

Abstract
Prunus mongolica Maxim, which is widely established in the Gobi Desert, shows extreme tolerance to drought. However, there is a lack of available transcriptomic resources for this species related to its response to water deficiency. To investigate the mechanisms that allow P. mongolica to maintain growth in extremely arid environments, the response of P. mongolica seedlings to drought stress was analyzed using morphological, physiological, biochemical and high-throughput sequencing approaches. We generated 28,713,735 and 26,650,133 raw reads from no-stress control and drought-stressed P. mongolica seedlings, respectively. In total, we obtained 67,352 transcripts with an average length of 874.44 bp. Compared with the no-stress control, 3,365 transcripts were differentially expressed in the drought-stressed seedlings, including 55.75% (1,876 transcripts) up-regulated and 44.25% (1,489 transcripts) down-regulated transcripts. The photosynthesis response showed a decreasing tendency under drought stress, but the changes in the levels of hormones (auxins, cytokinins and abscisic acid) resulted in the closing of stomata and decreased cell enlargement and division; these changes were effective for promoting P. mongolica survival in Gobi Desert. Next, we analyzed the aquaporin and superoxide dismutase gene families due to their importance in plant resistance to drought stress. We found that all of the plasma membrane intrinsic protein transcripts were down-regulated in the drought-stressed treatment, whereas drought did not affect the expression of nodulin intrinsic protein or small basic intrinsic protein transcripts in P. mongolica seedlings. In addition, activation of iron superoxide dismutase transcription and enhanced transcription of manganese superoxide dismutase were observed in P. mongolica to promote tolerance of drought stress. This study identified drought response genes in P. mongolica seedlings. Our results provide a significant contribution to the understanding of how P. mongolica responds to drought stress at the transcriptome level, which may help to elucidate molecular mechanisms associated with the drought response of almond plants.
Introduction
Most terrestrial ecosystems in nature are rain fed, and drought poses a major limitation to productivity in these ecosystems. Recurrent dry periods and scattered rainfall patterns have resulted in water shortages and the consequent loss of or damage to crop production in sufficiently rain-fed areas [1]. In addition, increased atmospheric CO2 concentrations may increase in the severity of drought conditions of some arid and semi-arid regions [2]. Atmospheric CO2 concentrations rose from 280 to 368 ppm during the 20th century and may rise to > 700 ppm by the end of the 21st century (IPCC, 2007). Elevated CO2 concentration leads to the increase of global mean temperature, reduces the rainfall and increases the evaporation especially in arid and semi-arid regions [3,4]. According to a recent estimation, the economic losses caused by drought may be as high as $80 billion per year worldwide, which may be due not only to the lack of water causing a decreased yield potential but also to the timing and duration of drought stress events in relation to plant phenological processes [5]. Droughts are expected to continue becoming longer and more severe in some regions. However, high relative yields of certain genotypes have been documented following exposure to drought stress [6]. Thus, it is important to reveal the unique molecular and biochemical mechanisms associated with drought tolerance in some extreme plants, such as desert plants.
Mongolian almond (Prunus mongolica Maxim, Fig 1) is distributed widely in the Gobi Desert of the Mongolian plateau. This species is an ancient relict flora that has been recorded as a rare plant on the China Plant Red List and adopted as a state key conservation species [7]. While P. mongolica grows well in water- and nutrient-limited desert areas and plays an important role in the local economy and ecological environment, no studies have been reported investigating the mechanisms (particularly the molecular mechanisms) underlying the drought tolerance of this shrub.
Fig 1. P. mongolica seedling, before (a) and after (b) the drought-stressed treatment.

The drought stress response in plants is followed by morphological and biochemical changes, such as a deeper root system, closure of stomata, shedding of older leaves [8], strengthening of reactive oxygen species (ROS) scavenging mechanisms and increases in the accumulation of osmoregulation substances [9]. These changes are effective for allowing survival under drought conditions. The mechanisms underlying the response to drought stress can be measured at many different levels, from the whole plant to the molecular level. Because stress responses are controlled by the plant genome, recent efforts have focused on the molecular response of the plants to drought stress [10]. The development of next-generation, high-throughput sequencing technologies has provided a significant and low-cost way to generate genome/transcriptome resources for non-model organisms at large scales [11]. Transcriptome research conducted in Agave deserti [12], Boehmeria nivea [13], Chenopodium quinoa [14], Manihot esculenta [8], Paulownia australis [15], Populus euphratica [16] and Pseudotsuga menziesii [17] has been used to successfully identify differentially expressed genes, implicating several biosynthetic pathways that assist in the overall tolerance to drought stress. For Prunus plants, Alimohammadi et al (2013) [18] identified the water-deficit resistance genes in wild almond (P. scoparia) using cDNA-AFLP technology, the drought stress-responsive miRNAs in peach (P. persica) [19] and the freezing stress-responsive genes in P. dulcis [20] were analyzed by the high-throughput sequencing technology.
In this study, we characterized the seedling transcriptome of P. mongolica during the response to drought stress. A core set of stress-related transcripts were determined, together with morphological changes (growth status), physiological process and biochemical functions, to provide a comprehensive analysis of drought acclimation in this species. The acquired information may provide new insights into the molecular mechanisms underlying P. mongolica’s response to drought stress.
Materials and Methods
Seedling cultivation and water stress treatments
The original seeds used in this study were collected from the wild P. mongolica which distributed in the Tengger Desert, Inner Mongolia, Northwest China (103°20′ E, 39°14′ N) on August 10, 2012. After their endocarp being removed by physical damage, the seeds were surface-sterilized with 10% H2O2 for 30 min and washed with sterilized water. Then they were germinated at 27°C in Petri dishes (diameter = 9 cm). The germinated seeds were transplanted into plastic pots (1 seedling per pot, cylindrical pots: 12×8×15 cm) containing 550 g of sterilized vermiculite (121°C, 90 min) and watered weekly with 150 mL of Hoagland’s solution for 45 days before being subjected to stress treatment in an experimental greenhouse under controlled environmental conditions (irradiation: 16.8 klx; day/night cycle: 14/10 h; temperature: 25/20°C).
In total, 72 uniform 45-day-old P. mongolica seedlings, including 36 controls and 36 water-deprived plants, were used to determine all parameters nondestructively throughout the experiment. The controls (WW) were watered daily to field capacity, with a 48.7% soil volumetric water content being recorded (measured using a Field Scout TDR 300 Soil Moisture Meter, Spectrum Technologies, USA). The drought stress treatment (DS) was implemented to simulate a gradual soil drying process, which was similar to a natural drought event. This treatment began on March 10, 2013 and was extended for 15 days without irrigation. The soil volumetric water content was 6.3% after drought stress.
All of the P. mongolica seedlings were harvested after being cultured in the greenhouse for a total of 60 days. Twelve seedlings from each treatment were thoroughly washed with sterile water. Rapid freezing was performed by submerging the samples in liquid nitrogen, followed by storage at -80°C until RNA extraction and high-throughput sequencing. The other 24 seedlings from each treatment were collected to assess growth and physiological and biochemical parameters as described below.
Growth, physiological and biochemical measurements
To evaluate the growth status of P. mongolica seedlings under drought conditions, growth-related parameters were determined in 24 seedlings from each treatment at the initial stage of water stress and at the final harvesting time, including the leaf number, length and width. After drought stress, the contents of auxin, abscisic acid and cytokinins were quantified via enzyme-linked immunosorbent assay (ELISA) [21] according to the manufacturer’s protocol, and total superoxide dismutase (SOD) (EC 1.15.1.1) activity was assessed according to the method described by Alvarez et al. (2009) [22].
All physiological measurements were performed daily at 11:00 during the process of drought stress. The net photosynthetic rate (Pn), stomatal conductance (Gs) and leaf water potential (LWP) were determined to identify the physiological adjustment of P. mongolica to the water stress treatment. Gs and Pn were measured using a LICOR-6400 portable photosynthesis system (LI-COR Inc., USA). The LWP (ψleaf) was measured on the abaxial leaf surface in intact plants using a PSYPRO Water Potential System (WESCOR Inc., USA).
All data obtained from the growth, physiological and biochemical measurements were subjected to one-way ANOVA at each harvesting time using SAS software (SAS Institute Inc., USA). The different treatment types were compared via Duncan’s multiple range tests at a 5% significance level (n = 12).
RNA extraction, mRNA library construction and sequencing
Total RNA was prepared using TRIzol (Invitrogen) from a mixture of 12 seedlings from each treatment. RNA integrity was analyzed using an Agilent bioanalyzer (Agilent Technologies 2000). mRNAs were purified with oligo (dT) magnetic beads, then fragmented and used to synthesize cDNA following the TruSeq RNA Sample Preparation v2 Guide (Illumina). Sequencing adaptors were ligated to cDNA fragments through PCR amplification. Raw sequencing data were generated using the Illumina HiSeq 2000 system (Illumina, USA) and the sequencing procedure was performed by CapitalBio Corporation (China). The raw data presented in this publication were deposited in the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) and are accessible using the following SRA accession number: SRP049799 (experiment accession numbers SRX759607 for the DS treatment and SRX759609 for the WW treatment).
Processing and assembly of sequencing reads
The raw data were processed before assembly. Low-quality reads were excluded according to their compliance with the following standards: (1) from the beginning base of each read, the quality value of each base cannot be less than 10—this base and the following bases were removed if the quality value was less than 10; and (2) the length of reads (2 reads of paired-end sequencing) must be greater than 30 bp after filtering. A de novo assembly method [23] was applied due to the lack of available genome information for P. mongolica. The obtained clean reads (Q20, calculate by CASAVA 1.8.2) were assembled with assembly software (trinityrnaseq-r2013-02-25) to construct unique consensus sequences [24]. The trimmed Solexa transcriptome reads were mapped onto the unique consensus sequences using Bowtie2 (Bowtie parameter: –v3—all—best—strata) [25]. A Perl script was written to process the mapping results and generate transcripts.
Functional annotation and classification
The obtained transcripts were compared with the NCBI non-redundant nucleotide database (NT, by Jan 2013) and the non-redundant protein database (NR, Nov, 2014) using BLASTN [26] and BLASTX [26], respectively, with the same E-value cutoff of ≤e-5. Transcripts were identified based on comparison of sequence similarity against SWISS-PROT (downloaded from the European Bioinformatics Institute: ftp://ftp.ebi.ac.uk/pub/databases/swissprot/ by Jan, 2013) using BLAST [26] at E-values ≤e-10. Transcripts were assigned functional annotation through comparison of sequence similarity against the Clusters of Orthologous Groups of proteins database (COG, http://www.ncbi.nlm.nih.gov/COG/) [27,28] with BLAST [26] at E-values ≤e-10, and a Perl script was written to assign functional classes to the transcripts. The transcripts were first compared with the Kyoto Encyclopedia of Genes and Genomes database (KEGG, http://www.genome.jp/kegg/, release 58) [29] using BLASTX [26] at E- values ≤e-10. A Perl script was employed to retrieve KEGG Orthology (KO) information from the BLAST results and subsequently establish pathway associations between the transcripts and database. InterPro domains [30] were annotated with InterProScan [31] Release 27.0, and functional assignments were mapped onto the gene ontology (GO, http://beta.geneontology.org) [32]; WEGO (http://wego.genomics.org.cn/) [33] was used for GO classification and to draw the GO tree.
Detection of differentially expressed transcripts
Similar to the credibility interval approaches reported for the analysis of SAGE [34] data, we employed IDEG6 [35] to identify transcripts showing statistically significant differences in relative abundance (gene expression levels were calculated using the transcript read number per million (TPM) method [36]) between the two libraries. The ratio was used to determine the differentially expressed transcripts. Transcripts exhibiting ratio values above 2.0 were regarded as up-regulated transcripts, while transcripts presenting values below 0.5 were regarded as down-regulated transcripts [16].
| (1) |
Note: 1 is added to the TPM value to prevent the inability to perform the calculations when the TPM value is equal to 0.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed to validate the results obtained from high-throughput sequencing. New RNA was isolated using TRIzol (Invitrogen) from a mixture of 12 seedlings from each treatment, purified with absolute alcohol and treated with DNase (EN0521, Thermo). The RNA was reverse-transcribed using the RevertAid First-Strand cDNA synthesis Kit (K1621, Thermo) following the manufacturer’s instructions. Quantitative reverse transcription PCR (qRT-PCR) was performed using the LightCycler 480 real-time PCR system (Roche). The reaction mixture (25 μL) contained 2× Maxima SYBR Green qPCR Master Mix (12.5 μL), 1 μM each of the forward and reverse primers (1 μL, the primers are listed in S1 Table), 2 μL of template cDNA and 9.5 μL of nuclease-free water. PCR amplification was conducted under the following conditions: 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 60 s. Three independent biological replicates for each sample and three technical replicates of each biological replicate were analyzed in the quantitative real-time PCR analysis. The gene expression of the selected transcripts was normalized against an internal reference gene, glyceraldehyde-3-phosphate dehydrogenase [37] (GAPDH) (comp67082_c0_seq1). Relative transcript expression was calculated using the 2-ΔΔCt method [38].
Results and Discussion
RNA-seq of P. mongolica transcriptome
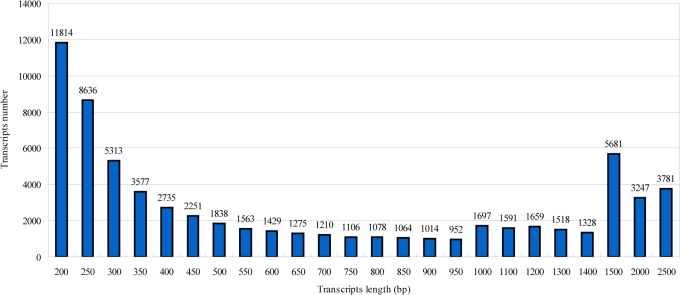
A total of 28,713,735 and 26,650,133 raw reads were generated from the WW and DS P. mongolica seedlings, respectively. After removing low-quality reads, a total of 26,851,249 clean reads were obtained for the WW treatment and 24,826,653 clean reads were obtained for the DS treatment. To generate more complete and representative information about the P. mongolica transcriptome, all of the clean reads from both libraries were mapped to transcripts. A total of 67,352 transcripts were obtained, ranging from 0.2 to 2.5 kb, with an average length 874.44 bp (Fig 2). Compared with previous studies [11,16,17] conducted using the 454 platform, both our results and the results of other studies [39,40] employing the same Solexa platforms show that Solexa sequencing technology can generate more clean reads mapped to transcripts and can be widely applied in non-model plant transcriptome sequencing. In addition, 63,613 transcripts were assembled in the DS library, which was a greater number than in the WW library, with 62,107 transcripts. Our results were consistent with previous conclusions indicating an activated transcriptome in plants in response to drought stress [17,41].
Fig 2. Size distribution of the total P. mongolica transcripts.
General features of the transcriptome of P. mongolica
The general features of the transcriptome of P. mongolica may yield potential information to understand the drought adaption of this shrub. The results for the 67,352 transcripts when matched with 7 databases are presented in Table 1. In total, 55,126 (81.8%) transcripts showed matches with the Nr database, 32,716 (48.6%) with the SwissProt database, and 29,719 (44.1%) with the InterPro database. Of the 55,126 transcripts displaying the best hits in the Nr database, 41,656 (75.6%) transcripts corresponded to known plant’s protein sequences, with 28,069, 2,236, 5,205, 3,238, 1,109, 1,027 and 772 transcripts matching sequences from P. persica, other Prunus plants (P. nume, P. dulcis and P. avium), Vitis vinifera, Populus trichocarpa, Glycine max, Malus spp. and Medicago truncatula, respectively (Fig 3). This result indicated that P. mongolica’s transcripts have a high level (50.9%) of annotation similarity with peach. When adding the same genus plants, including P. nume, P. dulcis and P. avium, this level will arise to 55.0%. In addition, we note that only 28,927 protein annotations appeared in the high quality draft genome of peach [42], but 28,069 transcripts properly hits peach protein sequence, accounted for 97.0% of peach protein sequences has been annotated. This could show high accuracy of our assembly and could reflect the presence of high similarity between Mongolian almond and peach genomes. The high level of homology between peach and almond plants as well as other Prunus genomes has been also reported in the previous reports [20,43,44]. These similarities could indicate that the quality of our assembly is good enough to proceed to the next steps of analysis.
Table 1. Summary of P. mongolica transcripts matched with the Nt (non-redundant nucleotide), Nr (non-redundant protein), SwissProt, COG (Clusters of Orthologous Groups of proteins), KEGG (Kyoto Encyclopedia of Genes and Genomes), InterPro and GO (Gene Ontology) databases.
| Database | Transcript annotation No. (Percentage) | E cut off | Database version |
|---|---|---|---|
| Nt | 47320 (70.3%) | 1.00×e-5 | 201301 |
| Nr | 55126 (81.8%) | 1.00×e-5 | 201411 |
| SwissProt | 32716 (48.6%) | 1.00×e-10 | 201301 |
| COG | 21176 (31.4%) | 1.00×e-10 | No version |
| KEGG | 47306 (70.2%) | 1.00×e-10 | Release 58 |
| InterPro | 29719 (44.1%) | — | InterProScan 4.8 |
| GO | 24553 (36.5%) | — | V36 |
| Total | 67357 (100%) | — | — |
Fig 3. The distribution of the top BLAST hits for the total P. mongolica transcripts.

For an overview of the transcripts that matched the COG database, see S2 Table. Detailed information, including the protein name in COG, BLAST E-value, function ID, COG ID, COG class definition and functional categories (first level and second level), is provided in S3 Table. Among the 21,176 transcripts, nearly one-third were annotated as function unknown or had only a general function prediction. Proteins with uncharacterized functions form a large part of many of the currently available biological databases [45], and this situation even exists in model plant species. The function of the proteins encoded by approximately 13% of the Arabidopsis thaliana genome is classified as completely unknown, and the functions of >30% of Arabidopsis proteins are poorly characterized [46] (http://www.arabidopsis.org/). However, many of these genes of unknown function have been demonstrated to play a key role in the response of plants to abiotic stresses [47]. The transcripts of unknown function found in P. mongolica might also play an important role in the response of P. mongolica to drought stress, and we will aim to elucidate the roles of these uncharacterized transcripts in our future research.
All of the transcripts were assigned a GO classification (Fig 4). In total, 31,357 transcripts significantly matched GO terms in the molecular function category; 23,093 in the cellular component category; and 36,684 in the biological process category. Among standard molecular functions, 12 types of functions were covered in our data. Among these functions, binding (13,917) and catalytic activity (12,324) were the most dominant categories. This result coincides with the previous conclusion that binding and catalytic activity operate universally at the transcriptome level [16,48]. Pathway-based analysis helps to further elucidate the roles that genes play in different biological functions. KEGG can assign different transcripts to different biochemical pathways using EC (enzyme commission) members. Overall, this analysis revealed the transcripts showed similarities to sequences in the KEGG database and assigned the transcripts to 232 different pathways (see S4 Table).
Fig 4. GO classification of the total P. mongolica transcripts.

General analysis of differentially expressed transcripts
To identify transcripts that are critical in the drought stress response, we analyzed the differentially expressed transcripts (DETs) between the DS and WW treatments. At a significance level of 0.01 (P value) and an absolute Log2(ratio) ≥1, we identified 3,365 reliable DETs, including 1,876 up-regulated transcripts and 1,489 down-regulated transcripts, in the DS treatment compared with the WW treatment.
Based on the GO annotations obtained using WEGO, the 3,365 reliable DETs were subjected to functional enrichment analysis. The GO functional categories of the up- and down-regulated transcripts, including biological processes, molecular functions and cellular components, are presented in Fig 5. The GO classification results showed that the up-regulated transcripts were mainly involved in the binding, catalytic activity, cellular process, metabolic process, molecular transducer activity, translation regulator activity, envelope, death and developmental process categories, whereas the exclusively down-regulated transcripts displayed functions in the binding, catalytic activity, cellular process, metabolic process, membrane-enclosed lumen, nutrient reservoir activity and macromolecular complex categories.
Fig 5. GO classification of the DETs found in P. mongolica.

The bottom leaves of the DS seedlings became brown and curled up, and 3.33 older leaves were shed per seedling on average (see Table 2) under drought stress. In the sequencing data, up-regulated transcripts with GO terms involved in death and developmental processes only appeared under the DS treatment. These two results indicate that a portion of the plant cells were dying and that the plants accelerated the developmental process to address drought stress [49] after an extended period without irrigation. Our findings were consistent with studies on Populus simonii [50] and Camellia sinensis [51].
Table 2. The growth status of well-watered (WW) and drought-stressed (DS) P. mongolica seedlings on March 10th and March 25th, 2013.
| Treatment | Leaf length (cm, 3/10/2013) | Leaf width (cm, 3/10/2013) | Leaf length (cm, 3/25/2013) | Leaf width (cm, 3/25/2013) | Number of leaves shed |
|---|---|---|---|---|---|
| WW | 3.28±0.16a | 1.31±0.08a | 3.78±0.23a | 1.52±0.21ab | 0.17±0.16b |
| DS | 3.26±0.22a | 1.33±0.15a | 3.37±0.19b | 1.37±0.16b | 3.33±1.33a |
Different letters in the same column indicate a significant difference between the WW and DS treatments: Duncan’s multiple range tests at the 5% level (n = 12). The presented data are the means of 12 replicates, and the standard errors of the means are shown.
The DETs were also matched with KEGG, and the top 20 KEGG pathways are shown in Table 3. Various significant pathways were observed to be related to P. mongolica drought tolerance, involving transcription factors, plant hormone signal transduction, starch and sucrose metabolism and cysteine and methionine metabolism. Transcription factors are major players in drought stress signaling, and some constitute major hubs in these signaling webs [52]. For adaptation to an environment showing a water deficit, transcription factors regulate the expression of special genes [53], and the zinc finger structure of transcription factors is composed of a polypeptide chain enriched in cysteine [54]. Thus, cysteine and methionine metabolism were increased in the P. mongolica’s transcriptome.
Table 3. Top 20 KEGG pathways of differentially expressed transcripts (DETs).
| KEGG pathway | A (B) | Percentage (%) | Up/Down |
|---|---|---|---|
| ko00194: Metabolism; Energy metabolism; Photosynthesis proteins | 168 (35) | 20.8 | 1/34 |
| ko04110: Cellular processes; Cell growth and death; Cell cycle | 224 (32) | 14.3 | 8/24 |
| ko03032: Genetic information processing; Replication and repair; DNA replication proteins | 315 (29) | 9.2 | 6/23 |
| ko03036: Genetic information processing; Replication and repair; Chromosome | 711 (27) | 3.8 | 8/19 |
| ko03000: Genetic information processing; Transcription; Transcription factors | 362 (25) | 6.9 | 16/9 |
| ko04111: Cellular processes; Cell growth and death; Cell cycle-yeast | 194 (24) | 12.4 | 7/17 |
| ko04075: Environmental information processing; Signal transduction; Plant hormone signal transduction | 225 (23) | 10.2 | 13/10 |
| ko04121: Genetic information processing; Folding, sorting and degradation; Ubiquitin system | 585 (22) | 3.8 | 18/4 |
| ko03110: Genetic information processing; Folding, sorting and degradation; Chaperones and folding catalysts | 575 (22) | 3.8 | 15/7 |
| ko00230: Metabolism; Nucleotide metabolism; Purine metabolism | 430 (21) | 4.9 | 13/8 |
| ko00500: Metabolism; Carbohydrate metabolism; Starch and sucrose metabolism | 301 (21) | 7.0 | 12/9 |
| ko04113: Cellular processes; Cell growth and death; Meiosis-yeast | 148 (20) | 13.5 | 8/12 |
| ko00040: Metabolism; Carbohydrate metabolism; Pentose and glucuronate interconversions | 122 (20) | 16.4 | 7/13 |
| ko00196: Metabolism; Energy metabolism; Photosynthesis-antenna proteins | 57 (20) | 35.1 | 0/20 |
| ko00010: Metabolism; Carbohydrate metabolism; Glycolysis/ Gluconeogenesis | 361 (18) | 5.0 | 9/9 |
| ko00680: Metabolism; Energy metabolism; Methane metabolism | 243 (18) | 7.4 | 9/9 |
| ko01003: Metabolism; Glycan biosynthesis and metabolism; Glycosyltransferases | 307 (17) | 5.5 | 8/9 |
| ko04812: Cellular Processes; Cell motility; Cytoskeleton proteins | 256 (16) | 6.3 | 0/16 |
| ko00270: Metabolism; Amino acid metabolism; Cysteine and methionine metabolism | 235 (16) | 6.8 | 12/4 |
| ko04114: Cellular processes; Cell growth and death; Oocyte meiosis | 214 (16) | 7.5 | 8/8 |
A is the number of total transcripts matched with the pathway; B is the number of total DETs matched with the pathway; Up is the number of up-regulated DETs; Down is the number of down-regulated DETs.
Validation of data reliability by qRT-PCR
To verify the RNA-seq data, we selected 20 DETs for validation using qRT-PCR. Although the fold changes obtained through qRT-PCR displayed some deviations compared with those obtained from high-throughput sequencing (Fig 6), most qRT-PCR-tested transcripts showed a concordant direction of change for both RNA-seq and qRT-PCR. Generally, the qRT-PCR analysis confirmed the transcript expression pattern detected through RNA-seq, indicating that our results were reliable.
Fig 6. Histogram presentation of the Lg(Fold) of 20 DETs.

Black column represents the Lg(fold change (DS/WW)) in transcript expression measured through RNA-seq. Red column is the Lg(fold change (DS/WW)) in transcript expression measured through qRT-PCR. The internal reference gene is glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (comp67082_c0_seq1). A is oxygen-evolving ehancer protein (comp47289_c0_seq1); B is function unknown protein (comp47789_c0_seq1); C is function unknown protein (comp56650_c0_seq1); D is function unknown protein (comp57915_c0_seq1); E is exopolyphosphatase (comp60350_c0_seq1); F is late embryogenesis abundant protein (comp60402_c0_seq1); G is chlorophyll a-b binding protein (comp61248_c0_seq1); H is RSI-1 protein precursor (comp61258_c0_seq1); I is temperature-induced lipocalin (comp62739_c0_seq1); J is protease (comp64242_c0_seq1); K is chlorophyll A/B binding protein (comp63546_c2_seq1); L is heat-shock protein (comp64264_c0_seq1); M is L-ascorbate oxidase (comp65578_c0_seq1); N is function unknown protein (comp67288_c0_seq3); O is light-harvesting complex I protein (comp67308_c0_seq2); P is thaumatin-like protein (comp67881_c0_seq1); Q is BAC insert containing Ma gene (comp68903_c0_seq1); R is dehydration-responsive protein (comp69940_c0_seq1); S is thaumatin-like protein (comp70848_c0_seq2); T is serine/threonine-protein kinase (comp73635_c0_seq2).
P. mongolica photosynthesis under drought conditions
The Pn and Gs of the WW and DS seedlings are shown in Fig 7. The direction of the change in stomatal conductance is the most important limiting factor for plant photosynthesis [55]. In the present study, when the P. mongolica seedlings were subjected extended drought conditions, the Pn and Gs showed the same decreasing tendencies observed in plants grown under water deficit conditions (on the 4th day since the last watering); hence, our results support the above conclusion regarding the change in stomatal conductance.
Fig 7. Net photosynthetic rate (Pn) and stomatal conductance (Gs) of well-watered (WW) and drought-stressed (DS) P. mongolica seedlings.
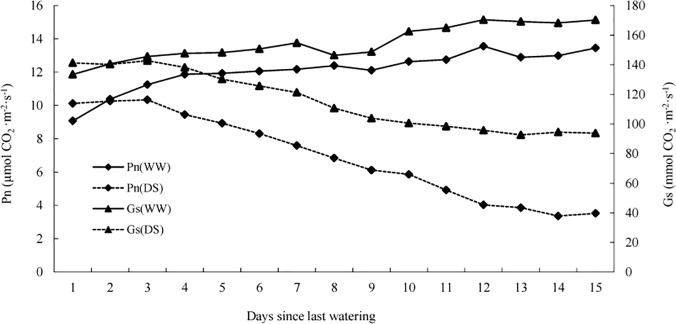
The core metabolism of plants consists of photosynthesis, which provides the energy and basic metabolic substrates for the other major metabolic pathways [56]. Thus, transcripts annotated with GO terms related to the membrane-enclosed lumen, nutrient reservoir activity and macromolecular complexes were down-regulated in the DS treatment (see Fig 5). Among the top 20 KEGG pathways of the DETs, most of the transcripts in the two energy metabolism categories, including photosynthesis proteins (ko00194, S1 Fig) and antenna proteins (ko00196, S2 Fig), were down-regulated, indicating that photosystem I (PSI), photosystem II (PSII) and the light-harvesting chlorophyll protein complex (LHC) also inhibited the photosynthesis of P. mongolica under the drought condition, similar to the findings of a previous study in wheat [57].
The balance of plant hormones and DETs in hormone signal transduction
Three types of plant hormones (auxins, abscisic acid (ABA) and cytokinins), were measured to evaluate the hormone balance of P. mongolica under drought conditions. Compared with the WW seedlings, the ABA content increased significantly under drought conditions, whereas the contents of auxin and cytokinin decreased significantly (Fig 8). The immediate cause of leaf shedding was the increased ABA contents [58]; the DS seedlings shed 3.3 old leaves over the 15-day period on average (see Table 2).
Fig 8. Auxin, abscisic acid (ABA) and cytokinin contents of well-watered (WW) and drought-stressed (DS) P. mongolica seedlings.
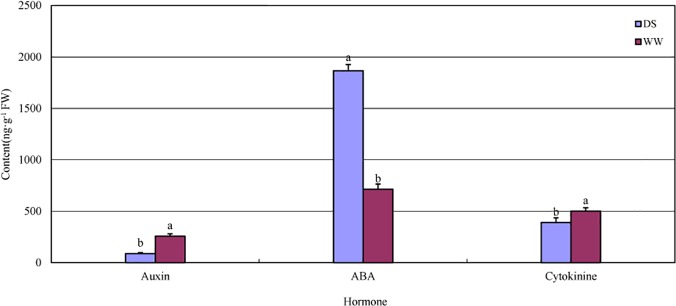
Different letters associated with same hormone indicate a significant difference between the WW and DS treatments: Duncan’s multiple range tests at the 5% level (n = 12).
Based on the enzymes reported by Davies [59], the most homologous transcripts involved in plant hormone signal transduction pathways in P. mongolica were examined in our RNA-seq data (ko04075, S3 Fig). The transcription of type 2C protein phosphate (PP2C), which is one of the most important genes involved in ABA signal transduction and is negatively regulated in the plant drought response [60], was up-regulated in the DS treatment. In contrast, the expression levels of AUX1, AUX/IAA, Gretchen Hagen 3 (GH3), small auxin up RNA (SAUR) and histidine-aspartate phosphorelays (AHP) were decreased; these transcripts are involved auxin and cytokinin signal transduction. As a direct result of the changes in these 6 transcripts, the stomata closed, and the enlargement and division of plant cells were inhibited. These results were verified by the lower values obtained for the leaf length, leaf width (Table 2) and Gs (Fig 7) of the DS seedlings. Generally, large leaves use more water. Thus, in water-limited environments, large leaves may be disadvantageous as long as reproductive success is not completely dependent on vegetative biomass [61,62]. Hence, the smaller shoots of P. mongolica observed during vegetative stages may reduce water use, which may be a factor related to the survival of this shrub in the Gobi Desert.
The leaf water potential and DETs in the aquaporin gene family
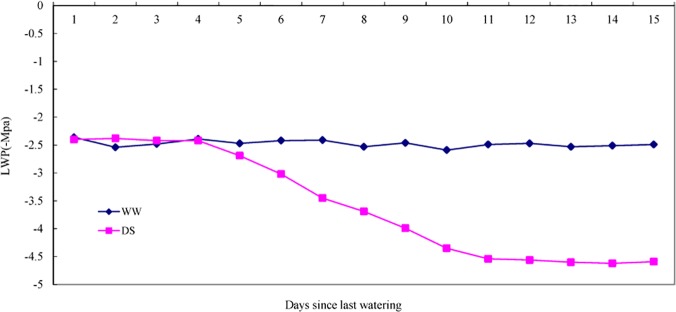
The water potential of P. mongolica leaves showed significant differences between the WW and DS seedlings (Fig 9). The control seedlings exhibited a stable, higher leaf water potential (LWP). In contrast, the plants grown under water-limited conditions displayed a decreased LWP as the drought strengthened compared with their early drought response, whereas the DS seedlings maintained a stable, lower LWP over the last 5 days. Aquaporins [63] are closely related to the maintenance of the water potential; thus, we investigated the related DETs in the aquaporin gene family.
Fig 9. The leaf water potential (LWP) of well-watered (WW) and drought-stressed (DS) P. mongolica seedlings.
Aquaporin proteins facilitate osmosis by forming water-specific pores as an alternative to water diffusion through the lipid bilayer, thus increasing the water permeability of the membrane under drought condition [64]. Phylogenetic analysis has revealed that aquaporin genes can be largely divided into at least six different subfamilies [65]: plasma membrane intrinsic proteins (PIPs), tonoplast intrinsic proteins (TIPs), nodulin intrinsic proteins (NIPs), small basic intrinsic proteins (SIPs), hybrid intrinsic proteins (HIPs) and unrecognized X intrinsic proteins (XIPs). Aquaporins influence plant drought resistance, but to the best of our knowledge, no comprehensive analysis of all of the aquaporin genes of a given plant species has been reported thus far.
In total, 42 transcripts belonging to five aquaporin subfamilies (excluding HIPs) were found in our RNA-seq data (S5 Table). We identified 16 possible PIP transcripts through BLAST [26] analysis; only one PIP transcript was significantly down-regulated at the p≤0.01 level, but the ratios of these transcripts were generally lower than 1 (except for some low-abundance (TPM≤10) transcripts). A previous study indicated that decreasing PIP expression reduces cellular water loss under drought conditions [66]. Thus, P. mongolica may adopt the same strategy to address water deficiency. PIP gene expression is regulated by the abscisic acid (ABA) concentration in plants during drought stress [67], and all of the identified PIPs were down-regulated in the DS treatment, which may be a result of the change in hormone (ABA) signal transduction in P. mongolica seedlings.
Pou et al. (2013) [66] examined the role of aquaporins in regulating leaf hydraulic conductance in Vitis vinifera and found that water stress increased the expression of VvTIP1;1 but decreased the expression of VvTIP2;1. Additionally, the expression of VvTIP2;1 was shown to be highly correlated with stomatal conductance, and the expression of P. mongolica TIP transcripts observed in the present work is in agreement with this previous study. In addition, the changes in TIP expression in the vacuoles of plant guard cells activate the coupled ion channel, resulting in ion outflow, cell shrinkage and stomatal closure [68]. Cell shrinkage and stomatal closure were also shown to result from hormone signal transduction in P. mongolica (see S3 Fig), which suggested that TIP transcript expression may be regulated by plant hormone signal transduction.
In addition, NOD-26, SIP1 and most NIPs [69] and SIPs [70] transport small molecules such as glycerol, urea, boric acid and silicon but show poor permeability to water. In the P. mongolica transcriptome, most of the calculated NIP and SIP ratios ranged form 0.573 to 1.292; the ratios of PmNIP9 (comp70470_c0_seq1) and PmNIP10 (comp70470_c0_seq2) were 0.352 and 0.354, respectively, but these two transcripts are low-abundance transcripts. In P. mongolica, the present results showed no obvious changes in NIP and SIP expression under drought conditions.
Total SOD activities and DETs in the SOD gene family
In total, 177 transcripts involved in antioxidant activity were defined in the RNA-seq data. These defined functions suggested that reactive oxygen species (ROS) were generated as a result of environmental stress, and an effective ROS scavenging and signaling system may exist in P. mongolica to address drought stress. SOD is a core enzyme in the ROS scavenging system. It is well known that SOD may be divided and grouped into four types of categories: manganese superoxide dismutase (Mn-SOD), copper/zinc superoxide dismutase (Cu/Zn-SOD), iron superoxide dismutase (Fe-SOD) and nickel superoxide dismutase (Ni-SOD); these divisions correspond to the metallic ion in the prosthetic group [71]. Ni-SOD has only been found in microorganisms to date [72]. We observed that total SOD activity in the DS treatment was 129% higher than in the WW treatment. Hence, the question arises of what contributions the three types of SOD (Mn-SOD, Cu/Zn-SOD and Fe-SOD) make to increasing total SOD activity in drought-stressed P. mongolica seedlings.
In total, 50 transcripts were predicted to show SOD activity, but most of them were low-abundance transcripts. The expression of 10 high-abundance transcripts is presented in Fig 10. The Cu/Zn-SOD ratios were lower than 1, which indicates that drought inhibited the expression of the Cu/Zn-SOD gene in P. mongolica. However, the expression of the Fe-SOD and Mn-SOD transcripts was up-regulated at this time; in particular, 3 transcripts (A, D and J) were significantly up-regulated in the DS treatment. In addition, we noted that many low-abundance Fe-SOD transcripts were only induced in the DS library (data not shown).
Fig 10. Histogram presentation of the ratios of 10 high-abundance SOD transcripts.

Fold a represents the fold change (DS/WW) in transcript expression measured through RNA-seq. Fold b is the fold change (DS/WW) in transcript expression measured through qRT-PCR. A is comp65789_c0_seq1; B is comp65789_c0_seq2; C is comp65789_c0_seq3; D is comp65789_c0_seq4; E is comp61947_c0_seq1; F is comp63868_c0_seq1; G is comp68247_c0_seq1; H is comp70463_c0_seq1; I is comp63735_c0_seq1; J is comp70328_c0_seq1.
Bhoomika et al. (2013) [73] indicated that the presence and elevated activity of Fe-SOD enhance the activities of Mn-SOD in aluminum-tolerant Oryza sativa, and Signorelli et al. (2013) [74] found that drought induces the accumulation of Fe-SOD and Mn-SOD to high levels, but not Cu/Zn-SOD in Lotus (Lotus corniculatus). Based on the above two studies and our results, we can deduce that P. mongolica activates Fe-SOD genes and enhances the expression of Mn-SOD genes to increase total SOD activity under drought stress conditions.
Conclusions
Mongolian almond (P. momgolica) is widely established in the Gobi Desert and shows extreme tolerance to drought. To investigate the mechanisms that allow P. mongolica to maintain growth in extremely arid environments, the response of P. mongolica seedlings to drought stress was analyzed using morphological, physiological, biochemical and high-throughput sequencing approaches. Compared with the no-stressed control, 3,365 transcripts were differentially expressed in drought-stressed P. momgolica seedlings, including 1,876 up-regulated transcripts and 1,489 down-regulated transcripts. After being treated with drought stress, the photosynthesis response showed a decreasing tendency, but the changes in the levels of hormones (auxins, cytokinins and abscisic acid) resulted in the closing of stomata and decreased cell enlargement and division; these changes were effective for promoting P. mongolica survival in the Gobi Desert. Then the aquaporin and superoxide dismutase gene families were analyzed, it is found that all of the plasma membrane intrinsic protein transcripts were down-regulated in the drought-stressed treatment, whereas drought did not affect the expression of nodulin intrinsic protein or small basic intrinsic protein transcripts in P. mongolica seedlings. In addition, activation of iron superoxide dismutase transcription and enhanced transcription of manganese superoxide dismutase were observed in P. mongolica to promote tolerance of drought stress. This study identified drought response genes in P. mongolica seedlings. Our results provide a significant contribution to the understanding of how P. mongolica responds to drought stress at the transcriptome level, which may help to elucidate the molecular mechanisms associated with the drought response of almond plants.
Supporting Information
A green box represents a down-regulated transcript in the DS treatment.
(TIF)
A green box represents a down-regulated transcript in the DS treatment.
(TIF)
A red box represents an up-regulated transcript in the DS treatment. A green box represents a down-regulated transcript in the DS treatment.
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Folda: Fold change (DS/WW) in transcript expression measured through RNA-seq. Foldb: Fold change (DS/WW) in transcript expression measured through qRT-PCR. “—” indicates not detected in the qRT-PCR analysis.
(XLSX)
Acknowledgments
We thank Dr. Xiwu Qi and Dr. Hongyi Nie of the State Key Laboratory of Silkworm Genome Biology for help in the submission of raw data.
Data Availability
The raw data presented in this publication have been deposited in NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) and is accessible through SRA accession number: SRP049799.
Funding Statement
This work was supported by the National Natural Science Foundation of China (Nos. 31060110 and 30860225). URL: http://www.nsfc.gov.cn/. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Xangsayasane P, Jongdee B, Pantuwan G, Fukai S, Mitchell J, Inthapanya P, et al. (2014) Genotypic performance under intermittent and terminal drought screening in rainfed lowland rice. Field Crops Research 156: 281–292. [Google Scholar]
- 2. Manea A, Leishman MR (2014) Leaf area index drives soil water availability and extreme drought-related mortality under elevated CO2 in a temperate grassland model system. PloS one 9: e91046 10.1371/journal.pone.0091046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keppel-Aleks G, Wolf AS, Mu M, Doney SC, Morton DC, Kasibhatla PS, et al. (2014) Separating the influence of temperature, drought and fire on interannual variability in atmospheric CO2. Global Biogeochemical Cycles: in press. [DOI] [PMC free article] [PubMed]
- 4. Arias R, Rodríguez-Blanco ML, Taboada-Castro MM, Nunes JP, Keizer JJ, Taboada-Castro MT (2014) Water resources response to changes in temperature, rainfall and CO2 concentration: a first approach in NW Spain. Water 6: 3049–3067. 10.3233/BME-141126 [DOI] [PubMed] [Google Scholar]
- 5. Jayanthi H, Husak GJ, Funk C, Magadzire T, Chavula A, Verdin JP (2013) Modeling rain-fed maize vulnerability to droughts using the standardized precipitation index from satellite estimated rainfall—Southern Malawi case study. International Journal of Disaster Risk Reduction 4: 71–81. [Google Scholar]
- 6.Okay S, Derelli E, Unver T (2014) Transcriptome-wide identification of bread wheat WRKY transcription factors in response to drought stress. Molecular Genetics and Genomics 10.1007/s00438-014-0849-x [DOI] [PubMed]
- 7.Yazbek MM, Al-Zein MS (2014) Wild almonds gone wild: revisiting Darwin’s statement on the origin of peaches. Genetic Resources and Crop Evolution: 1–10.
- 8. Utsumi Y, Tanaka M, Morosawa T, Kurotani A, Yoshida T, Mochida K, et al. (2012) Transcriptome analysis using a high-density oligomicroarray under drought stress in various genotypes of cassava: an important tropical crop. DNA research 19: 335–345. 10.1093/dnares/dss016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ajithkumar IP, Panneerselvam R (2014) ROS scavenging System, osmotic maintenance, pigment and growth status of Panicum sumatrense Roth. under drought dtress. Cell biochemistry and biophysics 68: 587–595. 10.1007/s12013-013-9746-x [DOI] [PubMed] [Google Scholar]
- 10. Nuruzzaman M, Sharoni AM, Satoh K, Kumar A, Leung H, Kikuchi S (2014) Comparative transcriptome profiles of the WRKY gene family under control, hormone-treated, and drought conditions in near-isogenic rice lines reveal differential, tissue specific gene activation. Journal of plant physiology 171: 2–13. 10.1016/j.jplph.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 11. Zhou Y, Gao F, Liu R, Feng J, Li H (2012) De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus . BMC Genomics 13: 266 10.1186/1471-2164-13-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gross SM, Martin JA, Simpson J, Abraham-Juarez MJ, Wang Z, Visel A (2013) De novo transcriptome assembly of drought tolerant CAM plants, Agave deserti and Agave tequilana. BMC genomics 14: 563 10.1186/1471-2164-14-563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu T, Zhu S, Tang Q, Yu Y, Tang S (2013) Identification of drought stress-responsive transcription factors in ramie (Boehmeria nivea L. Gaud). BMC plant biology 13: 130 10.1186/1471-2229-13-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raney JA, Reynolds DJ, Elzinga DB, Page J, A. Udall J, Jellen EN, et al. (2014) Transcriptome analysis of drought induced stress in Chenopodium quinoa . American Journal of Plant Sciences 5: 338–357. [Google Scholar]
- 15. Dong Y, Fan G, Zhao Z, Deng M (2014) Transcriptome expression profiling in response to drought stress in Paulownia australis . International Journal of Molecular Sciences 15: 4583–4607. 10.3390/ijms15034583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang S, Liang H, Yan D, Zhao Y, Han X, Carlson JE, et al. (2013) Populus euphratica: the transcriptomic response to drought stress. Plant molecular biology 83: 539–557. 10.1007/s11103-013-0107-3 [DOI] [PubMed] [Google Scholar]
- 17. Müller T, Ensminger I, Schmid KJ (2012) A catalogue of putative unique transcripts from Douglas-fir (Pseudotsuga menziesii) based on 454 transcriptome sequencing of genetically diverse, drought stressed seedlings. BMC genomics 13: 673 10.1186/1471-2164-13-673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alimohammadi A, Shiran B, Martínez-Gómez P, Ebrahimie E (2013) Identification of water-deficit resistance genes in wild almond (Prunus scoparia) using cDNA-AFLP. Scientia Horticulturae 159: 19–28. [Google Scholar]
- 19. Eldem V, Akçay UÇ, Ozhuner E, Bakır Y, Uranbey S, Unver T (2012) Genome-wide identification of miRNAs responsive to drought in peach (Prunus persica) by high-throughput deep sequencing. PloS one 7: e50298 10.1371/journal.pone.0050298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mousavi S, Alisoltani A, Shiran B, Fallahi H, Ebrahimie E, Imani A, et al. (2014) De novo transcriptome assembly and comparative analysis of differentially expressed genes in Prunus dulcis Mill. in response to freezing stress. PloS one 9: e104541 10.1371/journal.pone.0104541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tarkowská D, Novák O, Floková K, Tarkowski P, Turečková V, Grúz J, et al. (2014) Quo vadis plant hormone analysis? Planta: 1–22. [DOI] [PubMed]
- 22.Alvarez M, Huygens D, Fernandez C, Gacitúa Y, Olivares E, Saavedra I, et al. (2009) Effect of ectomycorrhizal colonization and drought on reactive oxygen species metabolism of Nothofagus dombeyi roots. Tree physiology: tpp038. [DOI] [PubMed]
- 23. Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, et al. (2010) De novo assembly and analysis of RNA-seq data. Nature methods 7: 909–912. 10.1038/nmeth.1517 [DOI] [PubMed] [Google Scholar]
- 24. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology 29: 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tatusov RL, Koonin EV, Lipman DJ (1997) A genomic perspective on protein families. Science 278: 631–637. [DOI] [PubMed] [Google Scholar]
- 28. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. (2003) The COG database: an updated version includes eukaryotes. BMC bioinformatics 4: 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, et al. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic acids research 34: D354–D357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Barrell D, Bateman A, et al. (2003) The InterPro Database, 2003 brings increased coverage and new features. Nucleic acids research 31: 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zdobnov EM, Apweiler R (2001) InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17: 847–848. [DOI] [PubMed] [Google Scholar]
- 32. Consortium GO (2004) The Gene Ontology (GO) database and informatics resource. Nucleic acids research 32: D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. (2006) WEGO: a web tool for plotting GO annotations. Nucleic acids research 34: W293–W297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vêncio RZ, Brentani H, Pereira CA (2003) Using credibility intervals instead of hypothesis tests in SAGE analysis. Bioinformatics 19: 2461–2464. [DOI] [PubMed] [Google Scholar]
- 35. Romualdi C, Bortoluzzi S, d’Alessi F, Danieli GA (2003) IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiological genomics 12: 159–162. [DOI] [PubMed] [Google Scholar]
- 36. Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN (2010) RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26: 493–500. 10.1093/bioinformatics/btp692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jain M (2009) Genome-wide identification of novel internal control genes for normalization of gene expression during various stages of development in rice. Plant science 176: 702–706. [Google Scholar]
- 38. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 39. Fan XD, Wang JQ, Yang N, Dong YY, Liu L, Wang FW, et al. (2013) Gene expression profiling of soybean leaves and roots under salt, saline–alkali and drought stress by high-throughput Illumina sequencing. Gene 512: 392–402. 10.1016/j.gene.2012.09.100 [DOI] [PubMed] [Google Scholar]
- 40. Varshney RK, Kudapa H, Roorkiwal M, Thudi M, Pandey MK, Saxena RK, et al. (2012) Advances in genetics and molecular breeding of three legume crops of semi-arid tropics using next-generation sequencing and high-throughput genotyping technologies. Journal of biosciences 37: 811–820. [DOI] [PubMed] [Google Scholar]
- 41. Peng S, Jiang H, Zhang S, Chen L, Li X, Korpelainen H, et al. (2012) Transcriptional profiling reveals sexual differences of the leaf transcriptomes in response to drought stress in Populus yunnanensis . Tree Physiology 32: 1541–1555. 10.1093/treephys/tps110 [DOI] [PubMed] [Google Scholar]
- 42. Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, et al. (2013) The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nature genetics 45: 487–494. 10.1038/ng.2586 [DOI] [PubMed] [Google Scholar]
- 43.Scalabrin S, Policriti A, Nadalin F, Pinosio S, Cattonaro F, Vendramin E, et al. A catalog of molecular diversity of Prunus germplasm gathered from aligning NGS reads to the peach reference sequence: bioinformatic approaches and challenges; 2011. pp. 169–176.
- 44. Shiran B, Amirbakhtiar N, Kiani S, Mohammadi S, Sayed-Tabatabaei B, Moradi H (2007) Molecular characterization and genetic relationship among almond cultivars assessed by RAPD and SSR markers. Scientia Horticulturae 111: 280–292. [Google Scholar]
- 45. Nadzirin N, Firdaus-Raih M (2012) Proteins of Unknown function in the Protein Data Bank (PDB): an inventory of true uncharacterized proteins and computational tools for their analysis. International journal of molecular sciences 13: 12761–12772. 10.3390/ijms131012761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, et al. (2012) The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic acids research 40: D1202–D1210. 10.1093/nar/gkr1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luhua S, Hegie A, Suzuki N, Shulaev E, Luo X, Cenariu D, et al. (2013) Linking genes of unknown function with abiotic stress responses by high‐throughput phenotype screening. Physiologia plantarum 148: 322–333. 10.1111/ppl.12013 [DOI] [PubMed] [Google Scholar]
- 48. Lu T, Lu G, Fan D, Zhu C, Li W, Zhao Q, et al. (2010) Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome research 20: 1238–1249. 10.1101/gr.106120.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Golldack D, Lüking I, Yang O (2011) Plant tolerance to drought and salinity: stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant cell reports 30: 1383–1391. 10.1007/s00299-011-1068-0 [DOI] [PubMed] [Google Scholar]
- 50. Chen J, Song Y, Zhang H, Zhang D (2013) Genome-wide analysis of gene expression in response to drought stress in Populus simonii . Plant Molecular Biology Reporter 31: 946–962. [Google Scholar]
- 51. Gupta S, Bharalee R, Bhorali P, Bandyopadhyay T, Gohain B, Agarwal N, et al. (2012) Identification of drought tolerant progenies in tea by gene expression analysis. Functional & integrative genomics 12: 543–563. [DOI] [PubMed] [Google Scholar]
- 52. Tripathi P, Rabara RC, Rushton PJ (2014) A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 239: 255–266. 10.1007/s00425-013-1985-y [DOI] [PubMed] [Google Scholar]
- 53. Nakashima K, Yamaguchi-Shinozaki K, Shinozaki K (2014) The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Frontiers in Plant Science 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Polozov R, Sivozhelezov V, Chirgadze YN, Ivanov V (2014) Recognition rules for binding of Zn-Cys2His2 transcription factors to operator DNA. Journal of Biomolecular Structure and Dynamics: 1–14. [DOI] [PubMed]
- 55. Chaves M, Flexas J, Pinheiro C (2009) Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Annals of botany 103: 551–560. 10.1093/aob/mcn125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blankenship RE (2014) Molecular mechanisms of photosynthesis: John Wiley & Sons. [Google Scholar]
- 57. Tian F, Gong J, Zhang J, Zhang M, Wang G, Li A, et al. (2013) Enhanced stability of thylakoid membrane proteins and antioxidant competence contribute to drought stress resistance in the tasg1 wheat stay-green mutant. Journal of experimental botany 64: 1509–1520. 10.1093/jxb/ert004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hancock JT, Neill S, Wilson ID (2011) Nitric oxide and ABA in the control of plant function. Plant Science 181: 555–559. 10.1016/j.plantsci.2011.03.017 [DOI] [PubMed] [Google Scholar]
- 59. Davies PJ (2004) Plant hormones: biosynthesis, signal transduction, action!: Springer. [Google Scholar]
- 60. Arshad M, Mattsson J (2014) A putative poplar PP2C-encoding gene negatively regulates drought and abscisic acid responses in transgenic Arabidopsis thaliana . Trees 28: 531–543. [Google Scholar]
- 61. Zadworny M, Jagodziński AM, Łakomy P, Ufnalski K, Oleksyn J (2014) The silent shareholder in deterioration of oak growth: common planting practices affect the long-term response of oaks to periodic drought. Forest Ecology and Management 318: 133–141. [Google Scholar]
- 62. Eilmann B, Dobbertin M, Rigling A (2013) Growth response of Scots pine with different crown transparency status to drought release. Annals of forest science 70: 685–693. [Google Scholar]
- 63. Maurel C, Verdoucq L, Luu D-T, Santoni V (2008) Plant aquaporins: membrane channels with multiple integrated functions. Annual Review of Plant Biology 59: 595–624. 10.1146/annurev.arplant.59.032607.092734 [DOI] [PubMed] [Google Scholar]
- 64. Verkman A (2013) Aquaporins. Current biology: CB 23: R52–55. 10.1016/j.cub.2012.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yan F, Qu D, Zhao Y, Hu X, Zhao Z, Zhang Y, et al. (2014) Effects of exogenous 5-aminolevulinic acid on PIP1 and NIP aquaporin gene expression in seedlings of cucumber cultivars subjected to salinity stress. Genetics and molecular research: GMR 13. [DOI] [PubMed] [Google Scholar]
- 66. Pou A, Medrano H, Flexas J, Tyerman SD (2013) A putative role for TIP and PIP aquaporins in dynamics of leaf hydraulic and stomatal conductances in grapevine under water stress and re‐watering. Plant, cell & environment 36: 828–843. [DOI] [PubMed] [Google Scholar]
- 67. Jang JY, Kim DG, Kim YO, Kim JS, Kang H (2004) An expression analysis of a gene family encoding plasma membrane aquaporins in response to abiotic stresses in Arabidopsis thaliana . Plant molecular biology 54: 713–725. [DOI] [PubMed] [Google Scholar]
- 68. Sarda X, Tousch D, Ferrare K, Legrand E, Dupuis J, Casse‐Delbart F, et al. (1997) Two TIP‐like genes encoding aquaporins are expressed in sunflower guard cells. The Plant Journal 12: 1103–1111. [DOI] [PubMed] [Google Scholar]
- 69. Chaumont F, Barrieu F, Wojcik E, Chrispeels MJ, Jung R (2001) Aquaporins constitute a large and highly divergent protein family in maize. Plant Physiology 125: 1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ishikawa F, Suga S, Uemura T, Sato MH, Maeshima M (2005) Novel type aquaporin SIPs are mainly localized to the ER membrane and show cell-specific expression in Arabidopsis thaliana . FEBS letters 579: 5814–5820. [DOI] [PubMed] [Google Scholar]
- 71. Jiang J, Su M, Chen Y, Gao N, Jiao C, Sun Z, et al. (2013) Correlation of drought resistance in grass pea (Lathyrus sativus) with reactive oxygen species scavenging and osmotic adjustment. Biologia 68: 231–240. [Google Scholar]
- 72. Gale EM, Narendrapurapu BS, Simmonett AC, Schaefer HF III, Harrop TC (2010) Exploring the effects of H-bonding in synthetic analogues of nickel superoxide dismutase (Ni-SOD): experimental and theoretical implications for protection of the Ni− SCys bond. Inorganic chemistry 49: 7080–7096. 10.1021/ic1009187 [DOI] [PubMed] [Google Scholar]
- 73. Bhoomika K, Pyngrope S, Dubey R (2013) Differential responses of antioxidant enzymes to aluminum toxicity in two rice (Oryza sativa L.) cultivars with marked presence and elevated activity of Fe SOD and enhanced activities of Mn SOD and catalase in aluminum tolerant cultivar. Plant Growth Regulation 71: 235–252. [Google Scholar]
- 74. Signorelli S, Casaretto E, Sainz M, Díaz P, Monza J, Borsani O (2013) Antioxidant and photosystem II responses contribute to explain the drought–heat contrasting tolerance of two forage legumes. Plant Physiology and Biochemistry 70: 195–203. 10.1016/j.plaphy.2013.05.028 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A green box represents a down-regulated transcript in the DS treatment.
(TIF)
A green box represents a down-regulated transcript in the DS treatment.
(TIF)
A red box represents an up-regulated transcript in the DS treatment. A green box represents a down-regulated transcript in the DS treatment.
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Folda: Fold change (DS/WW) in transcript expression measured through RNA-seq. Foldb: Fold change (DS/WW) in transcript expression measured through qRT-PCR. “—” indicates not detected in the qRT-PCR analysis.
(XLSX)
Data Availability Statement
The raw data presented in this publication have been deposited in NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) and is accessible through SRA accession number: SRP049799.