

Abstract
Rationale
Stable isotope analysis of archaeological charred plants has become a useful tool for interpreting past agricultural practices and refining ancient dietary reconstruction. Charred material that lay buried in soil for millennia, however, is susceptible to various kinds of contamination, whose impact on the grain/seed isotopic composition is poorly understood. Pre-treatment protocols have been adapted in distinct forms from radiocarbon dating, but insufficient research has been carried out on evaluating their effectiveness and necessity for stable carbon and nitrogen isotope analysis.
Methods
The effects of previously used pre-treatment protocols on the isotopic composition of archaeological and modern sets of samples were investigated. An archaeological sample was also artificially contaminated with carbonates, nitrates and humic acid and subjected to treatment aimed at removing the introduced contamination. The presence and removal of the contamination were investigated using Fourier transform infrared spectroscopy (FTIR) and δ13C and δ15N values.
Results
The results show a ca 1‰ decrease in the δ15N values of archaeological charred plant material caused by harsh acid treatments and ultra-sonication. This change is interpreted as being caused by mechanical distortion of the grains/seeds rather than by the removal of contamination. Furthermore, specific infrared peaks have been identified that can be used to detect the three types of contaminants studied. We argue that it is not necessary to try to remove humic acid contamination for stable isotope analysis. The advantages and disadvantages of crushing the grains/seeds before pre-treatment are discussed.
Conclusions
We recommend the use of an acid-only procedure (0.5 M HCl for 30 min at 80°C followed by three rinses in distilled water) for cleaning charred plant remains. This study fills an important gap in plant stable isotope research that will enable future researchers to evaluate potential sources of isotopic change and pre-treat their samples with methods that have been demonstrated to be effective. © 2014 The Authors. Rapid Communications in Mass Spectrometry published by John Wiley & Sons Ltd.
In recent years, increasing attention has been placed on involving archaeological plant material in stable isotope analysis, whether for better interpreting ancient human and animal diets or for reconstructing the scale and intensity of past agricultural practices.[1–10] Most of the studies attempted to remove contamination from the analyzed charred plant material, but no consensus yet exists for how it should be done. Reported investigations on how pre-treatment methods affect charred plant material involved comparing the stable isotopic measurements (δ13C and δ15N values)[3,9] and the structural composition[4] of untreated and pre-treated archaeological samples. However, no studies (that we are aware of) attempted to characterize the impact and removal of potential sources of contamination.
Most researchers apply a version of the acid-base-acid (ABA) protocol originally developed for radiocarbon dating,[11–13] using a variety of solution concentrations, temperatures and durations (see Table1). The effectiveness of this treatment for stable isotope analysis is unknown, and the degree of mass loss (leading to complete loss of some samples) is problematic. In addition, debate is still ongoing about the reliability/appropriateness of single-grain analysis, whether samples should be crushed prior to treatment, and how to assess the state of preservation/diagenetic alteration of the charred plant material.
Table 1.
Pre-treatment methods employed in the past to remove contamination from charred plant material prior to stable carbon and nitrogen isotope analysis. For comparison, the technique used at the Oxford Radiocarbon Accelerator Unit to clean non-woody plant material is also included
| Reference | Treatment description | Comment |
|---|---|---|
| DeNiro and Hastorf[29] | (1) 6 M HCl (aq.) for 24 h; (2) 1 M NaOH (aq.) for 24 h; (3) 6 M HCl (aq.) for 10 min; (4) shaking samples in 2 M KCl (aq.) for 60 min; (5) 5 M HF – 1 M HCl (aq.) for 24 h; all steps were carried out at room temperature and followed by rinsing in water | Adapted from Silva and Bremner;[38] Bremner and Keeney;[39] Stevenson[40] |
| Araus and Buxó[41] | H2O2(aq.), and “where necessary, acid treatment” | No source cited |
| Aguilera et al.;[34] Lightfoot and Stevens;[7] Ferrio et al.;[42,43] Fiorentino et al.[44] | 6 M HCl (aq.) at room temperature for 24 h; as many rinses in distilled water as it took to neutralize sample | Adapted from DeNiro and Hastorf[29] |
| Brock et al.[12] | (1) 1 M HCl (aq.) for 20 min (or until effervescence has stopped); (2) 0.2 M NaOH (aq.) for 20 min; (3) 1 M HCl (aq.) for 60 min; (4) 2.5% NaO2Cl (aq.) up to 30 min; all steps were carried out at room temperature to 80°C | Protocol used in radiocarbon dating laboratory |
| Kanstrup et al.[9] | (1) 1 M HCl (aq.) for 1 h; (2) 1 M NaOH (aq.) for 3 h (+additional hour for very dark samples); (3) 1 M HCl (aq.) for 16 h; first two steps were carried out at 80°C, last step at room temperature; samples were rinsed three times in distilled water only at the end | Adapted from Philippsen et al.:[45] carried out on food crusts; Kristiansen et al.:[46] carried out on soil organic matter; and Olsson[47] |
| Fraser et al.[30] | (1) 0.5 M HCl (aq.) for 30–60 min (or until effervescence has stopped); (2) 0.1 M NaOH (aq.) for 60 min; (3) 0.5 M HCl (aq.) for 25 min; the acid steps were followed by three rinses in Milli-U water; the base step was followed by as many rinses as it took to remove all brown material from solution; procedure was carried out at 70°C | No source cited; adopted by Bogaard et al.;[1] Wallace et al.;[48] Vaiglova et al.[10] |
| Styring et al.[3] | (1) 0.1 M HCl (aq.) for 40 min; (2) 0.1 M NaOH (aq.); (3) 0.1 M HCl (aq.); procedure was carried out at 80°C and samples were washed to neutrality | Goh[49] |
| Fiorentino et al.[50] | (1) 1 M HCl (aq.) for 10 h at room temperature; (2) 1 M NaOH (aq.) at 60°C (time unspecified); (3) 1 M HCl (aq.) for 10 h at room temperature | Adapted from D'Elia et al.[51] and Quarta et al.[52] |
| Heaton et al.;[31] Masi et al.[32] | No pre-treatment |
This study aims to identify the most appropriate pre-treatment method for archaeological charred plant remains. Given the variability in pre-treatment methods employed in the past, three questions were identified which formed the basis of the present study:
Do the different pre-treatment methods employed for cleaning archaeological grain/seeds for stable carbon and nitrogen isotope analysis produce the same results?
How can we detect contamination (carbonate, nitrate, and humic acid) in charred plant material?
How can we remove contamination (carbonate, nitrate, and humic acid) from charred plant material? Which of the methods employed in the past achieve this goal?
In order to address these questions, a series of experiments was carried out in two stages. The first stage involved the comparative assessment of the effects of different pre-treatment methods (three acid-base-acid procedures, two acid-only washes and one treatment involving thorough cleaning using ultra-sonication in purified (Milli-U) water) on the δ13C and δ15N values of two types of archaeological and two types of modern cereal and pulse samples. The second stage involved an attempt to detect and remove artificially introduced carbonate, nitrate and humic acid contamination from an archaeological sample using δ13C values, δ15N values and FTIR spectra. More attention was paid to humic acids, as their presence in and necessity for removal from charred plant material has received little attention in previous plant stable isotopic investigations.
Methodological Background: Contamination, Charring and Pre-Treatment
There are three major contaminants that may affect the stable isotopic ratios of charred plant material: carbonates, nitrates and humic acids. Carbonates are acid-soluble while nitrates are water-soluble salts and both are naturally present in different types of soils and can be adsorbed by archaeological material. Humic acids, a form of humus substance, are dark-colored, hydrophilic and chemically complex high molecular weight organic molecules that dissolve in alkali solutions.[14–17] Their capability to dissolve makes them more mobile in soils and, as a result, humic acids have a higher potential for contaminating buried samples (as opposed to other humus substances such as fulvic acids and humin).
As the degradation products of structurally organized organic matter (e.g. plants), humic acids are naturally present in soils and infrared analyses have shown that large variability exists in their structure and composition.[18–20] Assessment of their impact on the stable isotopic composition of buried plant material is potentially extremely complex: humic acids may be a degradation product of the plants themselves[21,22] and thus have a very similar isotopic composition to the material of interest, or they may have formed from an isotopically distinct organic source. There are as yet no methods for distinguishing between the endogenous and exogenous humic acids potentially contained in excavated charred plant material. In addition, even if charred plant material was exposed to exogenous humic acids, the effect on the bulk plant stable isotope values may be minimal. Fraser et al.[3] soaked modern and archaeological charred millet grains (δ13C = –12.4‰ and –10.8‰, respectively) in a humic acid (δ13C = –26.7‰) for 6–24 months and found no significant effects on the δ13C values of the plant samples.
Humic acids may also be difficult to distinguish structurally. Cohen-Ofri et al.[22] investigated the structural composition of modern and ancient charcoal using a range of analytical techniques and found that they contain two phases: a micro-crystalline graphitic structure and a non-organized phase. Humic acids were also found to contain both phases. The fossil charcoal was found to contain a higher proportion of the disorganized phase than modern charcoal and the authors inferred a process of " self-humification" as an integral part of diagenetic transformation. More specific to grains/seeds, Maillard reactions, which occur during charring, have been reported to create humic acids.[23,24] A chemical treatment aimed at removing humic acids may thus lead to the complete loss of the sample due to the base-soluble nature of the charred plant material itself.
The impact of carbonates and nitrates on the stable isotopic content of charred grain is less unpredictable. Carbonates affect the δ13C values while nitrates the δ15N values. Carbonates can reach δ13C values of +2‰ in some regions[25] and small amounts of contamination can have a significant impact on the measured δ13C values of charred C3 plants, which are usually between –22 and –30‰. The δ15N values of nitrates can range from –2 to +8‰; depending on the interplay of several factors such as the presence of chemical fertilizers and animal dung manure in soils.[26]
To survive in the archaeological record, grains/seeds need to undergo chemical transformation rendering them resistant to post-burial alterations (e.g. biological/microbial attacks). This can occur through charring under anaerobic conditions: for example, when plant material is discarded during food preparation and buried in the fill of a hearth that continues to be used, or when grain in storage containers becomes 'baked' when a building is destroyed by fire. The heat causes incomplete combustion of the organic material. Many of the original biomolecules are preserved[27] and, if charred under optimal combinations of times and temperatures, the grains/seeds/chaff can retain their morphological distinctiveness. Although the structural composition of both ancient charcoal (burnt wood) and charred grains/seeds starts to resemble that of humus substances over time, the two types of materials are inherently different due to their variable original composition; wood is mostly made of cellulose and grains/seeds primarily consist of starch/polysaccharides. For this reason, caution needs to be exercised when comparing the behavior of charcoal and charred grains/seeds during burning and pre-treatment.
Styring et al.[4] investigated the impact of charring on the chemical composition and structure of grain and highlighted the importance of the Maillard reactions that lead to the transformation of amino acids and sugars into a large variety of aromatic compounds. The authors showed, using solid-state 13C-NMR and FTIR data, that there is a clear difference in the chemical composition of experimentally charred modern grain and archaeological charred grain (the latter obtained from two sites situated in distinct environments). They suggested that possible reasons for this difference were the continuation of the Maillard reactions after burial and/or microbial activity in the soil, which transform the remaining alkyl-containing compounds into aromatic compounds.
The conventional method for pre-treating archaeological plant material for radiocarbon dating is to use an Acid-Base-Acid (ABA) protocol (sometimes referred to as acid-alkali-acid, or AAA).[11–13] The first acid wash removes exogenous carbonates and organic acids, the base wash removes humic acids and the second acid wash removes carbon that was adsorbed as CO2 during the base wash. In the Oxford Radiocarbon Accelerator Unit (Oxford, UK), all plant remains are also treated with 2.5% bleach (NaO2Cl).[28] Different permutations of the ABA technique (employing acids of varying concentrations and under variable conditions of time and temperature) have been used in the past to prepare charred plants for stable isotope analysis (see Table1).
Early work on plant stable isotopes was undertaken by DeNiro and Hastorf,[29] who cleaned charred and desiccated plant samples with 6 M hydrochloric acid (HCl (aq.)), 1 M sodium hydroxide (NaOH (aq.)), and some with 2 M potassium chloride (KCl (aq.)), and 5 M hydrofluoric acid (HF) mixed with 1 M HCl (aq.) at room temperature. Fraser et al.[3] employed a gentler ABA protocol, involving 0.5 M HCl (aq.) and 0.1 M NaOH (aq.) at 70°C, which was then replicated in a follow-up study[30] and adopted by Bogaard et al.,[1] Wallace and co-workers,[4] and Vaiglova et al.[10] Several stable isotope analyses of archaeobotanical remains were carried out without any pre-treatment at all.[31,32]
Investigations into the effects of ABA pre-treatment on charred archaeological grains/seeds were carried out by comparing the δ13C and δ15N values of samples that underwent chemical pre-treatment with matching untreated samples. Kanstrup et al.[9] cleaned their samples (n = 31) with 1 M HCl (aq.) and 1M NaOH (aq.) at 80°C (for 1–16h) and found an average offset in the δ15N value of +0.7 ± 1.0‰. No offset was found in the δ13C values and the mass loss was 30–80% (average of 43%). The treated samples were not rinsed in water between the chemical washes. The differences in δ15N values between the treated and untreated samples may have been caused by the removal of contamination (with lower δ15N values) or the removal of parts of the grains/seeds that were depleted in 15N. In another comparative experiment, Fraser et al.[3] reported no consistent changes to the δ13C and δ15N values caused by the ABA pre-treatment that employed 0.5 M HCl (aq.) and 0.1 M NaOH (aq.) at 70°C (for 0.5–1 h) (n = 17). The lack of any change indicates that the material that had been removed (whether contamination or part of the grain/seed) did not have a significant effect on the δ13C and δ15N values of the sample. Styring et al.[4] observed, using FTIR, 13C CP-MAS NMR and elemental composition analysis, that the 0.1 M HCl/0.1 M NaOH (aq.) ABA protocol causes structural changes in the form of carboxylate ions (R-COO–) being converted into carboxylic acid (R-COOH). Changes linked to the removal of particular contamination have not been observed and it was not demonstrated whether the material contained any contamination at the outset.
Experimental
Archaeological and modern samples
Three sets of archaeological samples and two sets of modern samples were used in this experiment. The archaeological samples were obtained from the Neolithic site of Çatalhöyük: PEAar (common pea, Pisum sativum L.; from a concentration of burnt seeds and fish remains from North Area, Hodder Level 4040G, building 77, unit number 16498), BARar (naked barley, Hordeum vulgare L. var. nudum; from a bin fill from the TPC area, Late Neolithic TP-L(?), unit number 30859, building B.122), and LENar (lentil, Lens culinaris Medik.; from a bin fill from the North area, unit number 1344, building 1, Hodder level G). This site was chosen because excavation has yielded an extremely rich archaeobotanical assemblage, which allowed for multiple sub-sampling of a single context. Initial analysis of crop samples from this site yielded %N values much higher than those of modern charred grains/seeds and archaeological samples (Fraser et al., unpublished). Previously, nitrates were detected in bones from Çatalhöyük using ion-exchange chromatography,[33] but since the archaeobotanical samples had been washed in water through pre-treatment, their contamination by nitrates is unlikely.
Samples from other sites, where the burial conditions vary and where the crop samples do not exhibit anomalously high %N values, would have allowed us to observe whether differently preserved samples respond the same way to pre-treatment. Unfortunately, such thorough sampling as was possible at Çatalhöyük was not possible at any other available site due to lack of preservation and access. For this reason, caution will need to be exercised in generalizing the findings to archaeobotanical assemblages from different burial environments.
The modern samples included BRWmo (bread wheat, Triticum aestivum) obtained from a long-term farming experiment archive (Bäd Lauchstädt, Germany; for details, refer to Fraser et al.[2]) and LENmo (lentil, Lens culinaris) obtained commercially from an organic farm in Sault Provence, France. The grains/seeds were charred in the laboratory in a reducing atmosphere. Fresh seeds were loosely wrapped in aluminum foil, buried in sand in glass beakers and placed in a pre-heated oven at 230°C for 24 h to replicate the likely conditions under which archaeological crop material is preserved (the conditions were identical to those used in previous experiments: see Fraser et al.[3] and work carried out by Michael Charles at the University of Oxford).
In the first part of this experiment, three sub-samples each of PEAar, BARar, BRWmo and LENmo were treated separately under each pre-treatment condition (crushed to a fine powder using a mortar and pestle after pre-treatment) and measured for δ13C values, δ15N values, %C, %N and C/N. Each bulk sample represents a homogenized batch of 10 grains/seeds (barley, bread wheat and lentil) or 5 seeds (pea), with average masses of 270 mg for PEAar, 143 mg for BARar, 284 mg for BRWmo and 155 mg for LENmo. In the second part of this experiment, 30 seeds of PEAar were first homogenized (also using a mortar and pestle) and then divided into sub-samples (of ca 20 mg), which underwent contamination and pre-treatment.
Pre-treatment methods
Six pre-treatment methods were used on the archaeological and three on the modern samples to investigate the effects of cleaning on the stable isotopic composition of charred grains/seeds. As discussed above, modern charred grain does not provide the ideal uncontaminated comparison with archaeological charred grain because the two are structurally different; its behavior could thus not be generalizable to archaeological samples. For this reason, the modern materials were only subjected to the 'harsh' treatments to observe how they would behave under the most extreme conditions.
ABA-full gentle: acid-base-acid treatment using aqueous 0.5 M HCl and 0.1 M NaOH at 70°C (for 30 min or until effervescence has ceased – first acid treatment; 60 min – base treatment; 25 min – second acid treatment). The acid steps were followed by three rinses in Milli-U water (Merck Millipore, division of Merck KGaA, Darmstadt, Germany) and the base step was followed by as many water rinses as it took for the solution to stop turning brown (adapted from Fraser et al.[3]).
ABA-neutrality: same as ABA-full gentle, except that the base step was followed by only three rinses in Milli-U water to neutralize the solution.
A-only gentle: treatment in aqueous 0.5 M HCl at 80°C for 30 min (or until effervescence has ceased) followed by three rinses in Milli-U water.
ABA-full harsh: acid-base-acid treatment using aqueous 1 M HCl and 1 M NaOH at 80°C (for 60 min – first acid treatment; 3 h – base treatment; 16 h – second acid treatment). Very brown samples underwent an additional base wash for an hour (adapted from Kanstrup et al.[9]).
A-only harsh: treatment in aqueous 6 M HCl at room temperature for 24 h, followed by as many rinses as it took to bring sample to neutrality (after DeNiro and Hastorf;[29] Aguilera et al.[34] and Lightfoot and Stevens[7]).
Ultra-sonication: treatment involved cleaning samples with Milli-U water in an ultra-sonic bath, performed for 30-min intervals for as long as it took for the solution to stop turning brown. The instrument was set at room temperature, and when the temperature of the water started to rise, the machine was switched off to allow it to cool. Some samples required 11 ultra-sonic washes until they stopped releasing brown material. The brown supernatant was collected, and studied under a high-magnification microscope to assess its organic/mineral content.
In order to minimize mass loss, at all stages of these experiments, samples were centrifuged before the solution/water was decanted. All samples were freeze-dried prior to analysis.
Contamination methods
In the second stage of this experiment, sub-samples of the homogenized PEAar were contaminated with three degrees of contamination (5%, 10% and 50% by dry weight), measured for IR spectra and stable isotope composition and subsequently subjected to pre-treatment aimed at removing the introduced contamination.
There are two reasons for contamination being achieved by dry weight mixing. First, one of our goals was to observe the IR spectra of each sample before and after contamination. The samples thus had to be crushed for the initial screening, and soaking crushed charred material would not replicate actual ground contamination conditions anyway. Secondly, we were more interested in testing the ability of the instrument to detect levels of contamination (having control over the exact percentage), rather than simulating the uptake of contamination by samples (where we could not control for the variability of uptake between the individual, differently preserved, grains/seeds). Because the nitrate contaminant could not be turned into powder, all nitrate-contaminated samples were mixed in water and subsequently freeze-dried.
Carbonate contamination
The carbonate used as a contaminant was the international standard IAEA-CO1: marble with an accepted δ13C value of +2.49‰ (International Atomic Energy Agency, Vienna International Centre, Vienna, Austria). As pre-treatment the contaminated samples were washed in acid (0.5 M HCl (aq.) at 80°C) and rinsed three times in Milli-U water; equivalent to A-only gentle treatment above. The same treatment was performed on a pure carbonate sample. Carbonate contamination should not have an effect on the δ15N values of the samples.
Nitrate contamination
As nitrate contaminant, we used a commercially available chemical fertilizer: Phostrogen NPK (MgO-SO3) fertilizer blend with a measured δ15N value of –1.8‰ and a δ13C value of –43.8‰ (%C = 6.8, %N = 10.7) (Bayer CropScience Ltd, Cambridge, UK). As pre-treatment, the contaminated samples were rinsed three times in Milli-U water. The same treatment was performed on a pure sample of the fertilizer. One sub-sample of 10% contamination was also treated with a gentle acid treatment (0.5 M HCl (aq.) followed by three rinses in Milli-U water). This contamination should mostly affect the δ15N values of the samples.
Humic acid contamination
The humic acid used as a contaminant was a commercially purchased humic acid sodium salt 50–60% with a measured δ15N value of 2.7‰ and a δ13C value of –25.9‰ (%C = 38.1, %N = 0.8) (Acros Organics, Thermo Fisher Scientific, Geel, Belgium). The salt was ground to a finer powder using a mortar and pestle before mixing. Due to its significantly higher %C composition, this contaminant should mostly affect the δ13C values of the samples.
There are reasons to believe that the first acid step of the ABA procedure is not necessary when dealing with plant material (which is not composed of an inorganic phase that would need to be initially demineralized, such as bones). The second acid step necessarily follows a base wash to remove the adsorbed atmospheric carbon, but it can also achieve the removal of carbonates and organic acids originally present in the samples. Thus, a BA treatment may be sufficient to remove the major types of contamination discussed in this paper. It may also incur a smaller mass loss, as was observed with fossil charcoal that underwent both ABA and BA washes (the range of mass loss was 50–90%).[35]
In this study, both ABA (0.5 M HCl (aq.) and 0.1 M NaOH (aq.) at 80°C; same as ABA-gentle from above) and BA (0.1 M NaOH (aq.) and 0.5 M HCl (aq.) at 80°C) procedures were used to remove the humic acid contamination from the samples in order to determine whether or not they produce the same results and to compare the mass loss between them. Both treatments were also performed on a pure humic acid sample. One sub-sample was contaminated with 50% humic acid and subsequently treated with a harsher BA protocol (1 M NaOH (aq.) and 0.5 M HCl (aq.) at 80°C).
For comparative purposes, two more archaeological samples (BARar and LENar) were crushed, contaminated with 10% humic acid and treated with both ABA and BA procedures.
Many of the contaminated+treated samples failed to give enough material to measure their δ13C and δ15N values, because the treatment dissolved the majority of the charred material. This loss is reflected in the mass loss calculations and discussed in the Discussion.
Stable isotope analysis
Stable isotope analyses were carried out at the Research Laboratory for Archaeology and the History of Art (University of Oxford, Oxford, UK) on a 20/22 continuous flow isotope ratio mass spectrometer coupled to a elemental analyzer (Sercon Ltd, Crewe, UK). Raw isotope ratios were normalized against international standards (IAEA-CH6, IAEA-CH7, USGS40 and IAEA-N2) using two-point normalization. Measurement uncertainties were calculated after Kragten.[36] The sample uncertainty over six independent C runs and six independent N runs ranged between 0.05 and 0.19‰ for δ13C values (average of 0.09‰) and between 0.11 and 0.39‰ for δ15N values (average of 0.21‰).
FTIR analysis
Crushed samples were analyzed by Fourier Transform Infrared Spectroscopy with Attenuate Total Reflectance (FTIR-ATR): Agilent Technologies (Stockport, UK) Cary 640 FTIR instrument with a GladiATR™ accessory from PIKE Technologies (Madison, WI, USA). Each sample was measured three times, the background was subtracted and a baseline correction was carried out using Agilent Resolution Pro. The spectra were normalized and all three spectra of each sample were averaged.
Results
Mass loss
All treatments caused a notable mass loss to the archaeological samples (see Table2). During ABA-gentle treatment, crushed samples incurred higher mass loss than uncrushed samples (87% vs 63%). The ABA-gentle and ABA-neutrality treatments (where the only difference was the number of water washes carried out after the base step) caused similar losses to uncrushed samples (63% and 58%, respectively). Crushed samples washed in BA-gentle lost smaller amounts of material than crushed samples washed in ABA-gentle (61% – uncontaminated and 89% – contaminated; vs 87%). The BA-harsh condition (employing 1 M NaOH (aq.)) was carried out in order to determine if humic acid contamination higher than 10% could be removed. This treatment, however, caused the highest loss of 98%. The residue left after this treatment was brown, and not black like all the other samples, which may mean that all the black charred material (humic acids, result of Maillard reaction) had been removed and what was left was the brown alkali-insoluble humin. Ultra-sonication caused similar losses to the acid-only treatments on archaeological uncrushed samples (34% vs 33% and 35%). Modern samples did not suffer as much mass loss as the archaeological samples, probably due to the structural differences between the two: higher robustness of modern samples and the fact that archaeological samples may have undergone further self-humification during burial.
Table 2.
Percentage mass loss suffered by archaeological and modern samples during different treatments used in this experiment
| Samples | Treatment | n | Mass loss (%) |
|---|---|---|---|
| ARCHAEOLOGICAL | |||
| Crushed | |||
| 1. ABA-full gentle | 3 | 87 | |
| 1. ABA-full gentle, humic acid contaminated | 1 | 99 | |
| 3. A-only gentle | 1 | 64 | |
| BA-gentle | 3 | 61 | |
| BA-gentle, humic acid contaminated | 1 | 89 | |
| BA-harsh, humic acid contaminated | 1 | 98 | |
| Uncrushed | |||
| 1. ABA-full gentle | 5 | 63 | |
| 2. ABA-neutrality | 5 | 58 | |
| 4. ABA-full harsh | 5 | 72 | |
| 3. A-only gentle | 5 | 35 | |
| 5. A-only harsh | 6 | 33 | |
| 6. Ultra-sonication | 6 | 34 | |
| MODERN | |||
| Uncrushed | |||
| 4. ABA-full harsh | 6 | 15 | |
| 5. A-only harsh | 6 | 5 | |
From these results, it is evident that no matter what method is used, a large portion of a given archaeological sample gets lost during pre-treatment. Part of this loss may result from the dissolution and removal of potential contaminants or parts of the grains/seeds (in the form of endogenous humic acids), part may result from accidental removal of fragments of the sample itself through repeated rinsing (something that happens even when utmost care is taken to carefully decant the solutions and even when a centrifuge is used). The losses observed in this study were consistent with mass loss suffered by fossil charcoal samples cleaned in ABA (1 M; temperature and times were not reported) which was interpreted to mean that the bulk of the samples were composed of the disorganized phase of charcoal.[22]
Overall, the results show that, in terms of mass loss, the most favorable treatments to crushed samples were the BA gentle and the A-only gentle protocols. For uncrushed samples, both A-only treatments (gentle and harsh) caused the smallest loss. This means that if a method that does not involve the full acid-base-acid procedure can be shown to be equally (or more) effective, an improvement will already be made towards reducing mass loss.
Pre-treatment comparisons
The data from the archaeological and modern samples subjected to six different cleaning protocols (ABA-full gentle, ABA-neutrality, A-only gentle, ABA-full harsh, A-only harsh) are presented in Table3 along with the untreated control. The differences in the δ13C and δ15N values between the sets of samples in each condition were assessed using a multiple linear regression, weighted by the sample measurement uncertainty using the programing language R (version 3.0.2, R Foundation for Statistical Computing, Vienna, Austria). In addition to investigating the offsets caused by the different treatments, a test was run to determine if the inclusion of the base step (i.e. an attempt to remove the humic acids) affected the resulting isotopic values. Coefficients representing the inclusion or exclusion of the base treatment were not significantly different from zero and so were excluded from subsequent analyses.
Table 3.
Stable isotope results of archaeological samples subjected to six pre-treatments and modern samples subjected to three pre-treatments in stage I of this experiment (for details of the treatments, refer to Experimental section). δ13C and δ15N values report the mean values of all the sub-samples in each condition. Reported standard deviations (SD) denote 1σ variation
| n | δ13CVPDB | δ13CVPDB SD | %C | %C SD | δ15NAIR | δ15NAIR SD | %N | %N SD | C/N | % mass loss | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PEAar (archaeological pea) | |||||||||||
| Untreated | 6 | –22.9 | 0.2 | 45.9 | 0.8 | 6.4 | 0.1 | 6 | 0.4 | 6 | / |
| 1. ABA-full gentle | 3 | –23.1 | 0.3 | 61.8 | 3.1 | 6.5 | 0.3 | 7.9 | 0.6 | 6.9 | 64 |
| 2. ABA-neutrality | 3 | –22.5 | 0.2 | 58.9 | 8.3 | 6.5 | 0.8 | 7.8 | 0.4 | 7.2 | 55 |
| 3. A-only gentle | 3 | –22.6 | 0.5 | 63.9 | 7 | 5.9 | 0.5 | 7 | 0.3 | 8.1 | 33 |
| 4. ABA-full harsh | 2 | –22.4 | – | 60.2 | – | 5.4 | – | 7.6 | – | 6.9 | 79 |
| 5. A-only harsh | 3 | –22.3 | 0.5 | 61.7 | 2.2 | 5 | 0.1 | 7.4 | 0.1 | 7.4 | 34 |
| 6. Ultra-sonicated | 3 | –22.5 | 0.1 | 52.2 | 2.5 | 5.5 | 0.1 | 6.2 | 0.3 | 6.5 | 32 |
| BARar (archaeological barley) | |||||||||||
| Untreated | 6 | –23 | 0.1 | 44.9 | 1.4 | 6.4 | 0.4 | 5.1 | 1 | 5.4 | / |
| 1. ABA-full gentle | 3 | –22.9 | 0.3 | 70.3 | 9.1 | 6.6 | 0.7 | 6.8 | 0.5 | 8.4 | 62 |
| 2. ABA-neutrality | 3 | –22.9 | 0.1 | 62.2 | 5.4 | 5.9 | 0.2 | 5.5 | 0.4 | 6.5 | 61 |
| 3. A-only gentle | 3 | –22.8 | 0.2 | 70.8 | 14 | 6.7 | 0.8 | 6.5 | 0.9 | 7.2 | 37 |
| 4. ABA-full harsh | 3 | –22.9 | 0.2 | 57.7 | 4.4 | 6.2 | 0.7 | 7 | 1.1 | 6.9 | 68 |
| 5. A-only harsh | 3 | –23 | 0.1 | 63.3 | 2.1 | 6.1 | 0.5 | 5.9 | 0.1 | 8 | 32 |
| 6. Ultra-sonicated | 3 | –23.1 | 0.2 | 56.2 | 2.3 | 5.5 | 0 | 6.5 | 3.6 | 5.5 | 31 |
| BRWmo (modern bread wheat) | |||||||||||
| Untreated | 3 | –27 | 0 | 61.4 | 1 | 1.6 | 0.3 | 2.5 | 0.1 | 7.9 | / |
| 4. ABA-full harsh | 3 | –26.7 | 0 | 66.4 | 1.9 | 1.7 | 0.3 | 2.3 | 0.3 | 8.7 | 16 |
| 5. A-only harsh | 3 | –27 | 0.2 | 64.9 | 2.6 | 1.4 | 0.3 | 2.1 | 0.3 | 8.6 | 13 |
| LENmo (modern lentil) | |||||||||||
| Untreated | 3 | –26.3 | 0.4 | 61.4 | 4.3 | 0.4 | 0.1 | 6.8 | 0.2 | 6.5 | / |
| 4. ABA-full harsh | 3 | –26.3 | 0.4 | 63.7 | 2.3 | –0.5 | 0.2 | 6.2 | 0.4 | 8.5 | 14 |
| 5. A-only harsh | 3 | –25.9 | 0.2 | 60.7 | 3.2 | 0.5 | 0.4 | 6.7 | 0.6 | 7.9 | 1 |
Figures1(a)–1(d) show the offsets in δ13C values between the means of the untreated and the experimentally treated archaeological and modern sets of samples. Taken individually, none of the treatments had a consistently significant effect on all the samples. The mean differences between the untreated and treated samples were less than 0.2‰ for the archaeological barley, less than 0.8‰ for the archaeological pea, less than 0.3‰ for the modern bread wheat and less than 0.5‰ for the modern lentil. For the archaeological samples, there were no statistically significant differences in the δ13C values between the 'gentle'-treatment samples and the untreated samples (p = 0.15), but those subjected to the 'harsh' treatments had significantly higher δ13C values, by +0.28‰ (p = 0.03). With all the uncertainties caused by climatic differences, mechanisms of carbon uptake during plant growth and interactions of soil fertility and soil water retention, this change is too small to be considered important for the interpretation of crop stable isotope results.
Figure 1.
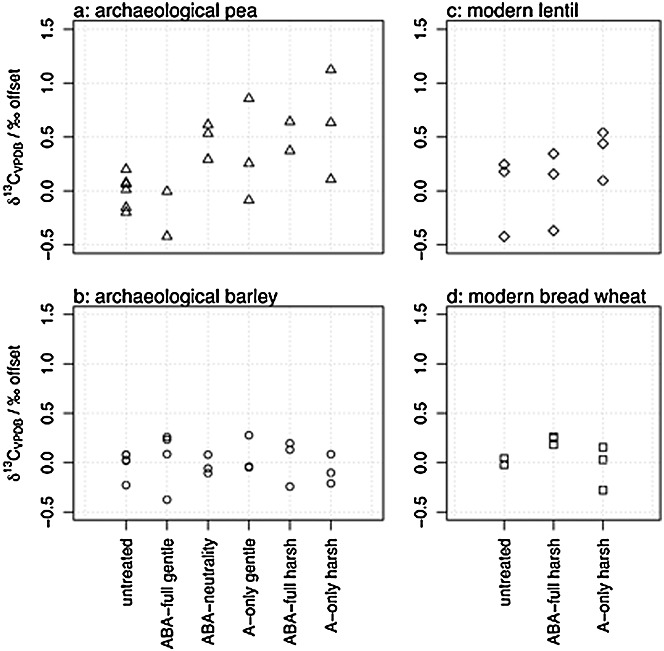
Offsets in δ13C values between untreated and chemically pre-treated archaeological (a: pea, b: barley) and modern (c: lentil, d: bread wheat) samples compared in stage I of this experiment. Offset was calculated as treated – untreated.
Figures2(a)–2(d) show the offsets in δ15N values between the means of the untreated and the experimentally treated archaeological and modern charred sets of samples. The 'harsh' treatments caused an average decrease of 1.0‰ (p = 0.001) in the archaeological samples, with no significant interaction between species and treatment. This offset is not consistent with the overall trend observed by Kanstrup et al.:[9] a positive mean δ15N-value offset of +0.7 ± 1.0‰ between 31 pairs of untreated and ABA-full harsh treated samples (where the offset was also calculated as treated – untreated). However, the standard deviation of this offset was large and some samples showed an equally large decrease in δ15N values to the two samples in this study. There were no significant differences in δ15N values between the modern untreated and treated samples.
Figure 2.
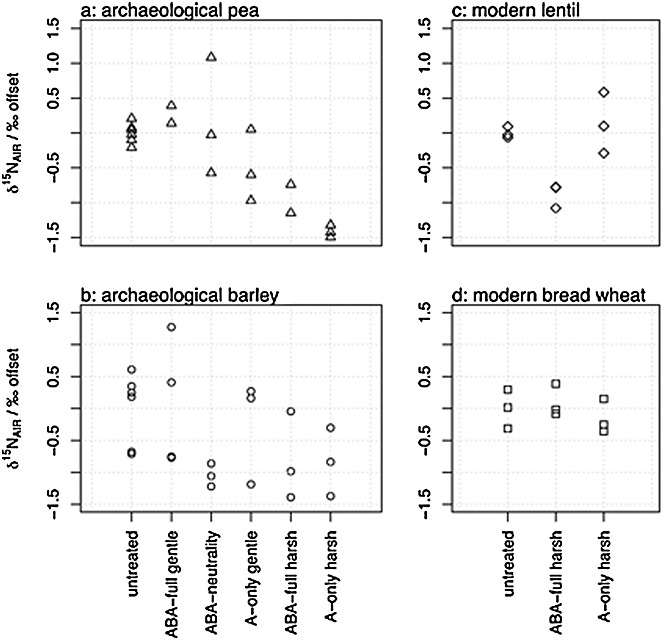
Offsets in δ15N values between untreated and chemically pre-treated archaeological (a: pea, b: barley) and modern (c: lentil, d: bread wheat) samples compared in stage I of this experiment. Offset was calculated as treated – untreated.
It is unlikely that the effects on the δ15N values were influenced by the (as yet unexplained) high N content of plant material from this archaeological site. All the samples were washed in water, and so any nitrates contained in the grains/seeds should have been removed. It is possible that some other N contamination was present or that the high %N values were caused by a biogenetic factor during the growth of the plants. In either case, it would be hard to explain why only the 'harsh' treatments had an impact on the isotopic composition of the samples.
Based on the comparisons carried out by Kanstrup et al.,[9] and in this part of the present experiment, it is not possible to determine whether the changes in the δ15N values were caused by the removal of contamination (that the gentler acid treatments failed to remove), due to destruction of the grains/seeds or to another reason entirely. In the modern samples, the A-only harsh treatment caused almost no change to both the wheat and the lentil samples, while the ABA-full harsh procedure caused opposite effects on the two plant species: increase of ca 1.0‰ in the wheat and decrease of less than 1.0‰ in the lentil.
All treatments caused an increase in the %C and %N of the archaeological samples and a small increase in the %C and either no change or a negligible decrease in the %N of the modern samples (see Table3). It is worth noting that the within-condition variability of the modern material is smaller, either due to the lack of contamination, greater robustness or the lack of diagenetically created humic acids in modern charred material. There are no statistically significant differences in %C or %N between the samples in the 'harsh' treatment groups and the 'gentle' treatment groups (p = 0.147 for %C offsets and p = 0.671 for %N). The C/N ratios of both archaeological samples increase during all treatments, because, proportionally, the increase in %C was higher than the increase in %N.
Figures3(a)–3(d) show the comparison between the untreated and ultra-sonicated archaeological samples. For both δ13C and δ15N values, the effect of ultra-sonication was examined using a two-way analysis of variance (ANOVA) comparing the species (PEAar vs BARar) and the effect of treatment (untreated vs ultra-sonicated). For δ13C values, there was a significant interaction between species and treatment; the effect of treatment was only significant for PEAar, increasing its δ13C value by 0.5‰ (95% CI = 0.14, 0.88‰, p = 0.006). For δ15N values, there was no significant interaction effect, and ultra-sonication affected both species similarly, decreasing their δ15N values by an average of 1.22‰ (95%CI = 0.81, 1.63‰, p <0.001). The confidence intervals and p-values were corrected for the 95% family-wise confidence level using the Tukey HSD adjustment, reducing any inflation of the type I error rate. No trend was observed in the %N results, which indicates that the differences in stable isotope values were not caused by differences in the amount of N available for measurement (see Table3).
Figure 3.
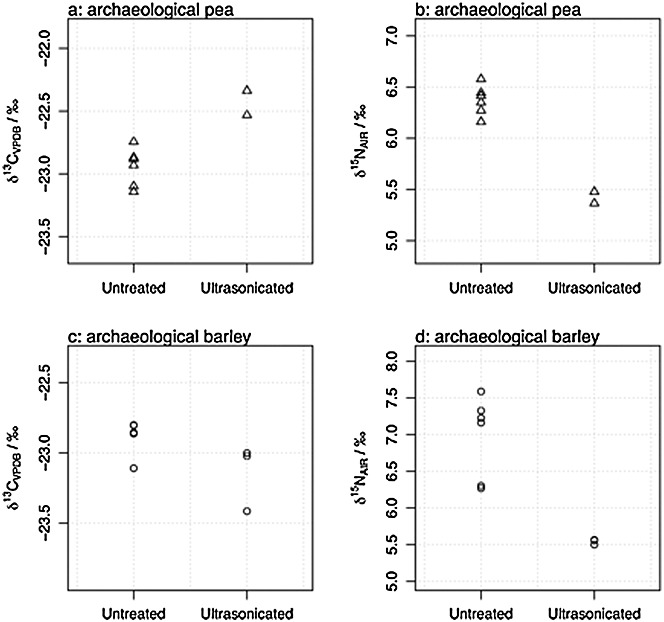
δ13C and δ15N values of untreated and ultra-sonicated archaeological samples (a, b: pea; c, d: barley) compared in stage I of this experiment.
When modern samples were subjected to this treatment, no brown material was released, which could suggest that what had been removed from the archaeological samples was dirt/soil. However, examination under the microscope revealed that the brown residue was organic/charred in nature. This suggests that the sonication had a damaging effect on the archaeological grain itself and that the change in the δ15N values was caused by the removal of 15N-enriched parts of the grain.
The lower variability in both chemical (%C, %N) and isotopic (δ15N, δ13C values) compositions of modern charred samples than of archaeological ones, as well as the higher robustness of modern grains/seeds observed during ultra-sonication, confirms that the two groups are not structurally equivalent (which had already been observed by Styring et al.[3]). For this reason, the second part of the experiment was carried out only on archaeological material.
Carbonate contamination
The FTIR results (Fig.4) show that the presence of carbonates in an archaeological sample causes the appearance of peaks at 720 and 870cm–1 (which increase with higher percentage contamination) and that these peaks are successfully removed by 0.5 M HCl (aq.). The removal of contamination by treatment is confirmed by the δ13C values (Table4): the contaminated samples show progressively less negative values with higher calcite content (–22.5‰, –22.2‰, –16.6‰) and the acid-treated samples have δ13C values within 0.1‰ of the uncontaminated value (treated: –22.9‰ and –23.0‰; uncontaminated: –22.9‰). The FTIR measurements confirm the observation made by Styring et al.:[4] acid treatment causes the COO– peak (at 1400 cm–1) to shift to COOH (at 1650 cm–1).
Figure 4.
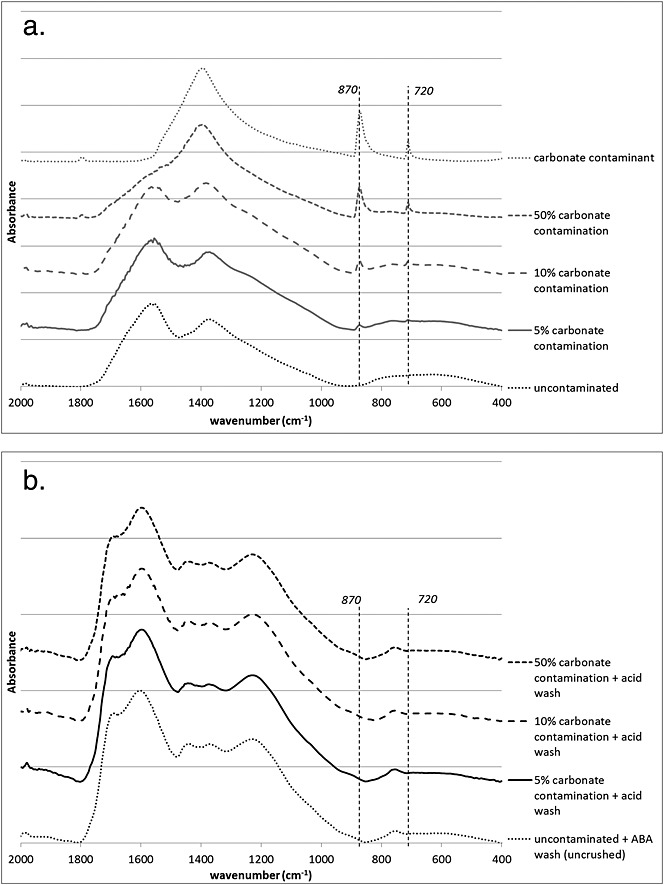
FTIR spectra of (a) untreated and carbonate contaminated (at 5%, 10% and 50% by dry mass) archaeological pea sample, and (b) carbonate contaminated samples from above treated with 0.5 M HCl.
Table 4.
δ13C and δ15N values of archaeological samples artificially contaminated with carbonate, nitrate and humic acids and subsequently treated for the removal of this contamination. Where n >1 (in the case of the untreated samples, taken from Table3), the reported values denote the means
| δ13CVPDB | %C | δ15NAIR | %N | C/N | |
| PEAar (archaeological pea) | |||||
| Untreated | –22.9 | 45.9 | 6.4 | 6.0 | 6.0 |
| 5% carbonate | –22.5 | 45.9 | 6.3 | 5.2 | 6.7 |
| 10% carbonate | –22.2 | 44.5 | 6.2 | 5.2 | 6.8 |
| 50% carbonate | –16.6 | 28.2 | 6.2 | 2.8 | 10.6 |
| 5% carbonate + acid wash | sample lost during pre-treatment | ||||
| 10% carbonate + acid wash | –22.9 | 57.1 | 5.2 | 7.4 | 7.7 |
| 50% carbonate + acid wash | –23.0 | 55.2 | 4.9 | 7.2 | 8.7 |
| 5% nitrate | –23.0 | 47.8 | 5.5 | 6.5 | 7.5 |
| 10% nitrate | –23.3 | 45.8 | 4.4 | 7.5 | 5.1 |
| 50% nitrate | –26.3 | 24.7 | 1.3 | 10.3 | 2.3 |
| 5% nitrate + water wash | –23.0 | 49.3 | 5.0 | 6.0 | 8.4 |
| 10% nitrate + water wash | sample lost during pre-treatment | ||||
| 50% nitrate + water wash | sample lost during pre-treatment | ||||
| 10% nitrate + acid wash | –23.1 | 52.2 | 5.1 | 7.6 | 5.0 |
| 5% humic acid | –23.2 | 46.3 | 6.2 | 6.0 | 5.9 |
| 10% humic acid | –23.6 | 44.4 | 5.9 | 5.0 | 7.9 |
| 50% humic acid | –24.4 | 42.2 | 5.6 | 3.2 | 11.3 |
| 5% humic acid + ABA | sample lost during pre-treatment | ||||
| 10% humic acid + ABA | sample lost during pre-treatment | ||||
| 50% humic acid + ABA | sample lost during pre-treatment | ||||
| 5% humic acid + BA | sample lost during pre-treatment | ||||
| 10% humic acid + BA | –24.0 | 59.1 | – | – | – |
| 50% humic acid + BA | sample lost during pre-treatment | ||||
| uncontaminated + BA | –23.5 | 57.8 | 6.5 | 7.6 | 6.9 |
| BARar (archaeological barley) | |||||
| Untreated | –23.0 | 44.9 | 6.4 | 5.1 | 5.4 |
| Uncontaminated + BA | –22.9 | 58.6 | 7.9 | 8.8 | 4.6 |
| LENar (archaeological lentil) | |||||
| Untreated | –23.6 | 42.7 | 4.5 | 6.4 | 6.1 |
| Uncontaminated + BA | –23.6 | 57.3 | 4.7 | 8.8 | 5.9 |
| Contaminants | |||||
| Pure carbonate | 2.5 | – | – | – | – |
| Pure nitrate | –43.8 | 6.8 | –1.8 | 10.7 | 0.5 |
| Pure humic salt | –25.9 | 38.1 | 2.7 | 0.8 | 24.3 |
| Pure humic acid + BA | –27.6 | 44.9 | – | – | – |
Unexpectedly, the δ15N values of the treated samples are lower than those of the uncontaminated sample (treated: 5.2‰ and 4.9‰; uncontaminated: 6.4‰), although acid treatment should not alter the δ15N values. It is hypothesized that gentle acid treatment on crushed archaeological samples causes a similar effect to ultra-sonication and harsh acid treatment on uncrushed samples (loss of parts of the grain more enriched in 15N).
Nitrate contamination
Nitrate contamination causes progressively lower δ15N values with increasing percent contamination (5.5‰, 4.4‰, 1.3‰), but is only detectable using FTIR when the contamination is 10% or higher (peaks at 1085, 1450, 3300 cm–1) (see Fig.5). After treatment with water, all samples become indistinguishable from the uncontaminated and 5% contaminated sample, suggesting that no more than 5% of nitrates remain in the samples after pre-treatment.
Figure 5.
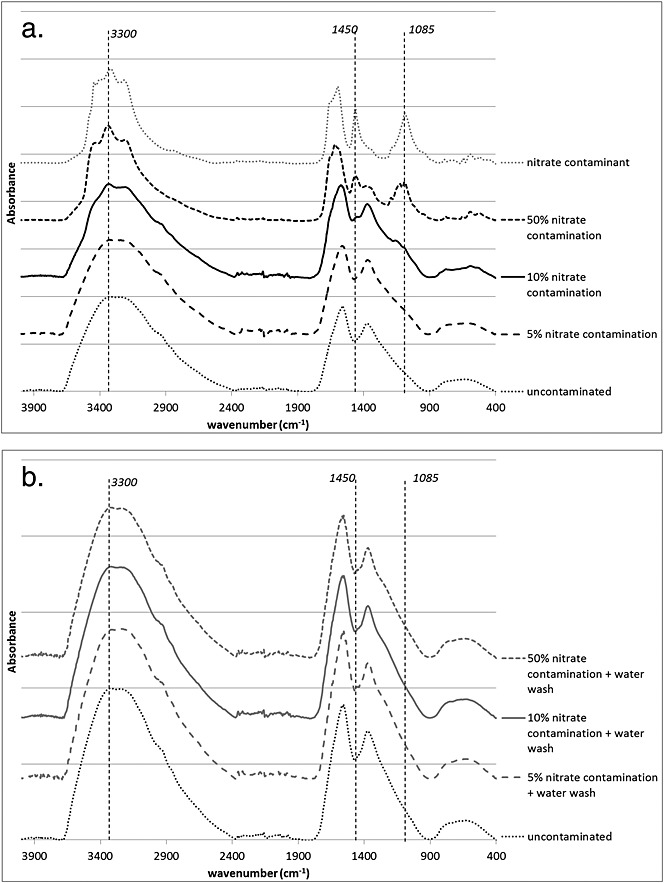
FTIR spectra of (a) untreated and nitrate contaminated (at 5%, 10% and 50% by dry mass) archaeological pea sample, and (b) nitrate contaminated samples from above washed with Milli-U water.
The one measured δ15N value of a treated sample (the other two samples did not yield enough material for stable isotope analysis) remains lower than that of the uncontaminated sample (treated: 5.0‰; uncontaminated: 6.4‰) (see Table4). This is probably due to the same phenomenon that caused the decrease in the δ15N values of the crushed samples that were treated for the removal of carbonates and the uncrushed samples that were ultra-sonicated.
The fertilizer that was used as the nitrate contaminant had a very negative δ13C value (–43.8‰) and its presence in the contaminated samples is detectable through decreasing δ13C values with increasing percentage contamination (–23.0‰, –23.3‰, –26.3‰). Both treatments (a water-only wash and a gentle acid wash followed by water rinsing) were successful at removing the fertilizer contamination on the δ13C values (treated: –23.0‰ and –23.0‰; uncontaminated: –22.9).
Humic acid contamination
Figure6(a) shows the IR spectra of the humic salt, the humic contaminated samples and the uncontaminated sample. The spectra of the archaeological samples and the pure humic salt are extremely similar, except for two regions, which produce higher peaks at 10% and 50% contamination (peaks at 1010, 1080 and 3690 cm–1). The undetectable 5% humic acid contamination does not significantly alter the δ13C and δ15N values of the sample (–23.2‰ and 6.2‰, respectively; compared with the uncontaminated δ13C value of –22.9‰ and δ15N value of 6.4‰). The presence of 10% and 50% contamination causes larger shifts in the δ13C values (–23.6‰ and –24.4‰, respectively) than in the δ15N values (5.9‰ and 5.6‰), which is not unexpected since the humic salt had a significantly higher C/N ratio than the sample (24.3 vs 6.0). It is important to note, however, that large variability exists in the C/N ratios of different humic acids (e.g.[37]) so their impact on the stable isotope composition of contaminated grain may be drastically different.
Figure 6.
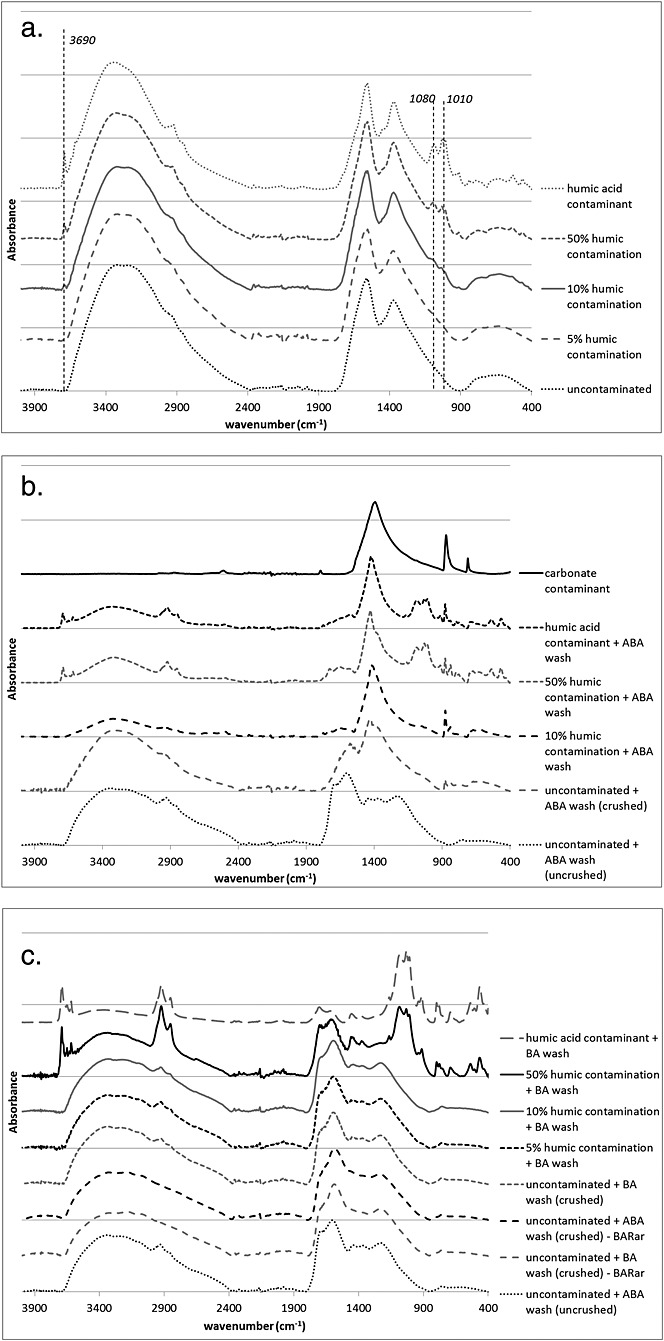
FTIR spectra of (a) untreated and humic salt contaminated (at 5%, 10% and 50% by dry mass) archaeological pea sample, (b) humic salt contaminated samples from above treated with an acid-base-acid protocol, and (c) humic salt contaminated samples from above treated with a base-acid protocol (for details on the treatment methods, refer to the text). All contaminated samples are PEAar unless specified otherwise.
Structurally, the ABA treatment had an unexpected effect on the archaeological material. Both the uncontaminated uncrushed (from the first stage of this experiment) and the uncontaminated crushed (from the second stage of this experiment) samples were analyzed using FTIR, and the resulting spectra were found to be quite different (see Fig.6(b)). The spectrum of the uncontaminated and treated (crushed) was similar to the spectra of the contaminated and treated, and the spectra of all these samples resemble the spectrum of pure calcite. This strongly suggests that during ABA treatment, the sample adsorbs a large amount of carbon dioxide from the atmosphere. In other words, it appears as if not much of the original grain is left after it has been crushed and treated with ABA (which also explains why most of the sample had been lost and its δ13C and δ15N values could not be measured).
The BA treatment did not have such a damaging impact on the structure of the charred sample. As shown in Fig.6(c), the base-acid treatment seems to have removed contamination from the 10% contaminated samples, as its spectrum looks very similar to that of the uncontaminated and 5% humic contaminated sample (where contamination cannot be detected). Furthermore, the calcite peaks observed in the ABA-treated samples (above) are not present in the BA-treated samples. When the sample was contaminated with 50% humic acid, the contamination was not completely removed. However, the likelihood of 50% exogenous humic contamination is quite low and this can be detected with FTIR, so samples that are so severely contaminated can be rejected for stable isotope analysis.
Only one sub-sample that had been contaminated with humic acid and pre-treated for the removal of this contamination yielded enough material for one isotopic measurement (10% contamination + BA, δ13C value = –24.0‰, see Table4). This value is ca 1.0‰ lower than that of the uncontaminated sample (–22.9‰). Pure humic acid treated with BA also yielded a more negative δ13C value (BA treated: –27.6‰; untreated: –25.9‰). As the %C increases in both instances, the shift can be attributed to the loss of parts of the grain that contain a lower proportion of C than the rest of the fraction. It cannot be explained by adsorption of atmospheric C, as that has a higher δ13C value of –8‰. Based on this, there is cause for concern that although the BA treatment may remove the humic acids, it may have an adverse effect on the δ13C value of the sample.
Whether or not ABA and BA treatments have varying effects on charred plant material (as was the case with PEAar above) is likely to vary between different samples. When two other archaeological samples (BARar and LENar) were treated with both ABA and BA treatments, the pairs of spectra for each sample looked quite similar to each other, and none of them resembled that of pure calcite (See Fig.6(c)). In addition, the δ13C values of these two uncontaminated samples did not decrease as a result of the BA treatment (as did the values of the contaminated+treated and uncontaminated+treated PEAar samples) (see Table4). However, PEAar may well be representative of many other archaeological samples and thus the choice of pre-treatment should assume the worst-case scenario. It is also important to remember that this experiment used only one type of humic salt; other forms may react with charred material and with pre-treatment methods in distinct ways.[16] Overall, it appears that, structurally, a BA treatment on crushed grains is the most appropriate way of removing humic acids, but it may have adverse effects on the isotopic composition of the sample.
Discussion and Conclusions
In order to assess the impact of contamination on archaeological charred plant material and to identify the most appropriate method of removing it prior to stable isotope analysis, two goals were pursued in the present study: (1) to test several different pre-treatment methods for cleaning ancient charred plant material with the aim to observe how they affect the δ13C and δ15N values of the samples, and (2) to investigate how three types of artificially introduced contaminants (carbonates, nitrates and humic acids) influence the stable isotopic and structural composition of charred plant material and how they can be removed.
The results of the first part of this experiment showed that the δ13C values of all the treated samples were indistinguishable from those of the untreated samples, given their within-treatment variability. The %C increased with all pre-treatments, indicating that some material had been removed, which was relatively low in C compared with the rest of the sample. The absence of a difference between the untreated and the treated samples was probably due to one of three reasons: (1) the material that was removed did not significantly influence the δ13C values of the samples, (2) none of the pre-treatment methods were successful at removing any significant carbon contamination, or (3) there was no contamination to start with.
The δ15N values, on the other hand, decreased with the use of harsher acid treatments (by ca 1.1‰) and with ultra-sonication in Milli-U water (by ca 1.0‰). Two explanations can be suggested for these effects: (1) the samples were contaminated with 15N-enriched material and both the harsher acid treatments and the ultra-sonication were successful at removing this contamination, or (2) both vigorous treatments had a directional effect on the stable isotopic composition of the charred plant material and caused the loss of grain/seed components that were enriched in 15N. The ultra-sonication results showed the effect of mechanical degradation of the sample, causing an alteration in δ15N values that was attributable (after microscopic examination) to the loss of material from the sample itself. This argues strongly for the latter hypothesis: that more vigorous treatments adversely affect the physical and chemical composition of the sample.
The results of the second part of this experiment showed that nitrates and (one type of) humic acid can be detected using FTIR when the contamination is 10% or higher, and that carbonates can be detected when the contamination is 5% or higher. All contamination caused significant changes to the δ13C and δ15N values of the archaeological sample, with higher contamination creating larger shifts. Humic acid contamination caused the smallest isotopic changes because humic acids, being derived from soil and organic material, have δ13C and δ15N values similar to those of the archaeological grains/seeds themselves. Although only one type of humic acid was used in this experiment, any kind of humic acid contamination would have to be substantial to make even a small change to the plant δ13C and δ15N values; and such high levels of exogenous humic acid contamination are unlikely.
Carbonate contamination was successfully removed using 0.5 M HCl (aq.) at 80°C and the nitrate contamination was removed using three rinses in Milli-U water. A base-acid wash was structurally less harmful to the samples than an acid-base-acid procedure. Based on the FTIR spectra, it was also more successful at removing humic acid contamination and caused a smaller mass loss than an acid-base-acid wash. However, the BA treatment caused a significant shift in the δ13C value of the archaeological sample (as well as of a pure humic acid sample).
The δ15N values of crushed samples that were subjected to pre-treatment (both a water-only wash and a gentle acid treatment followed by water wash, n = 4) decreased by ca 1.0‰. The same happened with the application of harsher acid treatments and ultra-sonication on uncrushed grains/seeds. It is hypothesized here that, in all these cases, the changes happened for the same reason, which was the removal of parts of the grain that are less tightly bound within the grain/seed structure, and which are also enriched in 15N. Cohen-Ofri et al.[22] described the parts of fossil charcoal samples that were removed with HCl (aq.) as the " degradation products". In the case of the archaeological pea samples, the decrease in δ15N values may have been caused by the removal of the testa, the seed coat, which had been preserved on the sampled seeds. It has not been demonstrated, however, whether and why the testa would be enriched in 15N, but different components of grains/seeds, having variable amino acid compositions, may conceivably have differing stable isotopic composition (cf.[4]). The decrease may, however, have been for another reason entirely, perhaps the removal of some contamination (undetectable by FTIR), which was only released through crushing and ultra-sonication, and more research with larger sample sizes needs to be carried out to explain this.
There are advantages and disadvantages to crushing charred grains/seeds prior to pre-treatment. As different archaeological samples may contain variable amounts of this less tightly bound material (creating variability caused by diagenesis), homogenizing the samples at the start may mean that all the freer material is removed and the resulting isotopic signatures are more comparable across different samples/sites. In addition to reducing some of this unwanted inter-sample variability, crushing will ensure that all the charred mass will get the same exposure to the chemical solutions. This is something that does not happen when only the surface area of grains/seeds is exposed to pre-treatment, and is another source of variability since some samples contain whole and some fragments of grains/seeds.
From the results of this experiment, we can see that pre-treatment is effective at removing contamination from crushed grain (the samples had to be crushed for FTIR and the artificial contamination), but we cannot deduce that it would be equally effective at cleaning whole grains/seeds. In some instances, the seed coat may be robust enough to protect the inside of the grain and needs to be broken in order to clean the charred material inside. After crushing, we can be more certain that the contamination that we aim to remove does in fact get removed. Some samples are also more fragile than others and end up disintegrating during the pre-treatment anyway; so they may end up having the freer material removed and the δ15N values affected unintentionally. For these reasons, it would be advisable to measure the δ13C and δ15N values on only the most robust part of the grain. In addition, if samples are to be screened for potential sources of contamination using FTIR, they would have to be crushed at the start regardless.
On the other hand, as the effect on the δ15N values of crushed grains is not yet fully understood, the removal of the freer material may cause unwanted changes to the original stable isotopic signatures of the charred samples (although note that the negative offset was observed in a very small sample size of four). It is worth pointing out that the direction of this δ15N-value offset is opposite to that caused by manuring[2] and charring[3] so it cannot become a confounding factor for explaining plant 15N enrichment in archaeobotanical samples (e.g.[1,10]).
Significant mass loss to the archaeological samples was incurred by all versions of pre-treatment; the result of the dissolution of contamination as well as the unavoidable removal of sample every time that a solution/water was decanted (even with the use of a centrifuge). Most of the material was lost during the base step aimed at removing humic acids. The base wash removes both exogenous humic acids (products of diagenesis from organic material present in the soil) and endogenous humic acids (formed in the grain during Maillard reactions and during diagenetic self-humification). As the entirety of a grain/seed sample may be composed of humic acids, the application of a base step may lead to the complete loss of a sample. However, loss of a sample through pre-treatment may also indicate that the grains/seeds are not well enough preserved for stable isotope analysis.
Taking all the findings of this study into consideration, we suggest the following procedure for preparing archaeological charred plant material for stable carbon and nitrogen isotope analysis:
Pre-screening using FTIR of at least a subset of all samples from a given site to assess the levels of contamination and the general state of preservation of the grain;
If carbonates and nitrates are present in detectable quantities (refer to Figs.4 and 5 for the characteristic IR peaks), they should be removed with 0.5 M HCl (aq.) at 80°C for 30min (or until effervescence stops) followed by three rinses in Milli-U water;
If the sediments at an archaeological site contain a high amount of organic matter, in which case one would expect the presence of significant amounts of exogenous humic substances, the charred plant material should be subjected to a BA treatment using 0.1 M NaOH (aq.) and 0.5 M HCl (aq.) at 80°C (for 1 h and 30 min, respectively), keeping in mind that the procedure may alter the δ13C values (and possibly the δ15N values) of the starting material (by 1.0‰ in the example used in this study).
The archaeological samples that were studied in this experiment represent only one type of burial environment. Material from other sites (where the conditions may be hotter, colder, wetter or drier) may contain different amounts of contamination, but, regardless of their concentration, the contaminants should still be removable using the technique above (especially if crushed). However, more work needs to be done on investigating whether more refined treatments would be appropriate for use on samples that have experienced a very different preservation history.
Acknowledgments
We are grateful to Ian Hodder and Arkadiusz Marciniak for permission to use archaeological samples from Çatalhöyük and to Peter Ditchfield, Amy Styring, Rick Schulting and Mike Charles for technical assistance and useful discussions of the data. The paper is much stronger thanks to the helpful feedback from the three anonymous reviewers. The study was made possible by an Oxford University Clarendon Fund PhD scholarship (P. Vaiglova) and funding for analyses from the AGRICURB project (ERC No. 312785, PI A. Bogaard). Materials from the TPC Area were made available thanks to the Polish Science Foundation Grant (DEC-2012/06/M/H3/00286, AMarciniak).
References
- [1].Bogaard A, Fraser R, Heaton THE, Wallace M, Vaiglova P, Charles M, Jones G, Evershed RP, Styring AK, Andersen NH, Arbogast R-M, Bartosiewicz L, Gardeisen A, Kanstrup M, Maier U, Marinova E, Ninov L, Schäfer M, Stephan E. Crop manuring and intensive land management by Europe's first farmers. Proc. Natl. Acad. Sci. 2013;110:12589. doi: 10.1073/pnas.1305918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fraser RA, Bogaard A, Heaton T, Charles M, Jones G, Christensen BT, Halstead P, Merbach I, Poulton PR, Sparkes D, Styring AK. Manuring and stable nitrogen isotope ratios in cereals and pulses: towards a new archaeobotanical approach to the inference of land use and dietary practices. J. Archaeol. Sci. 2011;38:2790. [Google Scholar]
- [3].Fraser R, Bogaard A, Charles M, Styring AK, Wallace M, Jones G, Ditchfield P, Heaton THE. Assessing natural variation and the effects of charring, burial and pre-treatment on the stable carbon and nitrogen isotope values of archaeobotanical cereals and pulses. J. Archaeol. Sci. 2013;40:4754. [Google Scholar]
- [4].Styring AK, Manning H, Fraser RA, Wallace M, Jones G, Charles M, Heaton THE, Bogaard A, Evershed RP. The effect of charring and burial on the biochemical composition of cereal grains: investigating the integrity of archaeological plant material. J. Archaeol. Sci. 2013;40:4767. [Google Scholar]
- [5].Araus J, Febrero A, Buxó R, Rodríguez-Ariza MO, Molina F, Camalich MD, Martin D, Voltas J. Identification of ancient irrigation practices based on the carbon isotope discrimination of plant seeds: a case study from the south-east Iberian Peninsula. J. Archaeol. Sci. 1997;24:729. [Google Scholar]
- [6].Riehl S, Bryson R, Pustovoytov K. Changing growing conditions for crops during the Near Eastern Bronze Age (3000–1200 BC): the stable carbon isotope evidence. J. Archaeol. Sci. 2008;35:1011. [Google Scholar]
- [7].Lightfoot E, Stevens RE. Stable isotope investigations of charred barley (Hordeum vulgare) and wheat (Triticum spelta) grains from Danebury Hillfort: implications for palaeodietary reconstructions. J. Archaeol. Sci. 2012;39:656. [Google Scholar]
- [8].Kanstrup M, Thomsen IK, Andersen AJ, Bogaard A, Christensen BT. Abundance of 13C and 15N in emmer, spelt and naked barley grown on differently manured soils: towards a method for identifying past manuring practice. Rapid Commun. Mass Spectrom. 2011;25:2879. doi: 10.1002/rcm.5176. [DOI] [PubMed] [Google Scholar]
- [9].Kanstrup M, Holst MK, Jensen PM, Thomsen IK, Christensen BT. Searching for long-term trends in prehistoric manuring practice. δ15N analyses of charred cereal grains from the 4th to the 1st millennium BC. J. Archaeol. Sci. 2013:1. [Google Scholar]
- [10].Vaiglova P, Bogaard A, Collins M, Cavanagh W, Mee C, Renard J, Lamb A, Gardeisen A, Fraser R. An integrated stable isotope study of plants and animals from Kouphovouno, southern Greece: a new look at Neolithic farming. J. Archaeol. Sci. 2014;42:201. [Google Scholar]
- [11].Goh K, Molloy B. Proceedings of the 8th International Conference on Radiocarbon Dating. Wellington: Royal Society Of New Zealand; 1972. Reliability of radiocarbon dates from buried charcoals; pp. 567–573. [Google Scholar]
- [12].Brock F, Higham T, Ditchfield P, Bronk Ramsey C. Current pretreatment methods for AMS radiocarbon dating at the Oxford Radiocarbon Accelerator Unit (ORAU) Radiocarbon. 2010;52:103. [Google Scholar]
- [13].Higham TFG, Barton HUW, Turney CSM, Barker G, Bronk Ramsey C, Brock F. Radiocarbon dating of charcoal from tropical sequences: results from the Niah Great Cave, Sarawak, and their broader implications. J. Quat. Sci. 2009;24:189. [Google Scholar]
- [14].Schnitzer M, Khan SU. Soil Organic Matter. Amsterdam: Elsevier; 1978. [Google Scholar]
- [15].Duchaufour P, Jacquin F. Comparaison des processus d'humification dans les principaux types d'humus forestiers. Bull. Appalach. For. Exp. Stn. 1975;1:29. [Google Scholar]
- [16].Kuwatsuka S, Tsutsuki K, Kumada K. Chemical studies on soil humic acids. Soil Sci. Plant Nutr. 1978;24:337. [Google Scholar]
- [17].Head MJ. Categorisation of organic sediments from archaeological sites. In: Ambrose WR, Mummery JMJ, editors. Archaeometry: Further Australian Studies. ANU, Canberra: Department Of Prehistory, Research School Of Pacific Studies; 1987. pp. 143–159. [Google Scholar]
- [18].Amir S, Hafidi M, Merlina G, Hamdi H, Revel J-C. Elemental analysis, FTIR and 13C-NMR of humic acids from sewage sludge composting. Agronomie. 2004;24:13. [Google Scholar]
- [19].González Pérez M, Martin-Neto L, Saab SC, Novotny EH, Milori DMBP, Bagnato VS, Colnago LA, Melo WJ, Knicker H. Characterization of humic acids from a Brazilian Oxisol under different tillage systems by EPR, 13C NMR, FTIR and fluorescence spectroscopy. Geoderma. 2004;118:181. [Google Scholar]
- [20].González-Pérez M, Vidal Torrado P, Colnago LA, Martin-Neto L, Otero XL, Milori DMBP, Gomes FH. 13C NMR and FTIR spectroscopy characterization of humic acids in spodosols under tropical rain forest in southeastern Brazil. Geoderma. 2008;146:425. [Google Scholar]
- [21].Vandenbroucke M, Largeau C. Kerogen origin, evolution and structure. Org. Geochem. 2007;38:719. [Google Scholar]
- [22].Cohen-Ofri I, Weiner L, Boaretto E, Mintz G, Weiner S. Modern and fossil charcoal: aspects of structure and diagenesis. J. Archaeol. Sci. 2006;33:428. [Google Scholar]
- [23].Maillard LC. Formation de matières humiques par action de polypeptides sur sucres. C. R. Acad. Sci. 1913;156:148. [Google Scholar]
- [24].Steelink C. Understanding Humic Substances. Cambridge: Royal Society Of Chemistry; 1999. What is humic acid? A perspective of the past forty years; pp. 1–8. [Google Scholar]
- [25].Candy I, Stephens M, Hancock J, Waghorne R. Palaeoenvironments of ancient humans in Britain: the application of oxygen and carbon isotopes to the reconstruction of Pleistocene environments. In: Ashton NM, Lewis SG, Stringer CB, editors. The Ancient Human Occupation of Britain. Oxford: Elsevier; 2011. pp. 23–38. [Google Scholar]
- [26].Rock L, Ellert BH, Mayer B. Tracing sources of soil nitrate using the dual isotopic composition of nitrate in 2 M KCl-extracts. Soil Biol. Biochem. 2011;43:2397. [Google Scholar]
- [27].Knicker H, Almendros G, González-Vila J, Martin F, Lüdemann H-D. 13C and 15N-NMR spectroscopic examination of the transformation of organic nitrogen in plant biomass during thermal treatment. Soil Biol. Biochem. 1996;28:1053. [Google Scholar]
- [28].Ascough PL, Bird MI, Meredith W, Wood RE, Snape CE, Brock F, Higham TFG, Large DJ, Apperley DC. Hydropyrolysis: implications for radiocarbon pretreatment and characterization of black carbon. Radiocarbon. 2010;52:1336. [Google Scholar]
- [29].DeNiro MJ, Hastorf CA. Alteration of 15N/14N and 13C/12C ratios of plant matter during the initial stages of diagenesis: Studies utilizing archaeological specimens from Peru. Geochim. Cosmochim. Acta. 1985;49:97. [Google Scholar]
- [30].Fraser R, Bogaard A, Schäfer M, Arbogast R, Heaton THE. Integrating botanical, faunal and human stable carbon and nitrogen isotope values to reconstruct land use and palaeodiet at LBK Vaihingen an der Enz, Baden-Württemberg. World Archaeol. 2013;45:492. [Google Scholar]
- [31].Heaton THE, Jones G, Halstead P, Tsipropoulos T. Variations in the 13C/12C ratios of modern wheat grain, and implications for interpreting data from Bronze Age Assiros Toumba. Greece. J. Archaeol. Sci. 2009;36:2224. [Google Scholar]
- [32].Masi A, Sadori L, Balossi Restelli F, Baneschi I, Zanchetta G. Stable carbon isotope analysis as a crop management indicator at Arslantepe (Malatya, Turkey) during the Late Chalcolithic and Early Bronze Age. Veg. Hist. Archaeobot. 2013:1. [Google Scholar]
- [33].Brock F, Wood R, Higham TFG, Ditchfield P, Bayliss A, Bronk Ramsey C. Reliability of nitrogen content (%N) and carbon: nitrogen atomic ratios (C:N) as indicators of collagen preservation suitable for radiocarbon dating. Radiocarbon. 2012;54:138. [Google Scholar]
- [34].Aguilera M, Araus JL, Voltas J, Rodríguez-Ariza MO, Molina F, Rovira N, Buxó R, Ferrio JP. Stable carbon and nitrogen isotopes and quality traits of fossil cereal grains provide clues on sustainability at the beginnings of Mediterranean agriculture. Rapid Commun. Mass Spectrom. 2008;22:1653. doi: 10.1002/rcm.3501. [DOI] [PubMed] [Google Scholar]
- [35].Rebollo NR, Cohen-Ofri I, Popovitz-Biro R, Bar-Yosef O, Meignen L, Goldberg P, Weiner S, Boaretto E. Structural characterization of charcoal exposed to high and low pH: implication for 14C sample preparation and charcoal preservation. Radiocarbon. 2008;50:289. [Google Scholar]
- [36].Kragten J. Calculating standard deviations and confidence intervals with a universally applicable spreadsheet technique. Analyst. 1994;11:2161. [Google Scholar]
- [37].de la Rosa JM, González-Pérez JA, González-Vila FJ, Knicker H, Araújo MF. Molecular composition of sedimentary humic acids from south west Iberian Peninsula: A multi-proxy approach. Org. Geochem. 2011;42:791. [Google Scholar]
- [38].Silva JA, Bremner JM. Determination and isotope-ratio analysis of different forms of nitrogen in soils: 5. Fixed ammonium. Soil Sci. Soc. Am. J. 1966;30:587. [Google Scholar]
- [39].Bremner JM, Keeney DR. Determination and isotope-ratio analysis of different forms of nitrogen in soils: 3. Exchangeable ammonium, nitrate, and nitrite by extraction-distillation methods. Soil Sci. Soc. Am. J. 1966;30:577. [Google Scholar]
- [40].Stevenson FJ. 1965. Methods of Soil Analysis. Part 2. Chemical and Microbiological Properties. American Society Of Agronomy, Soil Science Society Of America.
- [41].Araus JL, Buxó R. Changes in carbon isotope discrimination in grain cereals from the north-western Mediterranean basin during the past seven millennia. Aust. J. Plant Physiol. 1993;20:117. [Google Scholar]
- [42].Ferrio JP, Alonso N, Voltas J, Araus JL. Grain weight changes over time in ancient cereal crops: Potential roles of climate and genetic improvement. J. Cereal Sci. 2006;44:323. [Google Scholar]
- [43].Ferrio JP, Alonso N, Lopez JB, Araus JL, Voltas J. Carbon isotope composition of fossil charcoal reveals aridity changes in the NW Mediterranean Basin. Glob. Chang. Biol. 2006;12:1253. [Google Scholar]
- [44].Fiorentino G, Caracuta V, Casiello G, Longobardi F, Sacco A. Studying ancient crop provenance: implications from δ13C and δ15N values of charred barley in a Middle Bronze Age silo at Ebla (NW Syria) Rapid Commun. Mass Spectrom. 2012;26:327. doi: 10.1002/rcm.5323. [DOI] [PubMed] [Google Scholar]
- [45].Philippsen B. 2010. pp. 21–36. Landscapes and Human Development: The Contribution of European ArchaeologyTerminal mesolithic diet and radiocarbon dating at inland sites in Schleswig-Holstein, in. Proceedings of the International Workshop “Socio-Environmental Dynamics over the Last 12,000 Years: The Creation of Landscapes (1st-4th April 2009)” (Ed: Kiel Graduate School “Human Development in Landscapes”). Dr Rudolf Habelt GmbH, Bonn. [Google Scholar]
- [46].Kristiansen SM, Dalsgaard K, Holst MK, Aaby B, Heinemeier J. Dating of prehistoric burial mounds by 14C analysis of soil organic matter fractions. Radiocarbon. 2003;45:101. [Google Scholar]
- [47].Olsson IU. The importance of the pretreatment of wood and charcoal samples. In: Berger R, Suess HE, editors. Radiocarbon Dating – Ninth International Conference. Los Angeles and La Jolla: University Of California Press; 1976. pp. 135–146. [Google Scholar]
- [48].Wallace M, Jones G, Charles M, Fraser R, Halstead P, Heaton THE, Bogaard A. Stable carbon isotope analysis as a direct means of inferring crop water status and water management practices. World Archaeol. 2013;45:388. [Google Scholar]
- [49].Goh KM. Carbon Isotope Techniques. San Diego: Academic Press; 1991. [Google Scholar]
- [50].Fiorentino G, Caracuta V, Calcagnile L, D'Elia M, Matthiae P, Mavelli F, Quarta G. Third millennium B.C. climate change in Syria highlighted by carbon stable isotope analysis of 14C-AMS dated plant remains from Ebla. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2008;266:51. [Google Scholar]
- [51].D'Elia M, Calcagnile L, Quarta G, Sanapo C, Laudisa M, Toma U, Rizzo A. Sample preparation and blank values at the AMS radiocarbon facility of the University of Lecce. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms. 2004;223/224:278. [Google Scholar]
- [52].Quarta G, D'Elia M, Valzano D, Calcagnile L. New bomb pulse radiocarbon records from annual tree rings in the northern hemisphere temperate region. Radiocarbon. 2005;47:30. [Google Scholar]