

Abstract
Hepatitis D virus (HDV) is a defective circular shape single stranded HDV RNA virus with two types of viral proteins, small and large hepatitis D antigens, surrounded by hepatitis B surface antigen. Superinfection with HDV in chronic hepatitis B is associated with a more threatening form of liver disease leading to rapid progression to cirrhosis. In spite of some controversy in the epidemiological studies, HDV infection does increase the risk of hepatocellular carcinoma (HCC) compared to hepatitis B virus (HBV) monoinfection. Hepatic decompensation, rather than development of HCC, is the first usual clinical endpoint during the course of HDV infection. Oxidative stress as a result of severe necroinflammation may progress to HCC. The large hepatitis D antigen is a regulator of various cellular functions and an activator of signal transducer and activator of transcription (STAT)3 and the nuclear factor kappa B pathway. Another proposed epigenetic mechanism by which HCC may form is the aberrant silencing of tumor suppressor genes by DNA Methyltransferases. HDV antigens have also been associated with increased histone H3 acetylation of the clusterin promoter. This enhances the expression of clusterin in infected cells, increasing cell survival potential. Any contribution of HBV DNA integration with chromosomes of infected hepatocytes is not clear at this stage. The targeted inhibition of STAT3 and cyclophilin, and augmentation of peroxisome proliferator-activated receptor γ have a potential therapeutic role in HCC.
Keywords: Hepatitis D, Hepatocellular carcinoma, Necroinflammation, Epigenetic processes, Cirrhosis, Oxidative stress
Core tip: Role of hepatitis D virus (HDV) in the oncogenesis of hepatocellular carcinoma (HCC) has not been thoroughly investigated. Many epidemiological studies favour the increased risk of HCC with HDV superinfection. Oxidative stress as a result of severe necroinflammation may trigger the development of HCC. Epigenetic mechanisms like DNA methylation and histone modification may also be operating.
INTRODUCTION
Hepatitis D virus (HDV) is a small virus, often compared to viroids because of its unique characteristics. It is a defective virus with a circular shape single stranded HDV RNA and two types of viral proteins, small (sHDAg or p24) and large hepatitis D antigens (lHDAg or p27), surrounded by hepatitis B virus (HBV) surface antigen (HBsAg)[1]. The virus does not code any enzyme to replicate its genome and takes the help from hepatocyte RNA polymerase II for synthesizing its RNAs with positive and negative polarities. Both the smaller sHDAg, which is required for HDV genomic replication, and the larger lHDAg, which represses replication, colocalize with delta RNA throughout the nucleoplasm[2].
HDV is highly pathogenic. Whereas coinfection evolves to chronicity in only 2% of the cases, superinfection results in chronic infection in over 90% of the cases[3]. Superinfection with HDV in chronic hepatitis B is associated with a more threatening form of liver disease exacerbating the pre-existing liver damage leading to more rapid progression to cirrhosis in 70% to 80% of the cases[4]. It may lead to cirrhosis within 2 years in 10%-15% of patients[5]. HBV DNA levels are low in both hepatitis B e antigen (HBeAg)-negative and HBeAg-positive patients, suggesting suppressive effects of HDV on HBV irrespective of the phase of HBV infection. The clinical long-term outcome of HBeAg-positive patients is not different to HBeAg-negative patients infected with the HDV[6].
HEPATOCELLULAR CARCINOMA IN HDV INFECTION
Hepatocellular carcinoma (HCC) is the second most common cause of cancer-related death in men worldwide[7]. Persistent HDV replication and hepatic inflammation end up with cirrhosis and HCC formation[8]. Active replication of both HBV and HDV may be associated with a more progressive disease pattern leading to early cirrhosis and HCC[5]. Wu et al[9] described three phases of HDV superinfection: acute phase, active HDV replication and suppression of HBV with high alanine transaminase (ALT) levels; chronic phase, decreasing HDV and reactivating HBV with moderate ALT levels; and late phase, development of cirrhosis and hepatocellular carcinoma caused by replication of either virus or remission resulting from the marked reduction of both viruses. Therefore, HBV replication, in spite of being inhibited by HDV, appears to play a major role sustaining HDV pathogenicity.
Hepatic decompensation, rather than development of liver cancer, is the first clinical endpoint that develops during the course of HDV infection[10]. A clinical study has suggested that HCC in HDV infection may be a secondary effect of severe necroinflammation leading to cirrhosis. In this study, decreased liver size was noticed more in cases of HDV HCC compared to an HBV monoinfection group where the liver size was normal or increased. HDV patients had lower platelets and larger varices on endoscopy as an indirect evidence of more severe portal hypertension. HCC presented at an earlier TNM stage compared with HBV monoinfection[11].
EPIDEMIOLOGICAL STUDIES
Some controversy exists in the epidemiological studies on the role of HDV infection in increasing the risk of HCC. Early studies did not find an increased incidence of HCC in HDV co-infected individuals. But recent studies show an increased incidence of the tumor. The risk of HCC should be reconsidered according to the changing natural history of chronic HDV disease. Though the incidence of HDV infection has decreased in many Western countries, it is still very much prevalent in many parts of the world specially the Asia Pacific Region[12].
The European Concerted Action on Viral Hepatitis (Eurohep) study done on hepatitis B patients and published in 1995 failed to show any significance of HDV (anti-HDV) markers at presentation on prognosis. However, a later study done by the same group on 200 HDV patients with a median follow up of 6.6 years showed that the adjusted estimated five year risk for HCC was 13% for anti-HDV positive and 2%-4% in anti-HDV negative/HBsAg positive cirrhotics. HDV infection increases the risk for HCC threefold and for mortality two fold in patients with hepatitis B cirrhosis[13,14]. Analysis of retrospective data from South London showed that the risk of hepatocellular carcinoma was similar in anti-HDV positive and negative patients[15].
Two studies from Turkey show prevalence of anti-delta antibodies in 18.8% to 23.0% of HBsAg positive HCC[16,17]. In an older Jordanian study the prevalence of anti-HDV in a small group of HBsAg positive HCC patients was 67% (10/15). However, they were significantly older than patients without hepatitis D viral infection[18]. In another similar study from Greece done on 87 HBsAg positive HCC patients, 9 were positive for serum anti-delta (10%) whereas among the HBsAg positive controls none was positive for this antibody (P = 0.067)[19]. In a Romanian study, 166 consecutive patients with compensated HDV-related cirrhosis diagnosed since 1994 were followed up. HDV-related cirrhosis in Romania is an aggressive disease with a median time to decompensation less than 2 years and a median survival less than 5 years. Jaundice, the main clinical consequences of portal hypertension and HCC were the most frequent causes of decompensation. HCC developed in 12% cases[20].
A study from Mongolia considered the sero-epidemiological and social-historical background of the country, and compared HCV related and HDV related HCC prevalence[21]. In Mongolia co-infection with HBV and HDV had a stronger association with HCC development at a younger age while patients with HCV mono-infection were older. Their results demonstrated that the viruses had different epidemic dynamics in Mongolia; HCV was characterized by earlier epidemic expansion, whereas HDV spread with approximately 50 years lag. Keeping this in mind, there was a comparable contribution of the HCV-monoinfection and HBV + HDV co-infection in the current HCC rate.
In a study from the Kure district in Japan, where HDV infection of persons infected with HBV in 1990s was about 6%, such superinfection increases the risk of cirrhosis and HCC. The proportion of HCC per 1000 person years was 7.84 among cases with anti-HDV and 2.73 among those without anti-HDV. The overall relative risk of HCC was 2.87, 95%CI: 1.03-6.23[22]. A study from Taiwan failed to show any acceleration in the development of HCC in patients with HDV superinfection. Nevertheless, the numbers of patients in HDV group were small compared to HBV monoinfection group (42 vs 255)[23].
In a Spanish study, One hundred and fifty-eight patients with chronic HDV were followed for a median period of 158 mo. 18% had hepatic decompensation, 3% developed hepatocellular carcinoma[24]. Romeo et al[25] tracked the course of HDV infection in 299 patients over a mean period of 233 mo; 46 developed HCC. Persistent HDV replication led to cirrhosis and HCC at annual rates of 4% and 2.8%, respectively, and was the only predictor of liver-related mortality.
A recent study calculated the standardized incidence ratios (SIRs) for hepatitis D patients. The risk of hepatocellular carcinoma was greatly increased in patients with HBV and HDV (SIR = 137.17, 95%CI: 62.19 to 261.51) when compared with the general population. The risk of HCC among patients with HDV was increased (SIR = 6.11, 95%CI: 2.77 to 11.65) when patients with chronic HBV monoinfection were used as the reference population[26]. High levels of HDV viremia in non-cirrhotic patients were associated with a considerable likelihood of progression to cirrhosis and the development of HCC; multivariate analysis: OR = 1.42, 95%CI: 1.04-1.95; P = 0.03. Once cirrhosis has developed, the role of HDV replication as a predictor of a negative outcome lessens[27]. Table 1 summarizes the epidemiological studies on the role of HDV infection in increasing the risk of HCC.
Table 1.
The epidemiological studies on the role of hepatitis D virus infection in increasing the risk of hepatocellular carcinoma
| 1Romeo et al[27] | 193 patients with HDV co-infection were investigated for a median of 9.5 yr. HDV RNA levels appeared significantly associated with HCC |
| 1Romeo et al[25] | 299 HDV infected patients invstigated over 28 yr. Persistent HDV leads to cirrhosis and HCC at annual rates of 4% and 2.8% |
| 1Oyunsuren et al[28] | 292 chronic hepatitis patients were investigated retrospectively. HDV co-infection has a stronger association with HCC development at a younger age than HCV mono-infection |
| 1Fattovich et al[14] (EUROHEP study group) | A retrospective cohort study of 200 Western European patients was carried out with a follow-up median period of 6.6 yr. HDV infection increases the risk of HCC three-fold |
| 1Cenac et al[29] | 89 Sahelian African patients were tested alongside 47 controls. 55% of HDV patients had HCC compared to the 17% who had HBV mono-infection with HCC |
| 1Oliveri et al[30] | Patients with HDV co-infection developed HCC at a significantly younger age than those affected by HBV alone, by about 10 yr |
| 1Tamura et al[22] | 1127 patients were followed for atleast 3 yr. The prevalence was 4.05 per thousand person years in HDV co-infection patients compared to 2.73 in patients with HBV alone |
| 1Verme et al[31] | 62 patients were investigated. The findings suggest that HDV co-infection causes HCC at an earlier age |
| 1Smedile et al[32] | 85 patients were investigated. The outcome in patients with HDV co-infection was significantly worse than others |
| 1Trichopoulos et al[19] | 116 patients were investigated. There is a higher prevalence of HCC amongst HDV co-infected patients |
| 1Toukan et al[18] | The highest prevalence of HCC was found in those patients co-infected with HDV |
| 1Ji et al[26] | 650 out of 9160 HBV patients had HDV. The median follow up was 11 yr. The risk of HCC was increased. HDV was a strong risk factor |
| 2Huang et al[33] | 114 HCC patients were investigated prior to surgery. A higher prevalence of hepatic inflammation was observed in HCV patients and also, possibly, in HDV patients |
| 2Abbas et al[11] | 92 HDV positive and 92 negative patients with HCC were compared. HDV causes HCC in a different manner to HBV |
| 3Heidrich et al[6] | 71 out of 534 patients had HBV and HDV co-infection. The median follow-up period was 4.25 yr. The long-term outcome for HBeAg positive and negative was the same |
| 3Huo et al[23] | 42 HDV co-infected patients were compared to 255 HBV patients, all with HCC, over a period of 8 yr. HDV co-infection does not accelerate HCC development, and the outcomes are the same as HBV mono-infection |
| 3Fattovich et al[13] (EUROHEP study group) | 349 Western European patients were investigated for 5 yr. HDV co-infection had no prognostic value for the development of HCC |
| 3Realdi et al[34] (EUROHEP) | 366 caucasian patients were investigated for 6 yr. HDV infection did not influence the prognosis |
| 3Kage et al[35] | 58 patients were investigated. HDV is unlikely to have a role in the development of HCC |
| 3Tzonou et al[36] | 185 cases with HCC and 432 hospital controls were investigated. HDV was not a significant cause of HCC |
| 3Tassopoulos et al[37] | 47 patients with HCC were investigated. None of the 47 had any evidence of HDV infection |
| 3Chen et al[38] | 60 patients were investigated. However, the study indicated that HDV co-infection does not lead to a rise in HCC development amongst Chinese living in Taiwan |
| 3Govindarajan et al[39] | Sera from 39 patients with HBV associated with HCC were studied for the presence of HDV. Only one patient tested positive |
| 3Negro et al[40] | Liver tissues of 19 patients with chronic HDV were investigated and compared to tissues from 16 patients with chronic HBV, and 3 normal patients. Hepatocyte proliferation in HDV was similar to HBV, but higher than normal |
Studies favoring role of HDV in HCC;
Inconclusive;
Studies against role of HDV in HCC. HDV: Hepatitis D virus; HCC: Hepatocellular carcinoma; HBV: Hepatitis B virus; HBeAg: Hepatitis B e antigen; HCV: Hepatitis C virus.
HDV AND HBV GENOTYPES
Hepatitis D is an immune-mediated disease. Though it is more aggressive than HBV monoinfection, the rate of disease progression may vary, as with other immune mediated diseases. Active replication of both HBV and HDV may be associated with a more progressive disease pattern. HDV and HBV genotypes may play a role in various disease outcomes. Genotype II HDV infection is relatively less frequently associated with fulminant hepatitis at the acute stage and cirrhosis or HCC at the chronic stage as compared to genotype I[41,42]. The outcome of patients with genotype IV (IIb) HDV infection is more like of genotype II HDV infection. HBV of the genotype C is also a significant factor associated with adverse outcomes (cirrhosis, HCC or mortality) in patients with chronic hepatitis D in addition to genotype I HDV and age[42,43].
ONCOGENESIS
The mechanism by which HDV causes HCC remains to be elucidated, but recent advances seem to suggest a number of pathways that result in pathogenesis. HCC development itself is a complex process involving cumulative gain and loss of function mutations affecting tumor suppressor and oncogenic products[44].
HDV seems to exert epigenetic control over HBV transcription and replication. A possible explanation may be that p24 and p27 both repress HBV enhancers, pIIE1 and PIIE2 inhibit replication, thus accounting for the low serum levels of HBV DNA in co-infected patients[45]. P27 also inhibits interferon-α signaling by interfering with janus kinase, tyrosine kinase 2, signal transducer and activator of transcription (STAT)1 and STAT2, impairing the transcription of 2’, 5’ oligoadenylate synthase and protein kinase R but upregulating myxovirus resistance A gene transcription, which causes HBV replication inhibition[46,47]. In fact HDV has been shown to repress HCV replication as well and chronic HCV infection has been reported to be cleared in the presence of HBV and HDV superinfection[1]. This implies that HCC is caused by HDV alone in a conviction, but it may not be so, as the active proliferation of both HBV and HDV leads to more aggressive disease and HCC[5].
It is believed that the pathogenic effects of HDV arise from replication-associated cytopathogenecity rather than a direct effect, since there is little injury observed in liver tissues expressing HDAg alone[48]. An investigation by Taylor confirmed that the expression of the antigen alone had no cytopathic effect, however high levels of the antigen and viral RNA caused cell cycle arrest in the G1 phase within two days and cell death in six[49]. This experiment models the acute phase of infection wherein a high replicative rate is responsible for tissue injury. However, in chronic infection, wherein adequate levels of the large antigen are built up to suppress HDV RNA synthesis, the problem shifts to the development of HCC.
Oxidative stress
Oxidative stress as a result of severe necroinflammation in HDV infection may progress to HCC. Large hepatitis D antigens or p27 was shown by Williams et al[50] to be a regulator of various cellular functions and an activator of STAT3 and the nuclear factor kappa B (NF-κB) pathway (Figure 1). Studies on HCV and HBV have linked the activation of NF-κB and STAT3, via the overproduction of reactive oxygen species (ROS), to the pathology of the virus[51-58]. These proteins have been implicated in cell transformation and tumorigenesis, indeed STAT3 over expression is associated with leukemia, prostate cancer and melanoma[59-62]. The ROS are produced by endoplasmic reticulum (ER) stress, the NADPH oxidase (Nox) family (HCV induces Nox1 and Nox4 in hepatocytes)[63], the direct action of the HBV and HCV proteins and the ER overload response. Williams et al[50] found that in the presence of antioxidants (PDTC, NAC) or calcium inhibitors (TMB-8, BAPTA-AM, Ruthenium Red), p27-induced activation of STAT3 and NF-κB was dramatically reduced. They described that p27 caused an increase in ROS production, partly due to the isoprenylation process. P27 has a prenylation site on C211, which binds to farnesyl residues, and a nuclear export signal, which allows transport of the neosynthesized ribonucleoprotein to the ER[64,65]. HDV proteins also cause some ER stress, as p27 activates ER stress elements present in the promoter of target genes, GRP78 and GRP94, and the antigen also triggers Nox4 activity via transforming growth factor (TGF)b1. TGFb1 and c-Jun signaling cascades may also induce epithelial-mesenchymal transition and fibrogenesis[66,67] and cause cirrhosis. Isoprenylation inhibitors, still in early development, may play a key role in preventing these undesirable outcomes[11].
Figure 1.
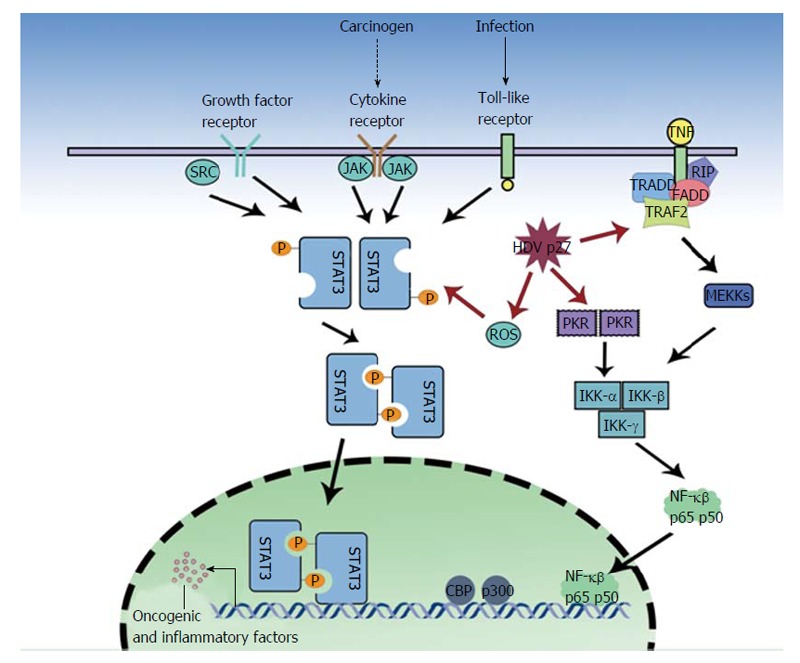
The influence of large hepatitis D antigen in activating oncogenic pathways. JAK: Janus kinase; SRC: Proto-oncogene tyrosine-protein kinase Src; TRADD: Tumor necrosis factor receptor type 1-associated DEATH domain protein; FADD: Fas-associated protein with death domain; TRAF2: TNF receptor associated factor 2; TNF: Tumor necrosis factor; RIP: Receptor-interacting protein; STAT3: Signal transducer and activator of transcription 3; NF-κβ: Nuclear factor kappa beta; ROS: Reactive oxygen species; MEKK: Mitogen-activated protein kinase kinase kinase (MEK kinase); PKR: Protein kinase R; IKK: IêB kinase; CBP: CREB-binding protein.
In a dose dependent manner, p27 also significantly increases (3.2 fold) NF-κB activity[50]. NF-κB complex activation requires the phosphorylation of the serine 32 and 36 (and possibly Tyr42) residues by an Inhibitor of kappa B kinases, IêB kinase (IKK)α and IKKβ, of I-κB (which is then proteosomally degraded), hence allowing the nuclear translocation and DNA binding of the active dimmer (p50/65)[50]. Park et al[68] demonstrated that p27 might also increase NF-κB activation via tumor necrosis factor α (TNF-α) induction. TNF-α is involved in a wide range of inflammation and immunity related actions[69-71]. The study also found that the large antigen increased TNF receptor associated factor (TRAF2), IKKβ and p65 mediated NF-κB activation. The investigators found TRAF2 (a protein involved in early signal transduction events) to interact with both SHDAg and LHDAg. An interesting parallel can be drawn to HCV, which via NS5A and NS5B proteins also modulates TNF-α induced NF-κB activation[72,73]. Furthermore, the HBX protein directly interacts with I-κB, preventing its association with NF-κB[74]. However Williams et al[50] showed that HDV proteins could not directly interact with NF-κB and STAT3 but could act to transcribe various unknown genes by binding to endoplasmic reticulum stress response element (ERSE) motifs in target genes.
The discussion above demonstrates some of the possible mechanisms by which the HDV induces HCC. Furthermore, clinical observations seem to reinforce the view that HCC in HDV infection may be a secondary to the necroinflammation and cirrhosis of the liver[11]. The investigators noted a decrease in liver size with HDV as opposed to HBV monoinfection and saw that HDV patients had lower platelets and larger varices.
DNA methylation
It has been suggested that another mechanism by which HCC forms is the aberrant silencing of tumor suppressor genes by DNA methyltransferases (DNMT1) and DNMT 3b[75]. DNMT1 is responsible for the maintenance of methylation patterns whereas DNMT 3a and 3b catalyze new methylation events[76]. Hence DNMT 3b is potentially oncogenic. Indeed, a study by Mota et al[77] noted that at least 32 proteins had differential expression in the presence of HDV components, pointing towards possible epigenetic links. The study did not identify the mechanism of pathogenesis, but noted that HMGB1 (over expression of which is associated with metastasis in various cancer types) was over expressed in Huh7-D12 cells while NASP, TPI and PABP2 (which interact with DNMT 3a and 3b) were found to be down regulated, hence promoting cell proliferation. Proteins involved in cellular metabolism, transport, signal transduction and growth (PCNA and FEN1 Endonuclease) were also found to be affected[77]. Indeed Negro et al[40] found that in the cirrhotic tissue of patients with HCC, HDV RNA occasionally co-localized with PCNA (a marker of hepatocyte proliferation).
It has been established that DNMT1 and DNMT 3b knockdown causes a global methylation reduction of over 95%, causing the loss of insulin-like growth factor 2 imprinting and the loss of silencing of the vital tumor suppressor p16INK4a[76]. Hence their roles in human cancers are clear. Benegiamo et al[75] went on to show the large antigen activates STAT3 via phosphorylation of Tyrosine 705 residue. STAT3 in turn regulates DNMT1 and causes the over expression of DNMT3b. Among the 24 genes investigated by the study, the promoter of E2F1, a vital regulator of the cell cycle (bound by the Retinoblastoma protein) was found to be hypermethylated. It has been proposed that E2F1 may also be responsible for Nox4 activation. E2F1 is often targeted by other small DNA and RNA viruses as well. The virus was thus found to cause cell cycle disruption and a 2-fold increase in G2/M phase arrest was observed[75]. It has been suggested by Kannan that following arrest, the cell acquires further mutations that allow it to proceed with the cycle, giving rise to cancerous cells[78].
Histone modification
HDAgs have also been associated with increased histone H3 acetylation of the clusterin promoter[79]. This enhances the expression of clusterin in infected cells, increasing cell survival potential. Histone acetyltransferases, CREB-binding protein and p300[80] are key to this process, as they interact with the antigens while the linker histone H1e binds to the small antigen[81]. Kang et al[82] reported that clusterin is over expressed in HCC, with the expression increasing with metastatic HCC[83]. Indeed, it has already been noted that increased levels of the protein is an important factor in determining the aggressiveness of a breast tumor[84]. It is believed that at least in human renal cell carcinoma clusterin contributes to a phenotype resistant to Fas-mediated apoptosis[85]. However, some conflicting results have been noted in the literature regarding the roles of clusterin, which has been involved in cell cycle arrest[86], cell death[87] and inhibition of proliferation[84]. An explanation suggested is that although clusterin may initially cause senescence in problematic cells, over time the molecule may be responsible for survival and with the accumulation of further mutations, may allow tumorigenesis[88].
Metabolic and autoimmune changes
Another factor to consider is the down-regulation of the Rho GDP dissociation inhibitor and guanine binding proteins[74]. These proteins are involved in the regulation of the mitogen activated protein kinase (MAPK) pathway, which is frequently implicated in cancer[89]. A lower availability of Triosephosphate Isomerase and Pyruvate Carboxylase, which lead to an abnormal retention of lipids may also be responsible for microvesicular steatosis during HDV infection[77].
Furthermore, Wedemeyer et al[45] suggest that hepatitis D is an immune mediated disease, noting a rise in CD4+ T cells in individuals with a HDV infection. Although the role of the host’s immune system seems unlikely, various autoantibodies have been detected in infected patients. Prominent amongst them is liver-kidney microsomal antibody type 3, directed against uridine diphosphate glucoronyl transferase[90]. The disruption of metabolism in this way could contribute to HCC. Indeed Hanahan et al[91] have already labeled some changes in cellular metabolism as hallmarks of cancer.
HBV DNA integration
It is interesting to note that the HBX product has been found to directly interact with p53 and has been associated with the MAPK pathway and hence causes HCC[92]. It was previously thought that HBV DNA integration with chromosomes of infected hepatocytes would be responsible for HCC. However, the process of integration has been noted to be entirely random rather than targeted to specific genes and the length and components of the integrant has found to vary considerably[93]. Interestingly, when Woodchuck hepatitis virus targets the intronless N-myc2 gene as a site of integration, it predisposes to HCC[94]. Together with the activity of the protein product, the increased expression of mechanistic of rapamycin (mTOR) and PI3K/Akt were found to be responsible for cancer development[95]. Indeed mTOR promotes cell proliferation, apoptosis resistance and vascularization of tumors[96] by regulating the transcriptional activity of FOXO1-3a and protein translation by pS6 and eIF-4E[95]. To the authors’ knowledge, no study has yet investigated the association of the HDV antigens with mTOR or the downregulation of MiR-101[97] (which is done by HBX protein and interacts with DNMT3A) and this could be a potential area of research.
Peroxisome proliferator-activated receptor and HCC
Peroxisome proliferator-activated receptor (PPAR) has been shown to play a role in the development of HCC[98]. PPARα (which normally has a role in lipid metabolism), found in the liver, kidney, heart, and small intestine, has been shown to be involved in the regulation of the cell cycle. In mice, knocking down PPARα led to HCC suppression[99]. However, conflicting reports of the role of PPARα exist. Meanwhile PPARγ, found in adipose tissue and macrophages, inhibits HCC[100-102]. These control epithelial-mesenchymal transition and prevent metastasis by increasing E-cadherin through TIMP3[103]. PPARγ is also involved in cell cycle arrest[103] and induces Fas dependent apoptosis, hence combating HCC. PPARδ (a gene derived from the TCF/β-catenin pathway) is found universally and has been reported to be involved in highly malignant colon cancer[104]. It is thus necessary to explore in the future whether PPAR are somehow exploited by HDV in the development of HCC. If so, thiazolidinediones, which act on PPARγ, could be used to treat HCC. Together with retinoic acid, PPAR agonists and antagonists could become the frontline therapeutic drugs in HCC treatment.
TOWARDS THERAPEUTICS AND A BETTER UNDERSTANDING OF HDV
A better understanding of the molecular events underlying HCC development following HDV infection is vital to not only the approach to the virus but also for the development of new drugs, which can target specific parts of the pathways involved if not the virus itself and prevent development of HCC in patients infected with HDV. For example the targeted inhibition of STAT3 with a decoy 15-mer double-stranded oligonucleotide, which corresponds to the STAT3 response element in the c-fos promoter region, has been experimentally proven to abrogate head and neck cancer growth[105] and could eventually be used to prevent or treat HCC as well.
Cyclophilins are a class of proteins localized in various cellular compartments, involved in metabolism and homeostasis and are upregulated during inflammation and cancer. Cyclophilin A (CypA), in the cytoplasm, is involved in the virus life cycle, while extracellular CypA and CypB are pro-inflammatory in nature. Cyclosporins are potential cyclophilin inhibitors and could have therapeutic potential for the treatment of virus induced liver diseases. Indeed cyclosporin A (CsA) has been shown to inhibit HBV and HDV entry via sodium taurocholate co-transporting polypeptide. There is a direct interaction between the drug and the NTCP receptor (which is also a bile salt transporter), with overlap at the preS1 domain (which mediates viral entry). CsA also has immunosuppressive effects, exercised via cyclophilin dependent inhibition of calcineurin[106].
Interestingly, HDV can, in vivo, infect the cells of hepadnavirus-induced hepatocellular carcinoma in Woodchucks[107]. Since it had been previously hypothesized that hepadnavirus-induced HCCs are resistant to reinfection, the experiment proves that the cells still have functioning woodchuck hepatitis virus receptors and if a resistance does exist, it occurs downstream of the receptor[108]. This information may facilitate development of novel strategies further dissecting the mechanism of liver carcinogenesis associated with HDV infection
The spread of HDV can be prevented by depriving the defective HDV of HBV necessary to propagate its infection. Countries with effective vaccination programs have shown a dramatic decrease in the incidence of HCC[109]. As there is no effective treatment for HDV and the only treatment available is interferon, which is of limited efficacy[110], vaccination against HBV should be stressed. Carriers of HBs should be informed of the risk of superinfection from carriers coinfected with HDV and educated about preventive practices.
Footnotes
P- Reviewer: Cavoli GL, Gara N, Lesmana CRA, Watanabe T
S- Editor: Gong XM L- Editor: A E- Editor: Liu SQ
Conflict-of-interest: Authors do not have any conflict of interest to declare.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 19, 2014
First decision: September 16, 2014
Article in press: January 19, 2015
References
- 1.Dastgerdi ES, Herbers U, Tacke F. Molecular and clinical aspects of hepatitis D virus infections. World J Virol. 2012;1:71–78. doi: 10.5501/wjv.v1.i3.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunha C, Monjardino J, Cheng D, Krause S, Carmo-Fonseca M. Localization of hepatitis delta virus RNA in the nucleus of human cells. RNA. 1998;4:680–693. doi: 10.1017/s135583829898013x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farci P, Niro GA. Clinical features of hepatitis D. Semin Liver Dis. 2012;32:228–236. doi: 10.1055/s-0032-1323628. [DOI] [PubMed] [Google Scholar]
- 4.Smedile A, Farci P, Verme G, Caredda F, Cargnel A, Caporaso N, Dentico P, Trepo C, Opolon P, Gimson A, et al. Influence of delta infection on severity of hepatitis B. Lancet. 1982;2:945–947. doi: 10.1016/s0140-6736(82)90156-8. [DOI] [PubMed] [Google Scholar]
- 5.Yurdaydın C, Idilman R, Bozkaya H, Bozdayi AM. Natural history and treatment of chronic delta hepatitis. J Viral Hepat. 2010;17:749–756. doi: 10.1111/j.1365-2893.2010.01353.x. [DOI] [PubMed] [Google Scholar]
- 6.Heidrich B, Serrano BC, Idilman R, Kabaçam G, Bremer B, Raupach R, Önder FO, Deterding K, Zacher BJ, Taranta A, et al. HBeAg-positive hepatitis delta: virological patterns and clinical long-term outcome. Liver Int. 2012;32:1415–1425. doi: 10.1111/j.1478-3231.2012.02831.x. [DOI] [PubMed] [Google Scholar]
- 7.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 8.Romeo R. [Role of the hepatitis Delta virus on the pathogenesis of hepatic cirrhosis and hepatocellular carcinoma. Recent advances] Recenti Prog Med. 2010;101:52–56. [PubMed] [Google Scholar]
- 9.Wu JC, Chen TZ, Huang YS, Yen FS, Ting LT, Sheng WY, Tsay SH, Lee SD. Natural history of hepatitis D viral superinfection: significance of viremia detected by polymerase chain reaction. Gastroenterology. 1995;108:796–802. doi: 10.1016/0016-5085(95)90453-0. [DOI] [PubMed] [Google Scholar]
- 10.Grabowski J, Wedemeyer H. Hepatitis delta: immunopathogenesis and clinical challenges. Dig Dis. 2010;28:133–138. doi: 10.1159/000282076. [DOI] [PubMed] [Google Scholar]
- 11.Abbas Z, Qureshi M, Hamid S, Jafri W. Hepatocellular carcinoma in hepatitis D: does it differ from hepatitis B monoinfection? Saudi J Gastroenterol. 2012;18:18–22. doi: 10.4103/1319-3767.91731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abbas Z, Jafri W, Raza S. Hepatitis D: Scenario in the Asia-Pacific region. World J Gastroenterol. 2010;16:554–562. doi: 10.3748/wjg.v16.i5.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fattovich G, Giustina G, Schalm SW, Hadziyannis S, Sanchez-Tapias J, Almasio P, Christensen E, Krogsgaard K, Degos F, Carneiro de Moura M. Occurrence of hepatocellular carcinoma and decompensation in western European patients with cirrhosis type B. The EUROHEP Study Group on Hepatitis B Virus and Cirrhosis. Hepatology. 1995;21:77–82. doi: 10.1002/hep.1840210114. [DOI] [PubMed] [Google Scholar]
- 14.Fattovich G, Giustina G, Christensen E, Pantalena M, Zagni I, Realdi G, Schalm SW. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep) Gut. 2000;46:420–426. doi: 10.1136/gut.46.3.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cross TJ, Rizzi P, Horner M, Jolly A, Hussain MJ, Smith HM, Vergani D, Harrison PM. The increasing prevalence of hepatitis delta virus (HDV) infection in South London. J Med Virol. 2008;80:277–282. doi: 10.1002/jmv.21078. [DOI] [PubMed] [Google Scholar]
- 16.Uzunalimoğlu O, Yurdaydin C, Cetinkaya H, Bozkaya H, Sahin T, Colakoğlu S, Tankurt E, Sarioğlu M, Ozenirler S, Akkiz H, et al. Risk factors for hepatocellular carcinoma in Turkey. Dig Dis Sci. 2001;46:1022–1028. doi: 10.1023/a:1010705910858. [DOI] [PubMed] [Google Scholar]
- 17.Değertekin H, Yalçin K, Yakut M. The prevalence of hepatitis delta virus infection in acute and chronic liver diseases in Turkey: an analysis of clinical studies. Turk J Gastroenterol. 2006;17:25–34. [PubMed] [Google Scholar]
- 18.Toukan AU, Abu-el-Rub OA, Abu-Laban SA, Tarawneh MS, Kamal MF, Hadler SC, Krawczynski K, Margolis HS, Maynard JE. The epidemiology and clinical outcome of hepatitis D virus (delta) infection in Jordan. Hepatology. 1987;7:1340–1345. doi: 10.1002/hep.1840070627. [DOI] [PubMed] [Google Scholar]
- 19.Trichopoulos D, Day NE, Tzonou A, Hadziyannis S, Kaklamani E, Sparos L, Muñoz N, Hatzakis A. Delta agent and the etiology of hepatocellular carcinoma. Int J Cancer. 1987;39:283–286. doi: 10.1002/ijc.2910390303. [DOI] [PubMed] [Google Scholar]
- 20.Gheorghe L, Iacob S, Simionov I, Vadan R, Gheorghe C, Iacob R, Parvulescu I, Constantinescu I. Natural history of compensated viral B and D cirrhosis. Rom J Gastroenterol. 2005;14:329–335. [PubMed] [Google Scholar]
- 21.Kurbanov F, Tanaka Y, Elkady A, Oyunsuren T, Mizokami M. Tracing hepatitis C and Delta viruses to estimate their contribution in HCC rates in Mongolia. J Viral Hepat. 2007;14:667–674. doi: 10.1111/j.1365-2893.2007.00864.x. [DOI] [PubMed] [Google Scholar]
- 22.Tamura I, Kurimura O, Koda T, Ichimura H, Katayama S, Kurimura T, Inaba Y. Risk of liver cirrhosis and hepatocellular carcinoma in subjects with hepatitis B and delta virus infection: a study from Kure, Japan. J Gastroenterol Hepatol. 1993;8:433–436. doi: 10.1111/j.1440-1746.1993.tb01543.x. [DOI] [PubMed] [Google Scholar]
- 23.Huo TI, Wu JC, Lai CR, Lu CL, Sheng WY, Lee SD. Comparison of clinico-pathological features in hepatitis B virus-associated hepatocellular carcinoma with or without hepatitis D virus superinfection. J Hepatol. 1996;25:439–444. doi: 10.1016/s0168-8278(96)80202-9. [DOI] [PubMed] [Google Scholar]
- 24.Buti M, Homs M, Rodriguez-Frias F, Funalleras G, Jardí R, Sauleda S, Tabernero D, Schaper M, Esteban R. Clinical outcome of acute and chronic hepatitis delta over time: a long-term follow-up study. J Viral Hepat. 2011;18:434–442. doi: 10.1111/j.1365-2893.2010.01324.x. [DOI] [PubMed] [Google Scholar]
- 25.Romeo R, Del Ninno E, Rumi M, Russo A, Sangiovanni A, de Franchis R, Ronchi G, Colombo M. A 28-year study of the course of hepatitis Delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology. 2009;136:1629–1638. doi: 10.1053/j.gastro.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 26.Ji J, Sundquist K, Sundquist J. A population-based study of hepatitis D virus as potential risk factor for hepatocellular carcinoma. J Natl Cancer Inst. 2012;104:790–792. doi: 10.1093/jnci/djs168. [DOI] [PubMed] [Google Scholar]
- 27.Romeo R, Foglieni B, Casazza G, Spreafico M, Colombo M, Prati D. High serum levels of HDV RNA are predictors of cirrhosis and liver cancer in patients with chronic hepatitis delta. PLoS One. 2014;9:e92062. doi: 10.1371/journal.pone.0092062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oyunsuren T, Kurbanov F, Tanaka Y, Elkady A, Sanduijav R, Khajidsuren O, Dagvadorj B, Mizokami M. High frequency of hepatocellular carcinoma in Mongolia; association with mono-, or co-infection with hepatitis C, B, and delta viruses. J Med Virol. 2006;78:1688–1695. doi: 10.1002/jmv.20755. [DOI] [PubMed] [Google Scholar]
- 29.Cenac A, Pedroso ML, Djibo A, Develoux M, Pichoud C, Lamothe F, Trepo C, Warter A. Hepatitis B, C, and D virus infections in patients with chronic hepatitis, cirrhosis, and hepatocellular carcinoma: a comparative study in Niger. Am J Trop Med Hyg. 1995;52:293–296. doi: 10.4269/ajtmh.1995.52.293. [DOI] [PubMed] [Google Scholar]
- 30.Oliveri F, Brunetto MR, Actis GC, Bonino F. Pathobiology of chronic hepatitis virus infection and hepatocellular carcinoma (HCC) Ital J Gastroenterol. 1991;23:498–502. [PubMed] [Google Scholar]
- 31.Verme G, Brunetto MR, Oliveri F, Baldi M, Forzani B, Piantino P, Ponzetto A, Bonino F. Role of hepatitis delta virus infection in hepatocellular carcinoma. Dig Dis Sci. 1991;36:1134–1136. doi: 10.1007/BF01297460. [DOI] [PubMed] [Google Scholar]
- 32.Smedile A, Rosina F, Saracco G, Chiaberge E, Lattore V, Fabiano A, Brunetto MR, Verme G, Rizzetto M, Bonino F. Hepatitis B virus replication modulates pathogenesis of hepatitis D virus in chronic hepatitis D. Hepatology. 1991;13:413–416. [PubMed] [Google Scholar]
- 33.Huang YH, Wu JC, Chau GY, Tsay SH, King KL, Sheng WY, Lui WY, Lee SD. Detection of serum hepatitis B, C, and D viral nucleic acids and its implications in hepatocellular carcinoma patients. J Gastroenterol. 1998;33:512–516. doi: 10.1007/s005350050124. [DOI] [PubMed] [Google Scholar]
- 34.Realdi G, Fattovich G, Hadziyannis S, Schalm SW, Almasio P, Sanchez-Tapias J, Christensen E, Giustina G, Noventa F. Survival and prognostic factors in 366 patients with compensated cirrhosis type B: a multicenter study. The Investigators of the European Concerted Action on Viral Hepatitis (EUROHEP) J Hepatol. 1994;21:656–666. doi: 10.1016/s0168-8278(94)80115-0. [DOI] [PubMed] [Google Scholar]
- 35.Kage M, Kosai K, Shimamatsu K, Nakashima O, Haramaki M, Yasunaga M, Iha H, Kojiro M, Govindarajan S. Incidence of hepatitis D virus infection in Japanese patients with hepatocellular carcinoma--immunohistochemical investigation of the delta antigen. Kurume Med J. 1992;39:231–234. doi: 10.2739/kurumemedj.39.231. [DOI] [PubMed] [Google Scholar]
- 36.Tzonou A, Trichopoulos D, Kaklamani E, Zavitsanos X, Koumantaki Y, Hsieh CC. Epidemiologic assessment of interactions of hepatitis-C virus with seromarkers of hepatitis-B and -D viruses, cirrhosis and tobacco smoking in hepatocellular carcinoma. Int J Cancer. 1991;49:377–380. doi: 10.1002/ijc.2910490311. [DOI] [PubMed] [Google Scholar]
- 37.Tassopoulos NC, Theodoropoulos G, Sjogren MH, Engle R, Purcell RH. Serological markers of hepatitis B virus and hepatitis D virus infections in Greek adults with primary hepatocellular carcinoma. Infection. 1989;17:17–19. doi: 10.1007/BF01643493. [DOI] [PubMed] [Google Scholar]
- 38.Chen DS, Lai MY, Sung JL. Delta agent infection in patients with chronic liver diseases and hepatocellular carcinoma--an infrequent finding in Taiwan. Hepatology. 1984;4:502–503. doi: 10.1002/hep.1840040324. [DOI] [PubMed] [Google Scholar]
- 39.Govindarajan S, Hevia FJ, Peters RL. Prevalence of delta antigen/antibody in B-viral-associated hepatocellular carcinoma. Cancer. 1984;53:1692–1694. doi: 10.1002/1097-0142(19840415)53:8<1692::aid-cncr2820530812>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Negro F, Papotti M, Taraglio S, Rubbia-Brandt L, Giostra E, Pacchioni D, Rizzetto M, Hadengue A. Relationship between hepatocyte proliferation and hepatitis delta virus replication in neoplastic and non-neoplastic liver tissues. J Viral Hepat. 1997;4:93–98. doi: 10.1111/j.1365-2893.1997.tb00210.x. [DOI] [PubMed] [Google Scholar]
- 41.Wu JC, Choo KB, Chen CM, Chen TZ, Huo TI, Lee SD. Genotyping of hepatitis D virus by restriction-fragment length polymorphism and relation to outcome of hepatitis D. Lancet. 1995;346:939–941. doi: 10.1016/s0140-6736(95)91558-3. [DOI] [PubMed] [Google Scholar]
- 42.Wu JC. Functional and clinical significance of hepatitis D virus genotype II infection. Curr Top Microbiol Immunol. 2006;307:173–186. doi: 10.1007/3-540-29802-9_9. [DOI] [PubMed] [Google Scholar]
- 43.Su CW, Huang YH, Huo TI, Shih HH, Sheen IJ, Chen SW, Lee PC, Lee SD, Wu JC. Genotypes and viremia of hepatitis B and D viruses are associated with outcomes of chronic hepatitis D patients. Gastroenterology. 2006;130:1625–1635. doi: 10.1053/j.gastro.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 44.Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25:3834–3847. doi: 10.1038/sj.onc.1209562. [DOI] [PubMed] [Google Scholar]
- 45.Wedemeyer H, Manns MP. Epidemiology, pathogenesis and management of hepatitis D: update and challenges ahead. Nat Rev Gastroenterol Hepatol. 2010;7:31–40. doi: 10.1038/nrgastro.2009.205. [DOI] [PubMed] [Google Scholar]
- 46.Williams V, Brichler S, Radjef N, Lebon P, Goffard A, Hober D, Fagard R, Kremsdorf D, Dény P, Gordien E. Hepatitis delta virus proteins repress hepatitis B virus enhancers and activate the alpha/beta interferon-inducible MxA gene. J Gen Virol. 2009;90:2759–2767. doi: 10.1099/vir.0.011239-0. [DOI] [PubMed] [Google Scholar]
- 47.Pugnale P, Pazienza V, Guilloux K, Negro F. Hepatitis delta virus inhibits alpha interferon signaling. Hepatology. 2009;49:398–406. doi: 10.1002/hep.22654. [DOI] [PubMed] [Google Scholar]
- 48.Ottobrelli A, Marzano A, Smedile A, Recchia S, Salizzoni M, Cornu C, Lamy ME, Otte JB, De Hemptinne B, Geubel A. Patterns of hepatitis delta virus reinfection and disease in liver transplantation. Gastroenterology. 1991;101:1649–1655. doi: 10.1016/0016-5085(91)90404-9. [DOI] [PubMed] [Google Scholar]
- 49.Taylor JM. Hepatitis delta virus. Virology. 2006;344:71–76. doi: 10.1016/j.virol.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 50.Williams V, Brichler S, Khan E, Chami M, Dény P, Kremsdorf D, Gordien E. Large hepatitis delta antigen activates STAT-3 and NF-κB via oxidative stress. J Viral Hepat. 2012;19:744–753. doi: 10.1111/j.1365-2893.2012.01597.x. [DOI] [PubMed] [Google Scholar]
- 51.Waris G, Tardif KD, Siddiqui A. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem Pharmacol. 2002;64:1425–1430. doi: 10.1016/s0006-2952(02)01300-x. [DOI] [PubMed] [Google Scholar]
- 52.Bureau C, Bernad J, Chaouche N, Orfila C, Béraud M, Gonindard C, Alric L, Vinel JP, Pipy B. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J Biol Chem. 2001;276:23077–23083. doi: 10.1074/jbc.M100698200. [DOI] [PubMed] [Google Scholar]
- 53.Chan SW, Egan PA. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005;19:1510–1512. doi: 10.1096/fj.04-3455fje. [DOI] [PubMed] [Google Scholar]
- 54.Gong G, Waris G, Tanveer R, Siddiqui A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci USA. 2001;98:9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li S, Ye L, Yu X, Xu B, Li K, Zhu X, Liu H, Wu X, Kong L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology. 2009;391:257–264. doi: 10.1016/j.virol.2009.06.039. [DOI] [PubMed] [Google Scholar]
- 56.Meyer M, Caselmann WH, Schlüter V, Schreck R, Hofschneider PH, Baeuerle PA. Hepatitis B virus transactivator MHBst: activation of NF-kappa B, selective inhibition by antioxidants and integral membrane localization. EMBO J. 1992;11:2991–3001. doi: 10.1002/j.1460-2075.1992.tb05369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 58.Waris G, Huh KW, Siddiqui A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-kappa B via oxidative stress. Mol Cell Biol. 2001;21:7721–7730. doi: 10.1128/MCB.21.22.7721-7730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940–2954. doi: 10.1182/blood-2002-04-1204. [DOI] [PubMed] [Google Scholar]
- 60.Feng DY, Zheng H, Jiang HY. [Effects of Stat3 phosphorylation and expression of c-fos and c-jun proteins on hepatocarcinogenesis] Hunan Yike Daxue Xuebao. 2001;26:17–19. [PubMed] [Google Scholar]
- 61.Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, Bowman T, Falcone R, Fairclough R, Cantor A, et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002;62:6659–6666. [PubMed] [Google Scholar]
- 62.Niu G, Bowman T, Huang M, Shivers S, Reintgen D, Daud A, Chang A, Kraker A, Jove R, Yu H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene. 2002;21:7001–7010. doi: 10.1038/sj.onc.1205859. [DOI] [PubMed] [Google Scholar]
- 63.de Mochel NS, Seronello S, Wang SH, Ito C, Zheng JX, Liang TJ, Lambeth JD, Choi J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology. 2010;52:47–59. doi: 10.1002/hep.23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glenn JS, Watson JA, Havel CM, White JM. Identification of a prenylation site in delta virus large antigen. Science. 1992;256:1331–1333. doi: 10.1126/science.1598578. [DOI] [PubMed] [Google Scholar]
- 65.Hwang SB, Lai MM. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis B virus surface antigen. J Virol. 1993;67:7659–7662. doi: 10.1128/jvi.67.12.7659-7662.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choi SH, Jeong SH, Hwang SB. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cascades: implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology. 2007;132:343–357. doi: 10.1053/j.gastro.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 67.Shih HH, Sheen IJ, Su CW, Peng WL, Lin LH, Wu JC. Hepatitis D virus isolates with low replication and epithelial-mesenchymal transition-inducing activity are associated with disease remission. J Virol. 2012;86:9044–9054. doi: 10.1128/JVI.00130-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park CY, Oh SH, Kang SM, Lim YS, Hwang SB. Hepatitis delta virus large antigen sensitizes to TNF-alpha-induced NF-kappaB signaling. Mol Cells. 2009;28:49–55. doi: 10.1007/s10059-009-0100-5. [DOI] [PubMed] [Google Scholar]
- 69.Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 70.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109 Suppl:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 71.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 72.Choi SH, Park KJ, Ahn BY, Jung G, Lai MM, Hwang SB. Hepatitis C virus nonstructural 5B protein regulates tumor necrosis factor alpha signaling through effects on cellular IkappaB kinase. Mol Cell Biol. 2006;26:3048–3059. doi: 10.1128/MCB.26.8.3048-3059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park KJ, Choi SH, Lee SY, Hwang SB, Lai MM. Nonstructural 5A protein of hepatitis C virus modulates tumor necrosis factor alpha-stimulated nuclear factor kappa B activation. J Biol Chem. 2002;277:13122–13128. doi: 10.1074/jbc.M111599200. [DOI] [PubMed] [Google Scholar]
- 74.Weil R, Sirma H, Giannini C, Kremsdorf D, Bessia C, Dargemont C, Bréchot C, Israël A. Direct association and nuclear import of the hepatitis B virus X protein with the NF-kappaB inhibitor IkappaBalpha. Mol Cell Biol. 1999;19:6345–6354. doi: 10.1128/mcb.19.9.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benegiamo G, Vinciguerra M, Guarnieri V, Niro GA, Andriulli A, Pazienza V. Hepatitis delta virus induces specific DNA methylation processes in Huh-7 liver cancer cells. FEBS Lett. 2013;587:1424–1428. doi: 10.1016/j.febslet.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 76.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 77.Mota S, Mendes M, Freitas N, Penque D, Coelho AV, Cunha C. Proteome analysis of a human liver carcinoma cell line stably expressing hepatitis delta virus ribonucleoproteins. J Proteomics. 2009;72:616–627. doi: 10.1016/j.jprot.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 78.Kannan RP, Hensley LL, Evers LE, Lemon SM, McGivern DR. Hepatitis C virus infection causes cell cycle arrest at the level of initiation of mitosis. J Virol. 2011;85:7989–8001. doi: 10.1128/JVI.00280-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao FT, Lee YJ, Ko JL, Tsai CC, Tseng CJ, Sheu GT. Hepatitis delta virus epigenetically enhances clusterin expression via histone acetylation in human hepatocellular carcinoma cells. J Gen Virol. 2009;90:1124–1134. doi: 10.1099/vir.0.007211-0. [DOI] [PubMed] [Google Scholar]
- 80.Huang WH, Mai RT, Lee YH. Transcription factor YY1 and its associated acetyltransferases CBP and p300 interact with hepatitis delta antigens and modulate hepatitis delta virus RNA replication. J Virol. 2008;82:7313–7324. doi: 10.1128/JVI.02581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee CZ, Sheu JC. Histone H1e interacts with small hepatitis delta antigen and affects hepatitis delta virus replication. Virology. 2008;375:197–204. doi: 10.1016/j.virol.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 82.Kang YK, Hong SW, Lee H, Kim WH. Overexpression of clusterin in human hepatocellular carcinoma. Hum Pathol. 2004;35:1340–1346. doi: 10.1016/j.humpath.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 83.Lau SH, Sham JS, Xie D, Tzang CH, Tang D, Ma N, Hu L, Wang Y, Wen JM, Xiao G, et al. Clusterin plays an important role in hepatocellular carcinoma metastasis. Oncogene. 2006;25:1242–1250. doi: 10.1038/sj.onc.1209141. [DOI] [PubMed] [Google Scholar]
- 84.Redondo M, Villar E, Torres-Muñoz J, Tellez T, Morell M, Petito CK. Overexpression of clusterin in human breast carcinoma. Am J Pathol. 2000;157:393–399. doi: 10.1016/S0002-9440(10)64552-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miyake H, Hara S, Zellweger T, Kamidono S, Gleave ME, Hara I. Acquisition of resistance to Fas-mediated apoptosis by overexpression of clusterin in human renal-cell carcinoma cells. Mol Urol. 2001;5:105–111. doi: 10.1089/10915360152559585. [DOI] [PubMed] [Google Scholar]
- 86.Featherstone C, Jackson SP. Ku, a DNA repair protein with multiple cellular functions? Mutat Res. 1999;434:3–15. doi: 10.1016/s0921-8777(99)00006-3. [DOI] [PubMed] [Google Scholar]
- 87.French LE, Wohlwend A, Sappino AP, Tschopp J, Schifferli JA. Human clusterin gene expression is confined to surviving cells during in vitro programmed cell death. J Clin Invest. 1994;93:877–884. doi: 10.1172/JCI117043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Trougakos IP, Gonos ES. Clusterin/apolipoprotein J in human aging and cancer. Int J Biochem Cell Biol. 2002;34:1430–1448. doi: 10.1016/s1357-2725(02)00041-9. [DOI] [PubMed] [Google Scholar]
- 89.Arozarena I, Matallanas D, Crespo P. Maintenance of CDC42 GDP-bound state by Rho-GDI inhibits MAP kinase activation by the exchange factor Ras-GRF. evidence for Ras-GRF function being inhibited by Cdc42-GDP but unaffected by CDC42-GTP. J Biol Chem. 2001;276:21878–21884. doi: 10.1074/jbc.M011383200. [DOI] [PubMed] [Google Scholar]
- 90.Philipp T, Durazzo M, Trautwein C, Alex B, Straub P, Lamb JG, Johnson EF, Tukey RH, Manns MP. Recognition of uridine diphosphate glucuronosyl transferases by LKM-3 antibodies in chronic hepatitis D. Lancet. 1994;344:578–581. doi: 10.1016/s0140-6736(94)91966-6. [DOI] [PubMed] [Google Scholar]
- 91.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 92.Di Bisceglie AM. Hepatitis B and hepatocellular carcinoma. Hepatology. 2009;49:S56–S60. doi: 10.1002/hep.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kew MC, Miller RH, Chen HS, Tennant BC, Purcell RH. Mutant woodchuck hepatitis virus genomes from virions resemble rearranged hepadnaviral integrants in hepatocellular carcinoma. Proc Natl Acad Sci USA. 1993;90:10211–10215. doi: 10.1073/pnas.90.21.10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Renard CA, Fourel G, Bralet MP, Degott C, De La Coste A, Perret C, Tiollais P, Buendia MA. Hepatocellular carcinoma in WHV/N-myc2 transgenic mice: oncogenic mutations of beta-catenin and synergistic effect of p53 null alleles. Oncogene. 2000;19:2678–2686. doi: 10.1038/sj.onc.1203617. [DOI] [PubMed] [Google Scholar]
- 95.Wang Z, Jin W, Jin H, Wang X. mTOR in viral hepatitis and hepatocellular carcinoma: function and treatment. Biomed Res Int. 2014;2014:735672. doi: 10.1155/2014/735672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng X, Zhu Z, Jiao H, Lin J, Jiang K, et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. 2013;123:630–645. doi: 10.1172/JCI64265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wei X, Xiang T, Ren G, Tan C, Liu R, Xu X, Wu Z. miR-101 is down-regulated by the hepatitis B virus x protein and induces aberrant DNA methylation by targeting DNA methyltransferase 3A. Cell Signal. 2013;25:439–446. doi: 10.1016/j.cellsig.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 98.Kimura O, Kondo Y, Shimosegawa T. PPAR Could Contribute to the Pathogenesis of Hepatocellular Carcinoma. PPAR Res. 2012;2012:574180. doi: 10.1155/2012/574180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Invest. 2008;118:683–694. doi: 10.1172/JCI33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schaefer KL, Wada K, Takahashi H, Matsuhashi N, Ohnishi S, Wolfe MM, Turner JR, Nakajima A, Borkan SC, Saubermann LJ. Peroxisome proliferator-activated receptor gamma inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res. 2005;65:2251–2259. doi: 10.1158/0008-5472.CAN-04-3037. [DOI] [PubMed] [Google Scholar]
- 101.Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001;61:6213–6218. [PubMed] [Google Scholar]
- 102.Yu J, Shen B, Chu ES, Teoh N, Cheung KF, Wu CW, Wang S, Lam CN, Feng H, Zhao J, et al. Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro. Hepatology. 2010;51:2008–2019. doi: 10.1002/hep.23550. [DOI] [PubMed] [Google Scholar]
- 103.Shen B, Chu ES, Zhao G, Man K, Wu CW, Cheng JT, Li G, Nie Y, Lo CM, Teoh N, et al. PPARgamma inhibits hepatocellular carcinoma metastases in vitro and in mice. Br J Cancer. 2012;106:1486–1494. doi: 10.1038/bjc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takayama O, Yamamoto H, Damdinsuren B, Sugita Y, Ngan CY, Xu X, Tsujino T, Takemasa I, Ikeda M, Sekimoto M, et al. Expression of PPARdelta in multistage carcinogenesis of the colorectum: implications of malignant cancer morphology. Br J Cancer. 2006;95:889–895. doi: 10.1038/sj.bjc.6603343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC, Robbins PD, Gadiparthi S, Burke NA, Watkins SF, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci USA. 2003;100:4138–4143. doi: 10.1073/pnas.0534764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nkongolo S, Ni Y, Lempp FA, Kaufman C, Lindner T, Esser-Nobis K, Lohmann V, Mier W, Mehrle S, Urban S. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol. 2014;60:723–731. doi: 10.1016/j.jhep.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 107.Freitas N, Salisse J, Cunha C, Toshkov I, Menne S, Gudima SO. Hepatitis delta virus infects the cells of hepadnavirus-induced hepatocellular carcinoma in woodchucks. Hepatology. 2012;56:76–85. doi: 10.1002/hep.25663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Naoumov NV. Cyclophilin inhibition as potential therapy for liver diseases. J Hepatol. 2014;61:1166–1174. doi: 10.1016/j.jhep.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 109.Chang MH. Hepatitis B virus and cancer prevention. Recent Results Cancer Res. 2011;188:75–84. doi: 10.1007/978-3-642-10858-7_6. [DOI] [PubMed] [Google Scholar]
- 110.Abbas Z, Khan MA, Salih M, Jafri W. Interferon alpha for chronic hepatitis D. Cochrane Database Syst Rev. 2011;(12):CD006002. doi: 10.1002/14651858.CD006002.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]