

Abstract
Long-term preservation of live cells is critical for a broad range of clinical and research applications. With the increasing diversity of cells that need to be preserved (e.g. oocytes, stem and other primary cells, genetically modified cells), careful optimization of preservation protocols becomes tedious and poses significant limitations for all but the most expert users. To address the challenge of long-term storage of critical, heterogeneous cell types, we propose a universal protocol for cell vitrification that is independent of cell phenotype and uses only low concentrations of cryoprotectant (1.5 M PROH and 0.5 M trehalose). We employed industrial grade microcapillaries made of highly conductive fused silica, which are commonly used for analytical chemistry applications. The minimal mass and thermal inertia of the microcapillaries enabled us to achieve ultrafast cooling rates up to 4,000 K/s. Using the same low, non-toxic concentration of cryoprotectant, we demonstrate high recovery and viability rates after vitrification for human mammary epithelial cells, rat hepatocytes, tumor cells from pleural effusions, and multiple cancer cell lines.
Innovation
For the long-term preservation of live cells, strategies relying on the use of cryogenic temperatures are the most common. Various cryopreservation and vitrification protocols that use different cooling rates, cryoprotectant chemical compositions, and cryoprotectant concentrations are currently available. However, most often, careful optimization of these protocols is necessary for achieving high yield and survival of specific cells. During cryopreservation, slower than optimal freezing rates can damage cells through the formation of intracellular ice, while concentrations of cryoprotectant that are too high could be toxic to the cells. During vitrification, the need for very high cryoprotectant concentrations requires time consuming protocols for loading and unloading cryoprotectants in successive steps. In this study, first, we overcome the limitation of current cryopreservation and vitrification protocols by developing a universal preservation protocol with high yield. Second, we improved the quartz capillary technique by employing the precisely machined microcapillary for the cryopreservation with a low non-toxic concentration of cryoprotectants.
Introduction
Cells are a key component for a myriad of biomedical applications ranging from stem cell therapeutics to tissue engineering, and from drug screening to reproductive medicine. To assure off-the-shelf access to cellular therapies, two long-term preservation approaches are available, namely, cryopreservation and vitrification. During cryopreservation, cells are cooled slowly in the presence of a cryoprotectant [e.g., dimethylsulfoxide (DMSO), glycerol, etc.] to reduce the formation of ice crystals and their deleterious effects on cell viability. Because different cell types have unique biophysical and biological characteristics1,2, optimized protocols must be developed for each cell type of interest. This optimization process becomes practically prohibitive when considering the need to cryopreserve the very heterogeneous range of cells isolated from animals and humans3,4. Vitrification (or “ice-free” cryopreservation) is an alternative preservation approach to cryopreservation, which uses very high concentration of cryoprotectant cocktails to completely prevent ice formation. Cryoprotectant concentrations as high as 4–8 M are typically required. Because such concentrations are very toxic for most cell types5–8, individual protocols are optimized for each cell type depending on its sensitivity to the cryoprotectant. Alternative approaches to vitrification protocol optimization have focused on increasing the cooling rate, which could allow lower concentrations of cryoprotectant to be used to achieve glassy-state. Technologies to allow faster cooling rates have been described in which cells are placed inside thin open straws9, cryotops10, electron microscopy grids11, or cryoloops12. More recently, we validated the use of quartz micro-capillaries for vitrification13–16, which enabled ultra high cooling rates and required significantly lower concentrations of cryoprotectants than standard vitrification protocols (∼ 2.0 M). In the previous study we showed that using 2 M Propanediol (PROH) and 0.5 M trehalose was sufficient for vitrification of embryonic stem cells13 and the use of lower concentrations (1.5 M PROH and 0.5 M trehalose) was effective to vitrify mouse oocytes14. A significant drawback, however, was the variability in the diameter and the wall thickness of quartz capillaries, which resulted in inconsistent yield and viability of cells between successive runs in similar conditions.
To overcome the limitation of current cryopreservation and vitrification protocols, here we report the development of a vitrification protocol that can be easily standardized and implemented without changes for a wide variety of cell types. We improved on our previous quartz capillary technique and employed fused silica microcapillaries, which are industrially manufactured to have well controlled dimensions and physical properties. The fused silica microcapillaries functioned as a low-thermal-mass, high-heat conductivity cell container to increase the cooling rate by an order of magnitude compared to most current cell vitrification technologies. Here, we demonstrate the application of the technique to the preservation of nine different cell types in low, non-toxic concentration of cryoprotectant solution containing 1.5 M PROH and 0.5 M trehalose. The vitrification process is universal, simple and can be completed in less than 15 minutes, significantly faster when compared to conventional vitrification techniques.
Methods
Preparation of fused silica capillary
Fused silica capillaries (Postnova analytics Inc, UT, USA) with polyimide coating were cut to 7 cm length and the polyimide coating was selectively removed by a solvent, N-methylpyrrolidone (NMP) (Sigma-Aldrich, MO, USA) (Fig. 1b).
Figure 1.
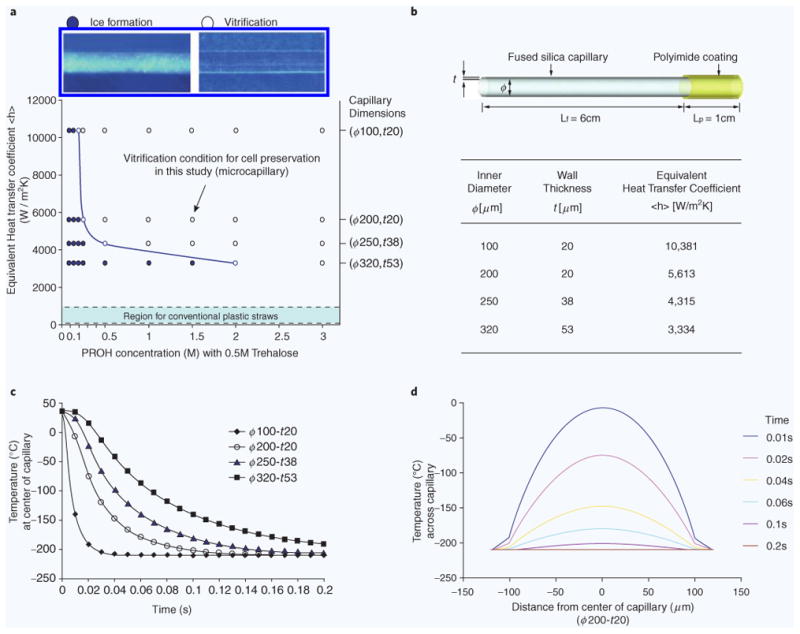
Evaluation of a fused silica microcapillary microcapillary as a tool for vitrification. (a) Map showing the heat transfer and cyoprotectant requirements for vitrification. The line indicated the relationship between the varying dimensions of microcapillary and the levels of CPAs, required to achieve “ice-free” conditions during freezing. Experimentally tested conditions are indicated as dots. (b) Equivalent heat transfer coefficients were calculated based on the inner diameter (ϕ) and wall thickness (t) of the microcapillaries, and used as a single parameter to evaluate the heat transfer rates. (c) The temperatures inside capillary filled with media decreases as the dimension of microcapillary increases — heat transfer analysis using computer simulations (COMSOL). (d) Estimated temperature profiles across a cylindrical model of a 200 μm(ϕ)−20 μm(t) capillary during freezing by plunging the capillary into slush nitrogen (−210 °C).
Calculation of equivalent heat transfer coefficient
Simple one dimensional heat transfer filled with water was assumed and the equivalent heat transfer coefficients were calculated by the following equation,
Here, r2, r1 are outer radius and inner radius of capillary, respectively. ks and kw are heat conductivity of fused silica and water, respectively.
Cell lines preparation (PC3, T24, SkBr-3, H1650, 3T3 and HepG2)
The cell lines PC3, T24, SkBr-3, NCI-H1650, HepG2 and NIH/3T3 were purchased from American Type Culture Collection (ATCC, USA). Cells were grown with the recommended medium in a cell culture incubator at 37 °C with 5% CO2 for at least 2 weeks to ensure optimal phenotype. Only one cell line at a time was chosen for experiments, and the following protocol was performed on different days for each cell type. All cell lines were handled using standard aseptic techniques. A T-75 flask of the desired cell line at 90% confluence (minimum 3 million cells) was removed from the incubator and placed in a sterile flow hood. The cells were first washed with 10 ml of warm 37 °C 1× phosphate buffered saline (1× PBS) without calcium and magnesium (Gibco). This was replaced by 6 ml of 0.05% trypsin with 0.2 g/L EDTA (Thermo Scientific). The flask was returned to the incubator with trypsin for the optimal time as suggested by original ATCC protocols (10 minutes for HepG2, 5 minutes for SkBr-3, NCI-H1650, PC3, T24, NIH/3T3). After the trypsin incubation, the flask was returned to the sterile flow hood and 12 ml of complete cell specific medium was added to the flask to neutralize the trypsin reaction. All trypsin, medium, and cells were removed with a 25 ml pipette and added to a 50 ml conical tube (Corning). This was centrifuged at 1050 rpm for 5 minutes, the supernatant aspirated, and the pellet re-suspended into 2 ml of complete cell specific medium using a 1ml pipette. The cells numbers in suspension were counted using a Beckman Coulter Counter. Cells were diluted with cell specific medium to reach a concentration of 1–1.5 million cells per milliliter, which was determined to be an optimal concentration for capillary loading (aiming for 800–1300 cells per capillary).
Cell preparation for primary rat hepatocytes
All animal procedure were performed in accordance with National Research Council guidelines and approved by the subcommittee on Research Animal Care at the Massachusetts General Hospital. Hepatocytes were isolated from adult female Lewis rats (Charles River Laboratories, Boston, MA) weighing 150 to 200 g, according to the two-step in situ collagenase digestion method as modified by Dunn et al.17,18 Following isolation, hepatocytes were prepared in cell specific medium at a cell concentration of 1 million cells per milliliter for pretreatment of cryoprotectants for vitrification. After vitrification and thawing, cells were plated for viability assay in the wells of a 384-well square clear flat bottom plate. The surfaces of plates were overnight treated by a mixture of one part of 1× PBS and nine parts of collagen (1.12 mg/mL, Type I collagen prepared from rat-tail tendon) before cell plating.
Albumin and urea assay for primary rat hepatocytes
Five sets of non-vitrified and vitrified cell suspension in microcapillary were seeded on each 96-well (surface was coated with collagen-gel configuration) for control and experiment sets, respectively and incubated in a humidified atmosphere of 90% air/10% CO2 at 37 °C for 24 hours. Analysis was repeated for four separate experiments. Collected medium samples were analyzed for rat albumin concentration by enzyme-linked immunosorbent assay, as previously described17. Urea concentration was determined from its specific reaction with diacetyl monoxime by using a commercially available assay kit (Sigma Chemical) scaled for use in a 96-well plate. The absorbance was measured in a Thermomax microplate reader (Molecular Devices).
Cell preparation for normal HMECs
Cells were purchased from the Lonza Walkersville, Inc (Walkersville, MD, USA). Cells were grown with the mammary epithelial growth media (MEGM) in a cell culture incubator at 37 °C with 5% CO2 for a week to ensure optimal phenotype. For the preservation procedure, cells were first trypsinized and washed for pretreatment of cryoprotectants for vitrification.
Cell culture from LAM patient pleural effusion
Pleural effusion fluid samples from a single LAM patient donor were provided through NDRI (National Development and Research Institutes Inc.). Fluid was delivered by courier service in 12–24 hours after evacuation, in a sterile plastic container on ice. Fresh fluid was filtered through 40 μm cell strainer nylon mesh (BD Falcon) to collect LAM clusters. The filter was then washed into a 10 cm tissue culture dish with media. Cells were grown for about a week in smooth-muscle growing media: SmGM (Lonza CC-3182 SmGM-2 BulletKit) containing supplements: 5% FBS, hEGF, insulin, hFGF-B. For the preservation procedure, cells were first trypsinized and washed.
Pretreatment with cryoprotectants for vitrification
Just before vitrification, the cells were divided into three 1.5 ml tubes containing 1 ml of cell suspension each. The tubes with their caps opened were returned to the incubator to allow gas exchange. Usually, the experimental conditions were completed first (instead of the control) so that artificial cell loss would be avoided as the cells settled over time. For each vitrification experiment, the cap of the tube was first sealed and the tube centrifuged for 5 minutes at 1050 rpm. After centrifugation, the supernatant was removed, and the cells re-suspended in a freezing equilibration solution (FES) containing 0.75 M PROH in 1× PBS. This tube of cells with 0.75 M PROH was placed in the incubator for 3 minutes with cap removed before centrifuging at 1050 rpm for 5 minutes. Once centrifugation was complete, the supernatant was removed and the pellet was re-suspended in the freezing vitrification solution (FVS) containing 1.5 M solution of PROH with 0.5 M trehalose in 1× PBS. This was placed in the incubator for 3 minutes before loading into capillaries. Vitrification was performed by submerging the capillaries in slush nitrogen (−208 °C ∼−210 °C). Thawing was accomplished by immediate submersion of the capillary in a thawing bath of 0.2 M trehalose solution in 1× PBS at 37 °C before plating; with one capillary designated per well. After each well was loaded, usually four per vitrification experiment, the remaining cells in the tube were counted on a Beckman Coulter Counter to verify a constant cell concentration. Cells from both controls and experiments were placed in the wells of a 384-well square clear flat bottom plate with 100 μl of complete cell specific medium.
For controls, first, the cap of the tube was sealed. Then, the cells were centrifuged at 1050 rpm for 5 minutes. The supernatant was subsequently removed, and the cells were re-suspended with 1 ml of complete cell specific medium. This was placed in the incubator with the cap removed for 3 minutes. After the 3 minutes, the cap was placed on again and cells were centrifuged for 5 minutes at 1050 rpm. After centrifugation, the supernatant was removed, and complete cell specific medium was again utilized to re-suspend the pellet. This was incubated for 3 minutes before loading into capillaries and immediately plating; with one capillary designated per well. After plating, usually four or five wells per control, the remaining cells in the tubes were counted using a Beckman Coulter Counter to ensure that no cell loss was incurred.
Vitrification and thawing
The fused silica microcapillaries have an inner diameter of 200 μm with a wall thickness of 20 μm. After re-suspension in the vitrification solution, the cells were then loaded into the 200 μm capillary, plunged into slush nitrogen and submerged for ∼ 60 seconds. Slush nitrogen at the temperature of 210 °C was produced using the VitMaster cooler (IMT International Ltd., UK). For warming, the microcapillaries with vitrified cells were immersed for ∼30 seconds into a medium containing 0.2 M trehalose in 1X Phosphate buffer saline (PBS) (Gibco) at 37 °C. The cells were then expelled in a well containing 100 μl of a complete culture media. The cells were cultured in an incubator at 37 °C and with 5% CO2 in air for 12–24 hours for the viability assay and 4–5 days for the growth rates assay.
Assays for viability and growth rates
For growth rate experiments, the cell medium was changed and the wells of all experimental and control groups imaged daily for 4–5 days. These images were utilized to compile a growth curve describing the conservation of growth rate after an initial recovery period for each vitrified cell line. Viability assays were performed using a Live/Dead Assay Kit (L-3224, Invitrogen), according to instructions from the manufacturer. First, the supernatant in each well was transferred into separate wells for separate counting of the non-adherent cells, assumed to be all dead. Each well was washed with serum free medium and then loaded with the solutions from the Live/Dead kit reconstituted in cell culture media. After incubating the plates for 20 minutes, images were acquired using a CCD camera (SPOT imaging/Diagnostic Instruments, USA) mounted on an inverted microscope (TE2000-S, Nikon, Japan). These images were used to calculate the percent yield and viability of living cells (Supplementary Fig. 2).
Yield calculation
We loaded a sample plug (L = 10∼20 mm in length) in a fused silica capillary (200 μm(ϕ)−20 μm(t)) from varying cell concentrations (C = 1∼1.5 × 106 cells/ml) as shown in Fig. 2 and measured the absolute cell numbers obtained after freezing and thawing. Estimated cell numbers loaded in capillary is calculated by the equation, No. of cells = π/4 × ϕ2 × L × C.
Figure 2.
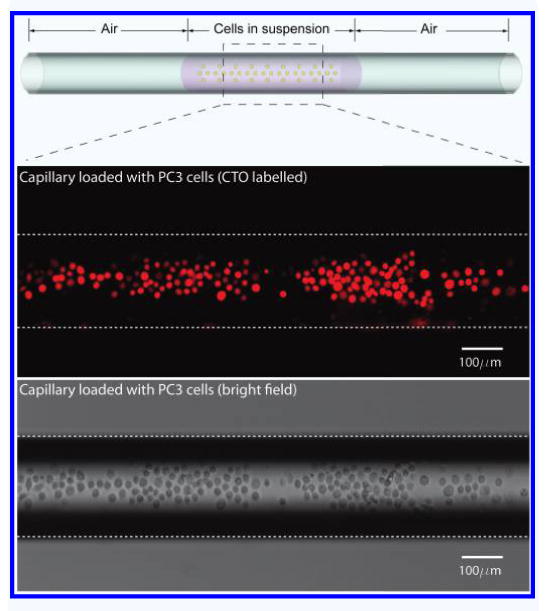
A schematic drawing of the loaded cells in capillary (Top). PC3 labeled with Cell Tracker Orange (CTO) epifluorescence in Red (Middle). Bright field image of the cells in capillary (Bottom).
Relative yield as calculated as the percentage of actual recovered cells over the estimated loaded cells into the freezing vessel.
Results
Optimization of the microcapillary dimensions
To better understand the heat transfer inside the microcapillary during vitrification, we employed a theoretical model13 to study the effect of the silica microcapillary dimensions on the cooling rate. In addition, we used this model to estimate the rate of cooling and the concentration of cryoprotectant (CPA) needed for vitrification (Fig. 1a). We introduced an equivalent heat transfer coefficient (W/m2K) by combining material and geometric properties [inner diameter (ϕ) and wall thickness (t) of capillary] in our model. Our simulations estimated an equivalent heat transfer coefficient between ∼3000 and ∼10,000 W/m2 K. The cooling rates achieved in silica microcapillaries were significantly higher compared to conventional plastic straws and vials, as indicated on the vitrification map (Fig. 1a)9,13. Simulations also predicted that the highest equivalent heat transfer coefficient and the fastest temperature drop, up to 4200 K/s, could be achieved inside the D100-δ20 microcapillaries (Fig. 1b,c). The temperature inside the microcapillary becomes quasi-uniform approximately 0.2 s after plunging the microcapillary in slush nitrogen (63K — Fig. 1d).
In addition to the simulations, we experimentally verified some of the model predictions. The viability of prostate cancer cells (PC3 cell line) increased from 73.0 ± 8.0 (N = 7) to 92.6 ± 4.9% (N = 6) when the size of the microcapillary was reduced from 320 μm(ϕ)−53 μm(t) to 200 μm(0)−20 μm(t), in the presence of 1.5 M PROH and 0.5 M trehalose. The viability decreased to 17.2 ± 12.7% (N = 8) in the absence of CPAs inside the 200 μm(ϕ)−20 μm(t) microcapillary. For a good balance between heat transfer properties and ease of cell loading, we chose to use the 200 μm(ϕ)−20 μm(t) microcapillary (equivalent heat transfer coefficient, 5613 W/m2K) in all subsequent experiments.
Vitrification of cell lines
We first tested the efficiency of the vitrification protocol on five human epithelial cancer cell lines (PC3: prostate cancer, T24: bladder cancer, SkBr-3: breast cancer, H1650: non-small-cell lung cancer, and HepG2: liver cancer) and one fibroblast cell line (mouse 3T3 fibroblasts). Cells were loaded inside capillaries in the presence of 1.5 M PROH and 0.5 M trehalose, as shown in Fig. 2. The overall yield across all the five cell lines after vitrification and thawing was 106.8 ± 28.0% (N = 30), indicating that most of the cells were recovered after the vitrification procedure (Fig. 3a). The viability of the cancer cell lines after vitrification was higher than 90% (Fig. 3b, and Table 1). The viability of the 3T3 fibroblasts was after vitrification 82.1 ± 3.3% (N = 5), compared to 93.6 ± 0.5% for the control cells (N = 5). Growth rates for each cell type were measured every 24 hours for up to 5 days and showed no evidence of cell damage after vitrification (Fig. 4a–e). It has been known that low cell recovery rate after cryopreservation is largely caused by apoptosis, rather than necrosis, although the details of the mechanisms of apoptosis are still not clear19. In Fig. 4, one can doubt that the lags in growth rates at 1∼2 days after vitrification may be caused by apoptosis but the slopes are gradually recovered to the levels of controls at later days, which indicate that the apoptosis may not be a significant factor to affect the growth rates of cells after vitrification with the proposed protocol. There were no significant differences in cytokeratin expression between the vitrified and control epithelial cell lines. We were not able to count the HepG2 cells for an accurate growth rate, because of the formation of cell aggregates. Nevertheless, HepG2 cells grew well and the sizes of the cell aggregates were comparable to those of the control. During the culture period, the morphologies of the five different cancer cell lines, fibroblasts and non-cancer cell types were similar to those of the control (Supplementary Fig. 1,2).
Figure 3.
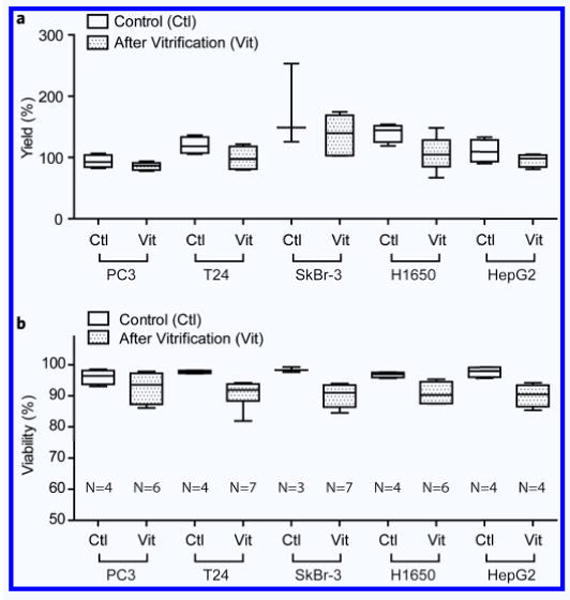
Yields and viability. (a) Yields of the loaded cell after vitrification. Yields were obtained by comparing the dispensed cell numbers with the calculated loaded cell numbers. (b) Viability of different cell types after vitrification using 200 μm(ϕ)−20 μm(t) fused silica capillary.
Table 1.
Viability of different cell types after vitrification and thawing.
| Cells | Relative viability (%) (Ratio of vitrification over Control) |
|---|---|
| PC3 | 96.4 ± 4.7 (N = 6) |
| T24 | 92.3 ± 4.4 (N = 7) |
| SkBr-3 | 91.5 ± 3.6 (N = 7) |
| H1650 | 93.9 ± 3.6 (N = 6) |
| HepG2 | 92.3 ± 3.7 (N = 4) |
| 3T3 | 87.7 ± 3.5 (N = 5) |
| Primary rat hepatocytes | 70.3 ± 12.1 (N = 4) |
| Normal HMECs | 92.7 ± 1.7 (N = 6) |
| LAM cells | 84.2 ± 6.6 (N = 10) |
Figure 4.

Comparisons of growth rates. (a–e) Growth rates of PC3, T24, SkBr-3, H1650, 3T3 and Normal HMEC in control cells and after vitrification using 200 μm(ϕ)−20 μm(t) fused silica capillary.
Vitrification of primary rat hepatocytes
Primary rat hepatocytes were vitrified inside 200 μm(ϕ)−20 μm(t) silica microcapillaries, in the presence of 1.5 M PROH and 0.5 M trehalose. The overall yield after vitrification was 98.3 ± 25.0% (N = 4), comparable to 115.5 ± 8.1% (N = 4) for control, unfrozen cells. Absolute viability for primary rat hepatocytes after vitrification was 45.4 ± 7.8% (N = 4) compared to 64.6 ± 4.8% (N = 4) for control cells (70.3 ± 12.1% relative viability). The levels of albumin secretion and urea synthesis decreased only slightly at 24 hours after vitrification compared to the fresh controls, from 3.6 ± 0.5 μg/ml to 3.1 ± 0.2 μg/ml (albumin) and from 27.9 ± 1.2 μg/ml to 20.7 ± 3.7 μg/ml (urea) (Fig. 5).
Figure 5.

Efficacy of the vitrification with hepatocytes. (a) Yields of the loaded cells. (b) Viability of hepatocytes after vitrification. (c) The levels of albumin secretion are 3.6 ± 0.5 μg/ml (Control) and 3.1 ± 0.2 μg/ml (After vitrification). (d) The levels of urea synthesis are 27.9 ± 1.2 μg/ml (Control) and 20.7 ± 3.7 μg/ml (After vitrification). Five sets of control and vitrified cell suspension in microcapillary were loaded each 96-well for the measurements of four separate experiments.
Vitrification of primary human cells
Human mammary epithelial cells (HMEC) were vitrified using our universal cryoprotectant solution (1.5 M PROH and 0.5 M trehalose) and microcapillary size (200 μm(ϕ)−20 μm(t)). The absolute viability of the human cells after vitrification was 80.8 ± 1.5% (N = 6), comparable to that of unfrozen control cells (87.2 ± 0.4%, N = 4), representing 92.7 ± 1.7% relative viability. We also measured the yield and viability after vitrification of tumor cells from pleural effusions of a patient with lymphangioleiomyomatosis (LAM)20,21. We achieved an overall yield up to 92.3 ± 38.4% (N = 10) after vitrification, comparable to 95.4 ± 30.8% (N = 5) for control, unfrozen cells. The viability of cells from pleural effusion after freezing and thawing was 84.2 ± 6.6% (N = 10) compared to that of the control with absolute viabilities of 76.5 ± 2.6% for control cells (N = 5) and 64.4 ± 5.4 after vitrification (N = 10). Growth rates (N = 5) were measured every 24 hours for up to 5 days and showed no evidence of damage (Fig. 6). We examined the HMB45 expression (a 100 kDa glycoprotein expressed in 20%–70% of LAM cells20) after vitrification over the course of five days and found expression levels comparable to control (Supplementary Fig. 1).
Figure 6.

Efficacy of the vitrification with cells from LAM patient pleural effusion. (a) Yield, (b) viability of cells at 18 hours after vitrification using 200 μm(ϕ)−20 μm(t) fused silica capillary, and (c) growth rates.
Discussion
We developed a universal vitrification protocol using silica microcapillaries and low cryoprotectant concentrations. We validated this protocol using five cancer cell lines, one fibroblast cell line, primary rat hepatocytes, normal human mammary epithelial cells (HMECs), and tumor cells obtained from the pleural effusion of one patient with lymphangioleio-myomatosis (LAM). For all nine types of cells, we measured high yield and high viability after vitrification.
The performance of the new vitrification protocol can be explained in part by the high cooling rates inside the microcapillaries. While it is known that the vitrification of pure water requires cooling rates of at least 106 K/s 22, slower cooling rates could also be used for vitrification after the addition of various solutes or CPAs to water 23. The addition of carbohydrates to penetrating cryoprotectants aids in the dehydration of cells and it is suggested that sugars are capable of preserving the structural and functional integrity of membranes. Hence, vitrification solutions containing trehalose or sucrose are widely used for cryopreservation of mammalian oocytes and embryos. Although the developed vitrification solutions may differ in the amount and type of additives (permeable and nonpermeable cryoprotectants), the total solute concentration should be sufficiently high to prevent ice formation at the given cooling and warming rate. Risco et al.16 extensively studied the thermal performance of quartz capillary for vitrification, in which the similar dimensions of capillary and cooling rate were used to vitrify the solution. Briefly, the critical cooling rate to achieve vitrification is a function of the concentration of cryoprotectant and the estimated critical cooling rates are between 100,000 K/min (or 1666 K/s) and 1,000,000 K/min (or 16,666 K/s) for the 1.8 M solution (1.5 M PROH and 0.3 M sucrose) based on the Boutron's semiemprical theory of glass forming tendency23. It was also experimentally evaluated that the quartz capillary with cooling rate of 250,000 K/min (4167 K/s) can vitrify the 1.8 M solution. For our protocol (2 M solution), the high cooling rates inside the microcapillaries, estimated to reach up to 4 × 103 K/s, enabled us to lower the concentrations of CPAs to levels that are non-toxic and easy to load and remove. The fast heat transfer through the thin microcapillary walls in our study was further augmented by the use of slush nitrogen, which reduced the vaporization16 at the interface between micro-capillary and the cryogenic immersion media24. We hypothesize that maximizing the heat transfer had an additional positive impact during warming after vitrification, preventing the recrystallization of water and subsequent formation of large intracellular ice crystals that would damage the cells25.
The cooling rates inside microcapillaries are at least one order of magnitude higher compared to other vitrification techniques previously reported. Most of these techniques have been developed specifically for the long-term storage of oocytes, and rely on the use of thin plastic straws9, electron microscopy grids11, cryotops10 or cryoloops12 to physically hold oocytes before plunging them directly into liquid nitrogen. Cooling rates for these protocols has been reported in the range of 0.03–0.5 × 103 K/s and high CPAs concentrations (4–8 M) were required in order to achieve vitrification conditions. The unavoidable multi-step CPA loading and unloading procedures, before and after vitrification, made these procedures cumbersome and time consuming. More recently, quartz capillaries were used for the vitrification of oocytes14,15 urine embryonic stem (ES) cells13. Because of the faster cooling rates, lower CPA concentrations were sufficient for vitrification conditions. Murine embryonic stem cells (2 M PROH + 0.5 M trehalose) and mouse oocytes (1.5 M PROH + 0.5 M trehalose) were vitrified with high viability rates (70% and 90%, respectively) using this technique13,14. The approach is also successfully validated for the cryopreservation of immature and mature human oocytes26. Despite the promising results however, quartz capillaries have one important practical drawback, in that their diameter and wall thickness might vary between capillaries and even along the same capillary, resulting in significant variability of the vitrification results. Our study addresses this limitation through the use of industrial grade microcapillaries; whose optimal geometric parameters were identified through our heat transfer model.
One important practical aspect of the new vitrification technique is its ease of implementation and reproducibility. Fused silica capillaries are commonly used as analytical chemistry tools for applications such as capillary electrophoresis and chromatography27. These capillaries are manufactured under strict quality control for precise diameter, wall thickness, and straightness characteristics. Wider use of the easily available fused silica microcapillaries could eventually lead to standardized cell vitrification protocols across various laboratories, for clinical and research applications.
The silica microcapillaries can only be loaded with small volume samples and the cells have to be in suspension. Though vitrification technique has been applied to preserve organs28, tissues29,30, and cell aggregates31,32, the translation of this approach for commercial applications might not be applicable to the larger patient-derived cells/tissues due to the small number of cells preserved in each capillary. However, in many instances, such requirements are easily satisfied, for example in applications related to assisted reproduction, when preserving sperm cells, oocytes, embryos, or follicles. In other instances, relatively high cell concentrations are needed for efficiency. For example, to load an estimated 1000 cells in one 200 μm inner diameter, 30 mm long microcapillary, the cells would have to be at an equivalent concentration of 1 × 106 cells/mL. The need for high cell concentration may become problematic during the preliminary cell preparation steps, in particular for epithelial cells that could adhere to each other and form large aggregates. The integration of cell preparation and sample loading procedures, with the goal of minimizing the time when cell aggregates could form, could become important for the successful vitrification of these cells.
In summary, we have developed a universal vitrification protocol and validated its use on nine different primary human and animal cells and cell lines. This is the first report of the fused silica capillary originally manufactured for chromatography applications, as a tool for cell vitrification. The protocol can be easily implemented in research and clinical laboratories and provides a practical approach for high recovery and viability rates for different cell types after vitrification.
Supplementary Material
Supplementary Figure 1 Images of viability and HMB 45 expression. (a) Viability of cells from LAM patient pleural effusion at 18 hours after vitrification using 200 μm(ϕ)−20 μm(t) fused silica capillary. (b) HMB45 expression in cells from a LAM patient pleural effusion. On Day 5 of growth rate experiments, the wells of control and experiment groups were fixed with 4% paraformaldhyde (PFA, Electron Microscopy Sciences) diluted in 1× PBS and were permeabilized with 0.2% Triton-X (Sigma) diluted in 1× PBS. After the permeabilization solution was removed, the cells were washed with 1× PBS and the cell membranes were blocked for 30 minutes in 2% normal goat serum (Vector Laboratories) and 3% bovine serum albumin (Sigma) in 1× PBS. Blocking agents were replaced by a primary antibody solution containing a 1:20 dilution of HMB45 antibody in 3% BSA in 1× PBS (w/o Ca2+, Mg2+). Following the primary antibody incubation, the patient samples were washed in 1× PBS before applying secondary antibodies (AlexaFluor488 IgG1) diluted at 1:500 in 3% bovine serum albumin in 1× PBS with 1:500 4′, 6-diamidino-2-phenylindole, dihydrochloride (DAPI, Invitrogen) for 1 hour for imaging. Scale bar for 100 μm.
Supplementary Figure 2 Images of viability after vitrification. Viability images of five cancerous epithelial cell lines (a–e), normal HMECs (f) at 12–18 hours after vitrification using 200 μm(ϕ)−20 μm(t) fused silica capillary. Entire plate (3.3 mm × 3.3 mm) images which contain all the cells dispensed from the capillary.
Acknowledgments
This research was partially supported by the National Institutes of Health (EB002503, HD061297), Folkman Fellowship from LAM Treatment Alliance (to SN), Harvard Catalyst Grant (to SN) NRF-2014R1A1A2056425 and NRF-2014R1A5A2010008. We also thank Dr. David J. Kwiatkowski and Izabela A. Malinowska for providing the pleural effusion fluid samples.
References
- 1.Mazur P. Cryobiology — Freezing of biological systems. Science. 1970;168:939–949. doi: 10.1126/science.168.3934.939. [DOI] [PubMed] [Google Scholar]
- 2.Stacey GN, Masters JR. Cryopreservation and banking of mam malian cell lines. Nat Protoc. 2008;3:1981–1989. doi: 10.1038/nprot.2008.190. [DOI] [PubMed] [Google Scholar]
- 3.Li AP. Human hepatocytes: Isolation, cryopreservation and applications in drug development. Chem Biol Interact. 2007;168:16–29. doi: 10.1016/j.cbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Linnebacher M, et al. Cryopreservation of human colorectal carcinomas prior to xenografting. BMC Cancer. 2010;10:9. doi: 10.1186/1471-2407-10-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fahy GM, Levy DI, Ali SE. Some emerging principles underlying the physical-properties, biological actions, and utility of vitrification solutions. Cryobiology. 1987;24:196–213. doi: 10.1016/0011-2240(87)90023-x. [DOI] [PubMed] [Google Scholar]
- 6.Karlsson JOM, Toner M. Long-term storage of tissues by cryopreservation: Critical issues. Biomaterials. 1996;17:243–256. doi: 10.1016/0142-9612(96)85562-1. [DOI] [PubMed] [Google Scholar]
- 7.Fahy GM, Wowk B, Wu J, Paynter S. Improved vitrification solutions based on the predictability of vitrification solution toxicity. Cryobiology. 2004;48:22–35. doi: 10.1016/j.cryobiol.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Hunt CJ, Pegg DE, Armitage SE. Optimising cryopreservation protocols for haematopoietic progenitor cells: A methodological approach for umbilical cord blood. Cryoletters. 2006;27:73–83. [PubMed] [Google Scholar]
- 9.Vajta G, et al. Open Pulled Straw (OPS) vitrification: A new way to reduce cryoinjuries of bovine ova and embryos. Mol Reprod Dev. 1998;51:53–58. doi: 10.1002/(SICI)1098-2795(199809)51:1<53::AID-MRD6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 10.Kuwayama M, Vajta G, Kato O, Leibo SP. Highly efficient vitrification method for cryopreservation of human oocytes. Reprod Biomed Online. 2005;11:300–308. doi: 10.1016/s1472-6483(10)60837-1. [DOI] [PubMed] [Google Scholar]
- 11.Martino A, Songsasen N, Leibo SP. Development into blastocysts of bovine oocytes cryopreserved by ultra-rapid cooling. Biol Reprod. 1996;54:1059–1069. doi: 10.1095/biolreprod54.5.1059. [DOI] [PubMed] [Google Scholar]
- 12.Lane M, Bavister BD, Lyons EA, Forest KT. Containerless vitrification of mammalian oocytes and embryos — Adapting a proven method for flash-cooling protein crystals to the cryopreservation of live cells. Nat Biotechnol. 1999;17:1234–1236. doi: 10.1038/70795. [DOI] [PubMed] [Google Scholar]
- 13.He XM, Park EYH, Fowler A, Yarmush ML, Toner M. Vitrification by ultra-fast cooling at a low concentration of cryoprotectants in a quartz micro-capillary: A study using murine embryonic stem cells. Cryobiology. 2008;56:223–232. doi: 10.1016/j.cryobiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HJ, et al. Ultra-rapid vitrification of oocytes in low cryoprotectant concentrations. Reprod Biomed Online. 2010;20:201–208. doi: 10.1016/j.rbmo.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Criado E, et al. Human oocyte ultravitrification with a low concentration of cryoprotectants by ultrafast cooling: A new protocol. Fertil Steril. 2011;95:1101–1103. doi: 10.1016/j.fertnstert.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 16.Risco R, Elmoazzen H, Doughty M, He XM, Toner M. Thermal performance of quartz capillaries for vitrification. Cryobiology. 2007;55:222–229. doi: 10.1016/j.cryobiol.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Dunn JC, Tompkins RG, Yarmush ML. Long-term in vitro function of adult hepatocytes in a collagen sandwich configuration. Biotechnol Prog. 1991;7:237–245. doi: 10.1021/bp00009a007. [DOI] [PubMed] [Google Scholar]
- 18.Sosef MN, et al. Cryopreservation of isolated primary rat hepatocytes — Enhanced survival and long-term hepatospecific function. Ann Surg. 2005;241:125–133. doi: 10.1097/01.sla.0000149303.48692.0f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, et al. The roles of apoptotic pathways in the low recovery rate after cryopreservation of dissociated human embryonic stem cells. Biotechnol Prog. 2010;26:827–837. doi: 10.1002/btpr.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crooks DM, et al. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Nat Acad Sci USA. 2004;101:17462–17467. doi: 10.1073/pnas.0407971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu J, Henske EP. mTOR activation, lymphangiogenesis, and estrogen-mediated cell survival: The “perfect storm” of pro-metastatic factors in LAM pathogenesis. Lymphat Res Biol. 2010;8:43–49. doi: 10.1089/lrb.2009.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debenedetti PG, Stanley HE. Supercooled and glassy water. Phys Today. 2003;56:40–46. [Google Scholar]
- 23.Boutron P. Comparison with the theory of the kinetics and extent of ice crystallization and of the glass-forming tendency in aqueous cryoprotective solutions. Cryobiology. 1986;23:88–102. doi: 10.1016/0011-2240(86)90022-2. [DOI] [PubMed] [Google Scholar]
- 24.Mazur P, Cole KW, Schreuders PD, Mahowald AP. Contributions of cooling and warming rate and developmental stage to the survival of drosophila embryos coo led to -205-Degrees-C. Cryobiology. 1993;30:45–73. doi: 10.1006/cryo.1993.1006. [DOI] [PubMed] [Google Scholar]
- 25.Rall WF, Reid DS, Farrant J. Innocuous biological freezing during warming. Nature. 1980;286:511–514. doi: 10.1038/286511a0. [DOI] [PubMed] [Google Scholar]
- 26.Criado EAE, Novara PV, Smeraldi A, Cesana A, Parini V, Levi-Setti PE. Human oocyte ultravitrification with a low concentration of cryoprotectants by ultrafast cooling: A new protocol. Fertil Steril. 2011;95:1101–1103. doi: 10.1016/j.fertnstert.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 27.Fonslow BR, Yates JR. Capillary electrophoresis applied to proteomic analysis. J Sep Sci. 2009;32:1175–1188. doi: 10.1002/jssc.200800592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahy GM, et al. Cryopreservation of organs by vitrification: Perspectives and recent advances. Cryobiology. 2004;48:157–178. doi: 10.1016/j.cryobiol.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Song YC, Khirabadi BS, Lightfoot F, Brockbank KGM, Taylor MJ. Vitreous cryopreservation maintains the function of vascular grafts. Nat Biotechnol. 2000;18:296–299. doi: 10.1038/73737. [DOI] [PubMed] [Google Scholar]
- 30.Song YC, et al. Vitreous preservation of articular cartilage grafts. J Invest Surg. 2004;17:65–70. doi: 10.1080/08941930490422438. [DOI] [PubMed] [Google Scholar]
- 31.Kuleshova LL, et al. Effective cryopreservation of neural stem or progenitor cells without serum or proteins by vitrification. Cell Transplant. 2009;18:135–144. [PubMed] [Google Scholar]
- 32.Chong YK, et al. Cryopreservation of neurospheres derived from human glioblastoma multiforme. Stem Cells. 2009;27:29–39. doi: 10.1634/stemcells.2008-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Images of viability and HMB 45 expression. (a) Viability of cells from LAM patient pleural effusion at 18 hours after vitrification using 200 μm(ϕ)−20 μm(t) fused silica capillary. (b) HMB45 expression in cells from a LAM patient pleural effusion. On Day 5 of growth rate experiments, the wells of control and experiment groups were fixed with 4% paraformaldhyde (PFA, Electron Microscopy Sciences) diluted in 1× PBS and were permeabilized with 0.2% Triton-X (Sigma) diluted in 1× PBS. After the permeabilization solution was removed, the cells were washed with 1× PBS and the cell membranes were blocked for 30 minutes in 2% normal goat serum (Vector Laboratories) and 3% bovine serum albumin (Sigma) in 1× PBS. Blocking agents were replaced by a primary antibody solution containing a 1:20 dilution of HMB45 antibody in 3% BSA in 1× PBS (w/o Ca2+, Mg2+). Following the primary antibody incubation, the patient samples were washed in 1× PBS before applying secondary antibodies (AlexaFluor488 IgG1) diluted at 1:500 in 3% bovine serum albumin in 1× PBS with 1:500 4′, 6-diamidino-2-phenylindole, dihydrochloride (DAPI, Invitrogen) for 1 hour for imaging. Scale bar for 100 μm.
Supplementary Figure 2 Images of viability after vitrification. Viability images of five cancerous epithelial cell lines (a–e), normal HMECs (f) at 12–18 hours after vitrification using 200 μm(ϕ)−20 μm(t) fused silica capillary. Entire plate (3.3 mm × 3.3 mm) images which contain all the cells dispensed from the capillary.