

Abstract
Background
The dramatic worldwide expansion of Aedes albopictus (the Asian tiger mosquito) and its vector competence for numerous arboviruses represent a growing threat to public health security. Molecular markers are crucially needed for tracking the rapid spread of this mosquito and to obtain a deeper knowledge of population structure. This is a fundamental requirement for the development of strict monitoring protocols and for the improvement of sustainable control measures.
Methods
Wild population samples from putative source areas and from newly colonised regions were analysed for variability at the ribosomal DNA internal transcribed spacer 2 (ITS2). Moreover, a new set of 23 microsatellite markers (SSR) was developed. Sixteen of these SSRs were tested in an ancestral (Thailand) and two adventive Italian populations.
Results
Seventy-six ITS2 sequences representing 52 unique haplotypes were identified, and AMOVA indicated that most of their variation occurred within individuals (74.36%), while only about 8% was detected among populations. Spatial analyses of molecular variance revealed that haplotype genetic similarity was not related to the geographic proximity of populations and the haplotype phylogeny clearly indicated that highly related sequences were distributed across populations from different geographical regions. The SSR markers displayed a high level of polymorphism both in the ancestral and in adventive populations, and FST estimates suggested the absence of great differentiation. The ancestral nature of the Thai population was corroborated by its higher level of variability.
Conclusions
The two types of genetic markers here implemented revealed the distribution of genetic diversity within and between populations and provide clues on the dispersion dynamics of this species. It appears that the diffusion of this mosquito does not conform to a progressive expansion from the native Asian source area, but to a relatively recent and chaotic propagule distribution mediated by human activities. Under this scenario, multiple introductions and admixture events probably play an important role in maintaining the genetic diversity and in avoiding bottleneck effects. The polymorphic SSR markers here implemented will provide an important tool for reconstructing the routes of invasion followed by this mosquito.
Keywords: Aedes albopictus, Microsatellites, Simple sequence repeats (SSRs), Internal transcribed spacer 2 (ITS2), Mosquito, Virus vectors, Population genetics
Background
The Asian tiger mosquito, Aedes (Stegomyia) albopictus is a vector of numerous arboviruses including Dengue, Chikungunya, and a variety of epizootic viruses [1-3]. It is considered to be the most invasive mosquito species in the world [4], and it is listed among the top hundred most dangerous invasive species (http://www.issg.org/database/species/search.asp?st=100ss).
Originally a native of the tropical forests of Southeast Asia, it has spread during the last 30–40 years to both tropical and temperate regions worldwide [4,5]. Global warming can result in wetter and warmer conditions that favour the spread of this mosquito and its aquatic larvae [6,7]. The dramatic expansion of this aggressive daytime biting mosquito has increased public health concerns for arbovirus-related disease outbreaks [8-11]. Some biological traits such as feeding preferences [12], winter diapause [13], and vector competence [14] of Ae. albopictus vary according to its geographic origin. Given that these traits may be genetically determined and may influence pathogen transmission, it is of paramount importance to define the geographic origin of invading populations.
Furthermore, as the threat represented by this mosquito is growing due to the lack of sustainable control measures and of the progressive spread of insecticide-resistance [15,16], efficient surveillance methods may depend on the detailed knowledge of the population structure and dynamics of this vector [10,17]. Moreover, the potential presence of cryptic subgroups, that may be unaffected by control methods, should not be under-estimated. Indeed, great variation in terms of genome size has been previously identified in strains from different regions [18-21]. Variations in the rDNA cistron have also been reported within and among populations of this mosquito [22]. In this context, the Internal Transcribed Spacer 2 (ITS2) of nuclear ribosomal DNA has been shown to be a useful marker, because of its power in detecting differentiation, not only among cryptic species [23-25], but also among conspecific populations [26-28]. As ITS2 is one of the less functionally restricted regions of the rDNA, it can accumulate mutations within isolated populations relatively quickly and can thus be an indicator of genetic discontinuity between populations [29,30]. The genetic diversity and the phylogeographic relationships among populations have been extensively analysed using mitochondrial DNA (mtDNA) markers [31-35]. The data from populations collected in various regions of the world were consistent in displaying the presence of weak sequence variation at mtDNA loci, probably due to the recent spread of the species, but also due to the low mutation rate of these markers. Indeed, when a set of 13 microsatellite markers were used to analyse the invasion of the Australian region [36], they appeared to be more informative, compared to mtDNA markers, in resolving the population structure and the invasion dynamics. Moreover, considering that Ae. albopictus populations are infected by the symbiont Wolbachia [37], whose presence can impact mtDNA diversity within host populations, the utility of mtDNA as a neutral marker is reduced [38].
In the present study we used the ITS2 region to address the presence of intraspecific differentiation (genetic barriers) among groups of populations from different ancestral and recently colonized geographic areas. Moreover, we developed additional novel microsatellite markers for this mosquito species. We provide evidence of their usefulness for the analysis of the genetic composition of populations.
Methods
Mosquito samples
Nine wild population samples of Ae. albopictus were collected across the species range (Table 1). Two samples were from Thailand (Ban Rai, Lampang Province in the North and Phato, Chumphon Province in the South); two from Réunion (St. Denis in the North and St. Pierre in the South-West); and five from Northern Italy (Brescia, Castellanza, Pavia, Modena, Cesena). The Thai samples are from a region that pertains to the putative home range of the species, while Réunion and the Italian samples are representative of old (17th and 18th century) [39] and recently colonized (1990) [40] areas, respectively. For each of the samples, eggs were collected using ovitraps and the emerging adults were reared in the insectary at 27°C, with 60-80% relative humidity and a 12:12 h (L:D) photoperiod. The identities of all the samples were confirmed using the morphological keys of Rueda [41]. The nine population samples were screened for sequence variability of the ITS2 marker.
Table 1.
Aedes albopictus wild population samples
| Region | Sample | Latitude | Longitude | Collection date |
|---|---|---|---|---|
| Thailand | Ban Rai | 18.31 | 99.55 | November 2010 |
| Phato | 9.84 | 98.79 | October 2010 | |
| Réunion | St. Denis | −20.88 | 55.45 | March 2010 |
| St. Pierre | −21.33 | 55.47 | March 2010 | |
| Italy | Brescia | 45.54 | 10.22 | July 2010 |
| Pavia | 45.19 | 9.16 | October 2010 | |
| Castellanza | 45.61 | 8.89 | October 2010 | |
| Cesena | 44.20 | 12.40 | August 2010 | |
| Modena | 44.65 | 10.92 | October 2009 |
The Rimini laboratory strain (F35) was used for the construction of the microsatellite-enriched libraries. This strain was established from mosquitoes collected in Rimini (Emilia Romagna) at the Centro Agricoltura Ambiente ‘G. Nicoli’ (Crevalcore, Italy). Three of the nine wild samples previously described, Ban Rai, Cesena and Brescia, were also screened to assess the variability of the newly developed microsatellites markers.
ITS2 marker characterization
DNA was extracted according to Baruffi et al. [42] from individual females of the population samples. The extracted DNA was resuspended in TE (10 mM Tris–HCl, pH 8, 1 mM EDTA) and its concentration was quantified using a Nanodrop ND-1000 spectrophotometer (Nano-drop Technologies Inc., Wilmington, DE, USA). The ribosomal DNA ITS2 region was amplified from the DNA samples using the universal primers 5.8Sf 5′-atcactcggctcgtggatcg-3′ and 28Sr 5′-atgcttaaatttagggggtagtc-3′ [43]. These primers anneal to highly conserved sequences in the 5.8S and 28S rDNA genes flanking the ITS2 region. Reactions were performed in a volume of 25 μl with approximately 3 ng DNA, 1.5 mM MgCl2, Reaction buffer (10 mM Tris–HCl pH 9, 50 mM KCl), 200 µM dNTP, 10 pmol of each primer and 1 unit Taq DNA polymerase (Invitrogen). Amplifications were performed using an Eppendorf MasterCycler with the following cycle conditions: an initial denaturing step at 94°C for 2 min; twenty-five cycles of denaturation at 94°C for 20 s, annealing at 54°C for 15 s, extension at 72°C for 25 s, followed by a final extension at 72°C for 5 min.
The PCR products were separated on 1.5% agarose gels, stained with ethidium bromide and photographed under ultraviolet light. Amplification products were ligated into the TOPO 2.1 vector and transformed into competent One Shot cells using the TOPO TA cloning kit (Invitrogen). DNA inserts were sequenced on both strands.
ITS2 data analysis
The boundaries of the ITS2 region were determined by comparison with the available Ae. albopictus sequences in GenBank. The ITS2 sequences were aligned using MUSCLE 3.8 [44]. Nucleotide diversity and gene diversity parameters were determined using Arlequin 3.5.1.2 [45]. Geographic structure in the ITS2 data set was investigated by spatial analyses of molecular variance implemented in SAMOVA 1.0 [46]. This approach identifies groups of populations that are genetically and geographically homogeneous and maximally differentiated from each other. The method requires the a priori definition of the number of groups (K) of populations that exist and generates F-statistics (FSC, FST and FCT) using an AMOVA approach. Different numbers of groups (K) were tested, and a simulated annealing procedure permitted the identification of the composition of each of the K groups that maximizes the FCT index (proportion of total genetic variance due to differences between groups). The program was run for two to eight groups (K = 2 to K = 8) each time with the simulated annealing process repeated 100 times, starting each time with a different partition of the population samples into the K groups. A phylogenetic analysis was also performed including the unique ITS2 sequences or haplotypes identified in the nine population samples by Arlequin 3.5.1.2 [45]. The haplotype sequences together with an Ae. flavopictus ITS2 outgroup sequence (GenBank accession number: AF353559.1) were aligned using MUSCLE 3.8 [44]. The Kimura-2-parameter and Γ distribution model indicated by the Akaike Information Criterion (AIC = 3064) and the Bayesian Information Criterion (BIC = 3899), was used to perform Maximum Likelihood analyses in MEGA 5.2.2 [47]. The reliability of the resulting haplotype tree was determined by 1000 bootstrap replications. The mid-point rooted tree was drawn using FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Microsatellite isolation and characterization
Total genomic DNA was extracted individually from 64 mosquitoes (40 males and 24 females) from the Rimini strain using the method reported by Baruffi et al. [42]. The DNA was subsequently pooled and submitted to Genetic Identification Services, GIS (Chatsworth, CA, USA, http://www.genetic-id-services.com) for library construction. The library was enriched for four different di- and tri-nucleotide microsatellite motifs, namely (CA)n, (GA)n, (AAC)n, and (ATG)n. Insert DNA from individual clones was amplified by PCR following GIS guidelines. The PCR products were purified using QIAquick columns (Qiagen) and sequenced using the DYEnamic ET Terminator Cycle Sequencing Kit (Amersham Bioscience) with the universal M13 primers (M13f 5′-aggaaacagctatgaccatg-3′ and M13r 5′-acgacgttgtaaaacgacgg-3′). Clones with inserts less than 200 bp were not considered for sequencing. Sequencing products were resolved on the Applied Biosystems model 377 DNA Sequencer (Applied Biosystems). The resulting sequences were screened for the presence of microsatellite motifs using WebSat [48]. All the clone sequences were checked for redundancy using BLASTn [49].
Microsatellite sequences that shared one or both the 5′/3′ flanking regions in common were discarded to exclude potential multi-locus microsatellites families. The sequences that were found to be unique within the library were subjected to BLASTn analysis against the nr database to identify eventual hits with previously described sequences. Primer pairs were designed for the flanking regions of the identified SSR sequences using Primer 3 [50]. Preliminary PCR screenings were performed on 20 individuals (10 males and 10 females) from the Rimini strain to validate the primer efficiencies in single SSR locus amplifications. DNA amplifications were performed on a Mastercycler gradient (Eppendorf). The PCR reaction mixture consisted of 50 ng genomic DNA, 1x PCR buffer, 1.5 mM MgCl2, 270 µM dNTPs (Invitrogen), 1 U Taq polymerase (Invitrogen), and 10 μM of each primer, one of which was 5′ labelled with a fluorescent dye, in a final volume of 15 μl. PCR cycling conditions were 94°C for 3 min, 30 cycles of 94°C for 30 s, 57-60°C for 30 s, and 72°C for 30 s, followed by a final extension step of 72°C for 10 minutes. Aliquots of PCR products were separated by electrophoresis on 2% agarose gels stained with ethidium bromide, and visualized under UV light. Each PCR product was then diluted 1:10 in ddH2O water and 1 μl of this dilution was added to 9 μl of a mixture of deionized formamide and GeneScan-500 ROX size standard (Applied Biosystems). After denaturation for 4 min at 94°C the fragments were resolved on an ABI PRISM 310 Genetic Analyzer (Applied Biosystems) with the GENESCAN software package (Applied Biosystems). To avoid genotyping errors, the program TANDEM [51] was used to automate binning of the microsatellite allele length variants.
Microsatellite data analysis
The parameters of intra- and inter-population genetic diversity, such as observed allele size, number of actual alleles (na), number of effective alleles (ne), number of private alleles (np) and AMOVA were computed using GenALEx 6.5 [52]. Observed and expected heterozygosity (HO and HE, respectively), gene diversity (HS) and FST values were computed using MICROSATELLITE ANALYSER (MSA) V.4.05 [53]. Linkage disequilibrium, the frequency of null alleles (An) and the deviations from Hardy-Weinberg expectations were computed using GENEPOP version 4.2 [54,55]. For loci with fewer than five alleles, an exact test of Hardy-Weinberg proportions was performed. For loci with five alleles or more, an unbiased estimate of the exact probability was obtained using the Markov chain method of Guo & Thompson [56] for each combination of locus and population. The allelic Polymorphic Information Content (PIC) was derived using CERVUS [57].
Results
ITS2 sequence variability
ITS2 sequences were obtained from an average of three individuals from each of the nine population samples collected in Thailand, Réunion and Italy (Table 2). In addition, to determine whether multiple copies of the ITS2 region varied within individuals, up to four clones were sequenced from each of these individuals. The primers amplified a product of 502–588 bp in length that included part of 5.8S and 28S sequences. The ITS2 sequence itself ranged from 322 to 408 bp in length with an average GC composition of 51%. The 76 ITS2 sequences, obtained from a total of 26 individuals, represent 52 unique sequence-types or haplotypes (Table 2). The gene diversity estimates were very high across the analysed sequences and ranged from 0.893 ± 0.111 in St. Denis (Réunion) and Castellanza (Italy) to 1.000 ± 0.052 in Brescia (Italy). Nucleotide diversity (π), was very low, ranging from 0.005 ± 0.004 in St. Pierre (Réunion) to 0.0191 ± 0.012 in Cesena (Italy). These low nucleotide diversity estimates are expected, as in the 415 bp MUSCLE ITS2 alignment only 44 sites (~10%) contained substitutions.
Table 2.
Features of the internal transcribed spacer 2 (ITS2) sequences in the different Aedes albopictus population samples
| Region | Sample | Individuals analysed | Sequences analysed | Haplotypes present | Gene diversity ± SD | Nucleotide diversity (π ± SD) |
|---|---|---|---|---|---|---|
| Thailand | Ban Rai | 3 | 9 | 7 | 0.917 ± 0.092 | 0.0166 ± 0.0098 |
| Phato | 4 | 13 | 9 | 0.936 ± 0.051 | 0.0128 ± 0.0075 | |
| Réunion | St. Denis | 3 | 8 | 6 | 0.893 ± 0.111 | 0.0129 ± 0.0080 |
| St. Pierre | 3 | 8 | 6 | 0.929 ± 0.084 | 0.0050 ± 0.0035 | |
| Italy | Pavia | 3 | 9 | 7 | 0.944 ± 0.070 | 0.0065 ± 0.0043 |
| Brescia | 3 | 9 | 9 | 1.000 ± 0.052 | 0.0134 ± 0.0081 | |
| Castellanza | 3 | 8 | 6 | 0.893 ± 0.111 | 0.0095 ± 0.0061 | |
| Cesena | 2 | 7 | 6 | 0.952 ± 0.096 | 0.0191 ± 0.0116 | |
| Modena | 2 | 5 | 4 | 0.900 ± 0.161 | 0.0080 ± 0.0058 | |
| Overall | 26 | 76 | 52 | 0.986 ± 0.005 | 0.0134 ± 0.0072 |
Gene diversity: Equivalent to the expected heterozygosity for diploid data. It is defined as the probability that two randomly chosen haplotypes are different in the sample.
Nucleotide diversity: the probability that two randomly chosen homologous nucleotide sites are different.
An estimate of variability distribution in and among the nine tested population samples is reported in Table 3. AMOVA indicated that most of the variation occurs within individuals (74.36%) while only about 8% of total variation is detected among populations. The inter-individual variation within samples accounted for 17.51% (P < 0.05) of the total variance. When the three main geographical population groups (Thailand, Réunion and Italy) are accounted for (Table 4), similar and relatively low percentages of variation were observed among groups (8.12%) and among populations within groups (7.09%), indicating that the genetic similarity is not related to the geographic proximity of populations. Indeed, according to the SAMOVA analysis (Table 5) the best partitioning of genetic diversity was obtained when the nine samples were grouped into seven groups: FCT =0.186, P = 0.011. Here the genetic diversity is not only among geographical regions, as even the two ancestral Thai populations are separated into two groups, while the North-Italian populations are separated into three groups and the two Réunion populations into two groups.
Table 3.
Hierarchical analysis of ITS2 molecular variance (AMOVA) among the 9 Aedes albopictus samples (using Pairwise difference distance method)
| Source of variation | d.f | Sum of squares | Variance components | Percentage of variation | P |
|---|---|---|---|---|---|
| Among populations | 8 | 125.064 | 0.64510 Va | 8.13 | 0.0401 |
| Among individuals within populations | 17 | 166.936 | 1.38968 Vb | 17.51 | 0.0020 |
| Within individuals | 50 | 295.000 | 5.90000 Vc | 74.36 | <0.0001 |
| Total | 75 | 587.000 | 7.93478 |
Table 4.
Hierarchical analysis of ITS2 molecular variance (AMOVA) comparing three geographical population groups: Thailand, Réunion, and Italy (using Pairwise difference distance method)
| Source of variation | d.f. | Sum of squares | Variance components | Percentage of variation | P |
|---|---|---|---|---|---|
| Among groups | 2 | 55.591 | 0.66071 Va | 8.12 | 0.0088 |
| Among populations within groups | 6 | 69.474 | 0.57664 Vb | 7.09 | 0.0010 |
| Within populations | 67 | 461.936 | 6.89456 Vc | 84.78 | <0.0001 |
| Total | 75 | 587.000 | 8.13192 |
Table 5.
Spatial analysis of molecular variance (SAMOVA) for different population partitions
| Number of groups ( K ) | F CT | P | Population partition |
|---|---|---|---|
| 2 | 0.1756 | 0.1124 | (Ban Rai), (Phato, Pavia, Brescia, Castellanza, Cesena, Modena, St. Pierre, St. Denis) |
| 3 | 0.1369 | 0.0274 | (Ban Rai), (Phato, Brescia, Castellanza, Cesena, Modena, St. Pierre, St. Denis), (Pavia) |
| 4 | 0.1406 | 0.0117 | (Ban Rai), (Phato, Brescia, Castellanza, Cesena, Modena, St. Pierre), (St. Denis), (Pavia) |
| 5 | 0.1471 | 0.0117 | (Ban Rai), (Phato, Brescia, Castellanza, Cesena, Modena), (Pavia), (St. Pierre), (St. Denis) |
| 6 | 0.1731 | 0.0010 | (Ban Rai), (Phato, Cesena), (Brescia, Castellanza, Modena), (Pavia), (St. Pierre), (St. Denis) |
| 7* | 0.1861 | 0.0108 | (Ban Rai), (Phato), (Brescia, Castellanza, Modena), (Cesena), (Pavia), (St. Pierre), (St. Denis) |
| 8 | 0.1783 | 0.0655 | (Ban Rai), (Phato), (Brescia, Modena), (Castellanza), (Cesena), (Pavia), (St. Pierre), (St. Denis) |
*Number of groups (K) with the highest F CT value.
ITS2 haplotype phylogeny and population distribution
The phylogenetic analysis based on the 52 ITS2 haplotype sequences identified in the nine geographic populations resulted in a Maximum Likelihood tree with a log likelihood of −1428.3 (Figure 1). The ITS2 sequences form a compact cluster with very few nodes supported by bootstrap values greater than 50%. Highly related sequences are distributed across populations from different geographical regions; some evidence of clustering of tightly related sequences is present in Ban Rai, Phato (Thailand) and to a lesser degree in St. Pierre (Réunion).
Figure 1.
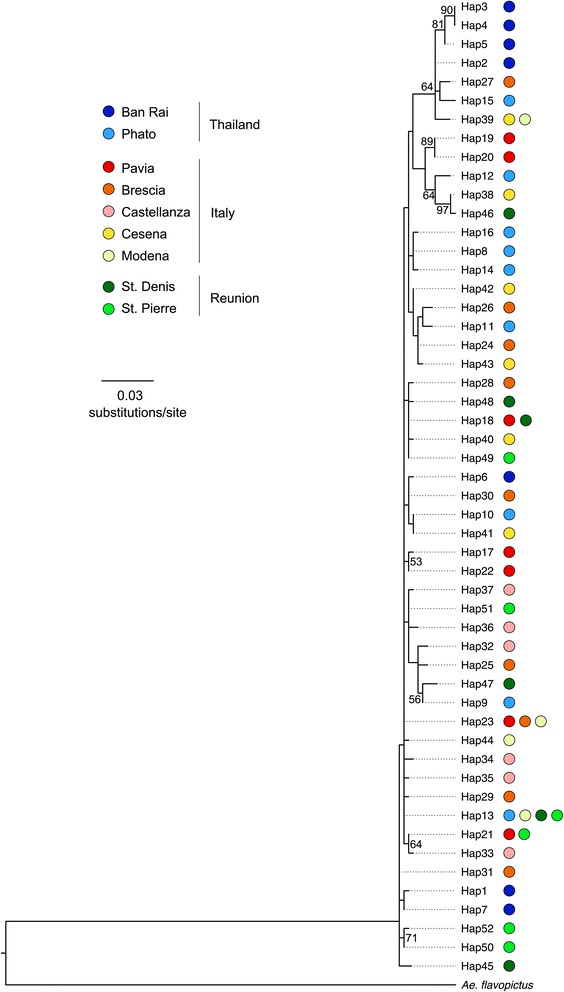
Maximum Likelihood phylogeny of ITS2 haplotypes derived from the wild Aedes albopictus population samples with an Ae. flavopictus sequence [GenBank:AF353559] as outgroup. The Kimura-2-parameter and Γ distribution model of molecular evolution was applied. Bootstrap values (1000 replicates) above 50% are indicated.
The majority (47of 52) of the haplotypes are present in only one population. Only three haplotypes are shared by two populations (haplotype 18 in Pavia and St. Denis, haplotype 21 in Pavia and St. Pierre, and haplotype 39 in Cesena and Modena). Haplotype 23 is shared by three Italian populations (Pavia, Brescia and Modena), and haplotype 13 is present in four populations (Phato, Modena, St. Denis and St. Pierre).
SSR: Isolation and characterization
Out of a total of 109 sequenced clones obtained from the Ae. albopictus library, 92 contained tandem DNA repeats with at least 5 repeat units. BLASTn analyses showed that one or both flanking regions were shared in 42 sequences and they were thus excluded from the subsequent characterization. Within the remaining 50 sequenced clones, five harboured more than one tandem DNA repeat, resulting in the identification of 55 microsatellite motifs. Among these, trinucleotide motifs were the most abundant (28), followed by dinucleotide (22), mononucleotide (3), heptanucleotide (1) and hendecanucleotide motifs (1). Among the trinucleotides motifs, CAA was highly abundant (17) followed by TGA (10). Among the dinucleotide motifs, TG was predominant (14) followed by CT (8). Thirty-seven of these microsatellite motifs contained perfect repeat arrays. Twelve microsatellites contained imperfect repeats and the other six microsatellites contained compound repeats.
Forty-five microsatellite sequences were considered for further validation, as the others lacked sufficient flanking regions to design primers. After the PCR screening on 20 individuals from the Rimini laboratory strain, 23 SSR sequences were validated as loci (Table 6). The nucleotide sequences of these validated SSR loci were submitted to GenBank [Genbank: KP859591-KP859613].
Table 6.
The 23 Aedes albopictus microsatellite loci validated in this study
| Locus | Type 1 | Repeat motif | Primer sequences (5’ to 3’) 2 | T a (°C) 3 |
|---|---|---|---|---|
| Aealbmic1 | P | (TC)10 | F: [6FAM]ACCTTGCGTAGGATGACGAT | 60 |
| R: GCCATGATCACGAGCCTATT | ||||
| Aealbmic2 | I | (GTT)5TGGAGTGAG(GTT)6 | F: [6FAM]ACGATGCGTAACCATTCGAT | 58 |
| R: AACACCGCCGAATATGAAAC | ||||
| Aealbmic3 | P | (AAC)9 | F: [HEX]ACCATACAGCCTGGAGTTCG | 58 |
| R: GGGGTTGTGTGAATTGTCGT | ||||
| Aealbmic4 | P | (CAA)8 | F: [6FAM]ATCGCGGGTTTTCTATTCCT | 58 |
| R: ATCAACGAAACCGAAAGCAT | ||||
| Aealbmic5 | P | (TGT)13 | F: [HEX]AACCCATCGAACACAGAAGG | 58 |
| R: GTACGGTTGACTCGCTGTGA | ||||
| Aealbmic6 | I | (GTT)3GCT(GTT)3 GGT(GTT)4 | F: [HEX]GATGGTCCGTATTTGGGTTG | 57 |
| R: ATCTTCACTCATCCGCCATC | ||||
| Aealbmic7 | I | (TTG)8ATG(TTG)4 | F: [HEX]ATAGACGGGAGTCGGTTCCT | 58 |
| R: TCCAACCGCTAGTGTCATCA | ||||
| Aealbmic8 | P | (GT)8 | F: [HEX]TTGTTGTTCGGTTGTTGTTTG | 57 |
| R: CGGGTTCCAACTATGTACGA | ||||
| Aealbmic9 | P | (GAT)7 | F: [HEX]GCGATGACAGTGGAACAAGA | 58 |
| R: GCTTGGCAGGGAACAAATTA | ||||
| Aealbmic10 | P | (ATC)9 | F: [6FAM]ATCGCCTTCACTCTTCTTCG | 60 |
| R: CCAATCCTGAGCCGTACATT | ||||
| Aealbmic11 | P | (TGT)5 | F: [6FAM]CTCTGCGTTCCGGTTCTATC | 58 |
| R: AGGCAACCTCTCGAATGAAA | ||||
| Aealbmic12 | P | (GAT)7 | F: [6FAM]AGAGCCCTCGAAAAGAGAGC | 58 |
| R: AGCACTCATTCTTGGCTTGG | ||||
| Aealbmic13 | I | (GAT)GAC(GAT)4 | F: [6FAM]TCACACCATGGTCAAAGCAT | 58 |
| R: TGCTGAGTTGAATGGAAACG | ||||
| Aealbmic14 | I | (CT)11 T(CT)2 | F: [6FAM]CAGAAGGTCTTGGATTCACTC | 58 |
| R: TTTCCAATTCGTTTCTGGTTC | ||||
| Aealbmic15 | P | (GTT)7 | F: [6FAM]GGAATGGTTCCCTGGCTAAT | 58 |
| R: CCAACTCCGAAGAAGCCATA | ||||
| Aealbmic16 | P | (CAT)7 | F: [HEX]CACAACAACGAGAGTGTCGAA | 60 |
| R: CCGAGGGCAACACGATATAC | ||||
| Aealbmic17 | P | (CAA)15 | F: TCACCACAACAAACGGAATC | 58 |
| R: AGAGTTTTGGGCGCAGTTTA | ||||
| Aealbmic18 4 | I | (TG)12TA(TG)7 | F: GGGCCGAATCTCATATACCA | 59 |
| R: CTCAAATCGGTTGACAGACG | ||||
| Aealbmic19 | P | (ATC)9 | F: ACATATAGGTAATCCTTGCGCTGAT | 59 |
| R: GTTCCCGTATTTGATATTTGCTTT | ||||
| Aealbmic20 | P | (CA)44 | F: GGAGGCCATCATAATTTCGAC | 58 |
| R: AGTGGACCACAGACAGACCATT | ||||
| Aealbmic21 | P | (ATG)7 | F: CCCTACAGCCCTGATTGAGA | 58 |
| R: CGAGTTGGGATGTGTGATTG | ||||
| Aealbmic22 5 | I | (AC)45AT(AC)2AT(AC)5 | F: AAATCGGTTGAGGTTTACTGC | 59 |
| R: ATTCTTAGCCGGCAACTACG | ||||
| Aealbmic23 | P | (ACA)8 | F: AACGGAGCGGAGTCGATTAT | 58 |
| R: CTACTACCCGCTGCCTTCTG |
1Motif type (P: perfect repeats; I: imperfect repeats).
2The fluorescent label (6FAM or HEX) on the forward primer is indicated for the 16 loci that were subsequently used for the population analysis.
3Annealing temperature (T a).
4,5SSR sequences that correspond to previously identified loci: (alb212) [57] and (AealbD2) [58], respectively.
Evaluation of microsatellite polymorphism in ancestral and adventive populations
On the basis of a preliminary screening performed on the Rimini strain, 16 (Aealbmic1 to Aealbmic16) of the 23 validated SSR loci (Table 6) were used to analyse the degree of polymorphism in 79 mosquitoes from three populations: Ban Rai (Thailand), Brescia and Cesena (Italy) (Table 7). The average Polymorphic Information Content (PIC) estimated across the 16 loci was 0.56 ± 0.29 in Ban Rai, 0.50 ± 0.26 in Cesena and 0.47 ± 0.21 in Brescia, suggesting that these loci are informative for population analyses. Heterozygous individuals were present in both sexes for all 16 loci, excluding any condition of sex linkage. Tests for Hardy-Weinberg equilibrium (HWE), after sequential Bonferroni correction [58], revealed that five loci did not meet the expectations of this model (Table 7): three (Aealbmic4, 11, 13) in the Ban Rai sample, one in Cesena (Aealbmic3) and two in Brescia (Aealbmic13, 16). However, the locus/population combinations that were not in HWE were not concentrated at any locus or in any population. As no evidence of linkage disequilibrium between loci was assessed, these 16 loci can be considered to be independent. In the three populations most of the loci display relatively high levels of gene diversity (Hs). The frequency of null alleles across the loci was generally relatively low apart from Aealbmic15 with an estimate of 0.98 in Cesena. Two alleles were detected in Cesena at the Aealbmic15 locus, but one was very rare. In the other populations this locus was monomorphic for the common allele. Aealbmic1 is monomorphic in Ban Rai and Cesena, while in Brescia two alleles were present. Across the loci a higher level of polymorphism was observed in Ban Rai, with a mean number of alleles per locus of 6.19 (Table 7). Private alleles (np) were detected in all populations, with the highest average value in the Ban Rai sample (2.25). Analysis of molecular variance (AMOVA) across the three populations indicates that the greatest portion of variance, 76.62%, is found within individuals, while 16.80% is among individuals and only the remaining 6.57% is among populations. The FST values between each pair of populations are statistically significant and indicate that the degree of differentiation between the two Northern Italian samples, Cesena and Brescia (FST = 0.061), is not very different from those between these two Italian samples and the Ban Rai sample (FST = 0.072 in both comparisons).
Table 7.
Microsatellite variation in three Aedes albopictus populations
| Locus | Ban Rai (n = 24) | Cesena (n = 31) | Brescia (n = 24) | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n a | n e | n p | H O | H E | Hs | F IS | PIC | A n | n a | n e | n p | H O | H E | Hs | F IS | PIC | A n | n a | n e | n p | H O | H E | Hs | F IS | PIC | A n | |
| Aealbmic1 | 1 | 1.00 | 0 | 0.00 | 0.00 | 0.00 | - | 0.00 | - | 1 | 1.00 | 0 | 0.00 | 0.00 | 0.00 | - | 0.00 | - | 2 | 1.28 | 1 | 0.25 | 0.22 | 0.22 | −0.14 | 0.20 | 0.00 |
| Aealbmic2 | 4 | 2.44 | 1 | 0.50 | 0.59 | 0.60 | 0.15 | 0.50 | 0.03 | 4 | 1.90 | 1 | 0.45 | 0.47 | 0.48 | 0.04 | 0.40 | 0.03 | 2 | 1.99 | 0 | 0.58 | 0.50 | 0.51 | −0.16 | 0.37 | 0.00 |
| Aealbmic3 | 13 | 8.35 | 7 | 0.75 | 0.88 | 0.90 | 0.15 | 0.87 | 0.09 | 8 | 4.49 | 1 | 0.45 | 0.78* | 0.80 | 0.42 | 0.76 | 0.21 | 4 | 2.53 | 0 | 0.70 | 0.60 | 0.62 | −0.17 | 0.55 | 0.00 |
| Aealbmic4 | 6 | 3.54 | 2 | 0.42 | 0.72* | 0.74 | 0.42 | 0.68 | 0.30 | 5 | 4.21 | 0 | 0.71 | 0.76 | 0.78 | 0.07 | 0.72 | 0.19 | 4 | 2.81 | 0 | 0.58 | 0.64 | 0.66 | 0.09 | 0.59 | 0.18 |
| Aealbmic5 | 6 | 3.89 | 3 | 0.50 | 0.74 | 0.76 | 0.32 | 0.70 | 0.14 | 8 | 2.27 | 2 | 0.39 | 0.56 | 0.57 | 0.30 | 0.54 | 0.13 | 6 | 3.59 | 3 | 0.44 | 0.72 | 0.74 | 0.39 | 0.67 | 0.22 |
| Aealbmic6 | 7 | 2.16 | 1 | 0.55 | 0.54 | 0.55 | −0.02 | 0.51 | 0.00 | 4 | 2.75 | 0 | 0.52 | 0.64 | 0.65 | 0.19 | 0.58 | 0.12 | 4 | 2.97 | 0 | 0.58 | 0.66 | 0.68 | 0.12 | 0.60 | 0.16 |
| Aealbmic7 | 6 | 3.87 | 2 | 0.79 | 0.74 | 0.76 | −0.07 | 0.70 | 0.00 | 5 | 2.44 | 2 | 0.73 | 0.59 | 0.60 | −0.24 | 0.53 | 0.00 | 4 | 2.77 | 2 | 0.63 | 0.64 | 0.65 | 0.02 | 0.57 | 0.00 |
| Aealbmic8 | 8 | 5.10 | 2 | 0.83 | 0.80 | 0.82 | −0.04 | 0.78 | 0.02 | 7 | 3.94 | 1 | 0.87 | 0.75 | 0.76 | −0.16 | 0.71 | 0.00 | 5 | 3.89 | 0 | 0.79 | 0.74 | 0.76 | −0.07 | 0.70 | 0.03 |
| Aealbmic9 | 10 | 4.52 | 6 | 0.67 | 0.78 | 0.80 | 0.14 | 0.76 | 0.10 | 8 | 4.73 | 0 | 0.81 | 0.79 | 0.80 | −0.03 | 0.76 | 0.11 | 3 | 2.11 | 0 | 0.54 | 0.53 | 0.54 | −0.02 | 0.42 | 0.00 |
| Aealbmic10 | 4 | 2.85 | 1 | 0.50 | 0.65 | 0.67 | 0.23 | 0.58 | 0.11 | 4 | 2.26 | 1 | 0.70 | 0.56 | 0.56 | −0.25 | 0.46 | 0.00 | 3 | 1.97 | 0 | 0.44 | 0.49 | 0.51 | 0.10 | 0.39 | 0.04 |
| Aealbmic11 | 9 | 4.18 | 4 | 0.39 | 0.76* | 0.79 | 0.49 | 0.73 | 0.23 | 6 | 3.11 | 0 | 0.58 | 0.68 | 0.69 | 0.15 | 0.63 | 0.06 | 4 | 2.58 | 0 | 0.41 | 0.61 | 0.63 | 0.33 | 0.54 | 0.15 |
| Aealbmic12 | 7 | 4.65 | 2 | 0.55 | 0.79 | 0.81 | 0.30 | 0.75 | 0.18 | 6 | 4.51 | 0 | 0.61 | 0.78 | 0.79 | 0.22 | 0.74 | 0.09 | 6 | 4.23 | 0 | 0.65 | 0.76 | 0.78 | 0.14 | 0.73 | 0.07 |
| Aealbmic13 | 8 | 3.95 | 3 | 0.46 | 0.75* | 0.77 | 0.39 | 0.72 | 0.17 | 8 | 2.74 | 2 | 0.55 | 0.63 | 0.65 | 0.13 | 0.60 | 0.10 | 4 | 2.31 | 0 | 0.21 | 0.57* | 0.59 | 0.63 | 0.52 | 0.24 |
| Aealbmic14 | 2 | 1.04 | 1 | 0.04 | 0.04 | 0.04 | 0.00 | 0.04 | 0.00 | 1 | 1.00 | 0 | 0.00 | 0.00 | 0.00 | - | 0.00 | - | 3 | 1.20 | 2 | 0.09 | 0.17 | 0.17 | 0.47 | 0.16 | 0.20 |
| Aealbmic15 | 1 | 1.00 | 0 | 0.00 | 0.00 | 0.00 | - | 0.00 | - | 2 | 1.07 | 1 | 0.00 | 0.06 | 0.07 | 1.00 | 0.06 | 0.98 | 1 | 1.00 | 0 | 0.00 | 0.00 | 0.00 | - | 0.00 | - |
| Aealbmic16 | 7 | 3.60 | 1 | 0.70 | 0.72 | 0.74 | 0.03 | 0.68 | 0.00 | 6 | 2.17 | 2 | 0.61 | 0.54 | 0.55 | −0.13 | 0.49 | 0.00 | 5 | 2.54 | 1 | 0.33 | 0.61* | 0.63 | 0.46 | 0.56 | 0.16 |
| Mean | 6.19 | 3.51 | 2.25 | 0.48 | 0.59 | 0.61 | 0.18 | 0.56 | 5.19 | 2.79 | 0.81 | 0.50 | 0.54 | 0.55 | 0.12 | 0.50 | 3.75 | 2.49 | 0.56 | 0.45 | 0.53 | 0.54 | 0.15 | 0.47 | |||
| SD | 3.25 | 1.85 | 1.98 | 0.27 | 0.30 | 0.31 | 0.29 | 2.40 | 1.27 | 0.83 | 0.28 | 0.27 | 0.28 | 0.26 | 1.39 | 0.92 | 0.96 | 0.22 | 0.22 | 0.22 | 0.21 | ||||||
Number of individuals (n), number of alleles (na), number of effective alleles (ne), number of private alleles (np), observed (HO), expected (HE) heterozygosities, gene diversity (HS), inbreeding index (FIS), Polymorphic Information Content (PIC) and null allele frequencies (An) per locus are reported. The asterisks indicate significant departures from Hardy-Weinberg equilibrium after Bonferroni correction at P < 0.05.
Discussion
In the absence of a fully annotated genome, molecular markers for Ae. albopictus are crucially needed to track the rapid spread of this mosquito that is causing increasing concern due to its status as a vector of several arboviruses of public health importance [3,9]. The two types of genetic markers here implemented give a picture of the distribution of genetic diversity within and between populations providing clues on the dispersion dynamics of this species. ITS2 sequence variation has been analysed in mosquitoes from the supposed native range, Thailand, from an old colonised area, Réunion, and from a newly invaded area, Italy. It appears that substantial variability, both in terms of haplotype number and gene diversity, is present in the native as well in the two areas of introduction. The haplotype variability is mostly concentrated within individuals (74.36%) or within populations (84.78%), while only a relatively small proportion into geographical differences, both at single population (8.13%) and at geographic area level (8.12%). This implies that, according to SAMOVA, the haplotype variation, besides being high, has a distribution that is independent from geography. Different haplotypes are present in each individual of the nine samples, and the tree clearly demonstrates that they are similar and dispersed across native and adventive populations (Figure 1). Substantially concordant conclusions can be obtained with the newly developed SSR markers, which display a high level of polymorphism both in the ancestral (Ban Rai, Thailand) and adventive populations (North Italian samples), previously scored for ITS2 variability. Although the chromosomal location of these markers remains unknown, the assessed linkage equilibrium between them suggests that they are statistically independent and their variability patterns might reflect genome wide patterns across populations. Based on the population samples here analysed, the SSR markers were, as expected, more informative than ITS2 in revealing the slight genetic diversity between native and derived populations both in terms of variability and differentiation. About 7% of total variability (AMOVA) is represented by the differences between the three populations, while the highest variability was found within populations. This is an agreement with a previous study on mosquitoes collected in Réunion [59]. The FST estimates indicate that the differentiation between the two Italian samples is slightly smaller (FST = 0.061) than that between the Italian and Thai samples (FST = 0.072), suggesting the absence of great differentiation between ancestral and derived populations. The ancestral nature of the Thai population is also corroborated by its high level of variability.
Taken together these genetic data raise questions on the way this mosquito is spreading and becoming established worldwide. Clearly, our data indicate that the diffusion of this mosquito does not conform to a progressive expansion away from the native Asian source area, but supports a relatively recent and chaotic propagule distribution mediated by human activities. Under this scenario, multiple introductions and admixture events probably play an important role in maintaining the genetic diversity and in avoiding bottleneck effects.
Conclusions
In this study we have exploited nuclear molecular markers that have allowed us to highlight the presence of high intraspecific variability and a low/moderate level of differentiation of Ae. albopictus populations collected in different eco-geographic areas. Most of the genetic variation was detected at the individual level and this contributed to the high genetic variation observed within populations. This genetic feature, also revealed using other markers [7,34,36,59] may have facilitated the characteristic ability of this invasive species to expand and adapt to novel environments.
The highly polymorphic SSR markers here implemented, together with the set previously described [36,59,60], represent an important tool for reconstructing the routes of invasion and for the identification of the origins of mosquito outbreaks.
Acknowledgements
This research is part of the EU-FP7 Research Infrastructures project (INFRAVEC) Grant N° 228421 and was partially supported by the Italian Ministry of Health (RF-2010-2318965) and the Fondazione Banca del Monte di Lombardia (Pavia, Italy). The study was performed within the FAO/IAEA research CRP programme “Exploring Genetic, Molecular, Mechanical and Behavioural Methods of Sex Separation in Mosquitoes (FS)”. Sincere thanks go to Dr. Romeo Bellini (Centro Agricoltura Ambiente “G. Nicoli”, Crevalcore, Italy), for providing Aedes albopictus specimens.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ARM, GG, CRG, MM and LMG contributed to the study design. PS, GT, LMG and FS contributed to field mosquito egg collection. MM, LMG, NA, FS and GT performed the experimental work. MM, LMG, CRG and ARM analysed the data. ARM, CRG, MM, LMG and GG wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Mosè Manni, Email: mose.manni01@universitadipavia.it.
Ludvik M Gomulski, Email: gomulski@unipv.it.
Nidchaya Aketarawong, Email: na_beau@hotmail.com.
Gabriella Tait, Email: gabriella.tait01@universitadipavia.it.
Francesca Scolari, Email: francesca.scolari@unipv.it.
Pradya Somboon, Email: psomboon@med.cmu.ac.th.
Carmela R Guglielmino, Email: c.r.guglielmino@gmail.com.
Anna R Malacrida, Email: malacrid@unipv.it.
Giuliano Gasperi, Email: gasperi@unipv.it.
References
- 1.Gratz NG. Critical review of the vector status of Aedes albopictus. Med Vet Entomol. 2004;18(3):215–27. doi: 10.1111/j.0269-283X.2004.00513.x. [DOI] [PubMed] [Google Scholar]
- 2.Paupy C, Delatte H, Bagny L, Corbel V, Fontenille D. Aedes albopictus, an arbovirus vector: from the darkness to the light. Microbes Infect Inst Pasteur. 2009;11(14–15):1177–85. doi: 10.1016/j.micinf.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Lambrechts L, Scott TW, Gubler DJ. Consequences of the expanding global distribution of Aedes albopictus for dengue virus transmission. PLoS Negl Trop Dis. 2010;4(5):e646. doi: 10.1371/journal.pntd.0000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Medlock JM, Hansford KM, Schaffner F, Versteirt V, Hendrickx G, Zeller H, et al. A Review of the invasive mosquitoes in Europe: ecology, public health risks, and control options. Vector Borne Zoonotic Dis. 2012;12(6):435–47. doi: 10.1089/vbz.2011.0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enserink M. A mosquito goes global. Science. 2008;320(5878):864–6. doi: 10.1126/science.320.5878.864. [DOI] [PubMed] [Google Scholar]
- 6.Erickson RA, Hayhoe K, Presley SM, Allen LJS, Long KR, Cox SB. Potential impacts of climate change on the ecology of dengue and its mosquito vector the Asian tiger mosquito (Aedes albopictus) Environ Res Lett. 2012;7:034003. doi: 10.1088/1748-9326/7/3/034003. [DOI] [Google Scholar]
- 7.Medley KA, Jenkins DG, Hoffman EA. Human-aided and natural dispersal drive gene flow across the range of an invasive mosquito. Mol Ecol. 2015;24(2):284–95. doi: 10.1111/mec.12925. [DOI] [PubMed] [Google Scholar]
- 8.Tatem AJ, Hay SI, Rogers DJ. Global traffic and disease vector dispersal. Proc Natl Acad Sci. 2006;103(16):6242–7. doi: 10.1073/pnas.0508391103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benedict MQ, Levine RS, Hawley WA, Lounibos LP. Spread of the tiger: global risk of invasion by the mosquito Aedes albopictus. Vector Borne Zoonotic Dis. 2007;7(1):76–85. doi: 10.1089/vbz.2006.0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gasperi G, Bellini R, Malacrida AR, Crisanti A, Dottori M, Aksoy S. A new threat looming over the Mediterranean basin: emergence of viral diseases transmitted by Aedes albopictus mosquitoes. PLoS Negl Trop Dis. 2012;6(9):e1836. doi: 10.1371/journal.pntd.0001836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaffner F, Medlock JM, Van Bortel W. Public health significance of invasive mosquitoes in Europe. Clin Microbiol Infect. 2013;19(8):685–92. doi: 10.1111/1469-0691.12189. [DOI] [PubMed] [Google Scholar]
- 12.Delatte H, Desvars A, Bouétard A, Bord S, Gimonneau G, Vourc’h G, et al. Blood-feeding behavior of Aedes albopictus, a vector of Chikungunya on La Réunion. Vector Borne Zoonotic Dis. 2010;10(3):249–58. doi: 10.1089/vbz.2009.0026. [DOI] [PubMed] [Google Scholar]
- 13.Poelchau MF, Reynolds JA, Elsik CG, Denlinger DL, Armbruster PA. Deep sequencing reveals complex mechanisms of diapause preparation in the invasive mosquito, Aedes albopictus. Proc Biol Sci. 2013;280(1759):20130143. doi: 10.1098/rspb.2013.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lourenço de Oliveira R, Vazeille M, de Filippis AM, Failloux AB. Large genetic differentiation and low variation in vector competence for dengue and yellow fever viruses of Aedes albopictus from Brazil, the United States, and the Cayman Islands. Am J Trop Med Hyg. 2003;69(1):105–14. [PubMed] [Google Scholar]
- 15.Tantely ML, Tortosa P, Alout H, Berticat C, Berthomieu A, Rutee A, et al. Insecticide resistance in Culex pipiens quinquefasciatus and Aedes albopictus mosquitoes from La Réunion Island. Insect Biochem Mol Biol. 2010;40(4):317–24. doi: 10.1016/j.ibmb.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Kawada H, Maekawa Y, Abe M, Ohashi K, Ohba S, Takagi M. Spatial distribution and pyrethroid susceptibility of mosquito larvae collected from catch basins in parks in Nagasaki city, Nagasaki, Japan. Jpn J Infect Dis. 2010;63(1):19–24. [PubMed] [Google Scholar]
- 17.Bonizzoni M, Gasperi G, Chen X, James AA. The invasive mosquito species Aedes albopictus: current knowledge and future perspectives. Trends Parasitol. 2013;29(9):460–8. doi: 10.1016/j.pt.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao PN, Rai K. Inter and intraspecific variation in nuclear DNA content in Aedes mosquitoes. Heredity. 1987;59(2):253–8. doi: 10.1038/hdy.1987.120. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Rai KS. Intraspecific variation in nuclear DNA content among world populations of a mosquito, Aedes albopictus (Skuse) Theor Appl Genet. 1990;79(6):748–52. doi: 10.1007/BF00224239. [DOI] [PubMed] [Google Scholar]
- 20.McLain DK, Rai KS, Fraser MJ. Intraspecific and interspecific variation in the sequence and abundance of highly repeated DNA among mosquitoes of the Aedes albopictus subgroup. Heredity. 1987;58(3):373–81. doi: 10.1038/hdy.1987.65. [DOI] [PubMed] [Google Scholar]
- 21.Black WC, Rai KS. Genome evolution in mosquitoes: intraspecific and interspecific variation in repetitive DNA amounts and organization. Genet Res. 1988;51(3):185–96. doi: 10.1017/S0016672300024289. [DOI] [PubMed] [Google Scholar]
- 22.Black WC, McLain DK, Rai KS. Patterns of variation in the rDNA cistron within and among World populations of a mosquito, Aedes albopictus (Skuse) Genetics. 1989;121(3):539–50. doi: 10.1093/genetics/121.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomulski LM, Meiswinkel R, Delécolle J-C, Goffredo M, Gasperi G. Phylogeny of the subgenus Culicoides and related species in Italy, inferred from internal transcribed spacer 2 ribosomal DNA sequences. Med Vet Entomol. 2006;20(2):229–38. doi: 10.1111/j.1365-2915.2006.00620.x. [DOI] [PubMed] [Google Scholar]
- 24.Young I, Coleman AW. The advantages of the ITS2 region of the nuclear rDNA cistron for analysis of phylogenetic relationships of insects: a Drosophila example. Mol Phylogenet Evol. 2004;30(1):236–42. doi: 10.1016/S1055-7903(03)00178-7. [DOI] [PubMed] [Google Scholar]
- 25.Zomuanpuii R, Ringngheti L, Brindha S, Gurusubramanian G, Senthil KN. ITS2 characterization and Anopheles species identification of the subgenus Cellia. Acta Trop. 2013;125(3):309–19. doi: 10.1016/j.actatropica.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Wörheide G, Hooper JN, Degnan BM. Phylogeography of western Pacific Leucetta “chagosensis” (Porifera: Calcarea) from ribosomal DNA sequences: implications for population history and conservation of the Great Barrier Reef World Heritage Area (Australia) Mol Ecol. 2002;11(9):1753–68. doi: 10.1046/j.1365-294X.2002.01570.x. [DOI] [PubMed] [Google Scholar]
- 27.Behbahani A, Dutton TJ, Davies N, Townson H, Sinkins SP. Population differentiation and Wolbachia phylogeny in mosquitoes of the Aedes scutellaris group. Med Vet Entomol. 2005;19(1):66–71. doi: 10.1111/j.0269-283X.2005.00542.x. [DOI] [PubMed] [Google Scholar]
- 28.Giessler S, Wolinska J. Capturing the population structure of microparasites: using ITS-sequence data and a pooled DNA approach. Mol Ecol Resour. 2013;13(5):918–28. doi: 10.1111/1755-0998.12144. [DOI] [PubMed] [Google Scholar]
- 29.Müller T, Philippi N, Dandekar T, Schultz J, Wolf M. Distinguishing species. RNA. 2007;13(9):1469–72. doi: 10.1261/rna.617107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coleman AW. Is there a molecular key to the level of “biological species” in eukaryotes? A DNA guide. Mol Phylogenet Evol. 2009;50(1):197–203. doi: 10.1016/j.ympev.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 31.Birungi J, Munstermann LE. Genetic structure of Aedes albopictus (Diptera: Culicidae) populations based on mitochondrial ND5 sequences: evidence for an independent invasion into Brazil and United States. Ann Entomol Soc Am. 2002;95(1):125–32. doi: 10.1603/0013-8746(2002)095[0125:GSOAAD]2.0.CO;2. [DOI] [Google Scholar]
- 32.Mousson L, Dauga C, Garrigues T, Schaffner F, Vazeille M, Failloux AB. Phylogeography of Aedes (Stegomyia) aegypti (L.) and Aedes (Stegomyia) albopictus (Skuse) (Diptera: Culicidae) based on mitochondrial DNA variations. Genet Res. 2005;86(1):1–11. doi: 10.1017/S0016672305007627. [DOI] [PubMed] [Google Scholar]
- 33.Maia RT, Scarpassa VM, Maciel-Litaiff LH, Tadei WP. Reduced levels of genetic variation in Aedes albopictus (Diptera: Culicidae) from Manaus, Amazonas State, Brazil, based on analysis of the mitochondrial DNA ND5 gene. Genet Mol Res. 2009;8(3):998–1007. doi: 10.4238/vol8-3gmr624. [DOI] [PubMed] [Google Scholar]
- 34.Ashfaq M, Hebert PD, Mirza JH, Khan AM, Zafar Y, Mirza MS. Analyzing mosquito (Diptera: culicidae) diversity in Pakistan by DNA barcoding. PLoS One. 2014;9(5):e97268. doi: 10.1371/journal.pone.0097268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamgang B, Brengues C, Fontenille D, Njiokou F, Simard F, Paupy C. Genetic structure of the tiger mosquito, Aedes albopictus, in Cameroon (Central Africa) PLoS One. 2011;6(5):e20257. doi: 10.1371/journal.pone.0020257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beebe NW, Ambrose L, Hill LA, Davis JB, Hapgood G, Cooper RD, et al. Tracing the tiger: population genetics provides valuable insights into the Aedes (Stegomyia) albopictus invasion of the Australasian Region. PLoS Negl Trop Dis. 2013;7(8):e2361. doi: 10.1371/journal.pntd.0002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minard G, Tran FH, Dubost A, Tran-Van V, Mavingui P, Moro CV. Pyrosequencing 16S rRNA genes of bacteria associated with wild tiger mosquito Aedes albopictus: a pilot study. Front Cell Infect Microbiol. 2014;4:59. doi: 10.3389/fcimb.2014.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graham RI, Wilson K. Male-killing Wolbachia and mitochondrial selective sweep in a migratory African insect. BMC Evol Biol. 2012;12:204. doi: 10.1186/1471-2148-12-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mattingly PF. The sub-genus Stegomyia (Diptera: Culicidae) in the Ethiopian Region II. Distribution of species confined to the East and South African sub-region. Bull Br Mus (Natural History) Entomol. 1953;3(1):1–65. [Google Scholar]
- 40.Sabatini A, Raineri V, Trovato G, Coluzzi M. Aedes albopictus in Italy and possible diffusion of the species into the Mediterranean area. Parassitologia. 1990;32(3):301–4. [PubMed] [Google Scholar]
- 41.Rueda LM. Pictorial keys for the identification of mosquitoes (Diptera: Culicidae) associated with Dengue virus transmission. Zootaxa. 2004;589:1–60. [Google Scholar]
- 42.Baruffi L, Damiani G, Guglielmino CR, Bandi C, Malacrida AR, Gasperi G. Polymorphism within and between populations of Ceratitis capitata: comparison between RAPD and multilocus enzyme electrophoresis data. Heredity. 1995;74(4):425–37. doi: 10.1038/hdy.1995.60. [DOI] [PubMed] [Google Scholar]
- 43.De Jong L, Moreau X, Dalia J, Coustau C, Thiéry A. Molecular characterization of the invasive Asian tiger mosquito, Aedes (Stegomyia) albopictus (Diptera: Culicidae) in Corsica. Acta Trop. 2009;112(3):266–9. doi: 10.1016/j.actatropica.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 44.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–7. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 46.Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11(12):2571–81. doi: 10.1046/j.1365-294X.2002.01650.x. [DOI] [PubMed] [Google Scholar]
- 47.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martins WS, Lucas DC, Neves KF, Bertioli DJ. WebSat - A web software for microsatellite marker development. Bioinformation. 2009;3(6):282–3. doi: 10.6026/97320630003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 51.Matschiner M, Salzburger W. TANDEM: integrating automated allele binning into genetics and genomics workflows. Bioinformatics. 2009;25(15):1982–3. doi: 10.1093/bioinformatics/btp303. [DOI] [PubMed] [Google Scholar]
- 52.Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research - an update. Bioinformatics. 2012;28(19):2537–9. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dieringer D, Schlötterer C. Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes. 2003;3(1):167–9. doi: 10.1046/j.1471-8286.2003.00351.x. [DOI] [Google Scholar]
- 54.Rousset F. genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8(1):103–6. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 55.Raymond M, Rousset F. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J Hered. 1995;86(3):248–9. [Google Scholar]
- 56.Guo SW, Thompson EA. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48(2):361–72. doi: 10.2307/2532296. [DOI] [PubMed] [Google Scholar]
- 57.Kalinowski ST, Taper ML, Marshall TC. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol. 2007;16(5):1099–106. doi: 10.1111/j.1365-294X.2007.03089.x. [DOI] [PubMed] [Google Scholar]
- 58.Rice WR. Analyzing Tables of Statistical Tests. Evolution. 1989;43(1):223–5. doi: 10.2307/2409177. [DOI] [PubMed] [Google Scholar]
- 59.Delatte H, Toty C, Boyer S, Bouetard A, Bastien F, Fontenille D. Evidence of habitat structuring Aedes albopictus populations in Réunion Island. PLoS Negl Trop Dis. 2013;7(3):e2111. doi: 10.1371/journal.pntd.0002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Porretta D, Gargani M, Bellini R, Calvitti M, Urbanelli S. Isolation of microsatellite markers in the tiger mosquito Aedes albopictus (Skuse) Mol Ecol Notes. 2006;6(3):880–1. doi: 10.1111/j.1471-8286.2006.01384.x. [DOI] [Google Scholar]