

Summary
Maintenance of a hematopoietic progenitor population requires extensive interaction with cells within a microenvironment or niche. In the Drosophila hematopoietic organ, niche-derived Hedgehog signaling maintains the progenitor population. Here we show that the hematopoietic progenitors also require a signal mediated by Adenosine deaminase growth factor A (Adgf-A) arising from differentiating cells that regulates extracellular levels of adenosine. The adenosine signal opposes the effects of Hedgehog signaling within the hematopoietic progenitor cells and the magnitude of the adenosine signal is kept in check by the level of Adgf-A secreted from differentiating cells. Our findings reveal signals arising from differentiating cells that are required for maintaining progenitor cell quiescence, and that function with the niche-derived signal in maintaining the progenitor state. Similar homeostatic mechanisms are likely to be utilized in other systems that maintain relatively large numbers of progenitors that are not all in direct contact with the cells of the niche.
Keywords: VEGF, PDGF, Drosophila, hemocyte, niche, Pvr, adenosine signaling, lymph gland, progenitor, quiescence
Introduction
The mammalian hematopoietic niche displays complex interactions between populations of HSCs and progenitors to maintain their numbers (Garrett and Emerson, 2009). The relative in vivo contributions of cues emanating from the microenvironment in regulating stem cell versus progenitor maintenance remains unclear (He et al., 2009; Martinez-Agosto et al., 2007; Orkin and Zon, 2008). Several (Jude et al., 2008; Tumbar et al., 2004), but not all (Li and Clevers, 2010) stem cell and progenitor populations demonstrate slow cell cycling and this property of “quiescence” is critical for maintaining their integrity over a period of time.
In vivo genetic analysis in Drosophila allows for the study of stem cell properties in their endogenous microenvironment (Losick et al., 2011). Drosophila blood cells, or hemocytes, develop within an organ called the lymph gland, where differentiating hemocytes, their progenitors, and the cells of the signaling microenvironment or niche, are found (Jung et al., 2005). Differentiated blood cells in Drosophila are all myeloid in nature and are located along the outer edge of the lymph gland, in a region termed the cortical zone (CZ, (Jung et al., 2005), Figure 1A). These arise from a group of progenitors located within an inner core of cells termed the medullary zone (MZ). The MZ cells are akin to the common myeloid progenitors (CMP) of the vertebrate hematopoietic system. They quiesce, lack differentiation markers, are multipotent, and give rise to all Drosophila blood lineages (Jung et al., 2005; Krzemien et al., 2010). MZ progenitors are maintained by a small group of cells, collectively termed the posterior signaling center (PSC), that function as a hematopoietic niche (Crozatier et al., 2004; Krzemien et al., 2007; Mandal et al., 2007). Clonal analysis has suggested the existence of a niche-bound population of hematopoietic stem cells (Minakhina and Steward, 2010), although such cells have not yet been directly identified.
Figure 1. Regulation of progenitor quiescence by differentiating hemocytes mediated by Pvf1/Pvr and Adgf-A signaling.
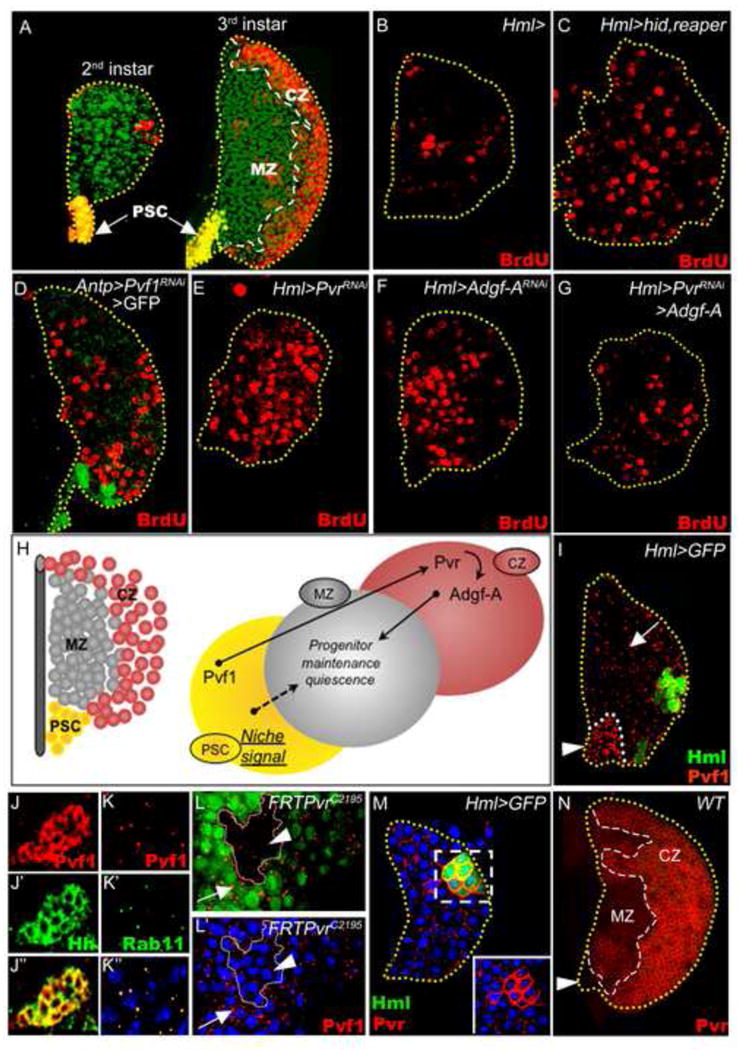
(A) A representative lymph gland primary lobe from a mid-second instar larva (left image) consisting of a posterior signaling center (PSC, yellow), progenitors (green), and a few differentiating cells (red). A primary lobe from a later-staged wandering third instar larva (right image) shows three distinct zones: the PSC (yellow), which functions as the niche for the maintenance of progenitors (green) within the medullary zone (MZ), and the peripheral cortical zone (CZ) region, comprised of differentiating blood cells (red).
(B-G) Cell proliferation profile in lymph glands from mid-second instar larvae analyzed by BrdU (red) incorporation. (B) Control lymph glands (genotype: Hml-gal4) at this stage have only a few proliferating cells (9 +/- 6 cells, N=6). (C) Induction of cell death in the differentiating cells by expression of Hid and Reaper (Hml-gal4 UAS-hid UAS-rpr) causes an increase in the number of BrdU positive cells (red), indicative of the loss of quiescence among the progenitors (compare with B). A similar loss of quiescence occurs upon (D) down-regulation of Pvf1 in the PSC using Pvf1RNAi (Antp-gal4 UAS-2xEGFP UAS-Pvf1RNAi), (E) down-regulation of Pvr in the differentiating cells using PvrRNAi (Hml-gal4 UAS-PvrRNAi; 39 +/- 7 cells, N=5, p<0.004 when compared to B), or (F) down-regulation of Adgf-A in the differentiating cells using Adgf-ARNAi (Hml-gal4 UAS-Adgf-ARNAi). (G) Over-expression of Adgf-A can suppress the proliferation phenotype due to loss of PvrRNAi (Hml-gal4 UAS-PvrRNAi UAS-Adgf-A; 17 +/- 7 cells, N=8, p<0.03 when compared with E).
(H) The color scheme of the model (right) corresponds to the zones represented in the schematic of the lymph gland (left). Maintenance of progenitor quiescence within the MZ requires Pvf1 from the PSC, Pvr function in the CZ, and Adgf-A from the CZ functioning downstream of Pvr.
(I-N) Expression of Pvf1 and Pvr in the developing lymph gland. TOPRO-3 (in K-M, blue) marks nuclei. (I and M) Mid-second instar lymph glands from control animals (Hml-gal4 UAS-2xEGFP); (J-L′ and N) Third instar lymph glands. (I) Pvf1 expression is seen as small punctae present at high levels in the PSC (arrowhead) and dispersed in the rest of the lymph gland (arrow). (J) Pvf1 (red, J) expression in the PSC co-localizes with Hedgehog (green, J′). The overlap is shown in yellow (J″). (K) Within the lymph gland proper, Pvf1 (red punctae, K) co-localizes with Rab11 (green punctae, K′), a marker for transcytosis vesicles. The overlap is shown in yellow (K″). (L) Pvr mutant clones (non-green) were generated within wild-type tissue (green) and stained for Pvf1 (red punctae). Pvf1 is present in the Pvr+ tissue (arrow) but is strongly reduced in the clones lacking Pvr (arrowhead). (M, N) Temporal analysis of Pvr expression. (M) The first differentiating cells (green, Hml-gal4 UAS-2xEGFP) show strong upregulation of Pvr expression (red, inset). (N) At later stages, Pvr expression is relatively high in the CZ as compared to the MZ, and is lacking in the PSC (arrowhead).
The PSC cells express Hedgehog (Hh), which is required for the maintenance of the MZ progenitors (Mandal et al., 2007). Cubitus interruptus (Ci) is a downstream effector of Hh signaling similar to vertebrate Gli proteins; it is maintained in its active Ci155 form in the presence of Hh and degraded to the repressor Ci75 form in the absence of Hh (Smelkinson and Kalderon, 2006). PSC-derived Hh signaling causes MZ cells to exhibit high Ci155 (Mandal et al., 2007).
Proliferation of circulating larval hemocytes is also regulated by Adenosine Deaminase Growth Factor-A (Adgf-A), which is similar to vertebrate adenosine deaminases (ADAs). Adgf-A is a secreted enzyme that converts extracellular adenosine into inosine by deamination (Dolezal et al., 2003; Maier et al., 2001). Two distinct adenosine deaminases, ADA1 and ADA2/CECR1, are found in humans. CECR1 is secreted by monocytes as they differentiate into macrophages (Zavialov et al., 2010). In Drosophila, mutation of Adgf-A causes an increased adenosine levels and increase in circulating blood cells (Dolezal et al., 2005; Zurovec et al., 2002). Extracellular adenosine is sensed by the single Drosophila adenosine receptor (AdoR) that generates a mitogenic signal through the G-protein/adenylate cyclase/cAMP-dependent Protein Kinase A (PKA) pathway (Dolezelova et al., 2007). A target of PKA is the transcription factor Ci, which also transduces the Hedgehog signal. We were intrigued by the potential link between adenosine and Hedgehog signaling, both through PKA mediated regulation of Ci, and propose a model that the niche signal and the CZ signal interact to maintain the progenitor population in a quiescent and undifferentiated state within the MZ of the lymph gland.
Results
Signals from differentiating hemocytes regulate hematopoietic progenitor quiescence
During the mid second instar, blood cells initiate differentiation in the larval lymph gland marking the beginning of cortical zone (CZ) formation (Figure 1A). The first cells that express differentiation markers appear stereotypically at the peripheral edge of the lymph gland (Figure 1A). These differentiating cells will eventually populate an entire peripheral compartment that will comprise the CZ. The timing of the first signs of differentiation matches closely with the onset of quiescence among the precursor population, eventually giving rise to the medullary zone (MZ).
The close temporal synchronization of CZ formation and the quiescence of MZ progenitors raised the intriguing possibility that the onset of differentiation might regulate the proliferation profile of the progenitors. To test this hypothesis, we induced cell death by expressing the pro-apoptotic proteins Hid and Reaper in the differentiating hemocytes and assayed for the effect of their loss on the progenitor population. We found that loss of CZ cells induces proliferation of the adjacent progenitor cells, which are normally quiescent at this stage (Figure 1B-C; S1A-B).
We knocked-down candidate ligands in the lymph gland by RNA interference (RNAi) and monitored for a loss of progenitor quiescence. This survey identified Pvf1 as a signaling molecule that is required for the maintenance of quiescence within the lymph gland. Expressing Pvf1RNAi using Gal4 drivers specific to either niche (PSC) cells using Antp-gal4, progenitor cells using dome-gal4, or differentiating cells using Hml-gal4 showed that PSC-specific knockdown is sufficient to induce progenitor proliferation (Figure 1D; S1C), whereas Pvf1 knockdown in progenitors or differentiating cells has no effect on the lymph gland (Figure S1G-I). These results indicate that Pvf1 synthesized in the PSC is required for progenitor quiescence.
To determine the site of Pvf1 function, its receptor Pvr was knocked down in the lymph gland using a similar approach. Interestingly, we found that PvrRNAi expressed under the control of drivers specific to differentiating cells (Hml-gal4 and pxn-gal4) causes a loss of progenitor quiescence (Figure 1E; S1D). The BrdU incorporating cells do not express differentiation markers (Figure S1A-F). Thus, differentiation follows the proliferative event. Lymph glands are not similarly affected when Pvr function is downregulated in the progenitors themselves (Figure S1J). These results indicate that Pvf1 originates in the niche and activates Pvr in maturing hemocytes, and that this signaling system is important for the quiescence of MZ progenitors. These results did not explain, though, how maturing cells might signal back to the progenitors causing them to maintain quiescence.
Given the previously known role of Adgf-A in the control of hemocyte number in circulation (Dolezal et al., 2005), we investigated whether this protein plays a similar role in the lymph gland. Remarkably, downregulation of the secreted Adgf-A protein in the differentiating hemocytes of the CZ, achieved by expressing Adgf-ARNAi under Hml-gal4 control, induces loss of quiescence of MZ progenitors (Figure 1F; S1E), similar to that seen with loss of Pvr in the CZ. This suggests that Adgf-A may act as a signal originating from differentiating hemocytes that is required for maintaining progenitor quiescence. In support of this idea, while overexpression of Adgf-A in differentiating hemocytes alone does not affect normal zonation (Figure S4G), it suppresses the induced progenitor proliferation caused by downregulation of Pvr (Figure 1G; S1F). Note that the phenotype in Figure 1C is similar to that seen in Figures 1E and F, but for different reasons. For loss of signaling molecules (Figures 1E-F), it is the break in the signaling network necessary for reducing adenosine that causes continued proliferation and eventual differentiation. For rpr/hid (Figure 1C) the signaling cell itself has been removed, thereby causing a lack in a backward signal. Quantitative analysis of the data (from Figure 1B, E, G, see legend) is consistent with a role for Adgf-A downstream of Pvr and leads us to the model shown in Figure 1H, the mechanistic details of which are genetically dissected in this study.
Step 1: Signaling from the niche to the differentiating cells
Pvf1 is expressed in the same cells as Hedgehog within the PSC in the primary lobe of the second instar lymph gland (Figure 1 I, J). Additionally, punctate dots that label with the anti-Pvf1 antibody can be seen throughout the lymph gland (Figure 1I) in Rab11-positive recycling vesicles (Figure 1K). When Pvf1 RNAi is targeted to the PSC, the Pvf1-positive punctae are eliminated throughout the lymph gland, suggesting that Pvf1 expression is limited to the PSC and the protein is transported by transcytosis from the PSC to the rest of the cells of the lymph gland. Additionally, clones of cells created within the lymph gland that lack the Pvr receptor are greatly reduced in Pvf1-positive punctae (Figure 1L,L′). The simplest explanation is that intercellular Pvf1 transport involves receptor-mediated endocytosis and the recycling of Pvf1 in Rab11 positive vesicles. With regard to Pvr expression, we found that it is absent from the PSC at all developmental stages. Pvr is initially expressed at low levels in early blood progenitors, but is then strongly up-regulated in the first differentiating hemocytes of the forming CZ during the second instar (Figure 1M). By the late third instar, all cells of the lymph gland (except PSC cells) express detectable levels of Pvr, although CZ cells continue to be higher in their expression (Figure 1N).
The loss of the progenitor cell quiescence phenotype upon loss of Pvf1/Pvr signaling apparent in the second instar (Figure 1D, E) is manifested in the third instar as a terminal phenotype in which all progenitor cells of the lymph gland differentiate. Cells occupying the position of the MZ in normal lymph glands all express differentiation markers in the mutants, leaving no progenitors behind. This is a correlation in every genetic background where the precursor population fails to quiesce in the second instar and continues to incorporate BrdU; the third instar phenotype is complete differentiation of the cells that would, in wild type, occupy the MZ and remain undifferentiated. This phenotype is observed when Pvf1RNAi is expressed in the PSC (Figure 2B) or when PvrRNAi is expressed in the differentiating hemocytes (Figure 2C). Pvr receptor activation is reduced when Pvf1 is downregulated in the PSC (Figure S3A-B). Importantly, TUNEL staining (Figure S1K-N′) and p35 misexpression studies (Figure S1O-P) show that PvrRNAi does not cause an increase in cell death levels compared to wild type when expressed in the developing CZ. Furthermore, cells lacking Pvr differentiate properly as assessed by retention of their phagocytosis function (Figure 2D). The phenotype caused by loss of Pvf1/Pvr signaling is reminiscent of the fully-differentiated phenotype seen upon loss of hedgehog function (Mandal et al., 2007). However, despite the similarity in loss of function phenotypes, Hedgehog expression in the PSC is not altered when Pvr is removed from the CZ (Figure 2C) or Pvf1 is removed from the PSC (Figure 2E). This suggests that the Pvf1-Pvr signaling process operates in parallel to the niche signal from Hh.
Figure 2. Role of STAT downstream of Pvr.
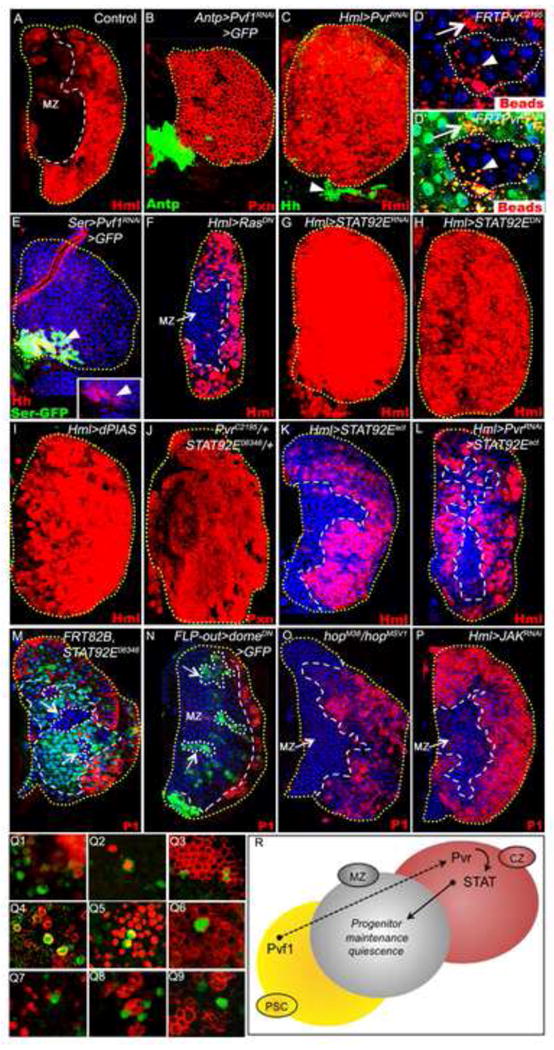
For uniformity, differentiating hemocytes are shown in red even if they are marked with EGFP (Hml-gal4 UAS-2xEGFP). Lymph glands shown are from wandering third-instar larvae. TOPRO3 marks nuclei (blue).
(A) Control. Normal Hml-gal4 expression pattern (Hml, red).
(B) Pvf1RNAi expression in the PSC (green; Antp-gal4 UAS-2xEGFP UAS-Pvf1RNAi). All non-PSC cells of the lymph gland express Peroxidasin (Pxn, red).
(C) PvrRNAi expression in the CZ (Hml-gal4 UAS-2xEGFP UAS-PvrRNAi) causes MZ progenitors to differentiate (Hml, red), although Hedgehog (green) expression in the PSC (arrowhead) remains normal.
(D and D′) Pvr-mutant clones (PvrC2195/PvrC2195, non-green cells) differentiate normally as judged by their ability to phagocytose FluoSphere beads (red), similar to neighboring wild type tissue (green).
(E) Hedgehog expression (red, inset) is unaffected when Pvf1RNAi is expressed in the PSC (green; Ser-gal4 UAS-2xEGFP UAS-Pvf1RNAi); yellow represents co-localization of Hedgehog and Ser-gal4 expression.
(F) Expression of RasDN in differentiating hemocytes (Hml-gal4 UAS-2xEGFP UAS-RasDN) does not affect progenitor fate.
(G-I) Loss of STAT function in differentiating cells causes progenitor differentiation (Hml, red). This phenotype can be induced by expressing either (G) Stat92ERNAi (Hml-gal4 UAS-2xEGFP UAS-Dcr-2 UAS-Stat92ERNAi), (H) Stat92EDN (Hml-gal4 UAS-2xEGFP UAS-Stat92EDN), or (I) dPIAS (Hml-gal4 UAS-2xEGFP UAS-dPIAS).
(J) Combined single-copy-loss of Pvr and Stat92E (PvrC2195/+; Stat92E06346/+). All progenitors express Pxn (red).
(K) As a control, expression of Stat92Eact (Ekas et al., 2010) in differentiating cells has no effect on progenitor fate (Hml-gal4 UAS-2xEGFP UAS-Stat92ΔNΔC) (L) Co-expression of Stat92Eact and PvrRNAi (Hml-gal4 UAS-2xEGFP UAS-Stat92Er UAS-PvrRNAi) suppresses the progenitor differentiation phenotype caused by PvrRNAi alone (compare with C).
(M) Stat92E mutant clones (Stat92E06346/Stat92E06346) within the MZ (lacking green, demarcated by white dots) do not cause ectopic differentiation (lack of red within the clones).
(N) Blocking Domeless function in MZ cells (FLP-out gal4 UAS-domeDN clones, green, see methods for details, demarcated by white dots), does not cause them to differentiate (P1, red).
(O) JAK mutant animals (hopM38/hopMSV1) do not exhibit ectopic differentiation (P1, red) of progenitors.
(P) Expression of JAKRNAi specifically in differentiating cells (Hml-gal4 UAS-2xEGFP UAS-hopRNAi) does not cause MZ progenitor differentiation (P1, red).
(Q1-9) In the third instar, lymph glands exhibit a small fraction of cells that express a reporter of STAT activity (green, 10X Stat92E-GFP). These cells are negative for domeless (dome-MESO-lacZ; Q1), proliferate (PH3; Q2), and express Pvract (Q3-4) and Hml (Hml-ga4, UAS-lacZ; Q5), but lack differentiation markers: Pxn (Q6), Lz (Q7), ProPO (Q8) and P1 (Q9). Cells appear yellow due to co-localization of the STAT reporter and Hml (anti-|3-gal), Pvract, or PH3.
(R) Schematic representation of STAT function in progenitor maintenance. Pvf1 from the PSC is transported to the differentiating cells in the CZ to activate its receptor Pvr, leading to the activation of STAT that, in turn, generates the CZ signal necessary for the maintenance of the progenitors in the MZ. Thus, loss of either Pvf1 from the PSC (in B), or loss of Pvr (in C) or STAT (in G-I) from the CZ results in proliferation and differentiation of MZ progenitors.
Knockdown of the second Pvr ligand Pvf2 by RNAi in the PSC does not cause a loss of progenitor quiescence and subsequent differentiation (Figure S2C-D). Rather, Pvf2 controls Shg (E-Cadherin) expression and prevents inappropriate migration of the progenitors into the CZ (Figure S2E-J, H).
Step 2: Signaling within the developing CZ
VEGFR-like receptors including Pvr function in the context of multiple effector molecules including Ras (Learte et al., 2008; Sims et al., 2009), Jun kinase (JNK) (Bond and Foley, 2009; Ishimaru et al., 2004; Mathieu et al., 2007), and STAT (Fu and Zhang, 1993; Li et al., 2002). Loss of Ras function in the CZ, achieved using a dominant negative form, does not phenocopy Pvr loss-of-function in these cells (Figure 2F). Also the bona fide marker of JNK activation, puc-lacZ, is not active in the CZ (not shown). In contrast, similar to Pvr, inactivation of Stat92E in the differentiating cells of the CZ using either Stat92ERNAi (Figure 2G), Stat92EDN (Figure 2H), or the STAT inhibitor, dPIAS (Figure 2I) causes loss of MZ progenitors. Furthermore, single copy loss of Pvr and Stat92E in the genetic combination PvrC2195/+; Stat92E06346/+ gives a similar loss of progenitor cell phenotype (Figure 2J; S3J). This second site non-complementation between Pvr and Stat92E suggests a close genetic relationship between them within a developmental pathway (see for example, (Giansanti et al., 2004; Rogge et al., 1991). In support of STAT functioning downstream of Pvr in differentiating cells, misexpression of Stat92Eact is sufficient to suppress the PvrRNAi phenotype (Figure 2L). Taken together, these data indicate that the developmental role of Stat92E in MZ progenitor cell maintenance is non-autonomous and attributable to a function of Stat92E in the CZ.
Stat92E loss-of-function clones generated outside of the CZ do not cause a differentiation phenotype (Figure 2M). Whole animal mutants in which the entire lymph gland lacks Stat92E do show differentiation of the progenitors (Krzemien et al., 2007) likely due to loss of Stat92E function in the CZ. Furthermore, Stat92E canonically functions downstream of the Domeless (Dome) receptor and the JAK homolog Hopscotch. However neither dome nor hopscotch loss of function causes a loss of progenitor quiescence and ectopic differentiation (Figure 2N-P). Domeless is not expressed in the CZ and as expected, expression of domeDN in the CZ has no effect on progenitor maintenance (Figure S3C-D). Therefore during normal development STAT functions downstream of Pvr in the CZ and the Dome/JAK-STAT pathway does not appear to have a role in this process. As previously proposed, Dome/JAK-STAT signaling in the MZ progenitors is required for their competency for immune-related responses (Krzemien et al., 2007; Makki et al., 2010).
Within the CZ, expression of a GFP reporter of active STAT signaling can be seen in a few scattered cells (Figure 2Q1-9). These cells are negative for MZ markers (Figure 2Q1), a subset actively proliferate (Figure 2Q2), and they also label with an antibody (Janssens et al., 2010) against the activated form of the Pvr receptor (Figure 2Q3-4). These cells express Hml (Figure 2Q5) but lack all markers of terminal differentiation (Figure 2Q6-9). The activity of this STAT reporter is reduced in a number of relevant genetic backgrounds involving components of this model (Figure S3E-H″). The reporter-expressing cells are likely “intermediate progenitors” (Krzemien et al., 2010) that receive an active Pvr/STAT signal and it is possible that these cells are the first to initiate the backward signal required for the Domeless positive progenitor quiescence (Figure 2R).
Step 3: A signal from differentiating cells to progenitor cells
As alluded to in Figure 1F, the primary candidate for a secreted signaling protein that regulates progenitor quiescence and is derived from differentiating cells is Adgf-A (Novakova and Dolezal, 2011). When Adgf-A is eliminated from differentiating cells, the progenitor population fails to quiesce, suggesting a central role for Adgf-A in CZ-derived signaling. Ultimately, the loss of quiescence associated with loss of Adgf-A function, either in whole-animal mutants (Figure 3B) or when Adgf-ARNAi is expressed in differentiating cells (Figure 3C), causes complete differentiation of the hematopoietic progenitor cell population. The loss of progenitors is not due to defects in PSC-derived signaling, as evidenced by intact Antp and Hh expression (Figure S4A-C). Also, the loss of progenitors in Adgf-ARNAi backgrounds is not associated with changes in reactive oxygen species levels, as they remain unchanged when Adgf-A function is downregulated in the differentiating hemocytes (Figure S4D-E). The phenotypic attributes due to loss of Adgf-A are the same as those associated with loss of Pvr or STAT function in differentiating cells, suggesting that Adgf-A may function in the same signaling pathway. In support of this idea, genetic interaction studies demonstrate that the combination of a single loss-of-function allele of Adgf-A with a single loss-of-function allele of Pvr (Figure 3D; S3J) or Stat92E (Figure 3E; S3J) is sufficient to cause a loss of progenitors in the developing lymph gland. These results are strongly indicative of a close functional relationship between Pvr, STAT, and Adgf-A signaling within the developing CZ. Additionally, the phenotypes caused by loss of Pvr or Stat92E in the differentiating cells can be suppressed by the simultaneous over-expression of Adgf-A in these cells (Figure 3F,G), further demonstrating that Adgf-A functions downstream of Pvr and STAT to regulate progenitor maintenance. Based upon similar mutant phenotypes, close genetic interaction, and the ability of Adgf-A to suppress Pvr and Stat92E phenotypes, we propose the signaling pathway schematized in Figure 3H.
Figure 3. Adgf-A functions downstream of STAT in hematopoietic progenitor maintenance.
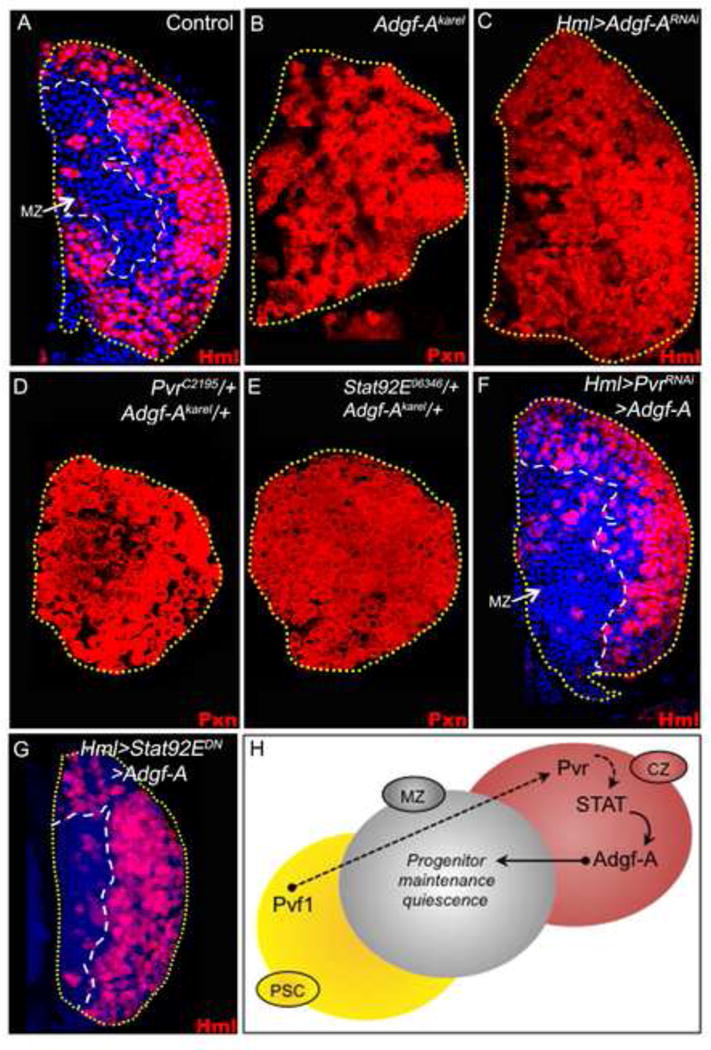
Third instar lymph glands are shown. Differentiating hemocytes are marked in red, with nuclei marked with TOPRO3 (blue). For uniformity, hemocytes in A, C, F and G are shown in red although they are genotypically Hml-gal4 UAS-2xEGFP.
(A) Normal expression pattern of Hml-gal4, showing differentiating cells (Hml, red). The MZ cells lack markers of differentiation and are marked by TOPRO3 (blue).
(B) Adgf-Akarel/Adgf-Akarel mutant lymph gland. All progenitors differentiate and express Pxn (red, compare pattern with A).
(C) Expression of Adgf-ARNAi in differentiating hemocytes (Hml-gal4 UAS-2xEGFP UAS-Adgf-ARNAi) causes differentiation of progenitors (Hml, red; compare with A).
(D) Combined single-copy-loss of Pvr and Adgf-A (PvrC2195/+; Adgf-Akarel/+). All progenitors differentiate and express Pxn.
(E) Combined single-copy-loss of STAT and Adgf-A (Stat92E06346/+ Adgf-Akarel/+). All progenitors differentiate and express Pxn.
(F) Co-expression of Adgf-A and PvrRNAi (Hml-gal4 UAS-2xEGFP UAS-PvrRNAi UAS-Adgf-A) suppresses the progenitor differentiation phenotype caused by PvrRNAi (compare with Fig. 2C).
(G) Co-expression of Adgf-A and STATDN (Hml-gal4 UAS-2xEGFP UAS-STATDN UAS-Adgf-A) suppresses the progenitor differentiation phenotype caused by STATDN (compare with Fig. 2H).
(H) Schematic representation of Adgf-A function in progenitor maintenance. Expression of Adgf-A from the differentiating cells, mediated by Pvf1/Pvr and STAT signaling, functions as the CZ signal. Therefore, loss of Adgf-A in the CZ (as in C) causes progenitor differentiation, and over-expression of Adgf-A in the CZ (in F and G) suppresses mutant effects of Pvr and STAT. The earlier steps are as described in Figure 2S.
As described previously, the primary function of Adgf-A is to inactivate adenosine, and loss of Adgf-A causes an increase in extracellular adenosine levels (Dolezal et al., 2005; Zurovec et al., 2002). Additionally, adenosine levels can also be regulated by intracellular uptake mediated by the nucleoside transporter ENT3 (Baldwin et al., 2005). The adenosine that remains extracellularly can bind the adenosine receptor AdoR to transmit a G protein/adenylate cyclase/PKA-mediated signal into the cell. Within the developing lymph gland, expression of ENT3RNAi in the progenitors causes them to differentiate (Figure 4B), presumably because loss of ENT3 among progenitors causes a local accumulation of extracellular adenosine that then signals through AdoR to cause them to proliferate and differentiate. This differentiation phenotype does not occur when ENT3 is downregulated in differentiating cells (Figure 4C). In contrast, expression of AdoRRNAi in the progenitor population causes an expansion of these cells at the expense of differentiation (Figure 4D) and over-expression of AdoR in progenitors causes a loss of progenitor maintenance (Figure 4E). These results indicate that adenosine signaling through AdoR in progenitor cells negatively impacts their maintenance, and that this signaling is critically controlled during development to maintain progenitors by the expression of Adgf-A in the newly differentiating cells.
Figure 4. Adenosine signaling regulates hematopoietic progenitor quiescence.
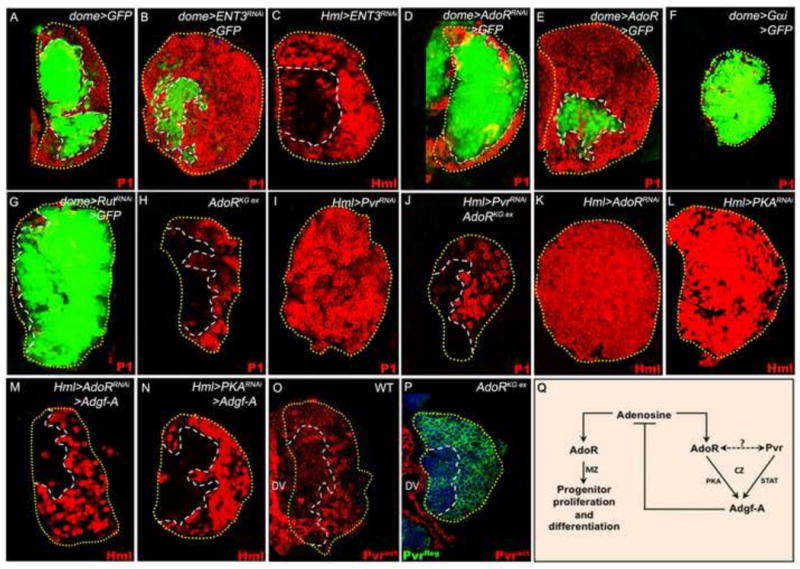
In A-B and D-G, the progenitor cell population is marked with dome-gal4, UAS-2xEGFP (green). In A-N, differentiating hemocytes are shown in red. All lymph glands shown are from wandering third instar larvae.
(A-G) Role of the adenosine transporter ENT3, the receptor AdoR, downstream signalling component Gα, and the adenylate cyclase Rutabaga.
(A) Control lymph gland showing the normal expression pattern of dome-gal4 UAS-2xEGFP (green) in progenitors and P1 (red) in differentiating cells.
(B) ENT3RNAi expression in progenitors (dome-gal4 UAS-2xEGFP UAS-ENT3RNAi) causes a reduction in MZ size (compare with A) and a corresponding increase in the CZ.
(C) ENT3RNAi expression in differentiating cells (Hml-gal4 UAS-2xEGFP UAS-ENT3RNAi) does not cause progenitor differentiation.
(D) AdoRRNAi expression in progenitors (dome-gal4 UAS-2xEGFP UAS-AdoRRNAi) causes their expansion and reduction of differentiating cells (compare with A).
(E) AdoR overexpression in progenitors (dome-gal4 UAS-2xEGFP UAS-AdoR) causes their differentiation (compare with A).
(F) Gαi overexpression in progenitor cells (dome-gal4 UAS-2xEGFP UAS-Gαi) blocks their differentiation.
(G) RutRNAi expression in progenitors (dome-gal4 UAS-2xEGFP UAS-RutRNAi) causes their expansion and reduces differentiating cells.
(H-J) AdoR function downstream of Pvr.
(H) Control AdoR mutants (AdoRKGex/AdoRKGex) develop a small lymph gland but with normal zonation.
(I) PvrRNAi expression in differentiating cells (Hml-gal4 UAS-2xEGFP UAS-PvrRNAi) causes loss of progenitors.
(J) Mutation in AdoR (AdoRKGex/AdoRKGex) partially suppresses the loss of progenitors caused by PvrRNAi in differentiating cells (Hml-gal4 UAS-2xEGFP UAS-PvrRNAi, compare with Fig 2C).
(K-N) AdoR and Pvr together control expression of Adgf-A in differentiating cells.
(K) AdoRRNAi expression in differentiating cells (Hml-gal4 UAS-2xEGFP UAS-AdoRRNAi) causes progenitors to differentiate (red).
(L) PKARNAi expression in differentiating cells (Hml-gal4 UAS-2xEGFP UAS-PKARNAi) causes progenitors to differentiate (red).
(M) Co-expression of Adgf-A with AdoRRNAi in differentiating cells (Hml-gal4 UAS-2xEGFP UAS-AdoRRNAi UAS-Adgf-A) suppresses the progenitor differentiation phenotype caused by AdoRRNAi alone (compare with K).
(N) Co-expression of Adgf-A with PKARNAi in differentiating cells (Hml-gal4 UAS-2xEGFP UAS-PKARNAi UAS-Adgf-A) suppresses the progenitor differentiation phenotype caused by PKARNAi alone (compare with L).
(O-P) Activation of Pvr is dependent upon the presence of AdoR.
(O) A wild-type (w1118) lymph gland showing the expression pattern of activated (phosphorylated) Pvr (Pvract, red). The red staining on the left is in the heart (dorsal vessel, DV).
(P) In AdoRKG03964ex/AdoRKG03964ex lymph glands expression of Pvr (total protein, Pvrreg, green) is normal. However, Pvract expression (red) is entirely missing from the lymph gland (compare with red staining in O). The red staining on the left is in the dorsal vessel (DV).
(Q) Adenosine signaling in the lymph gland. Adenosine signaling through AdoR in progenitors promotes their proliferation and differentiation. This process is kept in check by reduction of available extracellular adenosine. Therefore, in the progenitors, reduction of the adenosine transporter (in B) or increase in the adenosine receptor (in E) causes differentiation. Also in the progenitors, loss of the adenosine receptor (in O) or its downstream components (in F and G) causes expansion of this compartment at the cost of the CZ. In differentiating cells, Pvr collaborates with AdoR to maintain Adgf-A expression. Therefore, loss of adenosine receptor or its downstream components in the CZ (in K and L) causes progenitor differentiation and this phenotype is suppressed by overexpression of Adgf-A.
AdoR is a seven transmembrane domain receptor that signals through G-proteins to activate adenylate cyclase (Dolezelova et al., 2007). Consistent with this notion, over-expression of an inhibitory G-protein, Galphai (Yu et al., 2005) in progenitors blocks their differentiation (Figure 4F). Similarly, reduction of Drosophila adenylate cyclase function in progenitors using rutabagaRNAi (Figure 4G) or rutabaga mutants (Figure S4P) causes the larger number of cells to be maintained as progenitors when compared with wild type (Figure S4F), suggesting that cAMP signaling normally promotes differentiation. Finally, the phenotype associated with loss of Pvr (which in our model will block Adgf-A function and increase extracellular adenosine; Figure 4H-J) or Adgf-A can be suppressed in an AdoR mutant background (Figure S4H-L). In both cases, the loss of quiescence correlates with differentiation of cells that normally occupy the MZ compartment. These results further establishes a role for Pvr and Adgf-A in counteracting adenosine signaling through AdoR and adenylate cyclase in progenitor cells leading to their maintenance as quiescent hematopoietic progenitors.
Step 4: A feedback loop that limits adenosine levels
Expressing AdoRRNAi (Figure 4K) or PKARNAi (Figure 4L) in the CZ causes non-autonomous differentiation of MZ progenitors. This phenotype is suppressed by over-expression of Adgf-A in the CZ (Figure 4 M-N). In addition, although Pvr expression remains unaffected, no staining for activated Pvr is detected in cells that are mutant for AdoR (Figure 4P). The simplest model is that AdoR and Pvr collaborate together in a differentiating cell to activate Adgf-A, which is secreted and regulates the level of adenosine (Figure 4Q).
Step 5: Balancing PSC versus CZ signal
PKA occupies a unique position in being associated with both adenosine signaling (Dolezelova et al., 2007) and Hh signaling (Collier et al., 2004). Adenosine activates and Hh inhibits PKA, which promotes the formation of the repressor form of Ci, Ci75 (Collier et al., 2004), suggesting perhaps a link between the CZ and the niche-derived signals. PKA function was reduced in the MZ by expressing either the dominant negative PKAmR* or the dominant negative regulatory subunit PKA-R2 or PKARNAi (Figure 5A-D). In each case the result is a normal sized lymph gland in which there is a relative increase in the size of the MZ at the expense of the CZ. The proliferative profile of progenitors with loss of function PKA does not change (Figure S4N-O). Thus loss of PKA activity causes cells to be maintained in a progenitor state but does not cause an overall expansion in the total number of cells in the lymph gland. Similarly, overactivating PKA using a mutated mouse PKA catalytic subunit (PKAmC*) that is resistant to inhibition by its regulatory subunit causes differentiation of hematopoietic progenitors (Figure 5E). Most importantly, a single copy loss of PKA in an Adgf-A mutant background rescues the progenitor cell phenotype (Figure 5F-H). Adenosine levels are expected to be high in an Adgf-A mutant background, thus causing proliferation followed by differentiation. However, simultaneously reducing PKA function downstream of the adenosine receptor has an attenuating effect on this signal and hence suppresses the mutant phenotype. It is important to point out that the nature of the signal that initiates differentiation of the precursors remains unknown and is still operational in all of the above genetic backgrounds.
Figure 5. Interaction between PSC and CZ signals.
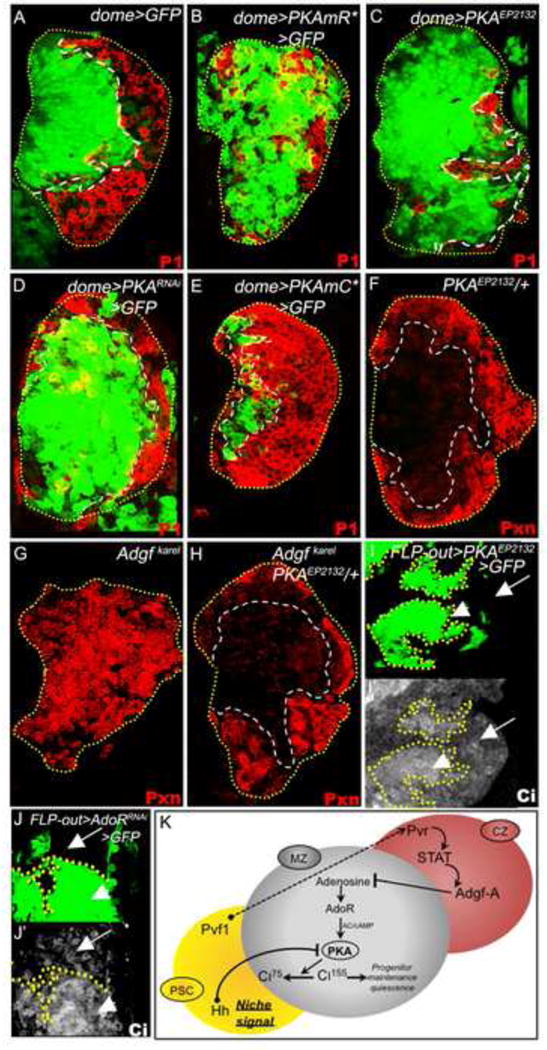
In A-E, progenitors are marked with dome-gal4 UAS-2xEGFP (green). In A-H, the differentiating cells are shown in red. All lymph glands shown are from wandering third instar larvae.
(A) Control lymph gland showing the normal expression pattern of dome-gal4 UAS-GFP (green) in progenitors and P1 (red) in differentiating cells.
(B-E) Role of PKA in progenitor maintenance. Loss of PKA function in progenitors causes their expansion and a corresponding reduction of differentiating cells, as shown by expressing (B) the regulatory domain of PKA (dome-gal4 UAS-2xEGFP UAS-PKmR*), which functions as a sink for cAMP, (C) PKA EP2132 (dome-gal4 UAS-2xEGFP UAS-PKAEP2132) which functions as a dominant-negative, and (D) PKARNAi (dome-gal4 UAS-2xEGFP UAS-PKARNAi). (E) Gain of PKA function in progenitors through the expression of the constitutively active PKAmC* (dome-gal4 UAS-2xEGFP UAS-PKAmC*) causes their differentiation. Compare with A.
(F-H) PKA opposes the function of Adgf-A in progenitor maintenance. (F) Normal control PKAEP2132/+. (G) Control Adgf-Akarel/Adgf-Akarel. Hematopoietic progenitors differentiate. (H) PKAEP2132/+; Adgf-Akarel/Adgf-Akarel. The progenitor differentiation phenotype due to loss of Adgf-A is suppressed by single copy loss of PKA (compare with G).
(I-J) Regulation of Ciactivated by PKA and AdoR. (I-I′) FLP-out cell clones (green in I, see materials and methods) that reduce PKA function (by expressing PKAEP2132) exhibit high levels of Ciactivated (Ciact) expression (shown in grayscale, I′). (J) FLP-out cell clones (green) that reduce AdoR function (by expressing AdoRRNAi) also exhibit elevated levels of Ciact (shown in grayscale) expression (J′).
(K) The niche and CZ signals function together to regulate the levels of Ciact necessary for progenitor maintenance in the MZ. Note that Ci is activated not only upon loss of PKA (in I) but also upon loss of the adenosine receptor (in J) thus creating a link between Hedgehog and adenosine signaling.
A direct readout of Hedgehog signaling is the expression level of the active form of Ci (Ci155) and the transcriptional target ptc. As expected, over-expression of PKA-R2, which inhibits PKA function, causes higher levels of active Ci and Ptc (Figure 5I, I′; S4M, M′), consistent with increased Hedgehog signaling. Additionally, staining for Ciact shows that compared with wild type, the Adgf-A/Adgf-A mutant shows lower Ci expression; this is enhanced upon PKA/+ heterozygosity (Figure S4S-T). The critical issue is to show whether Ci modulation is AdoR-dependent and we tested this possibility by decreasing the level of AdoR in MZ cells. This causes an increase in the level of activated Ci in the progenitor cell population showing that AdoR-mediated cAMP-dependent PKA activation modulates Ci activity (Figure 5J, J′). Ultimately, the Adgf-A-related signal limits the amount of PKA activated by adenosine signaling and therefore the degradation of active Ci. Similarly, active Hedgehog niche signaling also inhibits PKA, limiting degradation of Ci into its repressive form. Together, the CZ and the PSC signals maintain a balance of Ci activity within the MZ thereby controlling quiescence within the hematopoietic progenitor cell population (Figure 5K).
Discussion
The role of a niche signal is well established in many developmental systems that involve stem cell/progenitor populations (Fuchs et al., 2004; Losick et al., 2011; Orkin and Zon, 2008; Scadden, 2006). In the Drosophila lymph gland the niche expresses Hh and maintains a group of progenitor cells (Mandal et al., 2007). This current study establishes an additional mechanism, parallel to the niche signal that originates from differentiating cells, which also regulates quiescence of hematopoietic progenitors.
The cells of the lymph gland proliferate at early stages, from embryo to mid second instar. At this stage, cells farthest from the PSC initiate differentiation and the rest enter a quiescent phase defining a MZ. In wild type, the cells of the MZ remain quiescent and in progenitor form throughout the third instar and we show here that this process requires a combination of the PSC and CZ signals. If either signal is removed, the progenitor population will eventually be lost due to differentiation. In many different genetic backgrounds, if quiescence is lost, the progenitor population initially continues to incorporate BrdU during the second instar without expressing any maturation markers (Figure S1A-F). The differentiation phenotype, characterized by the expression of such markers, follows this abnormal proliferation. The net result is that whenever the progenitors accumulate BrdU (but not express any markers of differentiation) in the second instar, all cells of the lymph gland are differentiated and no MZ remains in the third instar. While we do not know the nature of the signal that triggers hemocyte differentiation, withdrawal of Wingless may play a role in this process (Sinenko et al., 2009).
Our experimental analysis has demonstrated a novel role for Pvr in maturing hemocytes and its ligand, Pvf1, in the cells of the PSC. Pvf1 expression increases at a stage when the lymph gland is highly proliferative. At this critical window in development, Pvf1 originating from the PSC is transported to the differentiating hemocytes, binds to its receptor Pvr, and activates a STAT-dependent signaling cascade. At this stage, Pvf1 is sensed by all cells but it is only in the differentiating hemocytes that it activates Adgf-A in an AdoR/Pvr-dependent manner. This secreted factor Adgf-A is required for regulating extracellular adenosine levels. High adenosine would signal through AdoR and PKA to inactivate Ci and reduce the effects of the niche-derived Hedgehog signal leading to differentiation of the progenitor cells. The function of the Adgf-A signal is to reduce this adenosine signal and therefore reinforce the maintenance of progenitors by the Hedgehog signal. Thus, the Adgf-A and Hh signals work in the same direction but Adgf-A does so by negating a proliferative signal due to adenosine. In wild type, equilibrium is reached through a signal not originating from the niche that opposes this proliferative process. The attractive step in this model is that the CZ and niche (in this case Hh-dependent) signals both impinge on common downstream elements allowing for control of the progenitor population relative to the niche and the differentiated cells. Most importantly, this is a mechanism for maintaining quiescence within a moderately large population of cells that is not in direct contact with a niche. By the time the three zone PSC/MZ/CZ system is set up in the late second instar all the cells of the MZ express high levels of E-cadherin, become quiescent and are maintained as progenitors and are capable of giving rise to all blood cell lineages (Jung et al., 2005; Krzemien et al., 2010). Under such circumstances, the interaction between a niche-derived signal and an equilibrium signal originating from differentiating cells can maintain homeostatic control of the progenitor population. Several vertebrate stem cell/progenitor scenarios such as during bone morphogenesis (Mendez-Ferrer et al., 2010) and hematopoiesis (He et al., 2009; Orkin and Zon, 2008) or in the Drosophila intestine (Mathur et al., 2010) have progenitors and differentiating cells in close proximity that could pose an opportunity for a similar niche and differentiating cell-derived signal interaction. In fact, evidence for such interactions have recently been provided for vertebrate skin cells (Hsu et al., 2011).
The role of small molecules such as adenosine has not yet been adequately addressed in vertebrate progenitor maintenance. A small molecule such as extracellular adenosine is unlikely to form a gradient over the population of cells and maintain such a gradient over a developmental time scale. It is much more likely that this system operates similar to the “quorum sensing” mechanisms described for prokaryotes (Ng and Bassler, 2009). A critical level of adenosine is required for proliferation and by expressing the Adgf-A signal this threshold amount is lowered, causing quiescence in the entire population.
We describe a developmental mechanism that is relevant to the generation of an optimal number of blood cells in the absence of any overt injury or infection. However, a system that utilizes such a mechanism to maintain a progenitor population could potentially sense a disruption upon induction of various metabolic stresses to cause differentiation of myeloid cells. Various mitochondrial and cellular stresses can cause an increase in extracellular adenosine (Fredholm, 2007), but whether they are relevant to this system remains to be studied. In the past, we have observed dual use of reactive oxygen species (ROS) as well as Hypoxia Inducible Factor-a (HIF-α) in both development and stress response of the Drosophila hematopoietic system (Mukherjee et al., 2011; Owusu-Ansah and Banerjee, 2009). Responses to injury have been described in the Drosophila intestine (Amcheslavsky et al., 2009; Jiang et al., 2009), and in satellite cells (Ten Broek et al., 2010) that respond during injury, a stress related signal could be the initiating factor that overrides a maintenance signal. Thus the equilibrium generated through developmental interactions is disrupted to promote a cellular response to stress signals.
Experimental Procedures
For sources of all stocks and antibodies please see Supplementary information. Progenitor and differentiated cell types for all genotypes are assessed by combining 3-5 middle optical sections of the lymph gland lobe. Flp-out clones within MZ were generated using a lineage tracing system controlled by hand-gal4 and HmlΔ-gal4 as described (Evans et al., 2009). Genotypes for, hs-flp derived clones: [hs-flp Ay-gal4 UAS-GFP UAS-domeDN]. FRT clones: [hs-flp FRT40A PvrC2195 FRT40A Ubi-GFP] and [hs-flp FRT82B Stat92E06346 FRT82B Ubi-GFP].
Supplementary Material
Acknowledgments
We thank members of the Banerjee lab for helpful discussions. We acknowledge VDRC, NIG, DHSB, BDSC, E. Bach, J. Calderon, J. Darnell, T. Dolezal, J. Hooper, D. Montell, P. Rorth, A. Sehgal, B. Shilo, M.Yoo, and M. Zeidler for reagents. Supported by NHLBI R01 HL067395 to UB and NHLBI K08 HL087026 to JM.
Contributor Information
Julian A. Martinez-Agosto, Email: julianmartinez@mednet.ucla.edu.
Utpal Banerjee, Email: banerjee@mbi.ucla.edu.
References
- Amcheslavsky A, Jiang J, Ip YT. Tissue Damage-Induced Intestinal Stem Cell Division in Drosophila. Cell Stem Cell. 2009;4:49–61. doi: 10.1016/j.stem.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin SA, Yao SY, Hyde RJ, Ng AM, Foppolo S, Barnes K, Ritzel MW, Cass CE, Young JD. Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. J Biol Chem. 2005;280:15880–15887. doi: 10.1074/jbc.M414337200. [DOI] [PubMed] [Google Scholar]
- Bond D, Foley E. A quantitative RNAi screen for JNK modifiers identifies Pvr as a novel regulator of Drosophila immune signaling. PLoS Pathog. 2009;5:e1000655. doi: 10.1371/journal.ppat.1000655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier LS, Suyama K, Anderson JH, Scott MP. Drosophila Costal1 Mutations Are Alleles of Protein Kinase A That Modulate Hedgehog Signaling. Genetics. 2004;167:783–796. doi: 10.1534/genetics.103.024992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozatier M, Ubeda JM, Vincent A, Meister M. Cellular immune response to parasitization in Drosophila requires the EBF orthologue collier. PLoS Biol. 2004;2:E196. doi: 10.1371/journal.pbio.0020196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolezal T, Dolezelova E, Zurovec M, Bryant PJ. A role for adenosine deaminase in Drosophila larval development. PLoS Biol. 2005;3:e201. doi: 10.1371/journal.pbio.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolezal T, Gazi M, Zurovec M, Bryant PJ. Genetic analysis of the ADGF multigene family by homologous recombination and gene conversion in Drosophila. Genetics. 2003;165:653–666. doi: 10.1093/genetics/165.2.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolezelova E, Nothacker HP, Civelli O, Bryant PJ, Zurovec M. A Drosophila adenosine receptor activates cAMP and calcium signaling. Insect Biochem Mol Biol. 2007;37:318–329. doi: 10.1016/j.ibmb.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Evans CJ, Olson JM, Ngo KT, Kim E, Lee NE, Kuoy E, Patananan AN, Sitz D, Tran P, Do MT, et al. G-TRACE: rapid Gal4-based cell lineage analysis in Drosophila. Nat Meth. 2009;6:603–605. doi: 10.1038/nmeth.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14:1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- Fu X, Zhang J. Transcription factor p91 interacts with the epidermal growth factor receptor and mediates activation of the c-fos gene promoter. Cell. 1993;74:1135–1145. doi: 10.1016/0092-8674(93)90734-8. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: stem cells and their niche. Cell. 2004;116:769–778. doi: 10.1016/s0092-8674(04)00255-7. [DOI] [PubMed] [Google Scholar]
- Garrett RW, Emerson SG. Bone and Blood Vessels: The Hard and the Soft of Hematopoietic Stem Cell Niches. Cell Stem Cell. 2009;4:503–506. doi: 10.1016/j.stem.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Giansanti MG, Farkas RM, Bonaccorsi S, Lindsley DL, Wakimoto BT, Fuller MT, Gatti M. Genetic dissection of meiotic cytokinesis in Drosophila males. Mol Biol Cell. 2004;15:2509–2522. doi: 10.1091/mbc.E03-08-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Nakada D, Morrison SJ. Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol. 2009;25:377–406. doi: 10.1146/annurev.cellbio.042308.113248. [DOI] [PubMed] [Google Scholar]
- Hsu YC, Pasolli HA, Fuchs E. Dynamics between Stem Cells, Niche, and Progeny in the Hair Follicle. Cell. 2011;144:92–105. doi: 10.1016/j.cell.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru S, Ueda R, Hinohara Y, Ohtani M, Hanafusa H. PVR plays a critical role via JNK activation in thorax closure during Drosophila metamorphosis. EMBO J. 2004;23:3984–3994. doi: 10.1038/sj.emboj.7600417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens K, Sung HH, Rorth P. Direct detection of guidance receptor activity during border cell migration. Proc Natl Acad Sci U S A. 2010;107:7323–7328. doi: 10.1073/pnas.0915075107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. Cytokine/Jak/Stat Signaling Mediates Regeneration and Homeostasis in the Drosophila Midgut. Cell. 2009;137:1343–1355. doi: 10.1016/j.cell.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jude CD, Gaudet JJ, Speck NA, Ernst P. Leukemia and hematopoietic stem cells: balancing proliferation and quiescence. Cell Cycle. 2008;7:586–591. doi: 10.4161/cc.7.5.5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SH, Evans CJ, Uemura C, Banerjee U. The Drosophila lymph gland as a developmental model of hematopoiesis. Development. 2005;132:2521–2533. doi: 10.1242/dev.01837. [DOI] [PubMed] [Google Scholar]
- Krzemien J, Dubois L, Makki R, Meister M, Vincent A, Crozatier M. Control of blood cell homeostasis in Drosophila larvae by the posterior signalling centre. Nature. 2007;446:325–328. doi: 10.1038/nature05650. [DOI] [PubMed] [Google Scholar]
- Krzemien J, Oyallon J, Crozatier M, Vincent A. Hematopoietic progenitors and hemocyte lineages in the Drosophila lymph gland. Developmental Biology. 2010;346:310–319. doi: 10.1016/j.ydbio.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Learte AR, Forero MG, Hidalgo A. Gliatrophic and gliatropic roles of PVF/PVR signaling during axon guidance. Glia. 2008;56:164–176. doi: 10.1002/glia.20601. [DOI] [PubMed] [Google Scholar]
- Li L, Clevers H. Coexistence of Quiescent and Active Adult Stem Cells in Mammals. Science. 2010;327:542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX, Agaisse H, Mathey-Prevot B, Perrimon N. Differential requirement for STAT by gain-of-function and wild-type receptor tyrosine kinase Torso in Drosophila. Development. 2002;129:4241–4248. doi: 10.1242/dev.129.18.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick Vicki P, Morris Lucy X, Fox Donald T, Spradling A. Drosophila Stem Cell Niches: A Decade of Discovery Suggests a Unified View of Stem Cell Regulation. Developmental Cell. 2011;21:159–171. doi: 10.1016/j.devcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier S, Podemski L, Graham S, McDermid H, Locke J. Characterization of the adenosine deaminase-related growth factor (ADGF) gene family in Drosophila. Gene. 2001;280:27–36. doi: 10.1016/s0378-1119(01)00762-4. [DOI] [PubMed] [Google Scholar]
- Makki R, Meister M, Pennetier D, Ubeda JM, Braun A, Daburon V, Krzemien J, Bourbon HM, Zhou R, Vincent A, et al. A short receptor downregulates JAK/STAT signalling to control the Drosophila cellular immune response. PLoS Biol. 2010;8:e1000441. doi: 10.1371/journal.pbio.1000441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal L, Martinez-Agosto JA, Evans CJ, Hartenstein V, Banerjee U. A Hedgehog- and Antennapedia-dependent niche maintains Drosophila haematopoietic precursors. Nature. 2007;446:320–324. doi: 10.1038/nature05585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Agosto JA, Mikkola HK, Hartenstein V, Banerjee U. The hematopoietic stem cell and its niche: a comparative view. Genes Dev. 2007;21:3044–3060. doi: 10.1101/gad.1602607. [DOI] [PubMed] [Google Scholar]
- Mathieu J, Sung HH, Pugieux C, Soetaert J, Rorth P. A Sensitized PiggyBac-Based Screen for Regulators of Border Cell Migration in Drosophila. Genetics. 2007;176:1579–1590. doi: 10.1534/genetics.107.071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur D, Bost A, Driver I, Ohlstein B. A transient niche regulates the specification of Drosophila intestinal stem cells. Science. 2010;327:210–213. doi: 10.1126/science.1181958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakhina S, Steward R. Hematopoietic stem cells in Drosophila. Development. 2010;137:27–31. doi: 10.1242/dev.043943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee T, Kim WS, Mandal L, Banerjee U. Interaction Between Notch and Hif-la in Development and Survival of Drosophila Blood Cells. Science. 2011;332:1210–1213. doi: 10.1126/science.1199643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng WL, Bassler BL. Bacterial quorum-sensing network architectures. Annu Rev Genet. 2009;43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakova M, Dolezal T. Expression of Drosophila Adenosine Deaminase in Immune Cells during Inflammatory Response. PLoS ONE. 2011;6:e17741. doi: 10.1371/journal.pone.0017741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge R, CA K, Banerjee U. Genetic dissection of neurodevelopmental pathway: Son of Sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell. 1991;64:39–48. doi: 10.1016/0092-8674(91)90207-f. [DOI] [PubMed] [Google Scholar]
- Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- Sims D, Duchek P, Baum B. PDGF/VEGF signaling controls cell size in Drosophila. Genome Biology. 2009;10:R20. doi: 10.1186/gb-2009-10-2-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinenko SA, Mandal L, Martinez-Agosto JA, Banerjee U. Dual Role of Wingless Signaling in Stem-like Hematopoietic Precursor Maintenance in Drosophila. Dev Cell. 2009;16:756–763. doi: 10.1016/j.devcel.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelkinson MG, Kalderon D. Processing of the Drosophila hedgehog signaling effector Ci-155 to the repressor Ci-75 is mediated by direct binding to the SCF component Slimb. Curr Biol. 2006;16:110–116. doi: 10.1016/j.cub.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Ten Broek RW, Grefte S, Von den Hoff JW. Regulatory factors and cell populations involved in skeletal muscle regeneration. J Cell Physiol. 2010;224:7–16. doi: 10.1002/jcp.22127. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Wang H, Qian H, Kaushik R, Bownes M, Yang X, Chia W. Locomotion defects, together with Pins, regulates heterotrimeric G-protein signaling during Drosophila neuroblast asymmetric divisions. Genes Dev. 2005;19:1341–1353. doi: 10.1101/gad.1295505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Zavialov AV, Lauvau G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. Journal of Leukocyte Biology. 2010;88:279–290. doi: 10.1189/jlb.1109764. [DOI] [PubMed] [Google Scholar]
- Zurovec M, Dolezal T, Gazi M, Pavlova E, Bryant PJ. Adenosine deaminase-related growth factors stimulate cell proliferation in Drosophila by depleting extracellular adenosine. Proc Natl Acad Sci U S A. 2002;99:4403–4408. doi: 10.1073/pnas.062059699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.