

Abstract
Background
Bats are amongst the natural reservoirs of many coronaviruses (CoVs) of which some can lead to severe infection in human. African bats are known to harbor a range of pathogens (e.g., Ebola and Marburg viruses) that can infect humans and cause disease outbreaks. A recent study in South Africa isolated a genetic variant closely related to MERS-CoV from an insectivorous bat. Though Madagascar is home to 44 bat species (41 insectivorous and 3 frugivorous) of which 34 are endemic, no data exists concerning the circulation of CoVs in the island’s chiropteran fauna. Certain Malagasy bats can be frequently found in close contact with humans and frugivorous bats feed in the same trees where people collect and consume fruits and are hunted and consumed as bush meat. The purpose of our study is to detect and identify CoVs from frugivorous bats in Madagascar to evaluate the risk of human infection from infected bats.
Methods
Frugivorous bats belonging to three species were captured in four different regions of Madagascar. We analyzed fecal and throat swabs to detect the presence of virus through amplification of the RNA-dependent RNA polymerase (RdRp) gene, which is highly conserved in all known coronaviruses. Phylogenetic analyses were performed from positive specimens.
Results
From 351 frugivorous bats, we detected 14 coronaviruses from two endemic bats species, of which 13 viruses were identified from Pteropus rufus and one from Eidolon dupreanum, giving an overall prevalence of 4.5%. Phylogenetic analysis revealed that the Malagasy strains belong to the genus Betacoronavirus but form three distinct clusters, which seem to represent previously undescribed genetic lineages.
Conclusions
Our findings suggest that CoVs circulate in frugivorous bats of Madagascar, demonstrating the needs to evaluate spillover risk to human populations especially for individuals that hunt and consume infected bats. Possible dispersal mechanisms as to how coronaviruses arrived on Madagascar are discussed.
Keywords: Coronavirus, Chiroptera, Pteropodidae, Madagascar
Background
Coronaviruses (CoVs) are enveloped viruses with single-stranded positive-sense RNA belonging to the subfamily Coronavirinae in the family Coronaviridae (order Nidovirales). Genomes of CoVs range from 25 to 32 kb and show high genetic diversity [1]. CoVs are classified into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus [2].
In mammals and birds, CoVs are associated with upper and lower respiratory illnesses or gastroenteritis. In humans, CoVs infections are commonly caused by HCoV-229E and HCoV-OC43 which generally cause mild respiratory illnesses [3]. A new CoV that causes severe acute respiratory syndrome (SARS-CoV) emerged in humans in 2002–2003 and infected more than 8,000 individuals with mortality rates estimated at around 10% [4]. The emergence of SARS-CoV and its mortality rate have raised the risk of a new pandemic that could threaten public health. For this reason, the scientific community invested considerable interest in the identification and characterization of CoVs especially within mammal reservoirs. Subsequently, two novel human CoVs were discovered: HCoV-NL63 in 2004 [5] and HCoV-HKU1 in 2005 [6]. In June 2012, a third novel coronavirus named HCoV-EMC/2012 (renamed MERS-CoV) was isolated from patients presenting with acute respiratory distress and pulmonary inflammation [7,8].
Studies which aimed to identify potential reservoirs of emerging human CoVs have revealed that the Betacoronavirus SARS-CoV was closely related to CoVs detected in bats, specifically members of the genus (Rhinolophus), which brought the hypothesis of a spillover of this virus to several animal species (including civet cats and raccoons) sold in Chinese markets as bushmeat for human consumption [9-11]. Bats have since become a particular focus and a number of Alphacoronavirus and Betacoronaviruses have been identified in many frugivorous and insectivorous bat species and in many countries worldwide in Asia, the Americas and Europe (see review from Drexler et al. 2014) [12]. Genomic characterization of the recently discovered MERS-CoV showed that this virus belongs to the genus Betacoronavirus and seems to be closely related to bat coronaviruses HKU4 and HKU5 isolated from bats (Tylonycteris and Pipistrellus) [13].
African bats are known to harbor a range of pathogens (e.g., Ebola and Marburg viruses) that can infect humans and cause disease outbreaks [14-16]. Some authors have reported the detection of bat CoVs from mainland Africa [17-21]. A recent study in South Africa detected a genetic variant closely related to MERS-CoV from Neoromicia zuluensis, an insectivorous bat [22]. The authors hypothesize that MERS-CoV may have a common ancestors with CoVs borne by bats from Africa. Though Madagascar is home to 44 bat species (41 insectivorous and 3 frugivorous) of which 34 are endemic [23-25], no data exists concerning the circulation of CoVs in Malagasy bats. Certain bat species on the island can be frequently found in close contact with humans, particularly members of the family Molossidae; these synanthropic species roost in human-occupied buildings, like houses, schools or hospitals [26], whilst frugivorous bats feed in the same fruit trees where people collect and consume fruits. Moreover, hunting and consumption of bat bush meat, especially the larger frugivorous species of the family Pteropodidae, is widespread on the island, bringing hunters, purveyors and consumers into contact with bats [27]. In this study, we report the detection of CoVs amongst frugivorous bats in Madagascar. Our results demonstrated for the first time that CoVs belonging to the genus Betacoronavirus are circulating amongst two endemic frugivorous bats species in Madagascar.
Results
Virus detection
A total of 351 bats belonging to 3 endemic bat species of the family Pteropodidae were captured and sampled: Rousettus madagascariensis (n = 179), Pteropus rufus (n = 76) and Eidolon dupreanum (n = 96) (Table 1). None of the throat swabs from any bat species (n = 265) tested positive for CoV, but 4.5% (14/313) of fecal specimens tested positive for CoV. Prevalence within P. rufus, E. dupreanum and R. madagascariensis was respectively 17.1% (13/76), 1.0% (1/96) and 0% (0/141). All positive specimens originated from bats captured in the Menabe Region (Figure 1).
Table 1.
Geographic location, number of bats tested and number of samples positive for coronavirus in Madagascar
| Sampling region/site | Geographic location | Period of capture | Nb. bats captured | Species | No. samples tested throat/fecal | No. samples positive throat/fecal |
|---|---|---|---|---|---|---|
| Toliara | Southwest | Feb. 2007 | 85 | Eidolon dupreanum | 0/85 | 0/0 |
| Anosibe An’ala | Northeast | Feb. 2010 | 55 | Rousettus madagascariensis | 55/19 | 0/0 |
| Menabe | Southwest | Jun. 2010 Jul. 2011 | ||||
| Manamby | 3 | Pteropus rufus | 3/3 | 0/0 | ||
| Befasy | 3 | Pteropus rufus | 3/3 | 0/0 | ||
| Ankiliabo | 48 | Pteropus rufus | 48/48 | 0/5 | ||
| 1 | Eidolon dupreanum | 1/1 | 0/0 | |||
| Mahabo | 9 | Pteropus rufus | 9/9 | 0/2 | ||
| 8 | Eidolon dupreanum | 8/8 | 0/1 | |||
| Bemanonga | 13 | Pteropus rufus | 13/13 | 0/6 | ||
| 2 | Eidolon dupreanum | 2/2 | 0/0 | |||
| Anjohibe | Northwest | Nov. 2010 Nov. 2011 | 124 | Rousettus madagascariensis | 123/122 | 0/0 |
| Total | 351 | 265/313 | 0/14 |
Figure 1.
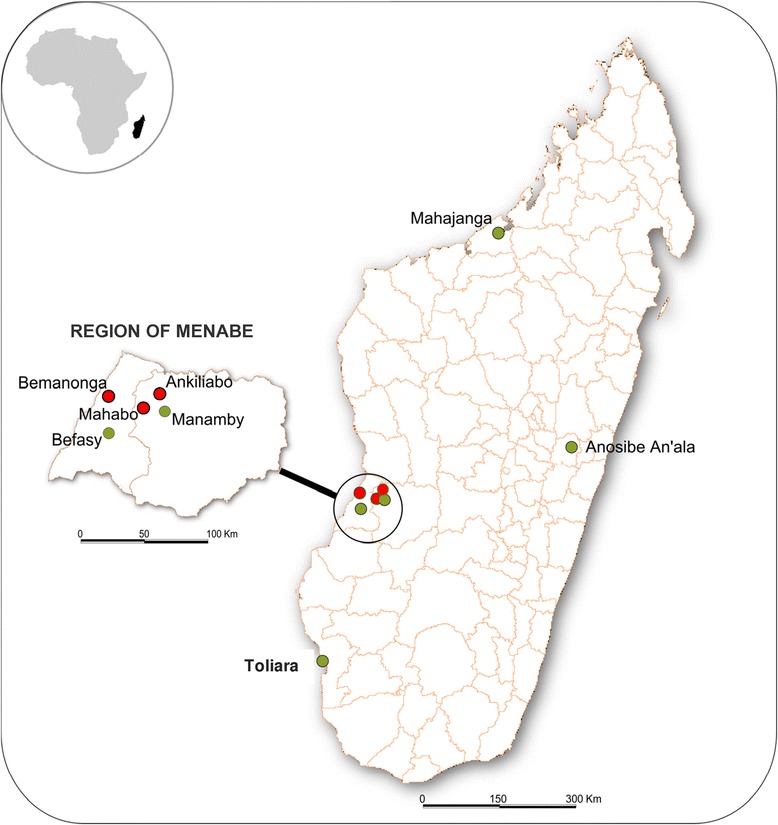
Location of sampling collection. Map of Madagascar showing sites of bats capture (green circle) and sites from the Region of Menabe where at least one CoV was detected from locally occurring bats (Red circles).
Short amplicon sequences of 329 bp in length of the RdRp gene were obtained for all PCR-positive animals, whereas larger fragment of 993 bp sequences could only be obtained from seven of the 14 PCR-positive animals.
Phylogenetic analysis
All amplicon sequences were aligned in-frame with a compilation of reference sequences from GenBank for which collection-date data was available [28], giving final alignments containing 51 different sequences of 993 bp in length and 64 different sequences of 329 bp in length. GTR + I + G was identified as the optimal substitution model using jModeltest v2.1.2 [29]. Multiple phylogenies were generated in BEAST using different combinations of model parameters, and the best models were selected using the Tracer [30]. Bayes factor analysis employing marginal likelihoods, as detailed in [12]. All parameter combinations produced identical, strongly supported tree topologies (data not shown). As has elsewhere been determined by Lau et al. [12], Bayesian skyline using a relaxed, exponentially distributed clock model was found to be the best fitting model for RdRp dated-tip phylogenies. The final phylogenetic analyses (Figure 2) revealed that strains from Madagascar are members of the Betacoronavirus genus, rooting with Hong Kong strain BtCoV-HKU9 (HM211100) and Kenyan strain KY77 (GU065421), with posterior probabilities of 1. These lineages could be described as SARS-like, and were uniquely affiliated with frugivorous bat hosts of the family Pteripodidae. Malagasy strains were sub-divided into three distinct clusters: two of which were closely related (clusters 1 and 2) and originating from P. rufus, and one more distantly related (cluster 3) containing a strain detected from E. dupreanum and sequences previously detected from E. helvum in Kenya [21].
Figure 2.
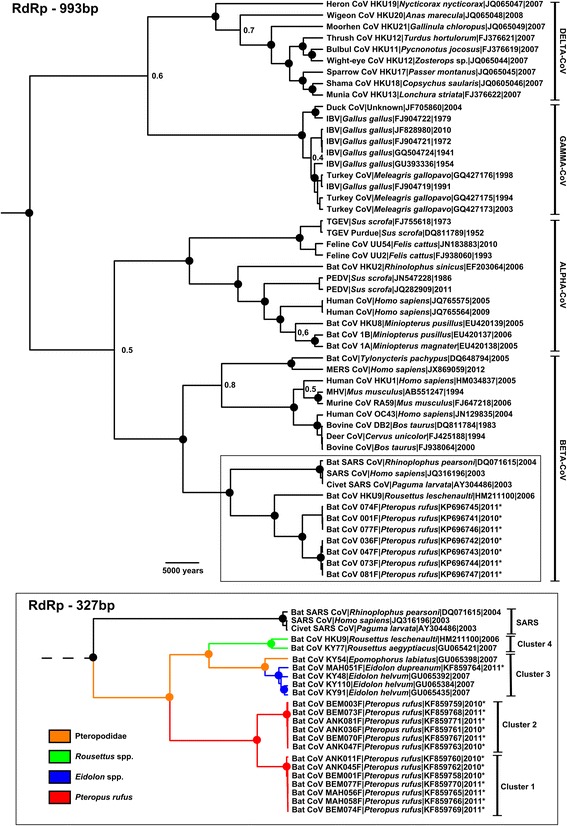
Bayesian phylogenetic tree generated using 993 nucleotides of the RdRp gene sequences of Malagasy bat CoVs and reference strains of CoVs group. Major viral genera are shown. Strains detected in this study are marked with an*. Posterior probabilities superior to 0.9 are indicated by a dot, other posterior probabilities are indicated in decimal form to the right of each node, and some have been left off for clarity. The scale bar is expressed in years. The inset figure shows the partial phylogeny of the highlighted region of the Betacoronavirus subgroup, including additional reference sequences and with a total alignment length of 329 nucleotides. Techniques used for generating each phylogenetic representation were identical, as detailed in the materials and methods section.
The Malagasy CoVs were detected from bats captured in three different sites of the Menabe region (West of Madagascar). Within cluster 1, strains were originated from Bemanonga, Mahabo and Ankiliabo. Within cluster 2, strains were originated from Bemanonga and Ankiliabo. The only virus detected belonging to cluster 3 was detected in one bat captured in Mahabo. Overall, identities among Malagasy CoVs ranged from 65 to 100% at the nucleotide level and 70.9 to 100% at the amino acid level (data not shown).
Molecular dating estimates based on the 993 bp fragment of the RdRp gene estimated the timescale of evolution of the coronavirus family to be thousands to tens of thousands of years, however dating estimates proved inaccurate, with broadly spanning HPD values at individual node positions.
Discussion
In the context of this study, we detected 14 coronaviruses forming nine genetically distinct strains in two endemic Malagasy frugivorous bat species. The overall prevalence (4.5%) is consistent with those identified in studies elsewhere [31-33]. Thirteen viruses were detected from Pteropus rufus and 1 virus from Eidolon dupreanum. We did not detect CoVs among the sampled R. madagascariensis. The detection of novel bat CoVs supports the observation that these viruses are diverse and have a nearly worldwide distribution [17,32,34,35].
We observed that all Malagasy bat CoVs detected in the present study belong to a SARS-related subgroup of the Betacoronaviruses, with relatively close homology to BtCoV-HKU9 [36]. Our strains displayed 3 distinct clusters: 2 clusters associated with P. rufus and 1 cluster associated with E. dupreanum. It can be inferred from the results that multiple clusters of CoVs occurring within Malagasy bat populations co-circulate and possibly in a syntopic manner. The high nucleotide and amino acid divergence between clusters and compared to the reference virus BtCoV-HKU9 suggests previously undescribed genetic lineages. Given the mobility of bats, and the especially long distances that can be travelled by colonies of fruit bats [37], these coronaviruses may be spread over a large region. However, host-genus-specific phylogenetic clustering (Figure 2, inset) suggests likely host-specificity which may limit viral spill-over. Thus, further molecular epidemiology studies would be required to fully understand the dispersal potential of CoVs amongst Malagasy bats species.
It is important to remember that, although all three of Madagascar’s fruit bat species were sampled, nothing is known of CoV dynamics in tropical fruit bats, and many factors such as seasonality, bioclimate and the presence of other host species may have important influences on CoV prevalence in these populations. More studies are needed in different locations including different species, particularly those with an insectivorous diet, to reveal a more comprehensive view of the diversity of these viruses in Madagascar. Since the strains of Betacoronavirus identified from Madagascar are closely related to those known from Africa, some preliminary biogeographic considerations are in order. All three bat species analyzed herein are endemic to Madagascar. The genus Pteropus has a broad distribution from the Australia-New Guinea region west across the Indian Ocean to offshore islands of Tanzania; it is unknown from the African continent. The genus Eidolon is composed of two species: E. helvum occurring on the African mainland and offshore islands and E. dupreanum restricted to Madagascar. Based on a phylogeographical study, both species show broad panmictic population structure [38]. Further, these two taxa are estimated to have diverged from one another sometime in the late Miocene or early Pliocene [39]. The genus Rousettus is broadly distributed across Africa and Asia and the ancestral origin of the Malagasy species, R. madagascariensis, is unresolved [40]. As with the other two Pteropodidae species occurring on Madagascar, R. madagascariensis shows little genetic population structure and presumably broadly disperses across the island, which in turn has important epidemiological implications for these bats transmitting different zoonoses.
Although estimates of the most recent common ancestors (MRCAs) proved inaccurate in our study, most likely as a result of a limited sequence availability from the identified viral strains, standard evolutionary analyses have estimated CoV origins to date to somewhere between 500–10,000 yrs. in the past [41-43]. Nevertheless, further investigations into the relevance of MRCA prediction methodologies are required and a great level of caution must be employed in the interpretation of MRCA data. .
Alternatively, viral lineages may have been imported to Madagascar in recent history: While the vast majority of the island’s bat fauna is endemic, a few species apparently disperse across the Mozambique Channel. Probably the best example is the Molossidae species Mops midas, for which, based on genetic data, Southern African animals are nested within Malagasy populations [44]. This bat makes its day roosts in rock crevices and may broadly occur synoptically at such sites with E. dupreanum and R. madagascariensis. These two fruit bat species are known to feed in the same fruiting trees with P. rufus [45], which would complete the cycle of how CoVs originated from Africa mainland could be carried to Madagascar and transmitted to different species of pteropodid bats.
Although we were not able to evaluate risk of human infection, the strains detected here may be considered as potential human pathogens, as bats are natural reservoirs of some pathogenic CoVs. Isolation of Malagasy CoVs using cell culture and molecular analysis of spike (S) gene could better evaluate risk of human infection. Also, a longitudinal study amongst people who frequently handle live bats (e.g. bat hunters, bat bushmeat purveyors, and scientists), and who represent populations at higher risk of infection, would be interesting to establish possible cases of transmission to humans and public health risks.
Conclusions
In our study, we confirm that CoVs are circulating in two species of endemic bats in Madagascar. Further work on CoV diversity amongst the island’s bat species, as well as aspects of the ecology and behavior of susceptible taxa, are needed to understand the origin, evolution and dispersal of these viruses across the island. To conclude, the results of our study demonstrate the need to develop research programs that aim at surveying viruses in the wild, especially in bats, in order to address possible emergence of zoonotic viruses within human populations.
Methods
Study sites
We sampled frugivorous bats in four different areas of Madagascar: Anjohibe, Anosibe An’Ala, Menabe and Toliara (Table 1/Figure 1) based on known accessible colonies of roosting bats and sites where bats are frequently hunted and eaten by people. In the region of Menabe, we selected 5 different sites situated at a mean distance of 28 km around Mahabo to capture and collect specimens, while in the three other regions, sampling occurred at a single site. Sampling was carried out under protocols approved and permitted by Ministry of Environment and Forest (Authorization # 301/08, 101/09, 163/10, 032/11 and 261/11).
Capturing and sampling
Fruit bats were captured by the use of mist-nets set near roosting sites (trees or caves) and with help of professional hunters [46]. Rectal and throat swabs were collected from each individual bat. All bats were identified according species specific morphological features well known by our field trained team (ecologist and veterinarian) and subsequently released. Swabs were placed in viral transport media, almost immediately conserved in liquid nitrogen in the field and stored at −80°C upon arrival at the laboratory in Antananarivo.
RNA extraction
RNA was extracted from specimens using the QIAamp Viral RNA minikit (QIAGEN, Courtaboeuf, France) according to the manufacturer’s protocol. Briefly, total RNA was extracted from 140 μL of each sample and eluted in 65 μL of Qiagen AVE elution buffer. The extracted RNA was either immediately analyzed or stored at −80°C until use.
Viral amplification and detection
Extracted RNA was reverse transcribed to generate cDNA by using the M-MLV Reverse Transriptase (Invitrogen, California, USA) into a 2 step reactions. First, 2 μL of RNA were mixed in a solution containing 0.5 μM of random hexamer primers (Roche Diagnostics, Mannheim, Germany), 1 U/μL of RNase inhibitor (10 000 units) and 8.5 μL of water, at 80°C for 5 min, 50°C for 5 min and 4°C for 15 min. Then, 8 μL of RNA issued from the first step was added to a mixture of 0.5 mM of each dNTP (deoxynucleotide triphosphates), 10 U/μL of reverse transcriptase M-MLV, 1 X of buffer and 0.01 M of DTT (dithiothreitol), and incubated at 42°C for 50 min and 95°C for 5 min.
PCR assay was performed to amplify the RNA-dependent RNA polymerase (RdRp) gene which is highly conserved in all known coronaviruses [47]. The primers pair (Forward 5′-GGTTGGGACTATCCTAAGTGTGA-3′; Reverse 5′-CCATCATCAGATAGAATCATCATA-3′) was designed to amplify a 440 bp fragment as described previously [47]. Reaction mixture was carried out using the GoTaq/dNTP mix, Custom kit (Promega Corporation, Madison, USA). Briefly, 5 μL of cDNA was mixed with 1 X of Green GoTaq Flexi Buffer, 1.5 mM of MgCl2, 0.2 mM of each dNTP, 0.2 μM of each primer, 0.025 U/μL of GoTaq DNA Polymerase and 28.75 μL of water nuclease-free giving a final volume of 50 μL. The thermocycling was performed under the following conditions: activation at 95°C for 10 min and 40 cycles of denaturation at 95°C for 1 min, annealing at 58°C for 1 min, extension at 72°C for 1 min, and a final extension at 72°C for 5 min. All negative samples were tested in a semi-nested PCR with the same PCR program and using the following pair of primers (Forward 5′-GGTTGGGACTATCCTAAGTGTGA-3′; Reverse 5′- ATCAGATAGAATCATCATAGAGA-3′). Amplicons products were subsequently electrophorezed on a 2.0% agarose gel and visualized using ethidium bromide under UV light. All specimens that showed a positive band at the expected size (440 bp) were sequenced on both strands by Beckman Coulter Genomics (Essex, UK).
From the sequences obtained from the 440 bp, fragment, we designed new primers (reverse) that were strain specific for Malagasy BatCoV (5′-GATGACCTGTATATTCCCA-3′ and 5′-ATGACCTATACATACCCATG-3). We then amplified a large fragment of the RdRp gene by using the consensus forward primer 5′-GTGTACGCTGCTGATCCTGCTATGCA-3′ [48]. The following conditions were performed: 95°C for 10 min and 40 cycles of denaturation at 95°C for 1 min, annealing 56°C for 1 min, extension at 72°C for 1 min, and a final extension at 72°C for 5 min. The final size of sequences used for molecular dating was 1086 bp.
Sequences and phylogenetic analysis
Sequences from the 440 bp or 1086 bp fragments of the RdRp gene were cleaned and aligned with reference sequences collected from a literature search. Alignment was performed using the translation alignment tool in Geneious pro™ v.6.3.2, created by Biomatters (available from http://www.geneious.com/), and the default ClustalW cost matrix. The final alignment was respectively 329 bp and 993 bp in length for fragments 440 bp and 1086 bp, and contained no free-end or internal gaps. From these alignments, the appropriate substitution model was identified in jModeltest v.2.1.2 [29,49]. Using the appropriate substitution model, 2*10^8 iterations were run with or without the use of a 3-base codon model, using different clock models, alternating between constant and Bayesian Skyline population size models, and seeding with uncorrelated log-normally distributed priors. Trees were sampled every 2x10^5 iterations, and analysis convergence was assessed in tracer v.1.4.0 [50] (available from http://beast.bio.ed.ac.uk/tracer). All analyses converged after a 10% burn-in to give Effective Sample Size values for all parameters superior to 200. Bayes factor analyses were performed in Tracer v1.4.0, with 1000 bootstrap replicates to assess the relative performance of model selections on the generated phylogenies. After identification of an optimal model for phylogenetic classification and dating, two further independent analyses (2*10^8 iterations, sampling every 2*10^5 iterations) were run in BEAST, and all analyses were combined in logCombiner (BEAST package) with a burn-in of 10%, leaving only converged parameter estimates. The final phylogeny and MRCA for fragments 993 bp in length dates were extracted using TreeCombiner (BEAST package) and FigTree v.1.4.0 (available at http://tree.bio.ed.ac.uk/software/figtree/).
Acknowledgments
This study was conducted in collaboration with the Association Madagasikara Voakajy and the Bangor University (Darwin Initiative Project), a project examining aspects of emerging viruses in small wild mammals. The work in the Toliara region was part of Action Concertée Inter-Pasteurienne (ACIP) research program. We would like to thank Felicien Herbert Randrianandrianina of Madagasikara Voakajy and local hunters for their help in capturing bats. David A. Wilkinson’s post-doctoral fellowship was funded by “RUN-Emerge”, a European project funded by European Commission under FP7 program.
Footnotes
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
NHR and DAW carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. LAN coordinated the fieldwork, participated to the sampling collection, participated to molecular testing and drafted the manuscript. JHR participated in the design of the study and coordinated the fieldwork SMG and RKJ helped to draft the manuscript. JPGJ participated in the design of the study and helped to draft the manuscript JMH: conceived, designed and coordinated the study and helped to draft the manuscript. All authors read and approved the final manuscript
Contributor Information
Norosoa H Razanajatovo, Email: norosoa@pasteur.mg.
Lalaina A Nomenjanahary, Email: lalaina@pasteur.mg.
David A Wilkinson, Email: dwilkin799@gmail.com.
Julie H Razafimanahaka, Email: hantajulie@gmail.com.
Steven M Goodman, Email: sgoodman@fieldmuseum.org.
Richard K Jenkins, Email: rkbjenkins@gmail.com.
Julia PG Jones, Email: julia.jones@bangor.ac.uk.
Jean-Michel Heraud, Email: jmheraud@pasteur.mg.
References
- 1.Woo PC, Lau SK, Huang Y, Yuen KY. Coronavirus diversity, phylogeny and interspecies jumping. Exp Biol Med (Maywood) 2009;234(10):1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- 2.Adams MJ, Carstens EB. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch Virol. 2012;157(7):1411–1422. doi: 10.1007/s00705-012-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heikkinen T, Jarvinen A. The common cold. Lancet. 2003;361(9351):51–59. doi: 10.1016/S0140-6736(03)12162-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO . Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. 2004. [Google Scholar]
- 5.van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, et al. Identification of a new human coronavirus. Nat Med. 2004;10(4):368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79(2):884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bermingham A, Chand MA, Brown CS, Aarons E, Tong C, Langrish C, et al. Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East. Euro Surveill. 2012;17(40):20290. [PubMed] [Google Scholar]
- 8.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 9.Kan B, Wang M, Jing H, Xu H, Jiang X, Yan M, et al. Molecular evolution analysis and geographic investigation of severe acute respiratory syndrome coronavirus-like virus in palm civets at an animal market and on farms. J Virol. 2005;79(18):11892–11900. doi: 10.1128/JVI.79.18.11892-11900.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310(5748):676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 11.Song HD, Tu CC, Zhang GW, Wang SY, Zheng K, Lei LC, et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci U S A. 2005;102(7):2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 2014;101:45–56. doi: 10.1016/j.antiviral.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Boheemen S, De Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio. 2012;3(6):e00473–12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banyard AC, Hayman D, Johnson N, McElhinney L, Fooks AR. Bats and lyssaviruses. Adv Virus Res. 2011;79:239–289. doi: 10.1016/B978-0-12-387040-7.00012-3. [DOI] [PubMed] [Google Scholar]
- 15.Biek R, Walsh PD, Leroy EM, Real LA. Recent common ancestry of Ebola Zaire virus found in a bat reservoir. PLoS Pathog. 2006;2(10):e90. doi: 10.1371/journal.ppat.0020090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Towner JS, Pourrut X, Albarino CG, Nkogue CN, Bird BH, Grard G, et al. Marburg virus infection detected in a common African bat. PLoS One. 2007;2(8):e764. doi: 10.1371/journal.pone.0000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geldenhuys M, Weyer J, Nel LH, Markotter W. Coronaviruses in South African Bats. Vector Borne Zoonotic Dis. 2013;13(7):516–519. doi: 10.1089/vbz.2012.1101. [DOI] [PubMed] [Google Scholar]
- 18.Quan PL, Firth C, Street C, Henriquez JA, Petrosov A, Tashmukhamedova A, et al. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. MBio. 2010;1(4):e00208–e00210. doi: 10.1128/mBio.00208-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfefferle S, Oppong S, Drexler JF, Gloza-Rausch F, Ipsen A, Seebens A, et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg Infect Dis. 2009;15(9):1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tao Y, Tang K, Shi M, Conrardy C, Li KS, Lau SK, et al. Genomic characterization of seven distinct bat coronaviruses in Kenya. Virus Res. 2012;167(1):67–73. doi: 10.1016/j.virusres.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg Infect Dis. 2009;15(3):482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, et al. Close relative of human middle East respiratory syndrome coronavirus in bat, South Africa. Emerg Infect Dis. 2013;19(10):1697–1699. doi: 10.3201/eid1910.130946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodman S. Association Vahatra, Antananarivo edition. 2011. [Les chauves-souris de Madagascar.] [Google Scholar]
- 24.Goodman SM, Puechmaille PJ, Friedli-Weyeneth N, Gerlach J, Ruedi M, Schoeman MC, et al. Phylogeny of the Emballonurini (Emballonuridae) with descriptions of a new genus and species from Madagascar. J Mammal. 2012;93:1440–1455. doi: 10.1644/11-MAMM-A-271.1. [DOI] [Google Scholar]
- 25.Goodman SM, Ramasindrazana B, Maminirina CP, Schoeman MC, Appleton B. Morphological, bioacoustical, and genetic variation in Miniopterus bats from eastern Madagascar, with the description of a new species. Zootaxa. 2011;2880:1–19. [Google Scholar]
- 26.Razafindrakoto N, Harwell A, Jenkins RKB. Bats roosting in public buildings: A preliminary assessment from Moramanga, eastern Madagascar. Madagascar Conserv Dev. 2010;5(2):85–88. [Google Scholar]
- 27.Jenkins RKB, Racey PA. Bats as bushmeat in Madagascar. Madagascar Conserv Dev. 2008;3(1):22–30. [Google Scholar]
- 28.Wertheim JO, Chu DK, Peiris JS, Kosakovsky Pond SL, Poon LL. A case for the ancient origin of coronaviruses. J Virol. 2013;87(12):7039–7045. doi: 10.1128/JVI.03273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suchard MA, Weiss RE, Sinsheimer JS. Bayesian selection of continuous-time Markov chain evolutionary models. Mol Biol Evol. 2001;18(6):1001–1013. doi: 10.1093/oxfordjournals.molbev.a003872. [DOI] [PubMed] [Google Scholar]
- 31.Lau SK, Woo PC, Li KS, Tsang AK, Fan RY, Luk HK, et al. Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports murine origin of Betacoronavirus 1 with implications on the ancestor of Betacoronavirus lineage A. J Virol. 2015;89(6):3076–3092. doi: 10.1128/JVI.02420-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Felix Drexler J, et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis. 2008;14(4):626–631. doi: 10.3201/eid1404.071439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, et al. Prevalence and genetic diversity of coronaviruses in bats from China. J Virol. 2006;80(15):7481–7490. doi: 10.1128/JVI.00697-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brandao PE, Scheffer K, Villarreal LY, Achkar S, Oliveira Rde N, Fahl Wde O, et al. A coronavirus detected in the vampire bat Desmodus rotundus. Braz J Infect Dis. 2008;12(6):466–468. doi: 10.1590/S1413-86702008000600003. [DOI] [PubMed] [Google Scholar]
- 35.Falcon A, Vazquez-Moron S, Casas I, Aznar C, Ruiz G, Pozo F, et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch Virol. 2011;156(10):1883–1890. doi: 10.1007/s00705-011-1057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol. 2007;81(4):1574–1585. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tidemann CR, Nelson JE. Long-distance movements of the grey-headed flying fox (Pteropus poliocephalus) J Zool. 2004;263:141–146. doi: 10.1017/S0952836904004960. [DOI] [Google Scholar]
- 38.Peel AJ, Sargan DR, Baker KS, Hayman DT, Barr JA, Crameri G, et al. Continent-wide panmixia of an African fruit bat facilitates transmission of potentially zoonotic viruses. Nat Commun. 2013;4:2770. doi: 10.1038/ncomms3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi JJ, Chan LM, Peel A, Lai R, Yoder AD, Goodman SM. A deep divergence time between sister species of Eidolon (Family Pteropodidae), with evidence for widespread panmixia. Acta Chiropterologica. 2014;16(2):279–292. doi: 10.3161/150811014X687242. [DOI] [Google Scholar]
- 40.Chan LM, Goodman SM, Nowak MD, Weisrock DW, Yoder AD. Increased population sampling confirms low genetic divergence among Pteropus (Chiroptera: Pteropodidae) fruit bats of Madagascar and other western Indian Ocean islands. PLoS Curr. 2011;3 doi: 10.1371/currents.RRN1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vijaykrishna D, Smith GJ, Zhang JX, Peiris JS, Chen H, Guan Y. Evolutionary insights into the ecology of coronaviruses. J Virol. 2007;81(8):4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woo PC, Huang Y, Lau SK, Yuen KY. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2(8):1804–1820. doi: 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86(7):3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ratrimomanarivo FH, Vivian J, Goodman SM, Lamb J. Morphological and molecular assessment of the specific status of Mops midas (Chiroptera: Molossidae) from Madagascar and Africa. Afr Zool. 2007;42:237–253. doi: 10.3377/1562-7020(2007)42[237:MAMAOT]2.0.CO;2. [DOI] [Google Scholar]
- 45.Hutcheon JM. The Natural History of Madagascar. Chicago: The University of Chicago Press; 2003. Frugivory by Malagasy bats. [Google Scholar]
- 46.Kunz TH, Hodgkison R, Weise CD. Methods of Capturing and Handling bats, In Ecological and Behavioral methods for the study of bats. Maryland (USA): The Johns Hopkins University Press; 2009. [Google Scholar]
- 47.Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, et al. Identification of a novel coronavirus in bats. J Virol. 2005;79(4):2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. 2010;84(21):11336–11349. doi: 10.1128/JVI.00650-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 50.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]