

Abstract
In this review, we discussed the findings and concepts underlying the potential role of Helicobacter pylori (H. pylori) infections in the initiation, development or persistence of atherosclerosis and coronary heart disease (CHD). This Gram-negative bacterium was described by Marshall and Warren in 1984. The majority of infected subjects carries and transmits H. pylori with no symptoms; however, in some individuals these bacteria may cause peptic ulcers, and even gastric cancers. The widespread prevalence of H. pylori infections and the fact that frequently they remain asymptomatic may suggest that, similarly to intestinal microflora, H. pylori may deliver antigens that stimulate not only local, but also systemic inflammatory response. Recently, possible association between H. pylori infection and extragastric disorders has been suggested. Knowledge on the etiology of atherosclerosis together with current findings in the area of H. pylori infections constitute the background for the newly proposed hypothesis that those two processes may be related. Many research studies confirm the indirect association between the prevalence of H. pylori and the occurrence of CHD. According to majority of findings the involvement of H. pylori in this process is based on the chronic inflammation which might facilitate the CHD-related pathologies. It needs to be elucidated, if the infection initiate or just accelerate the formation of atheromatous plaque.
Keywords: Helicobacter pylori, Coronary heart disease, Inflammation, Microbiota, Lipopolysaccharide
Core tip: Helicobacter pylori (H. pylori) is a Gram-negative spiral bacterium which colonizes gastric mucosa of nearly half of human population. A characteristic feature of H. pylori infection is an excessive inflammatory response. The majority of H. pylori infections remain asymptomatic. However, still it leads to the development of histological gastritis with the recruitment of immune cells. About 10% of infected subjects develop symptomatic gastritis, erosions or peptic ulcer. Gastric cancer is the most severe consequence of H. pylori infection. Recently, a possible association between chronic infections with H. pylori and extragastric disorders - including coronary heart disease, has been intensively investigated. Here we have revised recent studies confirming or excluding possible connections between chronic bacterial infections and the occurrence of coronary heart disease (CHD) within different populations, especially in the context of H. pylori infections. We have also presented various study approaches investigating direct and indirect interplay between H. pylori-driven consequences and CHD development to clarify already gained knowledge and suggest future directions. Considering the significance of already conducted research studies, the involvement of H. pylori infection in the process of CHD development is highly probably, however, still a lot need to be done to clarify whether this association is direct (with the involvement of H. pylori antigens and products) or indirect (with the involvement of inflammatory-related molecules accelerating/initiating CHD development).
INTRODUCTION
Since classic risk factors do not explain all cases of coronary heart disease (CHD) the concept that atherogenesis may have infectious background should be considered. The role of virus and bacterial pathogens including Helicobacter pylori (H. pylori) are now considered as factors implicated in the development of CHD. Chronic infections may influence the course of CHD via different mechanisms such as chronic inflammatory reactions, an autoimmune processes and modification of classic CHD risk factors. The pioneer finding of Mendall and co-workers, published in 1994, showed that CHD patients have elevated levels of serum anti-H. pylori antibodies. Following this finding, some authors confirm and some exclude the existence of this connection. Still there is no consensus on the role of H. pylori in either causation or progression of CHD. In order to describe the involvement of H. pylori in the development of CHD, it is necessary to find the largest number of reliable research studies confirming this relationship.
PATHOGENESIS OF CHD
CHD is one of the most severe chronic diseases of the coronary vessels - an important health and social problem - often life-threatening. It occurs due to endothelial dysfunction within the vessels, accompanied by an increased blood pressure, remodeling of vascular wall, local inflammation, platelet aggregation and blood clotting. These disorders promote the formation of atheromatous plaque, which is often unstable and subsequently ruptures. This might impair the blood flow leading to vascular blockage or myocardial infarction. Classic risk factors of CHD include cigarette smoking, hypertension, elevated levels of total cholesterol, triglycerides and low density lipoproteins (LDL) vs decreased high density lipoproteins (HDL) fraction, diabetes mellitus, as well as raised homocysteine and coagulation factors. Predisposing factors that increase the probability of CHD development are obesity, lack of physical activity, previous incidents of CHD in relatives, male gender, low socioeconomic status, as well as ethnic and behavioral factors that[1,2].
CHD is a group of symptoms resulting from chronic malnutrition and hypoxia of myocardial cells which is accompanied by oppression, burning, feeling the burden, discomfort and chest choking. These disorders are a consequence of atherosclerosis, which histologically is characterized by the accumulation of macrophages (MØ), LDL fractions, foam cells derived from macrophages filled with oxidized (ox) LDL and extracellular cholesterol complexes deposited within the vessels. On the inner surface of the vessel, lipid deposits are formed, which are gradually surrounded by a connective tissue and undergo fibrosis[3,4]. According to the statistics of World Health Organization (WHO), ischemia associated with atheromatous plaque is the main reason for CHD development, 70% of heart failure cases and 80% of sudden cardiac deaths. The natural history of atherosclerosis suggests that lesions in the arteries may occur already in the uterus or in early childhood. However, clinical manifestations of atherosclerosis are associated with the presence of atherosclerotic plaques, which in men usually develops after the age of 50 and in women postmenopausally[1].
DYSFUNCTION OF VASCULAR ENDOTHELIUM AS AN INITIATOR OF ATHEROMATOUS PLAQUE FORMATION
The interior of blood vessels is covered with a single layer of adjacent endothelial cells (size 0.2-0.3 mm) attached to the basal membrane and extracellular matrix molecules through integrin adhesion molecules[3]. Endothelium contacts with smooth muscle cells through gap junctions, which are permeable to the electric current, ions and low molecular weight compounds. Human vascular endothelium is a barrier that separates blood containing clotting proteins, platelets and inflammatory cells, from connective tissue and muscle layers of the blood vessel wall. The balance between the internal and external environment of the vessel depends on mechanical, chemical and immune reactions occurring within endothelial cells[1]. The endothelium is affected by physical pressure of blood flow (hemodynamic forces), various soluble substances and immune cells. Endothelium delivers many effector substances such as vasodilating and vasoconstrictioning factors (determining the proper tension of the vessel wall), cytokines and adhesion molecules (responsible for interactions with blood cells and the development of the inflammatory response), factors involved in blood coagulation and fibrinolysis. All together the endothelium plays a role in the maintenance of the vascular homeostasis which is determined by its large mass, distribution and the ability to receive and respond to signals from external environment (hemodynamic and chemical stimuli, pO2), by changing the expression of various active substances and proteins[1,5]. The endothelium expresses structures that are necessary for adhesion, migration, activation and diapedesis of immune cells and platelets, which allows for the development of inflammatory response[6,7]. These are mostly adhesion molecules (selectins) such as: P-selectin (platelet), E-selectin (endothelial) and L-selectin (leukocyte) and immunoglobulin-derived adressins, including: intracellular adhesion molecules (ICAM)-1 and -2, vascular cell adhesion molecule 1 (VCAM-1), platelet endothelial cell adhesion molecule 1, and macrophage chemotactic protein-1 (MCP-1). If endothelium is damaged it loses its functional integrity and homeostasis which initiates the occurrence of multiple lesions[5,8]. This dysfunction is usually leads to increased tension, vascular wall remodeling, vascular inflammation, increased platelet adhesion and aggregation. These processes contribute to the development of atherosclerosis or destabilization of existing atherosclerotic plaques[2].
CHD AS AN INFLAMMATORY PROCESS
In the late 90s we believed that the atherosclerotic process is a response to a mechanical trauma, resulting in the loss of endothelial cell lining in the vessels. Since the majority of CHD symptoms are induced by both local and systemic inflammatory responses, recently the attention is focused on the role of inflammation in the development of atherosclerosis[9-12]. Inflammatory markers, such as C-reactive protein (CRP) have been found to be higher in CHD patients than in controls, similarly to the concentration of interleukin (IL)-6 and tumor necrosis factor alpha (TNF-α) in plasma and supernatants of immune cells stimulated in vitro with bacterial lipopolysaccharide (LPS). Increased expression of E-selectin, L-selectin and P-selectin as well as higher expression of VCAM-1 and ICAM-1 was also noted in CHD cases[9,13]. It is difficult to identify factors that initiate cascade of inflammation and plaque formation. However, it is clear that endothelial dysfunction and raised cholesterol play a major role in the inflammation. Cholesterol contribute to the localization of atherosclerotic lesions, preferentially in the sites where it leads to the activation of endothelial NF-κB signal transduction pathway[14]. The inflammatory response is characterized by the influx of MØ and monocytes to the endothelium, with the latter being transformed first into MØ and subsequently to foam cells prior ingestion of oxLDL. Protein components of the LDL particles are processed by macrophages and dendritic cells and presented to T cells in the context of class II major histocompatibility complex[15]. Activated MØ and other inflammatory cells release chemokines that stimulate the migration of smooth muscle cells which together with foam cells, form a fibrous cap. This process is facilitated by interferon gamma (IFN-γ) and TNF-α secreted by T helper (Th)-1 lymphocytes, as well IL-12 produced by macrophages and foam cells[16]. The latter undergo apoptosis, and together with cholesterol crystals form lipid plaque cover[13,17]. It has been revealed that atherosclerotic lesions are associated with the increased reactivity of immune cells. The injured tissue releases IL-33 which alarms the immune system, induces expression of adhesion molecules and attracts Th2 lymphocytes delivering IL-4-considered anti-inflammatory cytokine[18-20]. However, a growing body of evidence indicates that IL-4 may play a role in atherosclerosis through induction of inflammatory responses, such as upregulation of VCAM-1 and MCP-1[21]. The main population of cells in newly formed atherosclerotic lesions are T lymphocytes, while in chronic lesions this proportion is reversed towards MØ that initiate immune processes by presenting antigens to T cells and the production of cytokines and chemokines[1,15,16,22].
MØ and neutrophils contains granules where myeloperoxidase and metalloproteinase are stored - the inflammatory markers correlated with a risk of atherosclerosis[23,24]. Myeloperoxidase contributes to leukocyte migration and the accumulation of foam cells. Indirectly it is involved in endothelial dysfunction and the induction of apoptosis with a consequence of plaque rupture and its destabilization. Due to the occurrence of vascular tissue factor is released and the activation of the blood coagulation cascade take place. Myeloperoxidase reduces the availability of endothelial nitric oxide and inhibits its diastolic and anti-inflammatory function. Moreover, it is involved in the oxidative modification of LDL to its atherogenous form, recognized by MØ receptors[3,10]. Prominent inflammation markers, activated by myeloperoxidase are delivered by macrophage-derived metalloproteinases (MMPs), hydrolyzing the components of extracellular matrix such as elastin and collagen, leading to the destabilization of atherosclerotic plaque. Metalloproteinases are also involved in the lipid peroxidation process and accelerated consumption of nitric oxide[22]. CRP belonging to the group of acute phase proteins which raises during infection or tissue damage, is an important marker of inflammation and is considered as an indicator of coronary events associated with endothelial damage. The upregulation of CRP is correlated with the elevation of IL-6, TNF-α, obesity and insulin resistance, which may indicate a link between chronic inflammation and endothelial dysfunction[12]. It has also been shown that CRP is more accurate marker of coronary events than the LDL cholesterol. This was based on the observation that women with the highest levels of CRP and low LDL were more susceptible to acute coronary insufficiency compared with those with high LDL and low CRP levels[25].
INFECTIOUS RISK FACTORS OF CHD
Classic risk factors do not explain all cases of CHD. Many data indicate that atherogenesis may be associated with chronic infections, accompanied by a long-term persistent inflammation[26-30]. Compelling evidence supports also the concept that gut microbiota actively promotes weight gain as well as fat accumulation, and indirectly sustains a condition of low-grade inflammation, thus escalating the risk of CHD[31-33]. The occurrence of microbiota favors not only intestinal but also the systemic exposure to the LPSs of Gram-negative bacteria. This microbiome-derived compound can cause a condition called “metabolic endotoxemia” characterized by low-grade inflammation, insulin resistance, and augmented cardiovascular risk. LPS is a powerful trigger for the cells of the innate immunity[34]. Variety of immune cells (monocytes, macrophages, Kupfer cells, and preadipocytes) and non-immune cells (adipocytes, hepatocytes, and endothelial cells) express Toll like receptor (TLR) 4 complex recognizing bacterial LPS[35]. Upon binding to TLR, it induces the release of proinflammatory molecules that interferes with metabolic paths of glucose and insulin, promotes development of the atherosclerotic plaque, and favors progression of fatty liver diseases[36,37].
Chronic infections may influence the development of CHD via various mechanisms such as chronic inflammatory reactions, an autoimmune responses and the modifications of classic risk factors for CHD[26,38]. They may pose direct effect on the vessel wall by inducing foam cell formation[39]. Therefore, Herpes simplex and Hepatitis C viruses as well as bacteria such as Chlamydophila pneumoniae, Mycoplasma pneumoniae, Porphyromonas gingivalis, Streptococcus mutans and H. pylori have been considered as factors involved in the development of CHD[40-45]. It has also been suggested, that Ch. pneumoniae promotes atherogenesis by inducing the synthesis of MCP-1, IL-8 and ICAM-1 in endothelial cells[44]. Among various pathogens possibly involved in atherogenesis H. pylori is particularly interesting, since it induces chronic long-term infection within gastric epithelium which leads not only to local but also systemic inflammation[45-48].
H. PYLORI A VERSATILE PATHOGEN
H. pylori is a Gram-negative bacterium demonstrating the affinity to gastric epithelial cells and perfect adaptation to the acidic environment of the stomach. In the majority of infected patients the interplay between H. pylori and the host cells are transformed into some sort of long lasting homeostasis. The majority of infected individuals (80%-90%) carry and transmit H. pylori with no symptoms, however, in some patients these bacteria induce pathological changes like gastroduodenal ulcers, as well as gastric cancers[49]. H. pylori are acquired early in life, and if not successfully treated persist for lifetime[50]. It is believed that the history and adaptation of H. pylori is associated with the evolution and migration of Homo sapiens. This bacterium has evolved to successfully colonize the hostile environment of the human stomach in the face of innate and adaptive immune responses[51]. In some ways, H. pylori resemble commensal bacteria. Contrary to this assumption, stays the fact that H. pylori expresses virulence factors with unquestionable pathogenic properties. For these reasons H. pylori infections should be monitored since, even if asymptomatic, they may cause systemic complications[52,53].
The interactions between H. pylori and gastric tissue cells determines the establishment and development of the disease[54]. Colonization of gastric epithelial cells by H. pylori via bacterial adhesins is followed by the occurrence of the acute phase of inflammation accompanied by the infiltration of gastric mucosa with granulocytes and MØ. H. pylori survives inside epithelial cells, also temporarily in MØ or in other niches within gastric tissues[55]. When infection becomes persistent, acute phase becomes chronic and is accompanied by an infiltration of lymphocytes. Inflammation is necessary for the proper recognition and elimination of infectious agents and tissue healing. But in case of H. pylori the inflammatory reaction is excessive and results in the development of pathological processes in gastric epithelium such as erosions, ulcers, modifications in the cells phenotype, their excessive proliferation as well as secretion of proinflammatory cytokines[56-58]. H. pylori possess an abundant composition of antigens[59]. Urease and vacuolating cytotoxin (VacA) stimulate inflammatory responses by damaging gastric epithelial cells, whereas cytotoxin-associated gene A (CagA) antigen, when introduced into the host cells through secretion system IV, evokes structural and functional changes. Also soluble forms of CagA may influence the activity of host gastric epithelial cells stimulating them to secrete IL-8 with chemotactic properties[60-62]. It inhibits proliferation of lymphocytes[63] and enhances expansion of gastric epithelial cells[64]. H. pylori modulates the activity of immune cells via different mechanisms such as molecular mimicry, antigen variation and immunomodulation of nonspecific and specific adaptive responses[65,66]. Some antigens of H. pylori enhance, while others inhibit the activity of immune cells. The first group includes surface lectins whereas CagA, VacA and LPS represents the second group[63,67-70]. H. pylori LPS shares some features common with human tissues. These are Lewis (Le) determinants: LeX, LeY, LeXY present in the O-specific chain of H. pylori LPS and on the surface of host cells: erythrocytes, granulocytes, monocytes, epithelial and vascular endothelial cells. In consequence, H. pylori can impair its recognition by host immune cells and pose a risk of autoreactive antibody production[71]. H. pylori LPS of LeXY type impairs phagocytic activity of granulocytes, cytotoxic activity of NK cells and lymphocyte proliferation[68,70]. It binds with dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) and may interfere with the development of specific immune response[72].
Recently, possible association between H. pylori infection and extragastric disorders, including iron deficiency anemia, chronic idiopathic thrombocytopenic purpura, growth retardation, diabetes mellitus and CHD is being considered[73]. Also, an inverse correlation between H. pylori prevalence and an increase in allergies, has been suggested. However, since the understanding of H. pylori-related pathologies continues to evolve, the idea that H. pylori might confer benefit to humans generates serious controversy. Postulated role of H. pylori in the pathogenesis of extragastric disorders is based on the following facts: (1) local inflammation induced by these pathogens has also systemic effects; (2) H. pylori infection induces chronic low grade process lasting for decades; and (3) persistent infection induces immune responses, which may have local and remote consequences.
The widespread prevalence of H. pylori infection and the fact that they are frequently asymptomatic may suggest that, similarly to intestinal microflora, H. pylori can be a source of antigenic components that stimulate not only local, but also systemic inflammatory response. Possibly H. pylori together with intestinal microbiota may enhance a risk of cardiovascular disorders, probably through a mechanism that involves an increased exposure to bacterial products translocated from the gut to the circulation[74,75]. Both H. pylori proteins and LPS demonstrate pro-inflammatory properties. Considering the role of H. pylori LPS as a proinflammatory compound, the different structure of its lipid A is taken into account[76,77]. This component of H. pylori LPS determines its diminished proinflammatory properties in comparison to other bacterial LPSs discussed in previous review[75]. Moreover the impact of Le determinants on the severity of H. pylori induced-inflammation has also been investigated. For instance, it has been shown that H. pylori LPS with or without LeXY determinants exhibit different effectiveness in stimulating the secretion of proinflammatory cytokines: IL-8 and TNF-α[78].
Recent knowledge on the pathoetiology of atherosclerosis together with current findings in the area of H. pylori infections constitute the background for the newly proposed hypothesis that those two processes may be related. To describe the involvement of H. pylori infection in the development of atherosclerosis, multiple study approaches have been undertaken. To discover a significance of H. pylori compounds, in the modulation of cell barrier function and its contribution to CHD development complex studies have to be undertake. The understanding of subsequent stages of H. pylori infections and the processes induced on the level of cellular barriers: gastrointestinal epithelium, vascular endothelium and the cells of innate immunity seem to be crucial.
Local chronic inflammation induced by H. pylori in the gastric epithelium, may be reflected on the periphery by the appearance of acute phase proteins and cytokines produced by immune cells and particular tissues[58,59,79]. These soluble systemic inflammatory markers may enhance the development of lesions within vascular endothelium. Also, it cannot be excluded that certain H. pylori components crossing the epithelial barrier in the stomach or intestines can have a direct influence on the vascular endothelial cells as well as circulating immune cells maintaining their constant activation (Figure 1). So far, it has been shown that H. pylori vacuolating toxin and urease contribute to the intercellular tight junction degradation[80]. If so, bacterial agents penetrating lamina propria may interact with immune cells or even enter the circulation. Although H. pylori colonize particularly the gastric epithelium its antigens are translocated to a deeper parts of gastrointestinal tract where they may be easily detected in feces[81]. In the jejunum components of H. pylori affect the expression of surface molecules, secretion of cytokines, epithelial permeability and its barrier function. Probably in Peyer’s patches H. pylori antigens initiate specific adaptive immunity and from this site could be spread into the circulation[79,82]. It has been hypothesized that H. pylori antigens may affect vascular endothelium by direct interactions with endothelium, indirectly in a form bound with leukocytes or as complexes with LDL/oxLDL fractions - classic risk factors of CHD[75]. The vascular endothelium can also be affected by H. pylori - driven cytokines and chemokines[57,78,83].
Figure 1.
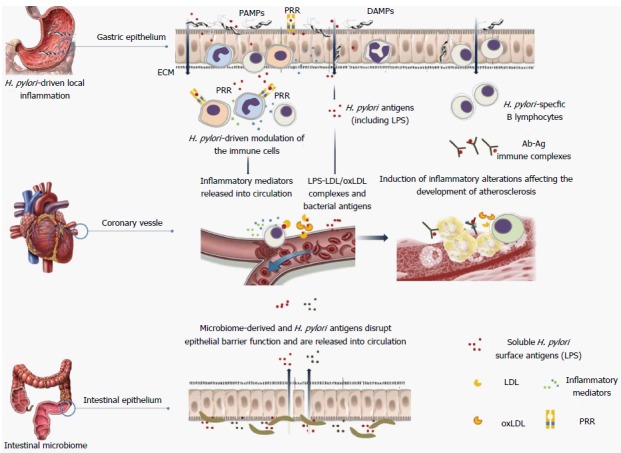
A possible link between local inflammation induced by Helicobacter pylori on surface of the gastric epithelium and the inflammatory response within vascular endothelium. H. pylori: Helicobacter pylori; LDL: Low density lipoproteins; LPS: Lipopolysaccharide; PRR: Pattern recognition receptor; PAMPs: Pathogen associated molecular patterns; DAMPs: Damage associated molecular patterns.
In order to evaluate the involvement of H. pylori infection in the development of CHD, it is necessary to find the largest number of research studies and possible connections confirming this relationship. The search for such connections should combine serological, biochemical, immunological as well as molecular markers. Serological and molecular studies on the material derived from patients with clinically confirmed CHD can provide markers helpful in defining individual susceptibility to chronic infections and extensive inflammation, predisposing to CHD. These cellular and molecular study approaches would describe the background of H. pylori-driven proinflammatory mechanisms directed towards epithelial and endothelial barrier functions, and innate immune cells, which would help to define their role in the atherogenesis.
H. PYLORI VS CHD - CURRENT STATE
Serological studies
The role of H. pylori infection in the development of CHD was suggested by Mendall et al[84] in 1994, where he observed for the first time the elevation of anti-H. pylori antibodies in the sera of CHD cases[84]. Following this pioneer finding, some authors made confirmed this association in several serological studies[85-88]. Searching for that connection other groups concentrated on the evaluation of bacteriological, biochemical, inflammatory and epidemiological parameters related with CHD and H. pylori infection (Table 1). The H. pylori seropositivity in CHD group varied from 40% up to 90%. Several studies also supported the association between CagA+ H. pylori infection and CHD prevalence. This relation is probably based on the increased levels of trombin - Factor VII and the prothrombin subunits: F1 + 2 or through the stimulation of low-grade persistent inflammatory response in CHD cases infected with H. pylori CagA+ strains[88-92]. However several authors obtained contrary data[93,94]. The findings coming from other studies showed no increase in the production of anti-H. pylori antibodies in CHD patients.
Table 1.
Major results of the clinical and basic research studies on the relationship between coronary heart disease and Helicobacter pylori infection
| Study characteristic | Ref. |
| Serological parameters | |
| Higher prevalence and concentrations of anti-H. pylori antibodies in CHD vs non-CHD individuals | [84-88] |
| Association between H. pylori CagA positive infections and CHD; exposure of endothelial and smooth muscle components within atherosclerotic plaques to the anti-CagA antibodies | [88-92] |
| Autoimmunity hypothesis: the presence of the immune complexes LeX/Y-anti-LeX/Y IgG in CHD patients infected with H. pylori | [47,127,134] |
| Bacteriological parameters | |
| Detection of H. pylori genomic material (16S rRNA) in the coronary arteries and atheromatous plaques from patients with cardiologic disorders | [43,91,103-105] |
| Presence of viable H. pylori bacteria in atherogenic plaques | [106] |
| Biochemical parameters | |
| Association of H. pylori infection with the increased biochemical and inflammatory parameters of CHD as well as coronary lumen reduction | [85,92,107-109] |
| Higher prevalence of LDL-hiperchlesterolemia, HDL-hypocholesterolemia and elevated levels of CRP in H. pylori infected than uninfected individuals | [110,127-129] |
| Lower activity of serum paraxonase-1 (a major anti-atherogenous component of HDL) and higher carotid-intima media thickness (one of the surrogate marker of atherosclerosis) in H. pylori positive in comparison to negative subjects | [108] |
| Positive correlation between raised LBP levels and the severity of CHD with co-existing H. pylori infection. The escalation of inflammatory process occurring via Toll-like receptors and LPS-LDL complexes | [127] |
| Increased levels of homocysteine in H. pylori infected individuals caused by malabsorption of vitamine B12 and foliate from diet, leading to obesity-related resistance to insulin | [48,130,131] |
| Inflammation and inflammation-related parameters | |
| Increased concentrations of IL-6, IL-8, TNF-α, plasminogen, activator inhibitor type-1, and von Willebrand factor in CHD patients infected with H. pylori | [3,38,69,83] |
| High levels of fibrinogen, a marker of systemic inflammation – putative link between H. pylori infections and pathophysiology of CHD | [133] |
| Recruitment of immune cells to the infectious foci and survival of H. pylori within the endothelium due to interaction of H. pylori LPS Le determinants with E- and L-selectins | [38,136] |
| Stimulation of Th1 lymphocytes to produce cytokines by H. pylori HspB | [47, 127,134] |
| Epidemiological studies | |
| Higher risk of CHD in ethnic groups of Central Africans and Mexican Americans with increased prevalence of H. pylori infections | [100-102] |
| Genetic susceptibility to infections and predisposition to strong inflammatory response | [140,145-146] |
CagA: Cytotoxin-associated gene A; CHD: Coronary heart disease; CRP: C-reactive protein; Ig: Immunoglobulines; HDL: High density lipoprotein; Hsp: Heat shock protein; LBP: Lipopolysaccharide binding protein; IL: Interleukin; LDL: Low density lipoprotein; Le: Lewis; TNF: Tumor necrosis factor.
Thus, still there is no consensus on the role of H. pylori infections in either causation or progression of CHD[95-98]. Possible reasons of these controversies may result from differences in: (1) magnitude of the study groups; (2) exclusion/inclusion criteria used in study groups selection; (3) the usage of serological tests for the H. pylori diagnostics; and (4) approaches for data analysis and statistical tests or insufficient knowledge on possible mechanisms involved. However, a new approach suggesting a role of gut microbiota in the development of chronic diseases prompts to continue the research[32]. Particularly stimulating are the results of research conducted in ethnic groups with low incidence of classic risk factors for CHD, and high prevalence of H. pylori infection. Recently published data[99] showed that high levels of anti-H. pylori IgG were significantly associated with the increased risk for CHD in a group of Central Africans. After adjusting with classic risk factors of CHD, H. pylori infection was found to be the only independent predictor of carotid plaque and stroke incidence in this group. Also Sealy-Jefferson et al[100] (2013) showed that the exposure to H. pylori in Mexican Americans may constitute a risk factor for stroke. Taking into consideration the increased prevalence of H. pylori in this population, the infection itself may contribute to the ethnic differences in stroke risk. It has also been suggested that H. pylori seroprevalence may influence long term prognosis for patients with unstable angina[101,102]. This finding is supported by several studies where genomic material (16S rRNA) of H. pylori was identified in the coronary arteries and atheromatous plaques from patients with cardiologic disorders including myocardial infarction and coronary artery disease - suggesting the direct involvement of H. pylori in CHD pathogenesis[43,91,103-105]. Some authors postulate the presence of via ble H. pylori in atherogenic plaques supporting their results by the culture of bacteria on solid media[106].
Inflammatory markers
It has been epidemiologically reported that H. pylori infections are associated with the changes in biochemical and inflammatory parameters as well as coronary lumen reduction[85,92,107-109]. In both H. pylori infected and CHD patients local inflammation occurring in gastric mucosa or in blood vessels, respectively turns into a chronic phase, which leads to a constitute presence of an inflammation-inducing agents. Increased concentrations of systemic inflammatory markers, both in patients with atherosclerosis and H. pylori infected individuals are usually considered a symptom or a result of a local inflammation. However, it has been claimed that systemic inflammation might be a cause and not a result of a local inflammatory reaction within atherosclerotic lesions[110]. Inflammation occurring in both, CHD and H. pylori infected individuals is determined by innate immune mechanisms with a participation of cell receptors called “alarmins”. They recognize conservative structures of infectious agents - pathogen associated molecular patterns (PAMPs) as well as host endogenous ligands - damage associated molecular patterns (DAMPs) appearing on MØ, dendritic cells (DC) and natural killer (NK) cells, as well as on epithelial and endothelial cells. It is supposed that the activation of immune or epithelial cells via pattern recognition receptors (PRRs) may be a reason for subacute inflammation in chronic diseases including CHD[11,69,111,112]. Local inflammation results with increased cytokine levels including IL–6 and TNF-α. Both stimulate the liver to produce acute phase proteins such as CRP, lipopolysaccharide binding protein (LBP) and MMP including MMP-9. Since, acute phase proteins are ligands for PRRs, they enhance the primary inflammation. However, chronic H. pylori infection lead to an excessive activation of inflammatory cells and a release of active radicals into the environment. This, due to oxidative stress, lead to tissue damage and apoptosis, therefore providing endogenous DAPMs such as heat shock protein (Hsp) 70, galectin-1, IL-1α, IL-33, mitochondrial damage motifs (mtDNA) and high mobility group box1 protein. Their probable role is a maintenance of inflammation, stimulation of tissue healing within the gastric ulcer niche, or removing damaged cells from the ischaemic niche, in the vascular endothelium. Mitochondrial DAMPs may increase endothelial permeability through neutrophil dependent and independent pathways[113]. Also specific microRNA expression is associated with the inflammatory response to damaged cells with possible deleterious implications[114]. Prolonged exposure to PAMPs and DAMPs is an apparent reason for a transformation of a local inflammation into a chronic form. The damage of vascular endothelium results in an increased production of reactive oxygen species and inactivation of nitric oxide, which has an anti-atherosclerotic properties. These changes lead to the activation of nuclear transcription factor NF-κB and result with a transformation of endothelium to a proinflammatory phenotype characterized by an increased expression of adhesins and chemokines, including MCP-1 and IL-8, with chemotactic activity towards inflammatory cells[1,14,22]. Proinflammatory phenotype of vascular endothelium exhibit an increased expression of PRR receptors including Toll-like receptors e.g., TLR4, CD14 and TLR2 recognizing bacterial LPS. The enhanced expression of these receptors also occurs on MØ accumulated in the atherosclerotic plaques[6,115,116].
Signaling pathways involving PRR receptors
In recent considerations recognizing CHD as an inflammatory disease, much attention has been paid to the role of signaling pathways involving PRR receptors present on MØ, DC and NK cells as well as endothelial and smooth muscle cells. There are different classes of PRR, including scavenger receptors, and the TLRs. Their role in the pathogenesis of CHD is still unclear and the results obtained in this issue vary greatly[112,117]. Toll-like receptors have been identified as molecules belonging to primary innate immunity. The studies on TLR4 and TLR2 knockout mice confirmed pro-atherogenous effect of TLR4/TLR2 signaling induction[118,119]. Although the expression of TLR2 and TLR4 on endothelial cells in normal arteries is rather low, it was found to be increased in the endothelium from atherosclerosis lesions[112]. Certain studies made an attempt to find a link between the susceptibility to CHD and TLR polymorphisms. Two single nucleotide polymorphisms of TLR4 - Asp299Gly and Thr399Ile were suspected to impair TLR signaling in response to LPS, in carriers of these alleles. It was suggested that both alleles were associated with the protection from carotid artery atherogenesis and the reduction of myocardial infarction risk up to 30%, in carriers of the Asp299Gly polymorphism[112,116,120]. Several TLR types: 1, 2, 4 and 5 are expressed in atherosclerotic plaques by resident cells and leukocytes that migrate into the arterial wall. The upregulation of TLR4 on MØ induced by proatherogenic oxidized LDL suggests that TLRs may provide a potential pathophysiological link between lipids, infection, inflammation and atherosclerosis[115]. The oxidized lipids may also serve as endogenous ligands of TLR2 and TLR4[121]. The study by Talreja et al[122] (2004) showed that mast cell-derived histamine up-regulates TLR4 and TLR2 expression on the host cells and by this enhances their sensitivity to cell wall components of Gram-positive and Gram-negative bacteria[122] - with Hsp and LPS considered as potential mediators linking bacterial infections with atherosclerosis. Moreover, it was shown that standard E. coli LPS induces the overexpression of TLR4, NF-κB, ICAM-1, VCAM-1 and the endothelial growth factor (VEGF), as well as the production of nitric oxide and IL-8[14,123].
The escalation of inflammatory process occurring in atherosclerosis does not exclude the participation of H. pylori LPS, which has low endotoxic activity, however, its proinflammatory potential is preserved. It stimulates MØ to secrete TNF-α, that inhibits lipoprotein lipase activity. This implies an increase in triglycerides and lower HDL cholesterol levels[124]. The recognition of H. pylori LPS by the immune cells and its interaction with vascular endothelium are not well understood. In the context of the correlation between the CHD incidence and H. pylori infection the interactions of LPS with TLR4 and TLR2 are taken into consideration, especially in regard to the variability of Le determinants in H. pylori LPS. It has been shown that H. pylori LPS without Le determinants (LeX-Y-) stimulates monocytes to produce lower concentrations of IL-8 and TNF-α than the LPS of LeX+Y+ type. Cytokine production induced by the latter type was inhibited by anti–CD14 and anti–LBP antibodies which confirms the involvement of both Le determinants and lipid A in those interactions[78].
H. pylori LPS exhibits weaker activity than the LPS of E. coli and express antagonistic properties towards TLR4. Current data do not rule out a role of TLR2 in the signaling induced by LPS of non-enterobacterial origin and its cooperation with TLR4[36]. It was shown that low stimulation of the TLR4 signaling by bacterial LPS may induce the expression of TLR2 in endothelial cells, probably via NADPH oxidase released by neutrophils[125]. Chronic H. pylori infection favors the formation of LPS-LDL complexes, directly or with the involvement of LBP. Such complexes, when deposited in the vascular endothelium, may enhance proinflammatory atherosclerotic processes[126]. It was shown that the presence of LBP is required for the LPS-dependent activation of intracellular TLR4 in endothelial cells. LBP acts as a catalyst of this process by the translocation of serum sCD14-LPS complexes into the cells[111]. In this context, the positive correlation between raised LBP and the severity of CHD with co-existing H. pylori infection seems to be of great importance[127]. It is also possible that H. pylori LPS contribute to CHD due to its anti-phagocytic, anti-cytotoxic and anti-proliferative properties, towards phagocytes, NK cells and lymphocytes respectively[68-70].
The expression of TLR4 and TLR2 is intensified in the inflamed endothelium. Recent data indicate that the binding of E. coli LPS with TLR4 may increase the permeability of the vascular epithelium[36]. Any kind of endothelial dysfunction, including a reduction of cell integrity may result in inflammatory cascade. The involvement of TLRs in the development of atherosclerosis is associated with the ability of those receptors to bind ox-LDL, which initiate atherogenesis. Binding of such complexes induces a cascade of signals that activate the transcription factor NF-κB and results in the upregulation of inflammasome components such as cytokines and acute phase proteins[14]. In the context of atherosclerosis the key NF-κB-dependent proteins include inflammatory cytokines: IL-1β and TNF-α, chemokines: IL-8, MCP-1 and MMPs hydrolyzing the extracellular matrix[1]. The role of IL–1β in the development of CHD is associated with the stimulation of endothelial cells to produce IL–6, fibrinogen, CRP and adhesins resulting in a activation of signal cascade leading to the destabilization of atherosclerotic plaques[8].
Acute phase response, lipid metabolism, homocysteine and fibrinogen related mechanisms
Significant association of H. pylori infection with LDL-hipercholesterolemia, HDL-hypocholesterolemia and elevated levels of CRP was found. This indicates a possible impact of chronic infection on a lipid metabolism, which is associated with the increased CHD risk[110,128,129]. It was also noted that seropositive patients with unstable angina develop diabetes more frequently than seronegative individuals. H. pylori infection increases obesity-related resistance to insulin causing malabsorption of vitamin B12 and foliate from diet, ultimately leading to an increase in circulating homocysteine levels[48,130,131]. Since raised homocysteine may disturb the function of vascular endothelium it might be implicated in the coronary slow flow phenomenon. However, there are also suggestions that homocysteine is a marker rather than a cause of CHD[132]. In H. pylori positive subjects the activity of serum paraoxonase-1 (a major anti-atherogenous component of HDL) was lower while carotid-intima media thickness (one of the surrogate marker of atherosclerosis) was higher[108]. The sera of H. pylori infected subjects contain increased concentrations of inflammatory cytokines, particularly IL-6, IL-8 and TNF-α, plasminogen, activator inhibitor type-1, and von Willebrand factor - a sensitive indicator of atherosclerosis and a predictive factor of acute coronary syndrome[133]. Certain studies also showed that high levels of fibrinogen, a marker of systemic inflammation can constitute a probable link between H. pylori infections and CHD pathophysiology[47]. The putative mechanism of this association might involve H. pylori-driven stimulation of mononuclear cells to produce a tissue-factor-like pro-coagulant that, converts fibrinogen to fibrin though the extrinsic blood coagulation pathway. Fibrinogen also stimulates macrophage chemokine secretion through TLR4, promoting immune surveillance at sites of inflammation[134]. However, there are also contradictory results and hypotheses that the occurrence of CHD is positively associated with age and lower social class[135]. It would be of great importance to check, whether H. pylori eradication is associated with the decrease in the level of the above markers and lower CHD incidence. To date, anti-H. pylori eradication therapy confirmed only some suggestions. Mean coronary artery lumen loss in patients undergoing percutaneous transluminal coronary angioplasty (PTCA) with stent and anti-H. pylori eradication therapy was significantly smaller compared to PTCA and placebo treated group. Similarly, cytokines such as TNF-α, IL-1β and IL-8 were significantly lower in plasma of PTCA patients after H. pylori eradication, while there were no changes in plasma lipids, homocysteine and clotting factors[85].
Autoimmunity hypothesis
Bacterial pathogens, including H. pylori might contribute to CHD pathogenesis. This approach is supported by the fact that CHD is starting to be considered as an autoimmune inflammatory process. The antigenic structures of infectious agents can induce the expansion of potentially autoreactive T and B cells, or B cells producing antibodies cross-reacting with host tissues. This phenomenon is defined as antigenic mimicry. For instance H. pylori HspB (60 kDa) might be implicated in CHD pathogenesis via stimulation of Th1 lymphocytes to secrete IFN-γ and IL-12 or activation of MØ to express metalloproteinases and adhesins[38]. Antigenic mimicry as a cause of inflammation in CHD is also related to Le determinants. In human tissues Le antigens serve as ligands for endothelial (E and P–selectin) and leukocytes (L–selectin) adhesins. This interaction drives cell migration into the inflammatory milieu and plays an important role in the accumulation of immune cells in peripheral lymph nodes. It was shown that H. pylori bearing Le antigens in their LPSs are able to bind E- and L-selectins. This linkage enables the recruitment of immune cells to the infectious foci and may promote survival of H. pylori within the endothelium[136]. The activity of H. pylori LPS is also manifested by the activation of monocytes, MØ and secretion of proinflammatory cytokines: IL-1, IL-6 and IL-8[69]. H. pylori strains bearing LeX or LeA attract circulating lymphocytes that express L–selectin. It was shown that H. pylori expressing Le determinants induce higher colonization rates and more excessive infiltration of gastric mucosa with neutrophils and lymphocytes - a phenomenon also observed in individuals infected with H. pylori expressing LeX determinants[137]. Due to the ongoing inflammation the endothelial and smooth muscle components within atherosclerotic plaque might be revealed and exposed to the anti-CagA antibodies. The formation of such immune complexes facilitates the risk for further damage of the endothelium caused by lytic complex of complement proteins[46].
INDIVIDUAL SUSCEPTIBILITY TO INFECTION AND INFLAMMATION IN RESPECT TO THE DEVELOPMENT OF CHD
The risk for cardiovascular diseases might also be considered on the genetic level-determining the susceptibility to CHD development related to inflammatory process[138]. For example, one of the explanations for elevated levels of CRP in CHD patients might lay in chronic, bacterial or viral infection. However, since viral as well H. pylori and Ch. pneumoniae infections, are very common, it is believed that an individual susceptibility to infections and accompanying inflammation could explain the role of infectious agents in the course of CHD. This individual predisposition to persistent infections and chronic inflammatory response can be determined, to some extent, by the Le antigens, receptors for PAMPs and proinflammatory cytokines. It is believed that Lewis antigens can play a key role in shaping the individual susceptibility to CHD development: by directing the adverse effects of infection and excessive inflammatory response[41]. There are also clear examples of protection against infectious diseases (particularly to H. pylori, norovirus, and Vibrio cholerae) based on polymorphisms in genes encoding and regulating the expression of ABH blood group and Lewis antigens[139]. There are two types of Lewis antigens in humans: Le a and Le b. Their expression depend on genes located on chromosome 1 encoding fucosyltransferases: FUT2 and FUT3. Depending on the genotype, and thus the expression of one or both Le antigens, in the Caucasian population, there are three dominating phenotypes: Lea+b-, Lea-b+, Lea-b-, and Lea+b+ which occurs very rarely. Le antigens expressed on cell surface and released in body fluids are associated with the susceptibility to infections especially related to the mucus layer, such as those caused by H. pylori[140]. It is assumed that Le antigens promote adhesion-dependent infections[141]. There are speculations on the link between the Lea-b- phenotype and several disorders constituting a risk factors for CHD development, with examples such as insulin resistant diabetes, elevated levels of clotting factor VIII and von Willebrand factor. This phenotype is considered a genetic marker for the risk for CHD development[142,143]. It is also believed that the polymorphism in FUT3 associated with the presence of point mutations 59T > G, 202T > C, 314 C > T, 1067 T > A, may determine the individual susceptibility to infections and the development of atherosclerotic lesions and strong inflammatory response[144].
The polymorphism of inflammation-related genes, may indirectly contribute to the development of CHD, and the dynamics of the disease. Such a possibility appears especially when the mutations accumulate in several genes related with inflammatory response. Thus, while searching for the relationship between H. pylori infections and their role in the development of CHD, mutations in the genes encoding TLR4/CD14 receptors (binding LPS), and IL–1β should be taken into consideration[145]. IL-1β acts as a stimulating mediator of IL-6, fibrinogen, CRP or adhesive molecules expression by endothelial cells within a cascade leading to the development and destabilization of the atherosclerotic plaques. In regard to IL-1β the most frequently considered gene mutations are: -511C > T and -31C > T[145,146]. It was showed that carriers of the two relatively frequent variants of IL-1β gene at -31 and -511 positions, i.e., -31 TT and -511 CC, are at a higher risk of developing CHD requiring surgical treatment or two-stage percutaneous angioplasty. In patients prone to the development of atherosclerosis, polymorphism of IL-1β gene cluster may be associated with the extent and dynamics of lesions in the coronary arteries[146]. For gene encoding TLR4: Asp299Gly and Thr399Ile and for CD14 gene: 159C > T mutations are considered to play a role in CHD and chronic infections[112,116]. Patients carrying Asp299Gly, a common variant of the TLR4 gene presented reduced prevalence of angiographic artery disease and low levels of CRP. This common variant of the TLR4 gene, probably attenuates receptor signalling and diminishes inflammatory response to Gram-negative pathogens[147].
Since neither infection nor the activation of TLR4/TLR2 is sufficient to induce atherosclerosis in animal models[148], it is rather unlikely that microbes and/or TLRs signaling play a causative role in this disease. Instead, it is thought that they may be important as associates of silent disease. For instance, microbial components such as LPS or lipoteichoic acid released during acute infection or exacerbation of chronic infection might activate plaque cells. It has been suggested that such local “echos” of infections could lead to increased local production of cytokines and initiate plaque activation and rupture. The expression of TLRs in plaques suggests a pathway through which such an echo effect could occur[6]. Because H. pylori infection is located in the stomach, the question arises why the possible inflammation should only be transferred to the heart blood vessels and not to other vessels of the body? Various activities of the immune cells are mediated by endothelial cells, which form specialized microcirculatory networks used by the immune cells under both physiological and pathological circumstances. Endothelial cells represent a highly heterogenous population of cells with the ability to interact with and modulate the function of immune cells[149]. Atherosclerotic lesions occur at distinct sites within the arterial tree, such as branches, bifurcations, and curvatures, where they cause characteristic alterations in the blood flow, including decreased shear stress and increased turbulence. The nature of the flow appears to determine whether lesions occur at these vascular sites. The low-shear hypothesis of atherosclerosis has been validated[150]. Decreasing shear stress at branches, bifurcations, and curvatures results in endothelial activation, adhesion molecule expression, and greater monocyte transmigration. It has been shown that atherosclerotic lesions appear first at lesion-prone sites, where activated endothelium expresses specific molecules, which favors the recruitment of monocytes and T cells. For instance, it has been hypothesized that the regiospecitifity of atherosclerotic lesions might be determined by the lower expression of TLR2 molecules[111]. The localization of atherosclerotic lesions could be also related to the local overexpression of NF-κB/IκB pathways[14].
FUTURE RESEARCH PERSPECTIVES
To describe the role of H. pylori in the initiation, acceleration or the development of CHD a few fundamental questions need to be addressed. It need to be elucidated, whether viable H. pylori or bacterial compounds are able to break the single layer of epithelial cells and have unimpeded access to the systemic circulation. Also, it is not clear, whether classic risk factors such as hypercholesterolemia may act synergistically with H. pylori or their compounds to destabilize or disrupt gastric epithelial barrier function. It is also interesting whether CHD as systemic disease can lead independently to the disruption of gastric epithelial barrier function. The use of well-defined cell lines which mimic the in vivo conditions and exclude the naturally occurring phenotypic variations or the influence of external agents will enable to clarify the relationship between H. pylori as effective colonizer of gastric mucosa and inflammatory response. Methodology of culturing the cells using trans-well systems can help to examine whether H. pylori antigens alone or in combination with classic CHD risk factors interfere with the integrity of gastric epithelial and endothelial cells, cytotoxicity, the cell cycle, chemokines as well as cytokines and cell signaling. Microfluidic culture systems enable to explain if H. pylori compounds might be delivered to the inflammatory sites within vascular endothelium and interact with both endothelial and the immune cells[151].
Since, H. pylori infection has been defined as class I gastric carcinogen and many epidemiological studies demonstrated positive correlation between serum lipids and the risk of gastrointestinal malignancies, it is tempting to evaluate the prevalence of malignancies in CHD patients infected with H. pylori. Although it has been shown that H. pylori infection is related with increased LDL level, the association between abnormal concentrations of serum lipid components, the infection with H. pylori and the risk of gastrointestinal cancer is unknown[152].
CHD patients are recommended for antitrombocythic therapy with aspirin, which can be beneficial to individuals who already have experienced a heart attack, stroke, angina or peripheral vascular disease, or have had certain procedures such as angiography or bypass. However, aspirin can be prescribed to prevent heart disease and stroke in same individuals who have not previously experienced these events. The United States Preventive Services Task Force recommends that men with no history of heart disease or stroke aged 45-79 years should use aspirin to prevent myocardial infractions and that woman with no history of heart disease or stroke aged 55-79 should use aspirin to prevent stroke[153]. On the other hand, NSAIDs such as aspirin is positively correlated with the incidence of gastrointestinal tract disorders. Such damage can take a form of mucosal erosions or ulcers. NSAIDs can stimulate leukocytes, particularly neutrophils, such that they adhere to the vascular endothelium within the gastrointestinal microcirculation. Moreover NSAIDs impair the rapid restitution that occurs through cell migration following damage to the superficial epithelium of the stomach, reduce rates of epithelial turnover and thus impair the healing process. It is necessary to elucidate, whether ulcers are more likely to develop in long-term NSAIDs users who have mucosal erosions or in individuals infected with H. pylori, or both – and what is the role of NSAIDs on the course of CHD and H. pylori-related pathologies[153,154].
Proinflammatory agents released directly due to damage induced by H. pylori or indirectly by neutrophils recruited to the site of infection break the epithelial barrier. An initial effect of H. pylori infection is amplified significantly and impair the proper action of cellular barrier. The question is whether inflammatory mediators generated in the stomach can reach and harm distant tissues, leading to systemic disorders related with CHD.
Tissue inflammation, cell injury or death result in the release of molecules that are endogenous PRR ligands. DAMPs stimulates cells to produce acute phase cytokines and activates other inflammatory compounds. Depending on the affected tissue, various stromal cells, including epithelial and endothelial cells, may function as sentinels for detection of DAMPs, which felicitate neutrophil recruitment. It was hypothesized that IL-33 may have protective effects during atherosclerosis by inducing a Th1-to-Th2 switch of immune responses[19]. However, many questions regarding the role of specific DAMPs during H. pylori infections and cardiovascular diseases remain to be solved.
Since initial moment of H. pylori infection is almost impossible to identify, little is known about the natural history and kinetics of infection and immune responses. There is an urgent need to establish and optimize the animal model mimicking human immune system, sensitive for H. pylori infection and CHD development. Immunologic similarities between guinea pigs and humans in regard to: leukocyte antigens, complement system, antigen presenting molecules, patterns of IFN-γ, IL-8, IL-12 release, as well as their receptors suggest that this animal model may be suitable for studies on the relation between H. pylori infection and the development of its extragastric consequences. Antigen-specific lymphocyte proliferation has been found a suitable marker of immune response in guinea pigs with sustained H. pylori infection. Recently guinea pigs were successfully used to show the role of endotoxemia in the myocardial injury and sepsis-associated dysfunction[42,72,155].
It is believed that an individual susceptibility to infections and accompanying inflammation could help to explain the role of infectious agents in the course of CHD. Using the samples from patients with clinically confirmed CHD infected or not with H. pylori in comparison with control group it is necessary to look for cellular and molecular markers which may determine an individual susceptibility to chronic infections and extensive inflammation, predisposing to CHD.
These cellular and molecular studies would help to understand the role of H. pylori infections in the pathogenesis of CHD. Describing the background of H. pylori - driven proinflammatory mechanisms directed towards epithelial and possibly endothelial barrier, would help to allocate their role in the process of atherogenesis. In case of proven causative role of this bacterium in the pathogenesis of CHD, its eradication will be important for diminishing one of CHD infectious risk factors.
CONCLUSION
CHD, one of the most severe chronic diseases of the coronary vessels is a multifactorial disorder. Since classic risk factors do not explain all cases of CHD it has been suggested that chronic infections and even commensal microorganisms may affect the development or maintenance of CHD. Among various pathogens possibly involved in atherogenesis H. pylori is particularly interesting, since it induces chronic long-term infection within gastric epithelium which leads to not only local but systemic inflammation. Recent knowledge on the pathogenesis of atherosclerosis together with current findings in the field of H. pylori related diseases constitute the background for the newly proposed hypothesis that those two processes may be related.
Footnotes
Conflict-of-interest: The authors have no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 24, 2014
First decision: December 26, 2014
Article in press: February 9, 2015
P- Reviewer: Chen W, Tosetti C S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 2.Kutuk O, Basaga H. Inflammation meets oxidation: NF-kappaB as a mediator of initial lesion development in atherosclerosis. Trends Mol Med. 2003;9:549–557. doi: 10.1016/j.molmed.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Rodella LF, Rezzani R. Endothelial and vascular smooth cell dysfunction: a comprehensive appraisal. In: Parthasarathy S. Artherogenesis. InTech under CC BY 3.0 licence; 2012. pp. 105–134. [Google Scholar]
- 4.Kawasaki M, Yoshimura S, Yamada K. Tissue characterization of carotid plaques. In: Rezzani R. Carotid artery disease-from bench to bedside and beyond. InTech under CC BY 3.0 licence; 2014. pp. 19–29. [Google Scholar]
- 5.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–III32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 6.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 7.Cybulsky MI. Morphing the topography of atherosclerosis: an unexpected role for PECAM-1. Arterioscler Thromb Vasc Biol. 2008;28:1887–1889. doi: 10.1161/ATVBAHA.108.174029. [DOI] [PubMed] [Google Scholar]
- 8.Bochner BS, Luscinskas FW, Gimbrone MA, Newman W, Sterbinsky SA, Derse-Anthony CP, Klunk D, Schleimer RP. Adhesion of human basophils, eosinophils, and neutrophils to interleukin 1-activated human vascular endothelial cells: contributions of endothelial cell adhesion molecules. J Exp Med. 1991;173:1553–1557. doi: 10.1084/jem.173.6.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Li JJ, Li Q, Li Z, Qian HY. A rational connection of inflammation with peripheral arterial disease. Med Hypotheses. 2007;69:1190–1195. doi: 10.1016/j.mehy.2007.02.043. [DOI] [PubMed] [Google Scholar]
- 10.Brevetti G, Giugliano G, Brevetti L, Hiatt WR. Inflammation in peripheral artery disease. Circulation. 2010;122:1862–1875. doi: 10.1161/CIRCULATIONAHA.109.918417. [DOI] [PubMed] [Google Scholar]
- 11.Chalubinski M, Wojdan K, Dorantowicz R, Jackowska P, Gorzelak P, Broncel M. Comprehensive insight into immune regulatory mechanisms and vascular wall determinants of atherogenesis - emerging perspectives of immunomodulation. Arch Med Sci. 2013;9:159–165. doi: 10.5114/aoms.2013.33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perkov S, Paro MMK, Vidjak V, Flegar-Mestric Z. The Evaluation of New Biomarkers of Inflammation and Angiogenesis in Peripheral Arterial Disease. In: Current trends in atherogenesis. Rezzani R. InTech under CC BY 3.0 license; 2013. pp. 97–120. [Google Scholar]
- 13.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–291. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 16.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 17.Milioti N, Bermudez-Fajardo A, Penichet ML, Oviedo-Orta E. Antigen-induced immunomodulation in the pathogenesis of atherosclerosis. Clin Dev Immunol. 2008;2008:723539. doi: 10.1155/2008/723539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demyanets S, Konya V, Kastl SP, Kaun C, Rauscher S, Niessner A, Pentz R, Pfaffenberger S, Rychli K, Lemberger CE, et al. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2011;31:2080–2089. doi: 10.1161/ATVBAHA.111.231431. [DOI] [PubMed] [Google Scholar]
- 19.Miller AM. Role of IL-33 in inflammation and disease. J Inflamm (Lond) 2011;8:22. doi: 10.1186/1476-9255-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caseli CH. Inflammation in cardiac disease: focus on interleukin-33/ST2 pathway. Inflamm Cell Signaling. 2014;1:118–126. Available from: http://www.smartscitech.com/index.php/ICS/article/view/149. [Google Scholar]
- 21.Lee YW, Kühn H, Hennig B, Neish AS, Toborek M. IL-4-induced oxidative stress upregulates VCAM-1 gene expression in human endothelial cells. J Mol Cell Cardiol. 2001;33:83–94. doi: 10.1006/jmcc.2000.1278. [DOI] [PubMed] [Google Scholar]
- 22.Sheikine Y, Hansson GK. Chemokines and atherosclerosis. Ann Med. 2004;36:98–118. doi: 10.1080/07853890310019961. [DOI] [PubMed] [Google Scholar]
- 23.Altieri P, Brunelli C, Garibaldi S, Nicolino A, Ubaldi S, Spallarossa P, Olivotti L, Rossettin P, Barsotti A, Ghigliotti G. Metalloproteinases 2 and 9 are increased in plasma of patients with heart failure. Eur J Clin Invest. 2003;33:648–656. doi: 10.1046/j.1365-2362.2003.01187.x. [DOI] [PubMed] [Google Scholar]
- 24.Tayebjee MH, Nadar S, Blann AD, Gareth Beevers D, MacFadyen RJ, Lip GY. Matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in hypertension and their relationship to cardiovascular risk and treatment: a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) Am J Hypertens. 2004;17:764–769. doi: 10.1016/j.amjhyper.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 25.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 26.Danesh J, Collins R, Peto R. Chronic infections and coronary heart disease: is there a link? Lancet. 1997;350:430–436. doi: 10.1016/S0140-6736(97)03079-1. [DOI] [PubMed] [Google Scholar]
- 27.Chiu B. Multiple infections in carotid atherosclerotic plaques. Am Heart J. 1999;138:S534–S536. doi: 10.1016/s0002-8703(99)70294-2. [DOI] [PubMed] [Google Scholar]
- 28.Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, Hafner G, Prellwitz W, Schlumberger W, Meyer J. Impact of infectious burden on progression of carotid atherosclerosis. Stroke. 2002;33:2581–2586. doi: 10.1161/01.str.0000034789.82859.a4. [DOI] [PubMed] [Google Scholar]
- 29.Calabrese F, van der Wal AC, Levi M. Infection and inflammation in the cardiovascular system. Cardiovasc Res. 2003;60:1–4. doi: 10.1016/s0008-6363(03)00569-8. [DOI] [PubMed] [Google Scholar]
- 30.Jafarzadeh A, Nemati M, Tahmasbi M, Ahmadi P, Rezayati MT, Sayadi AR. The association between infection burden in Iranian patients with acute myocardial infarction and unstable angina. Acta Med Indones. 2011;43:105–111. [PubMed] [Google Scholar]
- 31.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hattori M, Taylor TD. The human intestinal microbiome: a new frontier of human biology. DNA Res. 2009;16:1–12. doi: 10.1093/dnares/dsn033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. 2010;31:817–844. doi: 10.1210/er.2009-0030. [DOI] [PubMed] [Google Scholar]
- 34.Rabizadeh S, Sears C. New horizons for the infectious diseases specialist: how gut microflora promote health and disease. Curr Infect Dis Rep. 2008;10:92–98. doi: 10.1007/s11908-008-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beutler B, Hoebe K, Du X, Ulevitch RJ. How we detect microbes and respond to them: the Toll-like receptors and their transducers. J Leukoc Biol. 2003;74:479–485. doi: 10.1189/jlb.0203082. [DOI] [PubMed] [Google Scholar]
- 36.Faure E, Thomas L, Xu H, Medvedev A, Equils O, Arditi M. Bacterial lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol. 2001;166:2018–2024. doi: 10.4049/jimmunol.166.3.2018. [DOI] [PubMed] [Google Scholar]
- 37.de Kleijn D, Pasterkamp G. Toll-like receptors in cardiovascular diseases. Cardiovasc Res. 2003;60:58–67. doi: 10.1016/s0008-6363(03)00348-1. [DOI] [PubMed] [Google Scholar]
- 38.Matsuura E, Kobayashi K, Matsunami Y, Shen L, Quan N, Makarova M, Suchkov SV, Ayada K, Oguma K, Lopez LR. Autoimmunity, infectious immunity, and atherosclerosis. J Clin Immunol. 2009;29:714–721. doi: 10.1007/s10875-009-9333-5. [DOI] [PubMed] [Google Scholar]
- 39.Epstein SE. The multiple mechanisms by which infection may contribute to atherosclerosis development and course. Circ Res. 2002;90:2–4. [PubMed] [Google Scholar]
- 40.Ayada K, Yokota K, Kobayashi K, Shoenfeld Y, Matsuura E, Oguma K. Chronic infections and atherosclerosis. Clin Rev Allergy Immunol. 2009;37:44–48. doi: 10.1007/s12016-008-8097-7. [DOI] [PubMed] [Google Scholar]
- 41.Angiolillo DJ, Liuzzo G, Pelliccioni S, De Candia E, Landolfi R, Crea F, Maseri A, Biasucci LM. Combined role of the Lewis antigenic system, Chlamydia pneumoniae, and C-reactive protein in unstable angina. J Am Coll Cardiol. 2003;41:546–550. doi: 10.1016/s0735-1097(02)02899-1. [DOI] [PubMed] [Google Scholar]
- 42.Padilla C, Lobos O, Hubert E, González C, Matus S, Pereira M, Hasbun S, Descouvieres C. Periodontal pathogens in atheromatous plaques isolated from patients with chronic periodontitis. J Periodontal Res. 2006;41:350–353. doi: 10.1111/j.1600-0765.2006.00882.x. [DOI] [PubMed] [Google Scholar]
- 43.Kilic A, Onguru O, Tugcu H, Kilic S, Guney C, Bilge Y. Detection of cytomegalovirus and Helicobacter pylori DNA in arterial walls with grade III atherosclerosis by PCR. Pol J Microbiol. 2006;55:333–337. [PubMed] [Google Scholar]
- 44.Rosenfeld ME, Campbell LA. Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost. 2011;106:858–867. doi: 10.1160/TH11-06-0392. [DOI] [PubMed] [Google Scholar]
- 45.Huang WS, Tseng CH, Lin CL, Tsai CH, Kao CH. Helicobacter pylori infection increases subsequent ischemic stroke risk: a nationwide population-based retrospective cohort study. QJM. 2014;107:969–975. doi: 10.1093/qjmed/hcu117. [DOI] [PubMed] [Google Scholar]
- 46.Franceschi F, Sepulveda AR, Gasbarrini A, Pola P, Silveri NG, Gasbarrini G, Graham DY, Genta RM. Cross-reactivity of anti-CagA antibodies with vascular wall antigens: possible pathogenic link between Helicobacter pylori infection and atherosclerosis. Circulation. 2002;106:430–434. doi: 10.1161/01.cir.0000024100.90140.19. [DOI] [PubMed] [Google Scholar]
- 47.Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. JAMA. 1998;279:1477–1482. doi: 10.1001/jama.279.18.1477. [DOI] [PubMed] [Google Scholar]
- 48.Polyzos SA, Kountouras J, Zavos C, Deretzi G. The association between Helicobacter pylori infection and insulin resistance: a systematic review. Helicobacter. 2011;16:79–88. doi: 10.1111/j.1523-5378.2011.00822.x. [DOI] [PubMed] [Google Scholar]
- 49.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 50.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Monack DM. Helicobacter and salmonella persistent infection strategies. Cold Spring Harb Perspect Med. 2013;3:a010348. doi: 10.1101/cshperspect.a010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmad T, Sohail K, Rizwan M, Mukhtar M, Bilal R, Khanum A. Prevalence of Helicobacter pylori pathogenicity-associated cagA and vacA genotypes among Pakistani dyspeptic patients. FEMS Immunol Med Microbiol. 2009;55:34–38. doi: 10.1111/j.1574-695X.2008.00492.x. [DOI] [PubMed] [Google Scholar]
- 53.Ahmed N, Tenguria S, Nandanwar N. Helicobacter pylori--a seasoned pathogen by any other name. Gut Pathog. 2009;1:24. doi: 10.1186/1757-4749-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Posselt G, Backert S, Wessler S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal. 2013;11:77. doi: 10.1186/1478-811X-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allen LA. Phagocytosis and persistence of Helicobacter pylori. Cell Microbiol. 2007;9:817–828. doi: 10.1111/j.1462-5822.2007.00906.x. [DOI] [PubMed] [Google Scholar]
- 56.Naito Y, Yoshikawa T. Molecular and cellular mechanisms involved in Helicobacter pylori-induced inflammation and oxidative stress. Free Radic Biol Med. 2002;33:323–336. doi: 10.1016/s0891-5849(02)00868-7. [DOI] [PubMed] [Google Scholar]
- 57.Andersen LP, Holck S, Janulaityte-Günther D, Kupcinskas L, Kiudelis G, Jonaitis L, Janciauskas D, Holck P, Bennedsen M, Permin H, et al. Gastric inflammatory markers and interleukins in patients with functional dyspepsia, with and without Helicobacter pylori infection. FEMS Immunol Med Microbiol. 2005;44:233–238. doi: 10.1016/j.femsim.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 58.Algood HM, Cover TL. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin Microbiol Rev. 2006;19:597–613. doi: 10.1128/CMR.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Israel DA, Peek RM. pathogenesis of Helicobacter pylori-induced gastric inflammation. Aliment Pharmacol Ther. 2001;15:1271–1290. doi: 10.1046/j.1365-2036.2001.01052.x. [DOI] [PubMed] [Google Scholar]
- 61.Versalovic J. Helicobacter pylori. Pathology and diagnostic strategies. Am J Clin Pathol. 2003;119:403–412. [PubMed] [Google Scholar]
- 62.Backert S, Clyne M, Tegtmeyer N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun Signal. 2011;9:28. doi: 10.1186/1478-811X-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paziak-Domańska B, Chmiela M, Jarosińska A, Rudnicka W. Potential role of CagA in the inhibition of T cell reactivity in Helicobacter pylori infections. Cell Immunol. 2000;202:136–139. doi: 10.1006/cimm.2000.1654. [DOI] [PubMed] [Google Scholar]
- 64.Peek RM, Moss SF, Tham KT, Pérez-Pérez GI, Wang S, Miller GG, Atherton JC, Holt PR, Blaser MJ. Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst. 1997;89:863–868. doi: 10.1093/jnci/89.12.863. [DOI] [PubMed] [Google Scholar]
- 65.Baldari CT, Lanzavecchia A, Telford JL. Immune subversion by Helicobacter pylori. Trends Immunol. 2005;26:199–207. doi: 10.1016/j.it.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 66.Chmiela M, Michetti P. Inflammation, immunity, vaccines for Helicobacter infection. Helicobacter. 2006;11 Suppl 1:21–26. doi: 10.1111/j.1478-405X.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- 67.Chmiela M, Czkwianianc E, Wadstrom T, Rudnicka W. Role of Helicobacter pylori surface structures in bacterial interaction with macrophages. Gut. 1997;40:20–24. doi: 10.1136/gut.40.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grebowska A, Moran AP, Matusiak A, Bak-Romaniszyn L, Czkwianianc E, Rechciński T, Walencka M, Płaneta-Małecka I, Rudnicka W, Chmiela M. Anti-phagocytic activity of Helicobacter pylori lipopolysaccharide (LPS)--possible modulation of the innate immune response to these bacteria. Pol J Microbiol. 2008;57:185–192. [PubMed] [Google Scholar]
- 69.Grebowska A, Moran AP, Bielanski W, Matusiak A, Rechcinski T, Rudnicka K, Baranowska A, Rudnicka W, Chmiela M. Helicobacter pylori lipopolysaccharide activity in human peripheral blood mononuclear leukocyte cultures. J Physiol Pharmacol. 2010;61:437–442. [PubMed] [Google Scholar]
- 70.Rudnicka K, Włodarczyk M, Moran AP, Rechciński T, Miszczyk E, Matusiak A, Szczęsna E, Walencka M, Rudnicka W, Chmiela M. Helicobacter pylori antigens as potential modulators of lymphocytes’ cytotoxic activity. Microbiol Immunol. 2012;56:62–75. doi: 10.1111/j.1348-0421.2011.00399.x. [DOI] [PubMed] [Google Scholar]
- 71.Appelmelk BJ, Monteiro MA, Martin SL, Moran AP, Vandenbroucke-Grauls CM. Why Helicobacter pylori has Lewis antigens. Trends Microbiol. 2000;8:565–570. doi: 10.1016/s0966-842x(00)01875-8. [DOI] [PubMed] [Google Scholar]
- 72.Miszczyk E, Rudnicka K, Moran AP, Fol M, Kowalewicz-Kulbat M, Druszczyńska M, Matusiak A, Walencka M, Rudnicka W, Chmiela M. Interaction of Helicobacter pylori with C-type lectin dendritic cell-specific ICAM grabbing nonintegrin. J Biomed Biotechnol. 2012;2012:206463. doi: 10.1155/2012/206463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pacifico L, Osborn JF, Tromba V, Romaggioli S, Bascetta S, Chiesa C. Helicobacter pylori infection and extragastric disorders in children: a critical update. World J Gastroenterol. 2014;20:1379–1401. doi: 10.3748/wjg.v20.i6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 75.Chmiela M, Miszczyk E, Rudnicka K. Structural modifications of Helicobacter pylori lipopolysaccharide: an idea for how to live in peace. World J Gastroenterol. 2014;20:9882–9897. doi: 10.3748/wjg.v20.i29.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moran AP. Lipopolysaccharide in bacterial chronic infection: insights from Helicobacter pylori lipopolysaccharide and lipid A. Int J Med Microbiol. 2007;297:307–319. doi: 10.1016/j.ijmm.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 77.Moran AP. Relevance of fucosylation and Lewis antigen expression in the bacterial gastroduodenal pathogen Helicobacter pylori. Carbohydr Res. 2008;343:1952–1965. doi: 10.1016/j.carres.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 78.Rudnicka K, Grebowska A, Moran AP, Matusiak A, Walencka M, Miszczyk E, Bąk-Romaniszyn L, Czkwianianc E, Płaneta-Małecka I, Rudnicka W, et al. Different effectiveness of Helicobacter pylori lipopolysaccharides with or without LewisXY determinants in stimulating the secretion of proinflammatory cytokines IL-8 and TNF-α by peripheral blood mononuclear leukocytes. Przeg Gastroenterol. 2011;6:401–408. [Google Scholar]
- 79.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 80.Wroblewski LE, Peek RM. “Targeted disruption of the epithelial-barrier by Helicobacter pylori”. Cell Commun Signal. 2011;9:29. doi: 10.1186/1478-811X-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wiśniewska M, Nilsson HO, Bak-Romaniszyn L, Rechciński T, Bielański W, Płaneta-Małecka I, Płonka M, Konturek S, Wadström T, Rudnicka W, et al. Detection of specific Helicobacter pylori DNA and antigens in stool samples in dyspeptic patients and healthy subjects. Microbiol Immunol. 2002;46:657–665. doi: 10.1111/j.1348-0421.2002.tb02749.x. [DOI] [PubMed] [Google Scholar]
- 82.Pearson C, Uhlig HH, Powrie F. Lymphoid microenvironments and innate lymphoid cells in the gut. Trends Immunol. 2012;33:289–296. doi: 10.1016/j.it.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 83.Whittle BJ, Morschl E J, Moran AP, László F. Helicobacter pylori lipopolysaccharide provokes iNOS-mediated acute systemic microvascular inflammatory responses in rat cardiac, hepatic, renal and pulmonary tissues. J Physiol Paris. 2001;95:257–259. doi: 10.1016/s0928-4257(01)00035-3. [DOI] [PubMed] [Google Scholar]
- 84.Mendall MA, Goggin PM, Molineaux N, Levy J, Toosy T, Strachan D, Camm AJ, Northfield TC. Relation of Helicobacter pylori infection and coronary heart disease. Br Heart J. 1994;71:437–439. doi: 10.1136/hrt.71.5.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kowalski M, Konturek PC, Pieniazek P, Karczewska E, Kluczka A, Grove R, Kranig W, Nasseri R, Thale J, Hahn EG, et al. Prevalence of Helicobacter pylori infection in coronary artery disease and effect of its eradication on coronary lumen reduction after percutaneous coronary angioplasty. Dig Liver Dis. 2001;33:222–229. doi: 10.1016/s1590-8658(01)80711-8. [DOI] [PubMed] [Google Scholar]
- 86.Chmiela M, Kowalewicz-Kulbat M, Miszczak A, Wisniewska M, Rechcinski T, Kolodziej K, Kasprzak J, Wadstrom T, Rudnicka W. A link between Helicobacter pylori and/or Chlamydia spp. infections and atherosclerosis. FEMS Immunol Med Microbiol. 2003;36:187–192. doi: 10.1016/S0928-8244(03)00030-0. [DOI] [PubMed] [Google Scholar]
- 87.Park MJ, Choi SH, Kim D, Kang SJ, Chung SJ, Choi SY, Yoon DH, Lim SH, Kim YS, Yim JY, et al. Association between Helicobacter pylori Seropositivity and the Coronary Artery Calcium Score in a Screening Population. Gut Liver. 2011;5:321–327. doi: 10.5009/gnl.2011.5.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vafaeimanesh J, Hejazi SF, Damanpak V, Vahedian M, Sattari M, Seyyedmajidi M. Association of Helicobacter pylori infection with coronary artery disease: is Helicobacter pylori a risk factor? ScientificWorldJournal. 2014;2014:516354. doi: 10.1155/2014/516354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pasceri V, Cammarota G, Patti G, Cuoco L, Gasbarrini A, Grillo RL, Fedeli G, Gasbarrini G, Maseri A. Association of virulent Helicobacter pylori strains with ischemic heart disease. Circulation. 1998;97:1675–1679. doi: 10.1161/01.cir.97.17.1675. [DOI] [PubMed] [Google Scholar]
- 90.Gunn M, Stephens JC, Thompson JR, Rathbone BJ, Samani NJ. Significant association of cagA positive Helicobacter pylori strains with risk of premature myocardial infarction. Heart. 2000;84:267–271. doi: 10.1136/heart.84.3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kowalski M, Rees W, Konturek PC, Grove R, Scheffold T, Meixner H, Brunec M, Franz N, Konturek JW, Pieniazek P, et al. Detection of Helicobacter pylori specific DNA in human atheromatous coronary arteries and its association to prior myocardial infarction and unstable angina. Dig Liver Dis. 2002;34:398–402. doi: 10.1016/s1590-8658(02)80036-6. [DOI] [PubMed] [Google Scholar]
- 92.Khodaii Z, Vakili H, Ghaderian SM, Najar RA, Panah AS. Association of Helicobacter pylori infection with acute myocardial infarction. Coron Artery Dis. 2011;22:6–11. doi: 10.1097/MCA.0b013e3283402360. [DOI] [PubMed] [Google Scholar]
- 93.Stone AF, Risley P, Markus HS, Butland BK, Strachan DP, Elwood PC, Mendall MA. Ischaemic heart disease and Cag A strains of Helicobacter pylori in the Caerphilly heart disease study. Heart. 2001;86:506–509. doi: 10.1136/heart.86.5.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rogha M, Nikvarz M, Pourmoghaddas Z, Shirneshan K, Dadkhah D, Pourmoghaddas M. Is helicobacter pylori infection a risk factor for coronary heart disease? ARYA Atheroscler. 2012;8:5–8. [PMC free article] [PubMed] [Google Scholar]
- 95.Tsai CJ, Huang TY. Relation of Helicobacter pylori infection and angiographically demonstrated coronary artery disease. Dig Dis Sci. 2000;45:1227–1232. doi: 10.1023/a:1005522624004. [DOI] [PubMed] [Google Scholar]
- 96.Schöttker B, Adamu MA, Weck MN, Müller H, Brenner H. Helicobacter pylori infection, chronic atrophic gastritis and major cardiovascular events: a population-based cohort study. Atherosclerosis. 2012;220:569–574. doi: 10.1016/j.atherosclerosis.2011.11.029. [DOI] [PubMed] [Google Scholar]
- 97.Tan HJ, Goh KL. Extragastrointestinal manifestations of Helicobacter pylori infection: facts or myth? A critical review. J Dig Dis. 2012;13:342–349. doi: 10.1111/j.1751-2980.2012.00599.x. [DOI] [PubMed] [Google Scholar]
- 98.Christodoulou DK, Milionis HJ, Pappa P, Katsanos KH, Sigounas D, Florentin M, Elisaf M, Tsianos EV. Association of Helicobacter pylori infection with cardiovascular disease--is it just a myth? Eur J Intern Med. 2011;22:191–194. doi: 10.1016/j.ejim.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 99.Longo-Mbenza B, Nsenga JN, Mokondjimobe E, Gombet T, Assori IN, Ibara JR, Ellenga-Mbolla B, Vangu DN, Fuele SM. Helicobacter pylori infection is identified as a cardiovascular risk factor in Central Africans. Vasc Health Risk Manag. 2012;6:455–461. doi: 10.2147/VHRM.S28680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sealy-Jefferson S, Gillespie BW, Aiello AE, Haan MN, Morgenstern LB, Lisabeth LD. Antibody levels to persistent pathogens and incident stroke in Mexican Americans. PLoS One. 2013;8:e65959. doi: 10.1371/journal.pone.0065959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eskandarian R, Moosavi S, Babai M, Toussy J, Ghorbani R, Malek M, Shiasi M, Momeni B, Ghasemi A, Vatani A, et al. Impact of Helicobacter pylori on prognosis of patients with acute coronary syndrome. ARYA. 2005;1:164. [Google Scholar]
- 102.Kauglud RS, Kamath RL. Early prognosis of unstable angina patients with positive H. pylori IgG values. Int J Biom Res. 2013;4:3. [Google Scholar]
- 103.Farsak B, Yildirir A, Akyön Y, Pinar A, Oç M, Böke E, Kes S, Tokgözoğlu L. Detection of Chlamydia pneumoniae and Helicobacter pylori DNA in human atherosclerotic plaques by PCR. J Clin Microbiol. 2000;38:4408–4411. doi: 10.1128/jcm.38.12.4408-4411.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ameriso SF, Fridman EA, Leiguarda RC, Sevlever GE. Detection of Helicobacter pylori in human carotid atherosclerotic plaques. Stroke. 2001;32:385–391. doi: 10.1161/01.str.32.2.385. [DOI] [PubMed] [Google Scholar]
- 105.Iriz E, Cirak MY, Engin ED, Zor MH, Erer D, Ozdogan ME, Turet S, Yener A. Detection of Helicobacter pylori DNA in aortic and left internal mammary artery biopsies. Tex Heart Inst J. 2008;35:130–135. [PMC free article] [PubMed] [Google Scholar]
- 106.Kedzia A, Ciecierski M, Wierzbowska M, Kufel A, Kwapisz E. Isolation of Helicobacter pylori from femoral or iliac atherosclerotic plaques. Acta Angiol. 2010;16:129–134. [Google Scholar]
- 107.Rajasekhar D, Subramanyam G, Latheef SA, Vanajakshamma V, Srilatha A, Chaudhury A. Infectious aetiology in acute coronary syndromes. Indian J Med Microbiol. 2002;20:83–87. [PubMed] [Google Scholar]
- 108.Akbas HS, Basyigit S, Suleymanlar I, Kemaloglu D, Koc S, Davran F, Demir I, Suleymanlar G. The assessment of carotid intima media thickness and serum paraoxonase-1 activity in Helicobacter pylori positive subjects. Lipids Health Dis. 2010;9:92. doi: 10.1186/1476-511X-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Satoh H, Saijo Y, Yoshioka E, Tsutsui H. Helicobacter Pylori infection is a significant risk for modified lipid profile in Japanese male subjects. J Atheroscler Thromb. 2010;17:1041–1048. doi: 10.5551/jat.5157. [DOI] [PubMed] [Google Scholar]
- 110.Vahdat K, Jafari SM, Pazoki R, Nabipour I. Concurrent increased high sensitivity C-reactive protein and chronic infections are associated with coronary artery disease: a population-based study. Indian J Med Sci. 2007;61:135–143. [PubMed] [Google Scholar]
- 111.Dunzendorfer S, Lee HK, Soldau K, Tobias PS. Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: roles of LBP and sCD14 in mediating LPS responses. FASEB J. 2004;18:1117–1119. doi: 10.1096/fj.03-1263fje. [DOI] [PubMed] [Google Scholar]
- 112.Edfeldt K, Bennet AM, Eriksson P, Frostegård J, Wiman B, Hamsten A, Hansson GK, de Faire U, Yan ZQ. Association of hypo-responsive toll-like receptor 4 variants with risk of myocardial infarction. Eur Heart J. 2004;25:1447–1453. doi: 10.1016/j.ehj.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 113.Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One. 2013;8:e59989. doi: 10.1371/journal.pone.0059989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Unlu S, Tang S, Wang E, Martinez I, Tang D, Bianchi ME, Zeh HJ, Lotze MT. Damage associated molecular pattern molecule-induced microRNAs (DAMPmiRs) in human peripheral blood mononuclear cells. PLoS One. 2012;7:e38899. doi: 10.1371/journal.pone.0038899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu XP, Rajavashisth TB, Yano J, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 116.Unkelbach K, Gardemann A, Kostrzewa M, Philipp M, Tillmanns H, Haberbosch W. A new promoter polymorphism in the gene of lipopolysaccharide receptor CD14 is associated with expired myocardial infarction in patients with low atherosclerotic risk profile. Arterioscler Thromb Vasc Biol. 1999;19:932–938. doi: 10.1161/01.atv.19.4.932. [DOI] [PubMed] [Google Scholar]
- 117.Rechciński T, Grebowska A, Kurpesa M, Peruga Z, Dziuba M, Krzemińska-Pakuła M, Rudnicka W, Chmiela M. CD14 gene polymorphism 159C/T in a group of patients with coronary artery disease from a population with high morbidity of cardiovascular diseases. Kardiol Pol. 2007;65:237–244; discussion 245. [PubMed] [Google Scholar]
- 118.Hollestelle SC, De Vries MR, Van Keulen JK, Schoneveld AH, Vink A, Strijder CF, Van Middelaar BJ, Pasterkamp G, Quax PH, De Kleijn DP. Toll-like receptor 4 is involved in outward arterial remodeling. Circulation. 2004;109:393–398. doi: 10.1161/01.CIR.0000109140.51366.72. [DOI] [PubMed] [Google Scholar]
- 119.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–454. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 121.Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol. 2010;87:989–999. doi: 10.1189/jlb.1209775. [DOI] [PubMed] [Google Scholar]
- 122.Talreja J, Kabir MH, B Filla M, Stechschulte DJ, Dileepan KN. Histamine induces Toll-like receptor 2 and 4 expression in endothelial cells and enhances sensitivity to Gram-positive and Gram-negative bacterial cell wall components. Immunology. 2004;113:224–233. doi: 10.1111/j.1365-2567.2004.01946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Heo SK, Yun HJ, Noh EK, Park WH, Park SD. LPS induces inflammatory responses in human aortic vascular smooth muscle cells via Toll-like receptor 4 expression and nitric oxide production. Immunol Lett. 2008;120:57–64. doi: 10.1016/j.imlet.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 124.Ostos MA, Recalde D, Zakin MM, Scott-Algara D. Implication of natural killer T cells in atherosclerosis development during a LPS-induced chronic inflammation. FEBS Lett. 2002;519:23–29. doi: 10.1016/s0014-5793(02)02692-3. [DOI] [PubMed] [Google Scholar]
- 125.Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest. 2003;112:1234–1243. doi: 10.1172/JCI18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vreugdenhil AC, Snoek AM, van ‘t Veer C, Greve JW, Buurman WA. LPS-binding protein circulates in association with apoB-containing lipoproteins and enhances endotoxin-LDL/VLDL interaction. J Clin Invest. 2001;107:225–234. doi: 10.1172/JCI10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grebowska A, Rechciński T, Bak-Romaniszyn L, Czkwianianc E, Moran A, Druszczyńska M, Kowalewicz-Kulbat M, Owczarek A, Dziuba M, Krzemińska-Pakuła M, et al. Potential role of LPS in the outcome of Helicobacter pylori related diseases. Pol J Microbiol. 2006;55:25–30. [PubMed] [Google Scholar]
- 128.Niemelä S, Karttunen T, Korhonen T, Läärä E, Karttunen R, Ikäheimo M, Kesäniemi YA. Could Helicobacter pylori infection increase the risk of coronary heart disease by modifying serum lipid concentrations? Heart. 1996;75:573–575. doi: 10.1136/hrt.75.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Laurila A, Bloigu A, Näyhä S, Hassi J, Leinonen M, Saikku P. Association of Helicobacter pylori infection with elevated serum lipids. Atherosclerosis. 1999;142:207–210. doi: 10.1016/s0021-9150(98)00194-4. [DOI] [PubMed] [Google Scholar]
- 130.Tamura A, Fujioka T, Nasu M. Relation of Helicobacter pylori infection to plasma vitamin B12, folic acid, and homocysteine levels in patients who underwent diagnostic coronary arteriography. Am J Gastroenterol. 2002;97:861–866. doi: 10.1111/j.1572-0241.2002.05601.x. [DOI] [PubMed] [Google Scholar]
- 131.Evrengul H, Tanriverdi H, Kuru O, Enli Y, Yuksel D, Kilic A, Kaftan A, Kirac S, Kilic M. Elevated homocysteine levels in patients with slow coronary flow: relationship with Helicobacter pylori infection. Helicobacter. 2007;12:298–305. doi: 10.1111/j.1523-5378.2007.00505.x. [DOI] [PubMed] [Google Scholar]
- 132.Wierzbicki AS. Homocysteine and cardiovascular disease: a review of the evidence. Diab Vasc Dis Res. 2007;4:143–150. doi: 10.3132/dvdr.2007.033. [DOI] [PubMed] [Google Scholar]
- 133.Grabczewska Z, Nartowicz E, Szymaniak L, Wiśniewska E, Przybył R, Polak G, Kubica J, Dymek G, Giedrys-Kalemba S, Kotschy M, et al. Endothelial dysfunction in acute coronary syndrome without ST segment elevation in the presence of Helicobacter pylori infection. Kardiol Pol. 2002;57:533–534; discussion 541. [PubMed] [Google Scholar]
- 134.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 135.Faghihi AH, Agah S, Fereshtehnejad SM, Bahar MA. Assessment of the relationship between serum fibrinogen level and chronic Helicobacter pylori infection in patients with or without ischemic heart disease. Med J Isl Rep Iran. 2007;21:105–110. Available from: http://mjiri.iums.ac.ir/browse.php?a_code=A-10-1-170&slc_lang=en&sid=1. [Google Scholar]
- 136.Galustian C, Elviss N, Chart H, Owen R, Feizi T. Interactions of the gastrotropic bacterium Helicobacter pylori with the leukocyte-endothelium adhesion molecules, the selectins--a preliminary report. FEMS Immunol Med Microbiol. 2003;36:127–134. doi: 10.1016/S0928-8244(03)00021-X. [DOI] [PubMed] [Google Scholar]
- 137.Heneghan MA, McCarthy CF, Moran AP. Relationship of blood group determinants on Helicobacter pylori lipopolysaccharide with host lewis phenotype and inflammatory response. Infect Immun. 2000;68:937–941. doi: 10.1128/iai.68.2.937-941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Boekholdt SM, Peters RJ, Fountoulaki K, Kastelein JJ, Sijbrands EJ. Molecular variation at the apolipoprotein B gene locus in relation to lipids and cardiovascular disease: a systematic meta-analysis. Hum Genet. 2003;113:417–425. doi: 10.1007/s00439-003-0988-3. [DOI] [PubMed] [Google Scholar]
- 139.Anstee DJ. The relationship between blood groups and disease. Blood. 2010;115:4635–4643. doi: 10.1182/blood-2010-01-261859. [DOI] [PubMed] [Google Scholar]
- 140.Lindén S, Mahdavi J, Semino-Mora C, Olsen C, Carlstedt I, Borén T, Dubois A. Role of ABO secretor status in mucosal innate immunity and H. pylori infection. PLoS Pathog. 2008;4:e2. doi: 10.1371/journal.ppat.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Go MF. What are the host factors that place an individual at risk for Helicobacter pylori-associated disease? Gastroenterology. 1997;113:S15–S20. doi: 10.1016/s0016-5085(97)80005-4. [DOI] [PubMed] [Google Scholar]
- 142.Hein HO, Sørensen H, Suadicani P, Gyntelberg F. The Lewis blood group--a new genetic marker of ischaemic heart disease. J Intern Med. 1992;232:481–487. doi: 10.1111/j.1365-2796.1992.tb00620.x. [DOI] [PubMed] [Google Scholar]
- 143.Ellison RC, Zhang Y, Myers RH, Swanson JL, Higgins M, Eckfeldt J. Lewis blood group phenotype as an independent risk factor for coronary heart disease (the NHLBI Family Heart Study) Am J Cardiol. 1999;83:345–348. doi: 10.1016/s0002-9149(98)00866-2. [DOI] [PubMed] [Google Scholar]
- 144.Salomaa V, Pankow J, Heiss G, Cakir B, Eckfeldt JH, Ellison RC, Myers RH, Hiller KM, Brantley KR, Morris TL, et al. Genetic background of Lewis negative blood group phenotype and its association with atherosclerotic disease in the NHLBI family heart study. J Intern Med. 2000;247:689–698. doi: 10.1046/j.1365-2796.2000.00682.x. [DOI] [PubMed] [Google Scholar]
- 145.Rad R, Prinz C, Neu B, Neuhofer M, Zeitner M, Voland P, Becker I, Schepp W, Gerhard M. Synergistic effect of Helicobacter pylori virulence factors and interleukin-1 polymorphisms for the development of severe histological changes in the gastric mucosa. J Infect Dis. 2003;188:272–281. doi: 10.1086/376458. [DOI] [PubMed] [Google Scholar]
- 146.Rechciński T, Grebowska A, Kurpesa M, Sztybrych M, Peruga JZ, Trzos E, Rudnicka W, Krzemińska-Pakuła M, Chmiela M. Interleukin-1b and interleukin-1 receptor inhibitor gene cluster polymorphisms in patients with coronary artery disease after percutaneous angioplasty or coronary artery bypass grafting. Kardiol Pol. 2009;67:601–610. [PubMed] [Google Scholar]
- 147.Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 148.Wright SD, Burton C, Hernandez M, Hassing H, Montenegro J, Mundt S, Patel S, Card DJ, Hermanowski-Vosatka A, Bergstrom JD, et al. Infectious agents are not necessary for murine atherogenesis. J Exp Med. 2000;191:1437–1442. doi: 10.1084/jem.191.8.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Danese S, Dejana E, Fiocchi C. Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation, and inflammation. J Immunol. 2007;178:6017–6022. doi: 10.4049/jimmunol.178.10.6017. [DOI] [PubMed] [Google Scholar]
- 150.Caro CG, Fitz-Gerald JM, Schroter RC. Atheroma and arterial wall shear. Observation, correlation and proposal of a shear dependent mass transfer mechanism for atherogenesis. Proc R Soc Lond B Biol Sci. 1971;177:109–159. doi: 10.1098/rspb.1971.0019. [DOI] [PubMed] [Google Scholar]
- 151.Wulaningsih W, Garmo H, Holmberg L, Hammar N, Jungner I, Walldius G, Van Hemelrijck M. Serum Lipids and the Risk of Gastrointestinal Malignancies in the Swedish AMORIS Study. J Cancer Epidemiol. 2012;2012:792034. doi: 10.1155/2012/792034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Becker RC, Meade TW, Berger PB, Ezekowitz M, O’Connor CM, Vorchheimer DA, Guyatt GH, Mark DB, Harrington RA. The primary and secondary prevention of coronary artery disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition) Chest. 2008;133:776S–814S. doi: 10.1378/chest.08-0685. [DOI] [PubMed] [Google Scholar]
- 153.Perini R, Fiorucci S, Wallace JL. Mechanisms of nonsteroidal anti-inflammatory drug-induced gastrointestinal injury and repair: a window of opportunity for cyclooxygenase-inhibiting nitric oxide donors. Can J Gastroenterol. 2004;18:229–236. doi: 10.1155/2004/890585. [DOI] [PubMed] [Google Scholar]
- 154.Baek JH, Zhang X, Williams MC, Schaer DJ, Buehler PW, D’Agnillo F. Extracellular Hb enhances cardiac toxicity in endotoxemic guinea pigs: protective role of haptoglobin. Toxins (Basel) 2014;6:1244–1259. doi: 10.3390/toxins6041244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Smoot DT, Sewchand J, Young K, Desbordes BC, Allen CR, Naab T. A method for establishing primary cultures of human gastric epithelial cells. Methods Cell Sci. 2000;22:133–136. doi: 10.1023/a:1009846624044. [DOI] [PubMed] [Google Scholar]