

Abstract
To maintain copious insulin granule stores in the face of ongoing metabolic demand, pancreatic beta cells must produce large quantities of proinsulin, the insulin precursor. Proinsulin biosynthesis can account for up to 30–50% of total cellular protein synthesis of beta cells. This puts pressure on the beta cell secretory pathway, especially the endoplasmic reticulum (ER), where proinsulin undergoes its initial folding, including the formation of three evolutionarily conserved disulfide bonds. In normal beta cells, up to 20% of newly synthesized proinsulin may fail to reach its native conformation, suggesting that proinsulin is a misfolding-prone protein. Misfolded proinsulin molecules can either be refolded to their native structure or degraded through ER associated degradation (ERAD) and autophagy. These degraded molecules decrease proinsulin yield but do not otherwise compromise beta cell function. However, under certain pathological conditions, proinsulin misfolding increases, exceeding the genetically-determined threshold of beta cells to handle the misfolded protein load. This results in accumulation of misfolded proinsulin in the ER – a causal factor leading to beta cell failure and diabetes. In patients with Mutant INS-gene induced diabetes of Youth (MIDY), increased proinsulin misfolding due to insulin gene mutations is the primary defect operating as a “first hit” to beta cells. Additionally, increased proinsulin misfolding can be secondary to an unfavorable ER folding environment due to genetic and/or environmental factors. Under these conditions, increased wild-type proinsulin misfolding becomes a “second hit” to the ER and beta cells, aggravating beta cell failure and diabetes. In this article, we describe our current understanding of the normal proinsulin folding pathway in the ER, and then review existing links between proinsulin misfolding, ER dysfunction, and beta cell failure in the development and progression of type 2, type 1, and some monogenic forms of diabetes.
Keywords: Proinsulin folding and misfolding, Endoplasmic reticulum stress, Diabetes, beta cell, Insulin biosynthesis
Introduction
Insulin is a master hormone that regulates and maintains metabolic homeostasis in the body. In pancreatic β-cells, insulin is initially synthesized as the precursor molecule, preproinsulin, comprised sequentially of the signal peptide, insulin B domain, C domain flanked by dibasic cleavage sites, and insulin A domain. To make mature bioactive insulin, newly synthesized preproinsulin undergoes co- and post-translational translocation across the membrane of the endoplasmic reticulum (ER), where it is cleaved by signal peptidase to form proinsulin. Proinsulin then folds, forming three disulfide bonds that are conserved amongst the entire insulin/IGF superfamily. Proinsulin forms noncovalently-associated homodimers that undergo intracellular transport from the ER to the Golgi complex and into secretory granules, during which proinsulin forms hexamers and is proteolytically processed to C-peptide and mature insulin that is stored in granules (Dodson and Steiner, 1998; Liu et al., 2014b). Upon stimulation, insulin granule exocytosis rapidly releases insulin to the bloodstream to lower blood glucose.
Although insulin biosynthesis and secretion are both tightly regulated, the glucose concentration thresholds required to trigger insulin release are different from that for proinsulin biosynthesis. Insulin secretion is triggered by glucose concentrations above 5 mM, whereas its biosynthesis is most sensitive to fluctuations of glucose between 2–4 mM (Alarcón et al., 1993; Malaisse et al., 1979; Pipeleers DG et al., 1985; Schuit et al., 1988). Thus, insulin biosynthesis is constantly engaged to replenish insulin granule stores even at normal physiological glucose concentrations. Genetic analysis shows that, on average, approximately one third of total cellular proteins are targeted to the secretory pathway. However, in beta cells, insulin biosynthesis alone accounts for more than 10% of total protein synthesis under basal conditions, and this percentage can further increase up to 50% under stimulated conditions (Scheuner and Kaufman, 2008; Schuit et al., 1988; Van Lommel et al., 2006). Due to this large demand, proinsulin folding in beta cells is very sensitive to changes in the ER environment, and increasing demand for proinsulin synthesis and folding makes the beta cell one of the cell types that is most susceptible to ER stress (Eizirik et al., 2008; Papa, 2012; Vetere et al., 2014).
Over the past years, 30 different insulin gene mutations have been reported to cause a new syndrome named Mutant INS-gene-induced Diabetes of Youth [MIDY, for review, (Liu et al., 2010b; Liu et al., 2014a; Støy et al., 2010)]. Most of these mutations lead to proinsulin misfolding in the ER. These misfolded mutant proinsulin molecules generate a “first hit” causing ER stress and a decrease of insulin production that are responsible for the development of diabetes and progression of beta cell failure in MIDY patients. In other cases, even without any Ins gene mutation, a defective ER folding environment can generate a “first hit” to beta cells, affecting the folding pathway of wild-type proinsulin, leading to an increase of proinsulin misfolding. At or above a threshold level, these misfolded wild-type proinsulin molecules may further impair the ER folding environment in beta cells, providing a “second hit” that aggravates ER dysfunction and leads to beta cell failure and diabetes. In this article, we review the proinsulin folding pathway in the ER and current literature that focuses on links between proinsulin misfolding, ER dysfunction, and beta cell failure. The roles of proinsulin misfolding and ER stress in the development and progression of type 2 and type 1 diabetes, as well as some monogenic forms of diabetes, are discussed.
1. Proinsulin folding
1.1. Proinsulin disulfide maturation
Upon delivery to the ER lumen, preproinsulin signal peptide is immediately removed by signal peptidase on the luminal side of the ER. The efficiency and fidelity of signal peptide cleavage appears to be very important for subsequent proinsulin folding in the ER. The pathological consequence of a defect in signal peptide cleavage has been demonstrated both clinically and experimentally (Liu et al., 2012a; Stoy et al., 2007). After removal of the signal peptide, proinsulin undergoes rapid folding in the ER. Although some local folding, including formation of α-helical and β-strand segments in the insulin moiety, plays a role in the proinsulin folding pathway (Weiss, 2013; Yang et al., 2010b), correct disulfide pairing appears to be one of the most important events in determining whether proinsulin molecules can achieve their native folded structure.
Proinsulin contains six cysteines that form three evolutionally conserved disulfide bonds: B7-A7, B19-A20, and A6-A11 (Fig. 1). Non-native mispaired disulfide isomers have been observed both in vivo (Huang and Arvan, 1995; Liu et al., 2005; Liu et al., 2003; Zhang et al., 2003) and in vitro (Hua et al., 2002; Hua et al., 1996b; Weiss, 2009). Interestingly, assuming that all cysteine residues are engaged and randomly form disulfide pairings with other intramolecular cysteine residues, there would be fifteen possible disulfide combinations. However, the actual number of disulfide isomers observed from in vitro studies is relatively low: in one study only two major disulfide isomers were observed during proinsulin folding from a denatured precursor (Hua et al., 1995), and in another in vitro refolding study three human proinsulin disulfide isomers were recovered (Min et al., 2004). Presumably, other possible proinsulin disulfide isomers are either not formed or are very unstable. These in vitro studies suggest that proinsulin may form its three native disulfide bonds in a preferential order, with B19-A20 forming an initial one-disulfide folding intermediate (Hua et al., 2002; Qiao et al., 2003) that may kinetically facilitate formation of B7-A7 and A6-A11 bonds (Chang et al., 2003).
Figure 1. Structure of proinsulin.

Proinsulin is comprised sequentially of insulin B domain (blue), connecting C domain (which is disordered and therefore has no fixed structure), and insulin A domain (red). Six cysteines form three disulfide bonds that are labeled in yellow boxes. This figure was adapted from Liu, M., et al. 2010. PLoS ONE. 5(10):e13333.
The significance of the B19-A20 bond in the in vitro folding of insulin is consistent with studies of the folding pathway of IGF-1. The disulfide between Cys 18 and 61 of IGF-1, corresponding to the B19-A20 bond of insulin, is found in all detectable folding intermediates, suggesting that this is the most favorable disulfide bond, and the first to be formed (Hober et al., 1992; Miller et al., 1993). Among all IGF-1 isomers, the most abundant intermediate in the folding pathway contains an additional native disulfide bond between Cys 6 and 48, corresponding to the B7-A7 bond of insulin, and overall folding of the intermediates with those two disulfide bonds resembles that of the native protein except for local unfolding of helix 2, corresponding to the A1-A8 helix of insulin (Hua et al., 1996b). Similarly, by selectively disrupting individual disulfide bonds of insulin, studies find that deletion of B19-A20 produces the most significant impairment of in vitro refolding, resulting in loss of ordered secondary structure and markedly reduced compactness. By contrast, deletion of the A6-A11 bond causes the least structural perturbation that occurs only in the N-terminal A domain α-helix, suggesting that the native tertiary structure of insulin may be controlled independently of the local A1-A8 helical structure or the A6-A11 disulfide bond. Compared to deletion of the other two bonds, loss of the B7-A7 bond, which is exposed on the surface of the insulin molecule, leads to an intermediate disruption of the thermodynamic stability and overall structure (Chang et al., 2003; Guo and Feng, 2001; Hua et al., 1996a). Thus, based on in vitro folding studies, there are differences in the relative contribution of each of the three disulfide bonds to the native folded structure of insulin, with B19-A20 appearing to be the most important and perhaps the first disulfide bond formed.
Unlike in vitro folding, it is still an open question whether the three native disulfide bonds of proinsulin form randomly or in a particular order when expressed in the complex folding environment of the beta cell ER. The first published evidence that wild-type proinsulin can misfold in the ER came when two major disulfide isomers of newly synthesized proinsulin were observed in both yeast and mammalian cells (Liu et al., 2005; Liu et al., 2003; Zhang et al., 2003). Based on mobility shift in nonreducing SDS-PAGE, distinct disulfide isomers were proposed to be proinsulin folding intermediates that were mostly retained in the ER, where they were specifically recognized and bound by the ER member of the hsp70 chaperone family, known as BiP (Liu et al., 2005). Interestingly, although loss of the A6-A11 disulfide bond significantly impairs receptor binding and insulin bioactivity, proinsulin with the A6-A11 bond deleted was efficiently secreted from cells (Liu et al., 2005; Liu et al., 2009), suggesting that the overall structure of proinsulin lacking A6-A11 may still resemble that of wild-type proinsulin. In contrast, disruption of B19-A20 or B7-A7 bonds severely disturbs the normal folding pathway of proinsulin, leading to proinsulin misfolding and ER retention (Liu et al., 2005; Liu et al., 2003). These data highlight the importance of the two inter-domain disulfide bonds in the folding of proinsulin in the ER.
1.2. ER oxidoreductase and ER oxidoreducin family members involved in the folding of proinsulin
In the ER, protein folding is facilitated by enzymes that both catalyze the formation of disulfide bonds between cysteines and break improper disulfide bonds that may form during the folding process (Jansens et al., 2002). Thus in the ER lumen, there are oxidation and reduction events occurring simultaneously. Enzymes in the ER that play a central role in both oxidation and reduction of disulfide bonds are members of the protein disulfide isomerase (PDI) family of oxidoreductases; there are at least 20 members in this family (Bulleid, 2012; Ellgaard and Ruddock, 2005). Each enzyme contains as least one thioredoxin “C-X-X-C” motif, where C = cysteine and X = any amino acid. This motif shuttles between the reduced species C(SH)-X-X-C(SH) and the intramolecularly oxidized form:
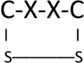 |
To drive disulfide bond formation in the reduced substrate, the thioredoxin motif of the ER oxidoreductase must itself be in the intramolecularly oxidized form. The enzyme then forms a transient mixed-disulfide intermediate with the substrate (an “adduct”) that is rapidly broken upon completion of the disulfide bond within the substrate. The product of the entire reaction leaves a new disulfide bond in the substrate as the thioredoxin motif in the ER oxidoreductase becomes reduced. In order to initiate another round of disulfide bond formation, the thioredoxin motif in the ER oxidoreductase must once again become oxidized. This is catalyzed by upstream ER oxidases, particularly ER oxidoreducin-1 (Ero1) (Fig. 2). Ero1 normally does not directly catalyze formation of substrate disulfide bonds, but rather uses PDI family members as key intermediates driving the electron flow from substrates (such as proinsulin) to Ero1 and ultimately to molecular oxygen, with hydrogen peroxide (H2O2) as a by-product (Sevier, 2010; Sevier and Kaiser, 2008).
Figure 2. Proinsulin disulfide maturation in the ER.

Upon delivery into the ER lumen, the signal peptide of newly synthesized preproinsulin (aqua) is removed by signal peptidase (not shown), forming proinsulin. This Figure posits that disulfide bond formation within proinsulin is catalyzed by one or more ER oxidoreductases. In its oxidized form (Oxi-Re, yellow), the oxidoreductase receives electrons from proinsulin and in the process, the ER oxidoreductase becomes reduced (Oxi-Re, beige). To initiate another round of disulfide bond formation, the reduced ER oxidoreductase must once again become oxidized. As shown in the diagram, this is catalyzed by one or more upstream Oxidases, particularly ER Oxidoreducin-1 (ERO1, which in the process converts from yellow to beige) that also results in production of one hydrogen peroxidase molecule (not shown) for each disulfide bond catalyzed; ERO1 itself is re-oxidized by electron transfer to FAD+ and molecular oxygen (not shown in the diagram). Well folded proinsulin with three native disulfide bonds undergoes anterograde export from the ER (green arrow). Up to 20 % of newly synthesized proinsulin may form mispaired disulfide bonds. These misfolded isomers may be isomerized by one or more ER oxidoreductases, such as PDI, in an attempt to refold to native proinsulin.
Although efforts have been made to better understand how ER oxidoreductases facilitate proinsulin disulfide oxidation and reduction, the specific pathways involved in these processes are far from clear. From in vitro studies, PDI has been shown to facilitate oxidative folding of reduced proinsulin and increase the yield of proinsulin with native disulfide bonds (Winter et al., 2002). The catalytic activity of PDI does not appear to depend on its peptide binding domain (Winter et al., 2011). However, the oxidation/reduction function of PDI in cells is complex (Phillip and Schreiber, 2013; Yon and Betton, 1991). PDI is found in both oxidized and reduced forms in the lumen of the ER. In the yeast ER, PDI is more oxidized compared to that in mammalian cells (Frand and Kaiser, 1999; Mezghrani et al., 2001). The mixture of redox states allows PDI to perform both oxidation and isomerization/reduction reactions in the ER. Surprisingly, a recent study shows that PDI knockdown actually improves proinsulin oxidative folding and accelerates proinsulin export from the ER (Rajpal et al., 2012). Moreover, overexpressing PDI decreases insulin secretion and causes proinsulin accumulation in the ER, with ER stress (Zhang et al., 2009). These studies suggest that PDI exhibits net unfoldase activity and serves as an ER retention factor for proinsulin in beta cells. Therefore, it is very likely that some other ER oxidoreductase family members – still to be discovered – play a role in oxidative folding of proinsulin in the ER.
The specific upstream ER oxidases required for proinsulin oxidative folding are also not clear. Ero1 has been identified as a primary protein oxidation source in the ER (Sevier and Kaiser, 2008). Pancreatic beta cells express two forms of Ero1: Ero1α and Ero1β. Unlike the ubiquitously expressed Ero1α, Ero1β expression is very limited among cell types but is well expressed in pancreatic beta cells, suggesting that it might play a role in proinsulin folding. Indeed, overexpressing Ero1β in beta cell lines improves proinsulin folding, facilitates its ER export, and increases insulin production and secretion (Liu et al., 2012b; Wright et al., 2013a). Conversely, Ero1β knockdown impairs proinsulin oxidative folding (Rajpal et al., 2012) and decreases insulin content in beta cells (Khoo et al., 2011). Interestingly, Ero1β knockout mice exhibit only a modest defect in proinsulin oxidation and processing, and Ero1α and Ero1β double knockout does not further aggravate the defect (Zito et al., 2010), suggesting that in addition to Ero1, other ER oxidases might provide a source of oxidizing equivalents for proinsulin disulfide maturation. Indeed, several ER oxidases, including peroxiredoxin IV (Tavender et al., 2010), glutathione peroxidases (Nguyen et al., 2011), Vitamin K epoxide reductase (Schulman et al., 2010), sulfhydryl oxidase QSOX (Alon et al., 2012), and oxidized glutathione (Appenzeller-Herzog, 2011), have been considered as additional potential sources of oxidizing equivalents for protein disulfide bond formation. However, the role of these oxidases in proinsulin oxidation in beta cells remains unexplored.
1.3. Proinsulin folding and dimerization in the ER
Proinsulin can form dimers both in vitro (Frank and Veros, 1970; Steiner, 1973) and in the ER of beta cells (Haataja et al., 2013; Huang and Arvan, 1995). Residues of the insulin B domain (B8-B29) are intimately involved in the dimerization interface (Blundell et al., 1972; Brange et al., 1988; Whittingham et al., 2006), and it is hypothesized that the proinsulin and insulin dimerization surfaces are identical (the C domain is in random coil and thus unlikely to contribute in any way to proinsulin dimerization). Mutating some residues involved in the dimerization interface allows the production of monomeric insulin — one example being insulin-lispro in which the proline and lysine at positions B28 and B29, respectively, are switched. To date, it is still unknown whether proinsulin oxidation/monomer folding and dimerization are events that occur sequentially or concurrently. However, given the fact that the bioactivities of monomeric insulin analogues, e.g., insulin-lispro or insulin-aspart have been largely preserved (Brange et al., 1988; Evans et al., 2011), monomeric insulins are thought to contain all the essential elements of the native structure of insulin. Additionally, the NMR structures of monomeric proinsulin [DKP proinsulin, analogue containing B domain substitutions H(B10)D, P(B28)K, and K(B29)P] and wild-type proinsulin are similar, both containing a native-like insulin moiety (Kiselar et al., 2011; Yang et al., 2010a). Thus, at least in vitro, dimerization is not required for proinsulin to make its native disulfide bonds. However, the relationship between proinsulin dimerization and its oxidation in the ER appears to be more complicated. While monomeric proinsulin S(B9)D has been shown to be efficiently exported from the ER and targeted to secretory granules (Quinn et al., 1991), a detailed analysis shows that S(B9)D, as well as two other mutants H(B10)D and V(B12)E that may disrupt proinsulin dimerization/hexamerization, form mispaired disulfide bonds, suggesting defective folding in the ER (Zhang et al., 2003). It remains unknown whether misfolding of these monomeric proinsulin mutants is a consequence of their failure to dimerize or because of a direct effect of the mutations on the oxidative folding of monomeric proinsulin.
1.4. Approaches to analyze proinsulin disulfide maturation, folding and misfolding
One of the challenges in the field of proinsulin folding is a lack of reliable approaches that can specifically detect misfolded proinsulin. Currently, there are only a few ways to analyze the folding and disulfide maturation of proinsulin in cells. One involves using nonreducing tris-tricine-urea-SDS-PAGE (Schägger and von Jagow, 1987). This gel system produces superior resolution of small proteins in the molecular weight range between 5 to 20 kDa (the molecular weights of preproinsulin, proinsulin, and insulin are about 11, 9, and 5.8 kDa, respectively). When newly synthesized preproinsulin and proinsulin are analyzed under nonreducing conditions such that preformed disulfide bonds in the cells are preserved, this gel system can discriminate between native proinsulin and misfolded disulfide isomers (Fig. 3A) (Hua et al., 2006; Huang and Arvan, 1995; Liu et al., 2005; Liu et al., 2003; Liu et al., 2009; Zito et al., 2010).
Figure 3. Approaches to analyze proinsulin folding and disulfide maturation in the ER.

A. HEK 293T cells were transfected with plasmids encoding human proinsulin wild-type (Proins-WT) or Akita mutant (Proins-AK), or empty vector (Control). The cells were metabolically pulse-labeled with 35S-Met/Cys for 0.5 hr. The folding of newly synthesized proinsulin in the cells was analyzed using tris-tricine-urea-SDS-PAGE under nonreducing conditions. Two misfolded disulfide isomers (isomer #1 and isomer #2) of Proins-WT were detected. The Proins-AK produced only misfolded isomer with a slower gel mobility. The figure is adapted from Liu M. et al., 2005. 280(14):13209–12. B. Isolated islets from WT and Akita mice were labeled with 35S-Met/Cys for 15 min followed by 2 h chase. The newly synthesized proinsulin was analyzed under both reducing (lanes 1–2) and nonreducing (lanes 3–8) conditions. Although the levels of proinsulin from WT and Akita islets were comparable under nonreducing conditions (lanes 3 and 6), when the identical samples were analyzed under reducing conditions, the synthesis of proinsulin was significantly increased in Akita islets (lanes 1 and 2). The lower recovery under nonreducing conditions was consistent with the idea that Akita proinsulin forms abnormal disulfide-linked complexes (open arrow). After 2 h chase, processed insulin was significantly decreased in Akita islets (compare lanes 4 and 7). The figure is adapted from Liu M. et al., 2007. PNAS 104(40), 15841–15846.
A second approach that involves gel electrophoresis examines the total recovery of native proinsulin under nonreducing conditions in comparison to total proinsulin recovered under reducing conditions (Fig. 3B) (Liu et al., 2010a; Liu et al., 2007; Wang et al., 2011; Wang and Osei, 2011). It has been shown that proinsulin can make abnormal intermolecular disulfide bonds with other proinsulin molecules or with other proteins (e.g. ER chaperones) in the ER. These adducts appear as higher molecular weight protein complexes when they are analyzed using SDS-PAGE under nonreducing conditions. However, when these complexes are examined under reducing conditions in which all intermolecular disulfide bonds are broken, proinsulin molecules are recovered as a monomeric species. Therefore, comparing the total recovery of monomeric proinsulin under nonreducing and reducing conditions can help to estimate the amount of proinsulin that has formed intermolecular disulfide bonds, including the formation of higher molecular weight disulfide-linked complexes in the ER (Liu et al., 2010a; Liu et al., 2012a; Wang et al., 2011).
2. Proinsulin misfolding in the ER
Over the past decade, increasing attention has been drawn to the possible role(s) of defective proinsulin folding and ER stress in the development and progression of beta cell failure and diabetes (Cavener et al., 2010; Fonseca et al., 2009; Fonseca et al., 2011; Liu et al., 2010b; Scheuner and Kaufman, 2008; Weiss, 2009). A growing body of evidence now indicates that proinsulin misfolding can be caused either by primary folding defects in proinsulin due to INS-gene mutations, or by an unfavorable ER folding environment in which the normal folding pathway of wild-type proinsulin is affected secondarily (Hua et al., 2006; Liu et al., 2010a; Liu et al., 2005; Wang et al., 2011; Wang and Osei, 2011; Weiss, 2013; Zhang et al., 2003).
2.1 Primary proinsulin misfolding
In MIDY, most experimentally tested mutants are found to cause proinsulin misfolding in the ER. More than half of these mutants generate unpaired cysteines either because of mutations eliminating one of the native cysteine residues, or mutations creating a new cysteine residue [for details of MIDY mutations, please see review (Liu et al., 2014a)]. These mutants disrupt the formation of correct disulfide pairs, leading to proinsulin misfolding. To date, among the six natural cysteine residues of proinsulin, five have already been reported to have a mutation that is linked to MIDY. In addition to those mutations that create an odd number of proinsulin cysteines, some MIDY mutations do not involve cysteine residues. These non-cysteine mutations are primarily amino acid substitutions of the most highly conserved (generally hydrophobic) residues that have been shown to be important for local folding and non-covalent intramolecular interactions (Hua et al., 2006; Liu et al., 2010c; Liu et al., 2009; Sohma et al., 2010; Weiss, 2009). Since this kind of misfolding is caused by a primary defect within proinsulin molecules, we refer to it as “primary proinsulin misfolding.” In this case, the misfolded mutant proinsulin can be considered as a “first hit” that is itself sufficient to trigger a cascading series of events leading to beta cell failure and diabetes in MIDY patients (Fig. 4).
Figure 4. Primary and secondary proinsulin misfolding and beta cell failure.
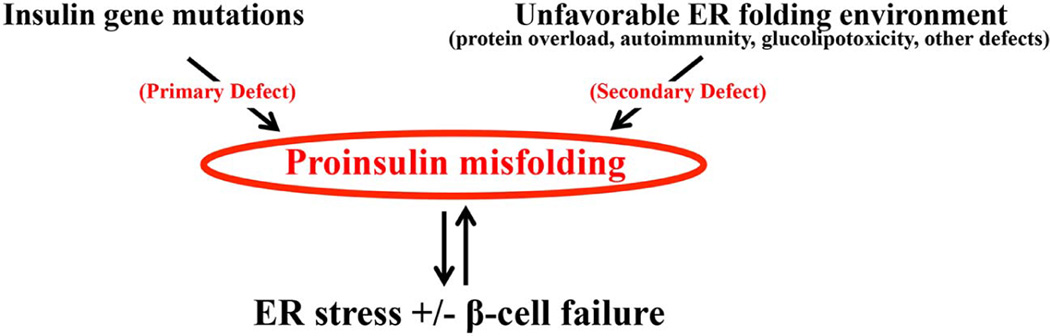
Proinsulin misfolding can be caused by Ins gene mutations, in which the misfolded proinsulin generates a “first hit” leading to ER stress and beta cell failure. In other cases, alterations in the ER folding environment can adversely affect the folding of wild-type proinsulin, leading to an increase of proinsulin misfolding. These misfolded proinsulin molecules may further impair the ER folding environment, providing a “second hit” that aggravates ER dysfunction and leads to beta cell failure.
Importantly, the folding pathway of newly synthesized wild-type proinsulin is also affected when it is co-expressed with MIDY proinsulin mutants in the ER. The direct evidence of this bystander effect comes from studies of Akita mice that carry a spontaneous missense insulin gene mutation encoding C(A7)Y in one of two Ins2 alleles. A mutation at the identical site has also been found to cause MIDY in humans. The mutation disrupts the essential B7-A7 inter-domain disulfide bond that triggers misfolding of mutant proinsulin (Liu et al., 2005). The mice develop diabetes shortly after weaning (Wang et al., 1999; Yoshioka et al., 1997). Accumulating evidence indicates that the underlying mechanism initiating insulin deficient diabetes in Akita mice and MIDY patients is an impairment of the ER export of wild-type proinsulin due to blockade by co-expressed mutant proinsulin (Liu et al., 2010b). Recent studies further elucidate that this blockade results from impaired oxidative folding of wild-type proinsulin in the ER. Using tris-tricine-urea SDS-PAGE that can distinguish wild-type and Akita proinsulin under both reducing and non-reducing conditions, it was found that more than 50% of wild-type proinsulin molecules (i.e. more than double compared to that in normal beta cells) are misfolded in Akita islets (Liu et al., 2010a). The effect of misfolded mutant proinsulin on the folding of wild-type proinsulin may be direct and/or indirect. It has been shown that mutant proinsulin can abnormally interact with wild-type proinsulin, forming intermolecular disulfide-linked protein complexes and directly impairing the folding of wild-type proinsulin (Liu et al., 2010a; Liu et al., 2012a). Additionally, misfolded mutant proinsulin may affect the folding of wild-type proinsulin indirectly by inducing ER stress and disturbing the ER protein folding environment.
2.2 Secondary proinsulin misfolding
Proinsulin appears to be a tough molecule to fold in the ER. As noted above, the folding pathway of proinsulin in the ER may be impacted adversely by an unfavorable ER folding environment. It has been recently shown that chemically synthetic proinsulin can robustly refold in vitro at pH 10; under these conditions the folding of some MIDY mutants can be accomplished successfully (Avital-Shmilovici et al., 2014). However, under condition of physiological pH within the ER, the folding environment seems to be less favorable for proinsulin folding. A study examining stability, secretion and aggregation of wild-type proinsulin in several non-beta cell lines found that none of them could efficiently fold proinsulin (Zhu et al., 2004). In fact, accumulating evidence suggests that, even in normal beta cells, up to 20% of newly synthesized wild-type proinsulin fails to achieve its native folding structure (Liu et al., 2010a; Liu et al., 2005; Wang et al., 2011; Wang and Osei, 2011), suggesting that during biosynthesis, a certain amount of misfolded proinsulin is expected as a by-product. Under normal physiological conditions, these misfolded wild-type proinsulin molecules can either be refolded to their native structure or degraded through ER associated degradation (ERAD) and autophagy (Bachar-Wikstrom et al., 2013a; Bachar-Wikstrom et al., 2013b; Tiwari et al., 2013).
Therefore, such misfolding does not compromise normal beta cell function. However, under certain pathological conditions (discussed in sections 3–5), the total amount of misfolded wild-type proinsulin increases, exceeding the genetically-determined capacity of beta cells to handle the misfolded protein load and leading to a build-up of misfolded wild-type proinsulin. At a certain threshold level, accumulated misfolded proinsulin can potentially propagate misfolding onto the remaining proinsulin, culminating in pancreatic beta cell toxicity. Since increased wild-type proinsulin misfolding is secondary to the defective ER folding environment, we call this “secondary proinsulin misfolding.” Indeed, increased misfolding of wild-type proinsulin can be considered as a “second hit” to beta cells, further aggravating ER dysfunction and leading to beta cell failure and diabetes (Fig. 4).
3. Unfolded protein response and proinsulin misfolding in a defective ER folding environment
3.1 ER homeostasis and ER stress response in beta cells
Disturbance of ER protein homeostasis is known to promote the development of neurodegenerative diseases (Hetz and Mollereau, 2014) and is linked to diabetes as well. In all eukaryotic cells, ER homeostasis is maintained by at least three known branches of unfolded protein response (UPR) signaling that are initiated by three distinct ER-localized transmembrane proteins: PERK [PKR-like ER kinase], IRE1 (inositol-requiring transmembrane kinase/endonuclease 1), and ATF6 (activating transcription factor 6) (Walter and Ron, 2011; Zhang and Kaufman, 2008). These three branches act in concert to monitor conditions in the ER, sense protein load and folding capacity, and coordinately regulate gene expression and protein modification to match the needs of ER protein folding (Papa, 2012; Ron and Walter, 2007; Ronald and Cavener, 2007; Scheuner and Kaufman, 2008). In the absence of ER stress, PERK, IRE1and ATF6 proteins are mostly sequestered in inactive complexes with the ER chaperone BiP. Although the precise UPR activation mechanism is not fully understood (Bernales et al., 2006; Credle et al., 2005; Gardner and Walter, 2011; Kimata et al., 2007), activation is correlated with dissociation of BiP from these proximal ER-stress sensor proteins (Todd et al., 2008). Upon the accumulation of unfolded proteins that preferentially bind BiP, each of the proximal ER stress sensors become activated in their own ways, including autophosphorylation of IRE1 and PERK, and trafficking of ATF6 to the Golgi complex.
The physiological function of UPR is to limit the accumulation of misfolded protein in the ER. Acute activation of UPR reduces general protein translation to decrease protein load, increases the surface area of ER membrane, and induces expression of ER chaperones that increase protein folding capacity and promote clearance of unfolded/misfolded proteins through ERAD (Guerriero, 2012; Schuck et al., 2009). These responses help cells maintain and re-establish protein homeostasis. However, if persistent ER stress leads to prolonged UPR activation, cell death signaling is activated and apoptosis is initiated (Shore et al., 2011). Thus, in cells experiencing ER stress, a life or death decision depends on whether ER stress can be alleviated (Papa, 2012; Tabas and Ron, 2011; Walter and Ron, 2011).
Impaired UPR pathways have been linked to beta cell failure and diabetes in both humans and animal models (Scheuner et al., 2005; Senée et al., 2004). PERK was the first of the ER stress-sensors found to be tightly linked to beta cell function (Harding and Ron, 2002; Harding HP, 2001). PERK is also known as eukaryotic translation initiation factor 2-alpha (eIF2alpha) kinase-3. PERK and other eIF2alpha kinases phosphorylate Ser51 of eIF2alpha; this phosphorylation is critically important for inhibition of general protein translation. PERK-mediated phosphorylation of eIF2alpha has emerged as an important negative regulatory step for proinsulin biosynthesis. Based on this, one might expect that less PERK activity would be better for beta cells, as it may lead to more proinsulin synthesis. However, results appear to be precisely the opposite, as absence of PERK function triggers human Wolcott-Rallison Syndrome characterized by permanent neonatal diabetes with beta cell failure and multiple other defects (Delépine et al., 2000; Senée et al., 2004). Perk knockout mice show phenotypes similar to those found in humans with Wolcott-Rallison Syndrome (Harding HP, 2001; Iida et al., 2007; Zhang et al., 2002), including early onset diabetes due to beta cell loss. This has been proposed to be a consequence of beta cell death following unsuppressed translation that leads to exuberant proinsulin biosynthesis (Harding HP, 2001), which is associated with increased proinsulin misfolding (Liu et al., 2005). A similar phenotype of increased proinsulin misfolding has been observed in islet beta cells of mice bearing mutation of eIF2alpha-S51A (Scheuner et al., 2005). Additionally, Perk knockout mice (Zhang et al., 2006) exhibit reduced beta cell proliferation (Cavener et al., 2010; Zhang et al., 2006). Altogether, there is general agreement that PERK-mediated phosphorylation of eIF2alpha plays an important role in regulating proinsulin biosynthesis, folding, trafficking, and ER quality control in pancreatic beta cells (Gupta et al., 2010; Harding et al., 2012).
The transmembrane kinase and endonuclease, IRE1, also appears to be vitally important for ER homeostasis in pancreatic beta cells. IRE1 represents the most evolutionarily conserved UPR branch. Mammals have two IRE1 homologs, α and β (Tirasophon et al., 1998; Wang et al., 1998), both of which are activated by the accumulation of unfolded proteins in the ER. Whereas the expression of Ire1β is limited to intestinal epithelial cells, IRE1α is expressed at particularly high levels in the pancreas and placenta (Tirasophon et al., 1998). Deletion of mouse IRE1 α results in embryonic lethality (Urano et al., 2000), but deletion of IRE1α and IRE1β does not prevent transcriptional activation of UPR target genes such as BiP and GRP94 (Urano et al., 2000; Wang et al., 1998), indicating overlap in the ultimate target gene activation by the different ER stress sensors. IRE1α possesses both endonuclease and kinase activities. Its endonuclease activity splices the mRNA that encodes active X-box binding protein 1 (XBP1). Specifically, upon activation of UPR, IRE1 cleaves a 26-nucleotide intron from unspliced XBP1 mRNA, leading to a translational frame shift to produce a potent active transcription factor that binds to ER stress-response elements of target genes involved in protein folding and degradation (Calfon et al., 2002; Harding and Ron, 2002; Kaufman, 2002; Shen et al., 2001). Unlike its endonuclease activity, the kinase activity of IRE1α appears to be regulated also by glucose in pancreatic beta cells (Lipson et al., 2006). Interestingly, glucose stimulated phosphorylation of IRE1α is not correlated with XBP1 splicing and dissociation from BiP, suggesting a specific regulating mechanism of IRE1α. Importantly, in pancreatic beta cells, acute elevation of glucose selectively activates IRE1α kinase activity that positively regulates insulin biosynthesis, whereas chronic exposure to high glucose results in severe stress, including activation of both kinase and endonuclease activities that may contribute to glucotoxicity and beta cell failure (Corbett, 2006; Lipson et al., 2006). Under high/prolonged ER stress, the catalytic activity of IRE1α is regulated in a complex manner to trigger divergent outcomes (Han et al., 2009) including ER stress-induced Ins mRNA decay and beta cell apoptosis. More recently, a family of small molecules called KIRAs (Kinase-inhibiting RNase Attenuators) was found to increase insulin content and secretion from Akita islets and improve blood glucose excursion during glucose tolerance testing (Ghosh et al., 2014). Thus, like PERK, IRE1 is also critical for maintaining beta cell function.
ATF6 is a type II ER transmembrane glycoprotein that undergoes intracellular transport from the ER to the Golgi complex in states of ER stress. At the Golgi complex, ATF6 is proteolytically processed to liberate its N-terminal 50kDa domain that migrates to the nucleus to function as a transcription factor regulating the expression of ER stress response genes including BiP, GRP94, ERO1β, and ERAD components such as HERP (Adachi et al., 2008; Chen et al., 2002; Shen et al., 2002; Yamaguchi et al., 2008). In pancreatic beta cells in culture, ATF6 knockdown leads to increased phosphorylation of JNK, p38, and c-Jun and renders cells susceptible to apoptosis. Inhibition of JNK or p38 kinases prevents the apoptosis associated with ATF6 deficiency, suggesting that ATF6 may play a role in maintaining β-cell survival even in the absence of ER stress (Teodoro et al., 2012). However, in mice with global ATF6α deletion, there was no detectable beta cell defect or diabetes phenotype when mice were fed normal chow, although the mice were less glucose tolerant, with a mild reduction of islet insulin content when fed with a high-fat diet (Usui et al., 2012; Yamamoto et al., 2007). The data suggest that ATF6 may play a more important role in beta cells under stress conditions.
3.2. Genetic examples of secondary proinsulin misfolding in pancreatic beta cells
As noted above, a growing body of evidence indicates that a defective ER folding environment impairs the folding pathway of wild-type proinsulin, leading to secondary proinsulin misfolding. Although it has been shown that mild acute ER stress (induced by low dose tunicamycin) does not significantly impair ER export of proinsulin (Liu et al., 2010a), chronic ER stress with dysfunction of ER homeostasis, including dysregulation of proinsulin biosynthesis in Perk deficient mice or mice bearing knock-in of eIF2alpha-S51, clearly impairs oxidative folding of wild-type proinsulin, leading to an increase in mispaired disulfide isomers that are recognized and bound by the ER chaperone BiP (Liu et al., 2005; Scheuner et al., 2005). Recently, increased wild-type proinsulin misfolding was also found in defective ER folding environments caused by defects in ER oxidoreductases (Rajpal et al., 2012; Zito et al., 2010) or by the expression of misfolded mutant proinsulin (Liu et al., 2010a; Wright et al., 2013a; Wright et al., 2013b). Of course, accumulation of misfolded proinsulin further aggravates ER stress and dysfunction. Thus, increased wild-type proinsulin misfolding may be a common pathway contributing to beta cell failure and diabetes from multiple initial insults.
4. ER stress and proinsulin misfolding in type 2 diabetes
Insulin resistance has long been considered a hallmark of type 2 diabetes. However, insulin resistance alone does not lead to the onset of diabetes. In fact, most individuals who are insulin resistant do not develop diabetes (Costes et al., 2013; Kitamura, 2013). Accumulating genetic and biological evidence indicates that in insulin resistant individuals, failure of beta cell compensation for increasing metabolic demand is linked to the development of overt diabetes (Costes et al., 2013; Scott et al., 2007; Zeggini et al., 2007). Factors that have been suggested to induce beta cell failure (as well as ER stress, see below) include hyperglycemia and hyperlipidemia, i.e., glucolipotoxicity [for review, please see (Back and Kaufman, 2012; Biden et al., 2014)], pro-inflammatory cytokines (Gurzov et al., 2009), and accumulation of islet amyloid polypeptide (Huang et al., 2007).
The process of normal beta cell compensation for insulin resistance has two components: an expansion of beta cell mass and an increase in beta cell insulin output (Bell and Polonsky, 2001; Leahy, 2005; Marchetti et al., 2010; Rhodes, 2005). Failure of compensatory expansion of beta cell mass involves a combination of increased dedifferentiation and apoptosis along with decreased proliferation/replication (Butler et al., 2003; Kitamura, 2013; Prentki and Nolan, 2006; Sachdeva and Stoffers, 2009; Saisho et al., 2012; Talchai et al., 2012; Weir et al., 2013). Inadequacy of beta cell insulin output includes insufficient compensatory increase of secretory capacity and inadequate insulin biosynthesis (Bell and Polonsky, 2001; Ma et al., 2012).
It remains unclear how these two primary components of beta cell compensation are mechanistically connected. However, both components can be affected by chronic activation of UPR, indicating that ER stress may play an important role in successful/unsuccessful beta cell compensation in type 2 diabetes (Fonseca et al., 2009; Fonseca et al., 2010b; Lipson et al., 2006; Oslowski and Urano, 2011; Scheuner and Kaufman, 2008). Indeed, it has been reported that beta cells exhibit elevated ER stress markers during the development and progression of type 2 diabetes both in humans (Huang et al., 2007; Laybutt et al., 2007) and in db/db diabetic mice (Laybutt et al., 2007). By comparing the pancreatic samples from non-diabetic controls and type 2 diabetes patients, Marchetti et al found an increase in both ER volume density and beta cell apoptosis in type 2 diabetes. Moreover, BiP, XBP1 and C/EBP homologus protein (also know as CHOP) were significantly induced when isolated islets from type 2 diabetes patients were incubated with high glucose, suggesting that beta cells in type 2 diabetes are more susceptible to high glucose-induced ER stress (Marchetti et al., 2007).
A key factor that may allow advance from ER stress to beta cell failure is protein overload. As the most abundant beta cell secretory protein, proinsulin is the primary driver of ER protein load. In response to insulin resistance and with typical postprandial blood glucose excursions, proinsulin biosynthesis can rise up to 50 fold (Goodge and Hutton, 2000), and the rate of insulin mRNA translation can approach one million proinsulin molecules per minute per cell. Folding at this rate means the formation of three million proinsulin disulfide bonds per minute (Liu et al., 2014b; Otero et al., 2010; Papa, 2012; Scheuner and Kaufman, 2008) and if 20% of newly synthesized proinsulin molecules fail to achieve native folding, then there is an additional need for isomerization of improper disulfide bonds from ER-retained proinsulin. Moreover, there may be a finite capacity of ERAD that may also contribute to proinsulin accumulation in the ER. As beta cells are susceptible to misfolding in states of oversynthesis of proinsulin, it is expected that the absolute amount of the misfolded proinsulin would rise in parallel with a compensatory increase in beta cell insulin production. Although this may explain the observation of ER stress activation in type 2 diabetes, no study has explicitly focused on detection of increased proinsulin misfolding in the beta cells of type 2 diabetes patients or animal models.
The importance of potential defects in ER function during the development and progression of type 2 diabetes may also be implied by Genome-Wide Association Studies (GWAS) in which many type 2 diabetes-associated genes, including CAMK1D, CDC123, NOTCH2, THADA, VEGFA, IRS1, WFS1, ADAMTS9, and SLC16A11, have been found to be involved in maintaining normal ER function and ER stress response (Bonnefond et al., 2010; Dombroski et al., 2010). Of interest, WFS1 (Wolfram syndrome 1) encodes an ER transmembrane protein that modulates ER calcium, which is an important factor necessary for normal ER function and secretory protein folding. In mice, WFS1 deficiency causes increased ER stress and beta cell death (Fonseca et al., 2010a; Riggs et al., 2005; Yamada et al., 2006); and in humans, mutations in WFS1 are associated with Wolfram Syndrome, including early onset insulin-dependent diabetes (Rigoli and Di Bella, 2012). SLC16A11 has been recently found to be a common risk factor for type 2 diabetes in Mexico. SLC16A11 protein localizes in the ER and is thought to play a role as an intracellular lipid transporter (Consortium, 2014). ADAMTS9 is a member of the secreted metalloprotease family that is known to digest extracellular matrix proteins outside of cells. A recent study shows that down-regulation of ADAMTS9 inhibits protein transport from the ER to the Golgi complex (Yoshina et al., 2012). Together, these studies suggest possible relationships between ER homeostasis and beta cell function in type 2 diabetes.
5. ER stress and proinsulin misfolding in type 1 diabetes
Type 1 diabetes is an autoimmune disease, in which pancreatic beta cells are destroyed by autoimmune attack, leading to absolute insulin deficiency (Atkinson et al., 2011; Jaberi-Douraki et al., 2014). Although it is still not completely understood how the autoimmunity against beta cells is initiated, islet autoantigens are thought to play a critical role in the development of type 1 diabetes (Nakayama et al., 2005; Pathiraja et al., 2014). Importantly, most of the known islet antoantigens are secretory and/or secretory-associated proteins [including (pro)insulin, zinc transporter-8, glutamic acid decarboxylase-65, IA2 (also known as ICA512), and IA2β (also known as phogrin)] (Arvan et al., 2012). When beta cell lines or isolated islets are incubated with proinflammatory cytokines [interleukin 1β (IL-1β), γ-interferon (IFN-γ), and tumor necrosis factor-α (TNF-α)], ER stress response pathways are activated, along with increased production of inducible nitric oxide synthase (iNOS) (Cardozo et al., 2005; Chambers et al., 2008). An in vivo study using NOD mice (an established mouse model of type 1 diabetes) found an age-dependent activation of ER stress signaling (that may be mediated by activation of NF-κB) during the development and progression of diabetes (Tersey et al., 2012). In a more recent study, defects in UPR signaling, especially in ATF6 and XBP1 branches, were not only confirmed in NOD mice, but also in beta cells of type 1 diabetes patients (Engin et al., 2013). These studies suggest that autoimmune attack by infiltration of macrophages and T cells into islets may directly target the beta cell secretory pathway, especially the ER, causing ER stress that may contribute to beta cell failure in type 1 diabetes.
In addition to autoimmunity, other factors may also contribute to aggravating beta cell ER stress after the onset of type 1 diabetes. At the time of initial clinical presentation of the type 1 diabetes phenotype, it is estimated that 60–90% of beta cells are already destroyed (Oldstone et al., 1984; Van Belle et al., 2011). To compensate for loss of these beta cells, the remaining beta cells need to produce more insulin. This increasing insulin biosynthesis along with metabolic stress caused by elevated blood glucose and fatty acids may further aggravate ER stress in residual beta cells. Moreover, in beta cell lines that are incubated with TNF-α, proinsulin folding, maturation, and ER export have been found to be adversely affected (Wang et al., 2011), although this observation has not yet been validated in vivo. Further, another recent study finds that the ratio of proinsulin to insulin is significantly elevated even in pre-diabetic NOD mice, suggesting that functional beta cell defects (involving proinsulin folding, intracellular trafficking and processing) may precede the clinical presentation of type 1 diabetes (Maganti et al., 2014; Tersey et al., 2012). Thus, for a variety of reasons, ER stress is likely to be an important contributor to the development and progression of type 1 diabetes, although it remains unknown whether the normal folding pathway of proinsulin is affected in these stressed beta cells.
6. Perspectives
The more we learn in this area, the more our studies point out how much we still do not know. For example, it has been established that misfolded proinsulin causes beta cell failure in a dose dependent manner (Hodish et al., 2011; Liu et al., 2012a; Renner et al., 2013), yet it remains unknown whether a specific amount of misfolded proinsulin must first be present before beta cell failure ensues. Given that increased misfolding of wild-type proinsulin occurs under some pathological conditions, further studies are needed to determine the threshold of misfolded proinsulin required to trigger beta cell failure in common forms of diabetes (Fig. 5), as well as to identify genetic modifiers that may render some individuals more susceptible than others.
Figure 5. Hypothetical model illustrating the progression of beta cell failure as a consequence of increased proinsulin misfolding.

During the development and progression of diabetes, increased proinsulin misfolding (red arrow) caused by genetic and environmental factors can induce ER stress and decrease insulin production (blue arrow). As misfolded proinsulin exceeds the genetically-determined threshold (dashed line) that beta cells can handle the misfolded protein load, diabetes occurs. Elevated blood glucose further stimulates proinsulin biosynthesis, producing more misfolded proinsulin load in the ER, aggravating ER stress and beta cell failure.
Another challenge to understanding the role of proinsulin misfolding in diabetes is the requirement for isolated live islets or beta cells to definitively measure proinsulin misfolding during its biosynthesis. Thus, suitable biomarkers are needed. Given the development of conformation-dependent antibodies for a-1 antitrypsin (Miranda et al., 2010) and IAPP (Kayed et al., 2007), it might be possible to develop antibodies that specifically recognize misfolded proinsulin in the plasma (perhaps, released from dead/dying beta cells) of living patients. Such antibodies might also be useful for the study of tissue sections from cohorts of cadaveric samples of diabetes patients at different stages of the disease. Such an approach could allow the development of diagnostic tools to improve our understanding of disease pathogenesis.
In addition, at this point we do not know how wild-type proinsulin “knows” how to form the correct disulfide pairings, and conversely, nor do we understand why non-cysteine MIDY mutants “become confused” in the proinsulin folding pathway. Presumably, the MIDY mutations alter the most sensitive folding steps involved in proper alignment of the B and A domains of proinsulin so as to perturb native proinsulin disulfide bond formation. It is possible that the collection of MIDY mutants may serve as nature’s best molecular models to highlight the most challenging kinetic obstacles to proinsulin folding, which are likely to involve those regions most in need of assistance by foldase enzymes and ER molecular chaperones. Indeed, it is still a central question to understand which ER oxidoreductases and chaperones are required for proper proinsulin folding, and to understand how those activities are upregulated in states of increased proinsulin load.
Further, the importance of proinsulin dimerization remains mysterious – is dimerization the basis by which misfolded proinsulin blocks the ER exit of bystander proinsulin molecules? If so, the dimerization interface might be a potential therapeutic target site that could prevent abnormal interactions between wild-type and mutant proinsulin and allow wild-type proinsulin export from the ER to produce mature bioactive insulin. Moreover, given the prominence of ER stress in the development and progression of type 1, type 2, and some monogenic diabetes, novel strategies focusing on alleviating ER stress by modulating UPR signaling might provide new therapeutic opportunities to prevent or delay beta cell failure and diabetes.
Finally, when proinsulin is misfolded, how do beta cells identify such molecules (compared to newly-synthesized wild-type proinsulin that must always begin as an unfolded species) and target these molecules for ERAD? Does degradation of misfolded proinsulin exhibit any unique beta cell-specific features? As we begin to better understand the functions of the beta cell ER, we hope that answers to these and related questions will clarify how defective proinsulin folding leads to beta cell failure and diabetes.
Acknowledgements
This work was supported by NIH RO1-DK088856 (to M.L.), NIH RO1-DK-48280 (to P.A.), the research grants from the National Natural Science Foundation of China 81070629 and 81370895. Jingqiu Cui was supported in part by research grants from the Key Project of applied basis research program of Tianjin Scientific and Technical Committee (11JCZDJC18500) and Novo Nordisk β-cell Academy. The authors thank Michael Weiss (Case-Western Reserve U.) and Donald Steiner (U. Chicago) for helpful discussions. The authors thank Yiran Liu for helping to edit the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 Is a Transcription Factor Specializing in the Regulation of Quality Control Proteins in the Endoplasmic Reticulum. Cell Structure and Function. 2008;33(1):75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- Alarcón C, Lincoln B, Rhodes CJ. The biosynthesis of the subtilisin-related proprotein convertase PC3, but no that of the PC2 convertase, is regulated by glucose in parallel to proinsulin biosynthesis in rat pancreatic islets. Journal of Biological Chemistry. 1993;268(6):4276–4280. [PubMed] [Google Scholar]
- Alon A, Grossman I, Gat Y, Kodali VK, DiMaio F, Mehlman T, Haran G, Baker D, Thorpe C, Fass D. The dynamic disulphide relay of quiescin sulphydryl oxidase. Nature. 2012;488(7411):414–418. doi: 10.1038/nature11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appenzeller-Herzog C. Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. Journal of Cell Science. 2011;124(6):847–855. doi: 10.1242/jcs.080895. [DOI] [PubMed] [Google Scholar]
- Arvan P, Pietropaolo M, Ostrov D, Rhodes CJ. Islet Autoantigens: Structure, Function, Localization, and Regulation. Cold Spring Harbor Perspectives in Medicine. 2012;2(8) doi: 10.1101/cshperspect.a007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, Pietropaolo M, Arvan PR, Von Herrath M, Markel DS, Rhodes CJ. How Does Type 1 Diabetes Develop?: The Notion of Homicide or β-Cell Suicide Revisited. Diabetes. 2011;60(5):1370–1379. doi: 10.2337/db10-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avital-Shmilovici M, Whittaker J, Weiss MA, Kent SBH. Deciphering a Molecular Mechanism of Neonatal Diabetes Mellitus by the Chemical Synthesis of a Protein Diastereomer, [d-AlaB8]Human Proinsulin. Journal of Biological Chemistry. 2014;289(34):23683–23692. doi: 10.1074/jbc.M114.572040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachar-Wikstrom E, Wikstrom J, Kaiser N, Cerasi E, Leibowitz G. Improvement of ER stress-induced diabetes by stimulating autophagy. Autophagy. 2013a;9(4):626–628. doi: 10.4161/auto.23642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachar-Wikstrom E, Wikstrom JD, Ariav Y, Tirosh B, Kaiser N, Cerasi E, Leibowitz G. Stimulation of Autophagy Improves Endoplasmic Reticulum Stress-Induced Diabetes. Diabetes. 2013b;62(4):1227–1237. doi: 10.2337/db12-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SH, Kaufman RJ. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annual Review of Biochemistry. 2012;81(1):767–793. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in [beta]-cell function. Nature. 2001;414(6865):788–791. doi: 10.1038/414788a. [DOI] [PubMed] [Google Scholar]
- Bernales S, Papa FR, Walter P. Intracellular Signaling by the Unfolded Protein Response. Annual Review of Cell and Developmental Biology. 2006;22(1):487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- Biden TJ, Boslem E, Chu KY, Sue N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends in Endocrinology & Metabolism. 2014;25(8):389–398. doi: 10.1016/j.tem.2014.02.003. [DOI] [PubMed] [Google Scholar]
- Blundell T, Dodson G, Hodgkin D, Mercola D. Insulin: The Structure in the Crystal and its Reflection in Chemistry and Biology by. In: Anfinsen CB, JTE, Frederic MR, editors. Advances in Protein Chemistry. Academic Press; 1972. pp. 279–402. [Google Scholar]
- Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends in Molecular Medicine. 2010;16(9):407–416. doi: 10.1016/j.molmed.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Brange J, Ribel U, Hansen JF, Dodson G, Hansen MT, Havelund S, Melberg SG, Norris F, Norris K, Snel L, Sorensen AR, Voigt HO. Monomeric insulins obtained by protein engineering and their medical implications. Nature. 1988;333(6174):679–682. doi: 10.1038/333679a0. [DOI] [PubMed] [Google Scholar]
- Bulleid NJ. Disulfide Bond Formation in the Mammalian Endoplasmic Reticulum. Cold Spring Harbor Perspectives in Biology. 2012;4(11) doi: 10.1101/cshperspect.a013219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-Cell Deficit and Increased β-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. 2002;415(6867):92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Cardozo AK, Ortis F, Storling J, Feng Y-M, Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz A, Eizirik DL. Cytokines Downregulate the Sarcoendoplasmic Reticulum Pump Ca2+ ATPase 2b and Deplete Endoplasmic Reticulum Ca2+, Leading to Induction of Endoplasmic Reticulum Stress in Pancreatic β-Cells. Diabetes. 2005;54(2):452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- Cavener DR, Gupta S, McGrath BC. PERK in beta cell biology and insulin biogenesis. Trends in Endocrinology & Metabolism. 2010;21(12):714–721. doi: 10.1016/j.tem.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers KT, Unverferth JA, Weber SM, Wek RC, Urano F, Corbett JA. The Role of Nitric Oxide and the Unfolded Protein Response in Cytokine-Induced β-Cell Death. Diabetes. 2008;57(1):124–132. doi: 10.2337/db07-0944. [DOI] [PubMed] [Google Scholar]
- Chang S, Choi K, Jang S, Shin H. Role of disulfide bonds in the structure and activity of human insulin. Molecules and Cells. 2003;16(3):323–330. [PubMed] [Google Scholar]
- Chen X, Shen J, Prywes R. The Luminal Domain of ATF6 Senses Endoplasmic Reticulum (ER) Stress and Causes Translocation of ATF6 from the ER to the Golgi. Journal of Biological Chemistry. 2002;277(15):13045–13052. doi: 10.1074/jbc.M110636200. [DOI] [PubMed] [Google Scholar]
- Consortium TSTD. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506(7486):97–101. doi: 10.1038/nature12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett JA. Insulin biosynthesis: The IREny of it all. Cell Metabolism. 2006;4(3):175–176. doi: 10.1016/j.cmet.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Costes S, Langen R, Gurlo T, Matveyenko AV, Butler PC. β-Cell Failure in Type 2 Diabetes: A Case of Asking Too Much of Too Few? Diabetes. 2013;62(2):327–335. doi: 10.2337/db12-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(52):18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop G, Julier C. EIF2AK3, encoding translation initiation factor 2- kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nature Genetics. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- Dodson G, Steiner DF. The role of assembly in insulin's biosynthesis. Curr Opin Struct Biol. 1998;8(2):189–194. doi: 10.1016/s0959-440x(98)80037-7. [DOI] [PubMed] [Google Scholar]
- Dombroski BA, Nayak RR, Ewens KG, Ankener W, Cheung VG, Spielman RS. Gene Expression and Genetic Variation in Response to Endoplasmic Reticulum Stress in Human Cells. The American Journal of Human Genetics. 2010;86(5):719–729. doi: 10.1016/j.ajhg.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr Rev. 2008;29(1):42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6(1):28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, Mathis D, Hotamisligil GS. Restoration of the Unfolded Protein Response in Pancreatic β Cells Protects Mice Against Type 1 Diabetes. Science Translational Medicine. 2013;5(211):211ra156. doi: 10.1126/scitranslmed.3006534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M, Schumm-Draeger PM, Vora J, King AB. A review of modern insulin analogue pharmacokinetic and pharmacodynamic profiles in type 2 diabetes: improvements and limitations. Diabetes, Obesity and Metabolism. 2011;13(8):677–684. doi: 10.1111/j.1463-1326.2011.01395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca SG, Burcin M, Gromada J, Urano F. Endoplasmic reticulum stress in [beta]-cells and development of diabetes. Current Opinion in Pharmacology. 2009;9(6):763–770. doi: 10.1016/j.coph.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic beta cell death. Trends in Endocrinology & Metabolism. 2011;22(7):266–274. doi: 10.1016/j.tem.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, Hayashi E, Ishihara H, Oka Y, Permutt MA, Urano F. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. The Journal of Clinical Investigation. 2010a;120(3):744–755. doi: 10.1172/JCI39678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca SG, Urano F, Burcin M, Gromada J. Stress hypERactivation in the β-cell. Islets. 2010b;2(1):1–9. doi: 10.4161/isl.2.1.10456. [DOI] [PubMed] [Google Scholar]
- Frand AR, Kaiser CA. Ero1p Oxidizes Protein Disulfide Isomerase in a Pathway for Disulfide Bond Formation in the Endoplasmic Reticulum. Molecular Cell. 1999;4(4):469–477. doi: 10.1016/s1097-2765(00)80198-7. [DOI] [PubMed] [Google Scholar]
- Frank B, Veros A. Interaction of zinc with proinsulin. Biochem Biophys Res Commun. 1970;38(2):284–289. doi: 10.1016/0006-291x(70)90710-2. [DOI] [PubMed] [Google Scholar]
- Gardner BM, Walter P. Unfolded Proteins Are Ire1-Activating Ligands That Directly Induce the Unfolded Protein Response. Science. 2011;333(6051):1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R, Wang L, Wang Eric S, Perera B, Gayani K, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, Weiberth Kurt F, Gliedt Micah J, Alavi Marcel V, Hari Sanjay B, Mitra Arinjay K, Bhhatarai B, Schürer Stephan C, Snapp Erik L, Gould Douglas B, German Michael S, Backes Bradley J, Maly Dustin J, Oakes Scott A, Papa Feroz R. Allosteric Inhibition of the IRE1α RNase Preserves Cell Viability and Function during Endoplasmic Reticulum Stress. Cell. 2014;158(3):534–548. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodge KA, Hutton JC. Translational regulation of proinsulin biosynthesis and proinsulin conversion in the pancreaticβ-cell. Seminars in Cell & Developmental Biology. 2000;11(4):235–242. doi: 10.1006/scdb.2000.0172. [DOI] [PubMed] [Google Scholar]
- Guerriero CJBJL. The Delicate Balance Between Secreted Protein Folding and Endoplasmic Reticulum-Associated Degradation in Human Physiology. Physiological Reviews. 2012;92(2):537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Feng Y. Effects of Cysteine to Serine Substitutions in the Two Inter-Chain Disulfide Bonds of Insulin. Biological Chemistry. 2001;382(3):443. doi: 10.1515/BC.2001.054. [DOI] [PubMed] [Google Scholar]
- Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) Regulates Proinsulin Trafficking and Quality Control in the Secretory Pathway. Diabetes. 2010;59(8):1937–1947. doi: 10.2337/db09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurzov EN, Ortis F, Cunha DA, Gosset G, Li M, Cardozo AK, Eizirik DL. Signaling by IL-1[beta]+IFN-[gamma] and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic [beta]-cell apoptosis. Cell Death Differ. 2009;16(11):1539–1550. doi: 10.1038/cdd.2009.99. [DOI] [PubMed] [Google Scholar]
- Haataja L, Snapp E, Wright J, Liu M, Hardy AB, Wheeler MB, Markwardt ML, Rizzo M, Arvan P. Proinsulin Intermolecular Interactions during Secretory Trafficking in Pancreatic β Cells. Journal of Biological Chemistry. 2013;288(3):1896–1906. doi: 10.1074/jbc.M112.420018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton J-P, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1[alpha] Kinase Activation Modes Control Alternate Endoribonuclease Outputs to Determine Divergent Cell Fates. Cell. 2009;138(3):562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Ron D. Endoplasmic Reticulum Stress and the Development of Diabetes: A Review. Diabetes. 2002;51(suppl 3):S455–S461. doi: 10.2337/diabetes.51.2007.s455. [DOI] [PubMed] [Google Scholar]
- Harding HPZH, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell. Mol Cell. 2001;7(6):1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zyryanova AF, Ron D. Uncoupling Proteostasis and Development in Vitro with a Small Molecule Inhibitor of the Pancreatic Endoplasmic Reticulum Kinase, PERK. Journal of Biological Chemistry. 2012;287(53):44338–44344. doi: 10.1074/jbc.M112.428987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15(4):233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Hober S, Forsberg G, Palm G, Hartmanis M, Nilsson B. Disulfide exchange folding of insulin-like growth factor I. Biochemistry. 1992;31(6):1749–1756. doi: 10.1021/bi00121a024. [DOI] [PubMed] [Google Scholar]
- Hodish I, Absood A, Liu L, Liu M, Haataja L, Larkin D, Al-Khafaji A, Zaki A, Arvan P. In Vivo Misfolding of Proinsulin Below the Threshold of Frank Diabetes. Diabetes. 2011;60(8):2092–2101. doi: 10.2337/db10-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Q-X, Gozani SN, Chance RE, Hoffmann JA, Frank BH, Weiss MA. Structure of a protein in a kinetic trap. Nat Struct Mol Biol. 1995;2(2):129–138. doi: 10.1038/nsb0295-129. [DOI] [PubMed] [Google Scholar]
- Hua Q-X, Hu S-Q, Frank BH, Jia W, Chu Y-C, Wang S-H, Burke GT, Katsoyannis PG, Weiss MA. Mapping the Functional Surface of Insulin by Design: Structure and Function of a Novel A-Chain Analogue. Journal of Molecular Biology. 1996a;264(2):390–403. doi: 10.1006/jmbi.1996.0648. [DOI] [PubMed] [Google Scholar]
- Hua Q-X, Jia W, Frank BH, Phillips NFB, Weiss MA. A Protein Caught in a Kinetic Trap: Structures and Stabilities of Insulin Disulfide Isomers†. Biochemistry. 2002;41(50):14700–14715. doi: 10.1021/bi0202981. [DOI] [PubMed] [Google Scholar]
- Hua Q-X, Narhi L, Jia W, Arakawa T, Rosenfeld R, Hawkins N, Miller JA, Weiss MA. Native and Non-native Structure in a Protein-folding Intermediate: Spectroscopic Studies of Partially Reduced IGF-I and an Engineered Alanine Model. Journal of Molecular Biology. 1996b;259(2):297–313. doi: 10.1006/jmbi.1996.0320. [DOI] [PubMed] [Google Scholar]
- Hua QX, Liu M, Hu SQ, Jia W, Arvan P, Weiss MA. A conserved histidine in insulin is required for the foldability of human proinsulin: structure and function of an ALAB5 analog. J Biol Chem. 2006;281(34):24889–24899. doi: 10.1074/jbc.M602617200. [DOI] [PubMed] [Google Scholar]
- Huang C-j, Lin C-y, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC. High Expression Rates of Human Islet Amyloid Polypeptide Induce Endoplasmic Reticulum Stress-Mediated β-Cell Apoptosis, a Characteristic of Humans With Type 2 but Not Type 1 Diabetes. Diabetes. 2007;56(8):2016–2027. doi: 10.2337/db07-0197. [DOI] [PubMed] [Google Scholar]
- Huang XF, Arvan P. Intracellular Transport of Proinsulin in Pancreatic β-Cells: STRUCTURAL MATURATION PROBED BY DISULFIDE ACCESSIBILITY. Journal of Biological Chemistry. 1995;270(35):20417–20423. doi: 10.1074/jbc.270.35.20417. [DOI] [PubMed] [Google Scholar]
- Iida K, Li Y, McGrath B, Frank A, Cavener D. PERK eIF2 alpha kinase is required to regulate the viability of the exocrine pancreas in mice. BMC Cell Biology. 2007;8(1):38. doi: 10.1186/1471-2121-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaberi-Douraki M, Schnell S, Pietropaolo M, Khadra A. Unraveling the contribution of pancreatic beta-cell suicide in autoimmune type 1 diabetes. Journal of Theoretical Biology. 2014;(0) doi: 10.1016/j.jtbi.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansens A, van Duijn E, Braakman I. Coordinated Nonvectorial Folding in a Newly Synthesized Multidomain Protein. Science. 2002;298(5602):2401–2403. doi: 10.1126/science.1078376. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease 10.1172/JCI200216886. J. Clin. Invest. 2002;110(10):1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Sarsoza F, Saing T, Cotman C, Necula M, Margol L, Wu J, Breydo L, Thompson J, Rasool S, Gurlo T, Butler P, Glabe C. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Molecular Neurodegeneration. 2007;2(1):18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo C, Yang J, Rajpal G, Wang Y, Liu J, Arvan P, Stoffers DA. Endoplasmic Reticulum Oxidoreductin-1-Like β (ERO1lβ) Regulates Susceptibility to Endoplasmic Reticulum Stress and Is Induced by Insulin Flux in β-Cells. Endocrinology. 2011;152(7):2599–2608. doi: 10.1210/en.2010-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. The Journal of Cell Biology. 2007;179(1):75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselar JG, Datt M, Chance MR, Weiss MA. Structural Analysis of Proinsulin Hexamer Assembly by Hydroxyl Radical Footprinting and Computational Modeling. Journal of Biological Chemistry. 2011;286(51):43710–43716. doi: 10.1074/jbc.M111.297853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T. The role of FOXO1 in [beta]-cell failure and type 2 diabetes mellitus. Nat Rev Endocrinol. 2013;9(10):615–623. doi: 10.1038/nrendo.2013.157. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Preston AM, Åkerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50(4):752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- Leahy JL. Pathogenesis of Type 2 Diabetes Mellitus. Archives of Medical Research. 2005;36(3):197–209. doi: 10.1016/j.arcmed.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, Rossini AA, Urano F. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metabolism. 2006;4(3):245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Liu M, Haataja L, Wright J, Wickramasinghe NP, Hua QX, Phillips NF, Barbetti F, Weiss MA, Arvan P. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS One. 2010a;5(10):e13333. doi: 10.1371/journal.pone.0013333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J, Arvan P. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends in Endocrinology & Metabolism. 2010b;21(11):652–659. doi: 10.1016/j.tem.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proceedings of the National Academy of Sciences. 2007;104(40):15841–15846. doi: 10.1073/pnas.0702697104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hua Q-x, Hu S-Q, Jia W, Yang Y, Saith SE, Whittaker J, Arvan P, Weiss MA. Deciphering the Hidden Informational Content of Protein Sequences. Journal of Biological Chemistry. 2010c;285(40):30989–31001. doi: 10.1074/jbc.M110.152645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Lara-Lemus R, Shan S-o, Wright J, Haataja L, Barbetti F, Guo H, Larkin D, Arvan P. Impaired Cleavage of Preproinsulin Signal Peptide Linked to Autosomal-Dominant Diabetes. Diabetes. 2012a;61(4):828–837. doi: 10.2337/db11-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Lara-Lemus R, Shan S-o, Wright J, Haataja L, Barbetti F, Guo H, Larkin D, Arvan P. Impaired Cleavage of Preproinsulin Signal Peptide Linked to Autosomal-Dominant Diabetes. Diabetes. 2012b doi: 10.2337/db11-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Li Y, Cavener D, Arvan P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J Biol Chem. 2005;280(14):13209–13212. doi: 10.1074/jbc.C400475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Ramos-Castañeda J, Arvan P. Role of the Connecting Peptide in Insulin Biosynthesis. Journal of Biological Chemistry. 2003;278(17):14798–14805. doi: 10.1074/jbc.M212070200. [DOI] [PubMed] [Google Scholar]
- Liu M, Sun J, Cui J, Chen W, Guo H, Barbetti F, Arvan P. INS-gene mutations: From genetics and beta cell biology to clinical disease. Molecular Aspects of Medicine. 2014a;(0) doi: 10.1016/j.mam.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wan ZL, Chu YC, Aladdin H, Klaproth B, Choquette M, Hua QX, Mackin RB, Rao JS, De Meyts P, Katsoyannis PG, Arvan P, Weiss MA. Crystal Structure of a "Nonfoldable" Insulin IMPAIRED FOLDING EFFICIENCY DESPITE NATIVE ACTIVITY. Journal of Biological Chemistry. 2009;284(50):35259–35272. doi: 10.1074/jbc.M109.046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wright J, Guo H, Xiong Y, Arvan P. In: Chapter Two - Proinsulin Entry and Transit Through the Endoplasmic Reticulum in Pancreatic Beta Cells. Gerald L, editor. Vitamins & Hormones: Academic Press; 2014b. pp. 35–62. [DOI] [PubMed] [Google Scholar]
- Ma ZA, Zhao Z, Turk J. Mitochondrial Dysfunction and β-Cell Failure in Type 2 Diabetes Mellitus. Experimental Diabetes Research. 2012;11 doi: 10.1155/2012/703538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maganti A, Evans-Molina C, Mirmira RG. From immunobiology to β-cell biology: The changing perspective on type 1 diabetes. Islets. 2014;6(1) doi: 10.4161/isl.28778. 0--1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaisse WJ, Sener A, Herchuelz A, Hutton JC. Insulin release: The fuel hypothesis. Metabolism. 1979;28(4):373–386. doi: 10.1016/0026-0495(79)90111-2. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, Filipponi F, Weir GC, Eizirik DL, Cnop M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50(12):2486–2494. doi: 10.1007/s00125-007-0816-8. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Lupi R, Del Guerra S, Bugliani M, Marselli L, Boggi U. The β-Cell in Human Type 2 Diabetes. In: Islam MS, editor. The Islets of Langerhans. Springer Netherlands; 2010. pp. 501–514. [Google Scholar]
- Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. The EMBO Journal. 2001;20(22):6288–6296. doi: 10.1093/emboj/20.22.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Narhi LO, Hua QX, Rosenfeld R, Arakawa T, Rohde M, Prestrelski S, Lauren S, Stoney KS. Oxidative refolding of IGF-1 yields two products of similar thermodynamic stability: A bifurcating protein-folding pathway. Biochemistry. 1993;32(19):5203–5213. doi: 10.1021/bi00070a032. [DOI] [PubMed] [Google Scholar]
- Min C-Y, Qiao Z-S, Feng Y-M. Unfolding of human proinsulin. European Journal of Biochemistry. 2004;271(9):1737–1747. doi: 10.1111/j.1432-1033.2004.04079.x. [DOI] [PubMed] [Google Scholar]
- Miranda E, Pérez J, Ekeowa UI, Hadzic N, Kalsheker N, Gooptu B, Portmann B, Belorgey D, Hill M, Chambers S, Teckman J, Alexander GJ, Marciniak SJ, Lomas DA. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with α1-antitrypsin deficiency. Hepatology. 2010;52(3):1078–1088. doi: 10.1002/hep.23760. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type[thinsp] 1 diabetes in NOD mice. Nature. 2005;435(7039):220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VD, Saaranen MJ, Karala A-R, Lappi A-K, Wang L, Raykhel IB, Alanen HI, Salo KEH, Wang C-c, Ruddock LW. Two Endoplasmic Reticulum PDI Peroxidases Increase the Efficiency of the Use of Peroxide during Disulfide Bond Formation. Journal of Molecular Biology. 2011;406(3):503–515. doi: 10.1016/j.jmb.2010.12.039. [DOI] [PubMed] [Google Scholar]
- Oldstone M, Southern P, Rodriquez M, Lampert P. Virus persists in beta cells of islets of Langerhans and is associated with chemical manifestations of diabetes. Science. 1984;224(4656):1440–1443. doi: 10.1126/science.6203172. [DOI] [PubMed] [Google Scholar]
- Oslowski CM, Urano F. The binary switch that controls the life and death decisions of ER stressed β cells. Current Opinion in Cell Biology. 2011;23(2):207–215. doi: 10.1016/j.ceb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JH, Liz√°k Bt, Hendershot LM. Life and death of a BiP substrate. Seminars in Cell & Developmental Biology. 2010;21(5):472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa FR. Endoplasmic Reticulum Stress, Pancreatic β-Cell Degeneration, and Diabetes. Cold Spring Harbor Perspectives in Medicine. 2012;2(9) doi: 10.1101/cshperspect.a007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathiraja V, Kuehlich JP, Campbell PD, Krishnamurthy B, Loudovaris T, Coates PTH, Brodnicki TC, O’Connell PJ, Kedzierska K, Rodda C, Bergman P, Hill E, Purcell AW, Dudek NL, Thomas HE, Kay TWH, Mannering SI. Proinsulin specific, HLA-DQ8 and HLA-DQ8 transdimer restricted, CD4+ T cells infiltrate the islets in type 1 diabetes. Diabetes. 2014 doi: 10.2337/db14-0858. [DOI] [PubMed] [Google Scholar]
- Phillip Y, Schreiber G. Formation of protein complexes in crowded environments - From in vitro to in vivo. FEBS Letters. 2013;587(8):1046–1052. doi: 10.1016/j.febslet.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipeleers DG, Schuit FC, In’T Veld PA, Maes E, Hooghe-Peters EL, Winkel MVD, Gepts W. Interplay of Nutrients and Hormones in the Regulation of Insulin Release. Endocrinology. 1985;117(3):824–833. doi: 10.1210/endo-117-3-824. [DOI] [PubMed] [Google Scholar]
- Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. The Journal of Clinical Investigation. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Z-S, Min C-Y, Hua Q-X, Weiss MA, Feng Y-M. In Vitro Refolding of Human Proinsulin: KINETIC INTERMEDIATES, PUTATIVE DISULFIDE-FORMING PATHWAY, FOLDING INITIATION SITE, AND POTENTIAL ROLE OF C-PEPTIDE IN FOLDING PROCESS. Journal of Biological Chemistry. 2003;278(20):17800–17809. doi: 10.1074/jbc.M300906200. [DOI] [PubMed] [Google Scholar]
- Quinn D, Orci L, Ravazzola M, Moore H. Intracellular transport and sorting of mutant human proinsulins that fail to form hexamers. J. Cell Biol. 1991;113(5):987–996. doi: 10.1083/jcb.113.5.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpal G, Schuiki I, Liu M, Volchuk A, Arvan P. Action of Protein Disulfide Isomerase on Proinsulin Exit from Endoplasmic Reticulum of Pancreatic β Cells. Journal of Biological Chemistry. 2012;287(1):43–47. doi: 10.1074/jbc.C111.279927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner S, Braun-Reichhart C, Blutke A, Herbach N, Emrich D, Streckel E, W√°nsch A, Kessler B, Kurome M, B√§hr A, Klymiuk N, Krebs S, Puk O, Nagashima H, Graw J, Blum H, Wanke R, Wolf E. Permanent Neonatal Diabetes in INSC94Y Transgenic Pigs. Diabetes. 2013;62(5):1505–1511. doi: 10.2337/db12-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes CJ. Type 2 Diabetes—a Matter of b-Cell Life and Death? Science. 2005;307:380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- Riggs AC, Bernal-Mizrachi E, Ohsugi M, Wasson J, Fatrai S, Welling C, Murray J, Schmidt RE, Herrera PL, Permutt MA. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia. 2005;48(11):2313–2321. doi: 10.1007/s00125-005-1947-4. [DOI] [PubMed] [Google Scholar]
- Rigoli L, Di Bella C. Wolfram syndrome 1 and Wolfram syndrome 2. Current Opinion in Pediatrics. 2012;24(4):512–517. doi: 10.1097/MOP.0b013e328354ccdf. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Ronald CW, Cavener RD. Translational Control and the Unfolded Protein Resonse. Antioxidants & Redox Signaling. 2007;9(12):2357–2372. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- Sachdeva MM, Stoffers DA. Minireview: Meeting the Demand for Insulin: Molecular Mechanisms of Adaptive Postnatal β-Cell Mass Expansion. Molecular Endocrinology. 2009;23(6):747–758. doi: 10.1210/me.2008-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-Cell Mass and Turnover in Humans. Diabetes Care. 2012 doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Analytical Biochemistry. 1987;166(2):368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Kaufman RJ. The Unfolded Protein Response: A Pathway That Links Insulin Demand with {beta}-Cell Failure and Diabetes. Endocr Rev. 2008;29(3):317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JW, Tsukamoto K, Ribick M, Schuit FC, Kaufman RJ. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11(7):757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. The Journal of Cell Biology. 2009;187(4):525–536. doi: 10.1083/jcb.200907074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuit FC, In't Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(11):3865–3869. doi: 10.1073/pnas.85.11.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman S, Wang B, Li W, Rapoport TA. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proceedings of the National Academy of Sciences. 2010;107(34):15027–15032. doi: 10.1073/pnas.1009972107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding C-J, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li X-Y, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M. A Genome-Wide Association Study of Type 2 Diabetes in Finns Detects Multiple Susceptibility Variants. Science. 2007;316(5829):1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senée V, Vattem KM, Delépine M, Rainbow LA, Haton C, Lecoq A, Shaw NJ, Robert J-J, Rooman R, Diatloff-Zito C, Michaud JL, Bin-Abbas B, Taha D, Zabel B, Franceschini P, Topaloglu AK, Lathrop GM, Barrett TG, Nicolino M, Wek RC, Julier C. Wolcott-Rallison Syndrome: Clinical, Genetic, and Functional Study of EIF2AK3 Mutations and Suggestion of Genetic Heterogeneity. Diabetes. 2004;53(7):1876–1883. doi: 10.2337/diabetes.53.7.1876. [DOI] [PubMed] [Google Scholar]
- Sevier CS. New insights into oxidative folding. The Journal of Cell Biology. 2010;188(6):757–758. doi: 10.1083/jcb.201002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2008;1783(4):549–556. doi: 10.1016/j.bbamcr.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Developmental Cell. 2002;3(1):99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Lee K, Liu C-Y, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, Kaufman RJ. Complementary Signaling Pathways Regulate the Unfolded Protein Response and Are Required for C. elegans Development. Cell. 2001;107(7):893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Current Opinion in Cell Biology. 2011;23(2):143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohma Y, Hua QX, Liu M, Phillips NB, Hu SQ, Whittaker J, Whittaker LJ, Ng A, Roberts CT, Jr, Arvan P, Kent SB, Weiss MA. Contribution of residue B5 to the folding and function of insulin and IGF-I: constraints and fine-tuning in the evolution of a protein family. J Biol Chem. 2010;285(7):5040–5055. doi: 10.1074/jbc.M109.062992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner DF. Cocrystallization of Proinsulin and Insulin. Nature. 1973;243(5409):528–530. doi: 10.1038/243528a0. [DOI] [PubMed] [Google Scholar]
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, Lipton RB, Greeley SAW, Patch A-M, Ellard S, Steiner DF, Hattersley AT, Philipson LH, Bell GI Neonatal Diabetes International Collaborative Group. Insulin gene mutations as a cause of permanent neonatal diabetes. Proceedings of the National Academy of Sciences. 2007;104(38):15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Støy J, Steiner D, Park S-Y, Ye H, Philipson L, Bell G. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord. 2010;11(3):205–215. doi: 10.1007/s11154-010-9151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β Cell Dedifferentiation as a Mechanism of Diabetic β Cell Failure. Cell. 2012;150(6):1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavender TJ, Springate JJ, Bulleid NJ. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. The EMBO Journal. 2010;29(24):4185–4197. doi: 10.1038/emboj.2010.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodoro T, Odisho T, Sidorova E, Volchuk A. Pancreatic β-cells depend on basal expression of active ATF6α-p50 for cell survival even under nonstress conditions. American Journal of Physiology - Cell Physiology. 2012;302(7):C992–C1003. doi: 10.1152/ajpcell.00160.2011. [DOI] [PubMed] [Google Scholar]
- Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, Mirmira RG. Islet B-Cell Endoplasmic Reticulum Stress Precedes the Onset of Type 1 Diabetes in the Nonobese Diabetic Mouse Model. Diabetes. 2012;61(4):818–827. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes & Development. 1998;12(12):1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A, Schuiki I, Zhang L, Allister EM, Wheeler MB, Volchuk A. SDF2L1 interacts with the ER-associated degradation machinery and retards the degradation of mutant proinsulin in pancreatic β-cells. Journal of Cell Science. 2013;126(9):1962–1968. doi: 10.1242/jcs.117374. [DOI] [PubMed] [Google Scholar]
- Todd DJ, Lee A-H, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8(9):663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Usui M, Yamaguchi S, Tanji Y, Tominaga R, Ishigaki Y, Fukumoto M, Katagiri H, Mori K, Oka Y, Ishihara H. Atf6α-null mice are glucose intolerant due to pancreatic β-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism. 2012;61(8):1118–1128. doi: 10.1016/j.metabol.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Van Belle TL, Coppieters KT, Von Herrath MG. Type 1 Diabetes: Etiology, Immunology, and Therapeutic Strategies. 2011 doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- Van Lommel L, Janssens K, Quintens R, Tsukamoto K, Vander Mierde D, Lemaire K, Denef C, Jonas J-C, Martens G, Pipeleers D, Schuit FC. Probe-Independent and Direct Quantification of Insulin mRNA and Growth Hormone mRNA in Enriched Cell Preparations. Diabetes. 2006;55(12):3214–3220. doi: 10.2337/db06-0774. [DOI] [PubMed] [Google Scholar]
- Vetere A, Choudhary A, Burns SM, Wagner BK. Targeting the pancreatic [beta]-cell to treat diabetes. Nat Rev Drug Discov. 2014;13(4):278–289. doi: 10.1038/nrd4231. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen Y, Yuan Q, Tang W, Zhang X, Osei K. Control of precursor maturation and disposal is an early regulative mechanism in the normal insulin production of pancreatic beta-cells. PLoS One. 2011;6(4):e19446. doi: 10.1371/journal.pone.0019446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Osei K. Proinsulin maturation disorder is a contributor to the defect of subsequent conversion to insulin in beta-cells. Biochem Biophys Res Commun. 2011 doi: 10.1016/j.bbrc.2011.06.119. [DOI] [PubMed] [Google Scholar]
- Wang J, Takeuchi T, Tanaka S, Kubo S-K, Kayo T, Lu D, Takata K, Koizumi A, Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the Mody mouse. J. Clin. Invest. 1999;103(1):27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. Journal Article. 1998;17(19):5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC, Aguayo-Mazzucato C, Bonner-Weir S. β-cell dedifferentiation in diabetes is important, but what is it? Islets. 2013;5(5):233–237. doi: 10.4161/isl.27494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MA. Proinsulin and the Genetics of Diabetes Mellitus. J. Biol. Chem. 2009;284(29):19159–19163. doi: 10.1074/jbc.R109.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MA. Diabetes mellitus due to the toxic misfolding of proinsulin variants. FEBS Letters. 2013;587(13):1942–1950. doi: 10.1016/j.febslet.2013.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittingham J, Záková L, Dodson E, Turkenburg P, Brange J, Dodson G. I222 crystal form of despentapeptide (B26-B30) insulin provides new insights into the properties of monomeric insulin. Acta Cryst. 2006:505–511. doi: 10.1107/S0907444906006871. [DOI] [PubMed] [Google Scholar]
- Winter J, Gleiter S, Klappa P, Lilie H. Protein disulfide isomerase isomerizes non-native disulfide bonds in human proinsulin independent of its peptide-binding activity. Protein Science. 2011;20(3):588–596. doi: 10.1002/pro.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J, Klappa P, Freedman RB, Lilie H, Rudolph R. Catalytic Activity and Chaperone Function of Human Protein-disulfide Isomerase Are Required for the Efficient Refolding of Proinsulin. Journal of Biological Chemistry. 2002;277(1):310–317. doi: 10.1074/jbc.M107832200. [DOI] [PubMed] [Google Scholar]
- Wright J, Birk J, Haataja L, Liu M, Ramming T, Weiss MA, Appenzeller-Herzog C, Arvan P. Endoplasmic Reticulum Oxidoreductin-1α (Ero1α) Improves Folding and Secretion of Mutant Proinsulin and Limits Mutant Proinsulin-induced Endoplasmic Reticulum Stress. Journal of Biological Chemistry. 2013a;288(43):31010–31018. doi: 10.1074/jbc.M113.510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J, Wang X, Haataja L, Kellogg AP, Lee J, Liu M, Arvan P. Dominant protein interactions that influence the pathogenesis of conformational diseases. The Journal of Clinical Investigation. 2013b;123(7):3124–3134. doi: 10.1172/JCI67260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, Tokita A, Satake C, Tashiro F, Katagiri H, Aburatani H, Miyazaki J-i, Oka Y. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic β-cells. Human Molecular Genetics. 2006;15(10):1600–1609. doi: 10.1093/hmg/ddl081. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Larkin D, Lara-Lemus R, Ramos-Castaneda J, Liu M, Arvan P. Endoplasmic reticulum (ER) chaperone regulation and survival of cells compensating for deficiency in the ER stress response kinase, PERK. J Biol Chem. 2008;283(25):17020–17029. doi: 10.1074/jbc.M802466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Developmental Cell. 2007;13(3):365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hua Q-x, Liu J, Shimizu EH, Choquette MH, Mackin RB, Weiss MA. Solution Structure of Proinsulin. Journal of Biological Chemistry. 2010a;285(11):7847–7851. doi: 10.1074/jbc.C109.084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hua Q-x, Liu J, Shimizu EH, Choquette MH, Mackin RB, Weiss MA. Solution Structure of Proinsulin: CONNECTING DOMAIN FLEXIBILITY AND PROHORMONE PROCESSING. Journal of Biological Chemistry. 2010b;285(11):7847–7851. doi: 10.1074/jbc.C109.084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yon J, Betton J-M. Protein folding in vitro and in the cellular environment. Biol. Cell. 1991;71(1–2):17–23. doi: 10.1016/0248-4900(91)90047-q. [DOI] [PubMed] [Google Scholar]
- Yoshina S, Sakaki K, Yonezumi-Hayashi A, Gengyo-Ando K, Inoue H, Iino Y, Mitani S. Identification of a novel ADAMTS9/GON-1 function for protein transport from the ER to the Golgi. Molecular Biology of the Cell. 2012;23(9):1728–1741. doi: 10.1091/mbc.E11-10-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. (non-insulin-dependent diabetes mellitus), (non-insulin-dependent diabetes mellitus) American Diabetes Association. 1997;(888):887. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JRB, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney ASF, Consortium TWTCC, McCarthy MI, Hattersley AT. Replication of Genome-Wide Association Signals in UK Samples Reveals Risk Loci for Type 2 Diabetes. Science. 2007;316(5829):1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BY, Liu M, Arvan P. Behavior in the eukaryotic secretory pathway of insulin-containing fusion proteins and single-chain insulins bearing various B-chain mutations. Journal of Biological Chemistry. 2003;278(6):3687–3693. doi: 10.1074/jbc.M209474200. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454(7203):455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lai E, Teodoro T, Volchuk A. GRP78, but Not Protein-disulfide Isomerase, Partially Reverses Hyperglycemia-induced Inhibition of Insulin Synthesis and Secretion in Pancreatic β-Cells. Journal of Biological Chemistry. 2009;284(8):5289–5298. doi: 10.1074/jbc.M805477200. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li Sa, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK Eukaryotic Initiation Factor 2α Kinase Is Required for the Development of the Skeletal System, Postnatal Growth, and the Function and Viability of the Pancreas. Molecular and Cellular Biology. 2002;22(11):3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic β cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metabolism. 2006;4(6):491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Zhu YL, Abdo A, Gesmonde JF, Zawalich KC, Zawalich W, Dannies PS. Aggregation and Lack of Secretion of Most Newly Synthesized Proinsulin in Non-{beta}-Cell Lines 10.1210/en.2003-1512. Endocrinology. 2004;145(8):3840–3849. doi: 10.1210/en.2003-1512. [DOI] [PubMed] [Google Scholar]
- Zito E, Chin KT, Blais J, Harding HP, Ron D. ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J Cell Biol. 2010;188(6):821–832. doi: 10.1083/jcb.200911086. [DOI] [PMC free article] [PubMed] [Google Scholar]