab. 2003;14(6):274–281. doi: 10.1016/s1043-2760(03)00111-5. [DOI] [PubMed] [Google Scholar]
- 78.Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 79.Mori J, Patel VB, Ramprasath T, et al. Angiotensin 1-7 mediates renoprotection against diabetic nephropathy by reducing oxidative stress, inflammation, and lipotoxicity. Am J Physiol Renal Physiol. 2014;306(8):F812–F821. doi: 10.1152/ajprenal.00655.2013. [DOI] [PubMed] [Google Scholar]
- 80.Zhang K, Meng X, Li D, et al. Angiotensin(1-7) attenuates the progression of streptozotocin-induced diabetic renal injury better than angiotensin receptor blockade. Kidney Int. 2015;87(2):359–369. doi: 10.1038/ki.2014.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miyata N, Park F, Li XF, Cowley AW., Jr Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol. 1999;277(3 Pt 2):F437–F446. doi: 10.1152/ajprenal.1999.277.3.F437. [DOI] [PubMed] [Google Scholar]
- 82.Ozono R, Wang ZQ, Moore AF, Inagami T, Siragy HM, Carey RM. Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension. 1997;30(5):1238–1246. doi: 10.1161/01.hyp.30.5.1238. [DOI] [PubMed] [Google Scholar]
- 83.Arima S, Ito S. Angiotensin II type 2 receptors in the kidney: evidence for endothelial-cell-mediated renal vasodilatation. Nephrol Dial Transplant. 2000;15(4):448–451. doi: 10.1093/ndt/15.4.448. [DOI] [PubMed] [Google Scholar]
- 84.Oh BH, Mitchell J, Herron JR, Chung J, Khan M, Keefe DL. Aliskiren, an oral renin inhibitor, provides dose-dependent efficacy and sustained 24-hour blood pressure control in patients with hypertension. J Am Coll Cardiol. 2007;49(11):1157–1163. doi: 10.1016/j.jacc.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 85.Nicholls SJ, Bakris GL, Kastelein JJ, et al. Effect of aliskiren on progression of coronary disease in patients with prehypertension: the AQUARIUS randomized clinical trial. JAMA. 2013;310(11):1135–1144. doi: 10.1001/jama.2013.277169. [DOI] [PubMed] [Google Scholar]
- 86.Erdos EG, Marcic BM. Kinins, receptors, kininases and inhibitors – where did they lead us? Biol Chem. 2001;382(1):43–47. doi: 10.1515/BC.2001.007. [DOI] [PubMed] [Google Scholar]
- 87.Sironi L, Nobili E, Gianella A, Gelosa P, Tremoli E. Anti-inflammatory properties of drugs acting on the renin-angiotensin system. Drugs Today. 2005;41(9):609–622. doi: 10.1358/dot.2005.41.9.899613. [DOI] [PubMed] [Google Scholar]
- 88.Kothari SA, Le MK, Gandhi PJ. Effects of angiotensin converting enzyme inhibitors on thrombotic mediators: potential clinical implications. J Thromb Thrombolysis. 2003;15(3):217–225. doi: 10.1023/B:THRO.0000011379.68077.64. [DOI] [PubMed] [Google Scholar]
- 89.Neutel JM. Effect of the renin–angiotensin system on the vessel wall: using ACE inhibition to improve endothelial function. J Hum Hypertens. 2004;18(9):599–606. doi: 10.1038/sj.jhh.1001714. [DOI] [PubMed] [Google Scholar]
- 90.Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med. 2003;115(1):41–46. doi: 10.1016/s0002-9343(03)00158-x. [DOI] [PubMed] [Google Scholar]
- 91.Hilgers KF, Mann JF. ACE inhibitors versus AT(1) receptor antagonists in patients with chronic renal disease. J Am Soc Nephrol. 2002;13(4):1100–1108. doi: 10.1681/ASN.V1341100. [DOI] [PubMed] [Google Scholar]
- 92.Ma J, Nishimura H, Fogo A, Kon V, Inagami T, Ichikawa I. Accelerated fibrosis and collagen deposition develop in the renal interstitium of angiotensin type 2 receptor null mutant mice during ureteral obstruction. Kidney Int. 1998;53(4):937–944. doi: 10.1111/j.1523-1755.1998.00893.x. [DOI] [PubMed] [Google Scholar]
- 93.Morrissey JJ, Klahr S. Effect of AT2 receptor blockade on the pathogenesis of renal fibrosis. Am J Physiol. 1999;276(1 Pt 2):F39–F45. doi: 10.1152/ajprenal.1999.276.1.F39. [DOI] [PubMed] [Google Scholar]
- 94.Couser WG, Riella MC, Joint International Society of Nephrology and International Federation of Kidney Foundations’ World Kidney Day Steering Committee World Kidney Day 2011: protect your kidneys, save your heart. Kidney Int. 2011;79(5):483–485. doi: 10.1038/ki.2010.522. [DOI] [PubMed] [Google Scholar]
- 95.American Diabetes Association Diabetic nephropathy. Diabetes Care. 1998;21:S50–S53. doi: 10.2337/diacare.21.2.315. [DOI] [PubMed] [Google Scholar]
- 96.Arauz-Pacheco C, Parrott MA, Raskin P, American Diabetes Association Treatment of hypertension in adults with diabetes. Diabetes Care. 2003;26(Suppl 1):S80–S82. doi: 10.2337/diacare.26.2007.s80. [DOI] [PubMed] [Google Scholar]
- 97.KDOQI KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis. 2007;49(2 Suppl 2):S12–S154. doi: 10.1053/j.ajkd.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 98.Klahr S, Levey AS, Beck GJ, et al. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of diet in Renal Disease Study Group. N Engl J Med. 1994;330(13):877–884. doi: 10.1056/NEJM199403313301301. [DOI] [PubMed] [Google Scholar]
- 99.Peterson JC, Adler S, Burkart JM, et al. Blood pressure control, proteinuria, and the progression of renal disease. The modification of diet in renal disease study. Ann Intern Med. 1995;123(10):754–762. doi: 10.7326/0003-4819-123-10-199511150-00003. [DOI] [PubMed] [Google Scholar]
- 100.Sarnak MJ, Greene T, Wang X, et al. The effect of a lower target blood pressure on the progression of kidney disease: long-term follow-up of the modification of diet in renal disease study. Ann Intern Med. 2005;142(5):342–351. doi: 10.7326/0003-4819-142-5-200503010-00009. [DOI] [PubMed] [Google Scholar]
- 101.Wright JT, Jr, Bakris G, Greene T, et al. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA. 2002;288(19):2421–2431. doi: 10.1001/jama.288.19.2421. [DOI] [PubMed] [Google Scholar]
- 102.Appel LJ, Wright JT, Jr, Greene T, et al. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med. 2010;363(10):918–929. doi: 10.1056/NEJMoa0910975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jafar TH, Stark PC, Schmid CH, et al. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann Intern Med. 2003;139(4):244–252. doi: 10.7326/0003-4819-139-4-200308190-00006. [DOI] [PubMed] [Google Scholar]
- 104.Bakris GL, Weir MR, Shanifar S, et al. Effects of blood pressure level on progression of diabetic nephropathy: results from the RENAAL study. Arch Intern Med. 2003;163(13):1555–1565. doi: 10.1001/archinte.163.13.1555. [DOI] [PubMed] [Google Scholar]
- 105.Pohl MA, Blumenthal S, Cordonnier DJ, et al. Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the irbesartan diabetic nephropathy trial: clinical implications and limitations. J Am Soc Nephrol. 2005;16(10):3027–3037. doi: 10.1681/ASN.2004110919. [DOI] [PubMed] [Google Scholar]
- 106.Ruzicka M, Quinn RR, McFarlane P, et al. Canadian Society of Nephrology commentary on the 2012 KDIGO clinical practice guideline for the management of blood pressure in CKD. Am J Kidney Dis. 2014;63(6):869–887. doi: 10.1053/j.ajkd.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 107.Taler SJ, Agarwal R, Bakris GL, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for management of blood pressure in CKD. Am J Kidney Dis. 2013;62(2):201–213. doi: 10.1053/j.ajkd.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Verbeke F, Lindley E, Van Bortel L, et al. A European Renal Best Practice (ERBP) position statement on the Kidney Disease: Improving Global Outcomes (KDIGO) clinical practice guideline for the management of blood pressure in non-dialysis-dependent chronic kidney disease: an endorsement with some caveats for real-life application. Nephrol Dial Transplant. 2014;29(3):490–496. doi: 10.1093/ndt/gft321. [DOI] [PubMed] [Google Scholar]
- 109.Roberts MA. Commentary on the KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease. Nephrology. 2014;19(1):53–55. doi: 10.1111/nep.12168. [DOI] [PubMed] [Google Scholar]
- 110.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329(20):1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 111.Hebert LA, Bain RP, Verme D, et al. Remission of nephrotic range proteinuria in type I diabetes. Collaborative Study Group. Kidney Int. 1994;46(6):1688–1693. doi: 10.1038/ki.1994.469. [DOI] [PubMed] [Google Scholar]
- 112.Patel A, ADVANCE Collaborative Group. MacMahon S, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet. 2007;370(9590):829–840. doi: 10.1016/S0140-6736(07)61303-8. [DOI] [PubMed] [Google Scholar]
- 113.de Galan BE, Perkovic V, Ninomiya T, et al. Lowering blood pressure reduces renal events in type 2 diabetes. J Am Soc Nephrol. 2009;20(4):883–892. doi: 10.1681/ASN.2008070667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Atkins RC, Briganti EM, Lewis JB, et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am J Kidney Dis. 2005;45(2):281–287. doi: 10.1053/j.ajkd.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 115.Jafar TH, Stark PC, Schmid CH, et al. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease. Kidney Int. 2001;60(3):1131–1140. doi: 10.1046/j.1523-1755.2001.0600031131.x. [DOI] [PubMed] [Google Scholar]
- 116.Imai E, Haneda M, Chan JC, et al. Reduction and residual proteinuria are therapeutic targets in type 2 diabetes with overt nephropathy: a post hoc analysis (ORIENT-proteinuria) Nephrol Dial Transplant. 2013;28(10):2526–2534. doi: 10.1093/ndt/gft249. [DOI] [PubMed] [Google Scholar]
- 117.Bakris GL, Mangrum A, Copley JB, Vicknair N, Sadler R. Effect of calcium channel or beta-blockade on the progression of diabetic nephropathy in African Americans. Hypertension. 1997;29(3):744–750. doi: 10.1161/01.hyp.29.3.744. [DOI] [PubMed] [Google Scholar]
- 118.Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345(12):851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 119.Barnett AH, Bain SC, Bouter P, et al. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med. 2004;351(19):1952–1961. doi: 10.1056/NEJMoa042274. [DOI] [PubMed] [Google Scholar]
- 120.Jacobsen P, Andersen S, Rossing K, Jensen BR, Parving HH. Dual blockade of the renin-angiotensin system versus maximal recommended dose of ACE inhibition in diabetic nephropathy. Kidney Int. 2003;63(5):1874–1880. doi: 10.1046/j.1523-1755.2003.00940.x. [DOI] [PubMed] [Google Scholar]
- 121.Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ. 2000;321(7274):1440–1444. doi: 10.1136/bmj.321.7274.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Song JH, Cha SH, Lee HJ, et al. Effect of low-dose dual blockade of renin-angiotensin system on urinary TGF-beta in type 2 diabetic patients with advanced kidney disease. Nephrol Dial Transplant. 2006;21(3):683–689. doi: 10.1093/ndt/gfi310. [DOI] [PubMed] [Google Scholar]
- 123.Fried LF, Emanuele N, Zhang JH, et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med. 2013;369(20):1892–1903. doi: 10.1056/NEJMoa1303154. [DOI] [PubMed] [Google Scholar]
- 124.Mann JF, Anderson C, Gao P, et al. Dual inhibition of the renin-angiotensin system in high-risk diabetes and risk for stroke and other outcomes: results of the ONTARGET trial. J Hypertens. 2013;31(2):414–421. doi: 10.1097/HJH.0b013e32835bf7b0. [DOI] [PubMed] [Google Scholar]
- 125.Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK, AVOID Study Investigators Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008;358(23):2433–2446. doi: 10.1056/NEJMoa0708379. [DOI] [PubMed] [Google Scholar]
- 126.Parving HH, Brenner BM, McMurray JJ, et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med. 2012;367(23):2204–2213. doi: 10.1056/NEJMoa1208799. [DOI] [PubMed] [Google Scholar]
- 127.Jorde UP, Vittorio T, Katz SD, Colombo PC, Latif F, Le Jemtel TH. Elevated plasma aldosterone levels despite complete inhibition of the vascular angiotensin-converting enzyme in chronic heart failure. Circulation. 2002;106(9):1055–1057. doi: 10.1161/01.cir.0000030935.89559.04. [DOI] [PubMed] [Google Scholar]
- 128.Sato A, Hayashi K, Naruse M, Saruta T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension. 2003;41(1):64–68. doi: 10.1161/01.hyp.0000044937.95080.e9. [DOI] [PubMed] [Google Scholar]
- 129.Young DB, Smith MJ, Jr, Jackson TE, Scott RE. Multiplicative interaction between angiotensin II and K concentration in stimulation of aldosterone. Am J Physiol. 1984;247(3 Pt 1):E328–E335. doi: 10.1152/ajpendo.1984.247.3.E328. [DOI] [PubMed] [Google Scholar]
- 130.Bianchi S, Bigazzi R, Campese VM. Antagonists of aldosterone and proteinuria in patients with CKD: an uncontrolled pilot study. Am J Kidney Dis. 2005;46(1):45–51. doi: 10.1053/j.ajkd.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 131.Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005;28(9):2106–2112. doi: 10.2337/diacare.28.9.2106. [DOI] [PubMed] [Google Scholar]
- 132.Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone in diabetic nephropathy. Kidney Int. 2005;68(6):2829–2836. doi: 10.1111/j.1523-1755.2005.00756.x. [DOI] [PubMed] [Google Scholar]
- 133.van den Meiracker AH, Baggen RG, Pauli S, et al. Spironolactone in type 2 diabetic nephropathy: Effects on proteinuria, blood pressure and renal function. J Hypertens. 2006;24(11):2285–2292. doi: 10.1097/01.hjh.0000249708.44016.5c. [DOI] [PubMed] [Google Scholar]
- 134.Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2641–2650. doi: 10.1681/ASN.2009070737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rachmani R, Slavachevsky I, Amit M, et al. The effect of spironolactone, cilazapril and their combination on albuminuria in patients with hypertension and diabetic nephropathy is independent of blood pressure reduction: a randomized controlled study. Diabet Med. 2004;21(5):471–475. doi: 10.1111/j.1464-5491.2004.01194.x. [DOI] [PubMed] [Google Scholar]
- 136.Epstein M, Williams GH, Weinberger M, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol. 2006;1(5):940–951. doi: 10.2215/CJN.00240106. [DOI] [PubMed] [Google Scholar]
- 137.Israili ZH, Hall WD. Cough and angioneurotic edema associated with angiotensin-converting enzyme inhibitor therapy. A review of the literature and pathophysiology. Ann Intern Med. 1992;117(3):234–242. doi: 10.7326/0003-4819-117-3-234. [DOI] [PubMed] [Google Scholar]
- 138.Matchar DB, McCrory DC, Orlando LA, et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann Intern Med. 2008;148(1):16–29. doi: 10.7326/0003-4819-148-1-200801010-00189. [DOI] [PubMed] [Google Scholar]
- 139.Ianiro G, Bibbo S, Montalto M, Ricci R, Gasbarrini A, Cammarota G. Systematic review: Sprue-like enteropathy associated with olmesartan. Aliment Pharmacol Ther. 2014;40(1):16–23. doi: 10.1111/apt.12780. [DOI] [PubMed] [Google Scholar]
- 140.Stirling C, Houston J, Robertson S, et al. Diarrhoea, vomiting and ACE inhibitors: – an important cause of acute renal failure. J Hum Hypertens. 2003;17(6):419–423. doi: 10.1038/sj.jhh.1001571. [DOI] [PubMed] [Google Scholar]
- 141.Yacoub R, Patel N, Lohr JW, Rajagopalan S, Nader N, Arora P. Acute kidney injury and death associated with renin angiotensin system blockade in cardiothoracic surgery: a meta-analysis of observational studies. Am J Kidney Dis. 2013;62(6):1077–1086. doi: 10.1053/j.ajkd.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 142.Rim MY, Ro H, Kang WC, et al. The effect of renin-angiotensin-aldosterone system blockade on contrast-induced acute kidney injury: a propensity-matched study. Am J Kidney Dis. 2012;60(4):576–582. doi: 10.1053/j.ajkd.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 143.Chernin G, Gal-Oz A, Ben-Assa E, et al. Secondary prevention of hyperkalemia with sodium polystyrene sulfonate in cardiac and kidney patients on renin-angiotensin-aldosterone system inhibition therapy. Clin Cardiol. 2012;35(1):32–36. doi: 10.1002/clc.20987. [DOI] [PMC free article] [PubMed] [Google Scholar]