

Abstract
Stem cell pluripotency and differentiation are global processes regulated by several pathways that have been studied intensively over recent years. Nitric oxide (NO) is an important molecule that affects gene expression at the level of transcription and translation and regulates cell survival and proliferation in diverse cell types. In embryonic stem cells NO has a dual role, controlling differentiation and survival, but the molecular mechanisms by which it modulates these functions are not completely defined. NO is a physiological regulator of cell respiration through the inhibition of cytochrome c oxidase. Many researchers have been examining the role that NO plays in other aspects of metabolism such as the cellular bioenergetics state, the hypoxia response and the relationship of these areas to stem cell stemness.
Keywords: Pluripotency, Differentiation, Nitric oxide, Metabolism, Hypoxia, Stem cell
Core tip: Increasing research interest has focused on the role of nitric oxide (NO) in regulating many physiological functions such as metabolism, the hypoxia response, pluripotency, and stem cell differentiation. NO has been proven to act as a powerful agent for promoting the maintenance of cell pluripotency and survival, thus explaining how it can act as an alternative factor for the maintenance of certain cultured cell lines.
INTRODUCTION
Nitric oxide (NO) is a short-lived free radical synthesised via the L-arginine to L-citrulline pathway, which is mediated by nitric oxide synthase, NOS1 (neuronal NOS), NOS2 (inducible NOS), and NOS3 (endothelial NOS)[1], in most animal cells[2]. NO reacts with molecules such as oxygen, superoxide or metals, nucleic acids, and proteins. NO is quickly oxidised into nitrate and nitrite, which are now considered as non-inert products because they are a source of NO through their reduction by reductase enzymes when the endogenous L-arginine/NOS pathway is dysfunctional[3]. In addition to serving as a germicide in the immune system as part of the inflammatory response and as a neurotransmitter in the central nervous system, NO acts as a second messenger and has multiple biological effects implicated in a variety of physiological functions in mammals, such as the regulation of blood pressure via smooth muscle relaxation and inhibition of platelet aggregation[4-7]. Moreover, it has been shown that it affects gene expression at the level of transcription and translation and regulates cell survival and proliferation in diverse cell types[8,9]. Furthermore, NO plays a role in growth, survival, proliferation, differentiation, as well as in the pathology of illnesses such as cancer, diabetes, and neurodegenerative diseases[10,11]. In addition, it has been reported that NO is involved in the control of heart functions and cardiac development[12,13].
The presence and concentration of other free radicals are critical factors that influence the effects of NO on cellular processes. For example, it has been described that low concentrations of NO inhibit cytochrome c oxidase (CcO), which catalyses the final step of the mitochondrial transport chain, competing with O2 in a reversible manner[14,15]. Nevertheless, high levels of NO may cause nitrosylation of protein thiols and perhaps the removal of iron from iron-sulphur centres[16,17]. CcO, as catalyst of the central step of oxidative phosphorylation and adenosinetriphosphate (ATP) generation, regulates cellular oxygen consumption. The physiological concentration of NO modulates CcO activity, depending on the concentration of intracellular oxygen and the redox state of CcO. This interaction between CcO and NO allows the detection of changes in oxygen concentration and the initiation of adaptive responses. This indicates that NO might be a physiological regulator of cellular respiration and metabolism. Furthermore, NO has been described to have an important role in regulating the hypoxia response[15,18,19].
On the other hand, it has been reported that low concentrations of NO have a direct effect on processes such as cell proliferation and survival[8]. In RINm5F cells homeostatic concentrations of NO (1-10 μmol/L) can initiate signalling pathways implicated in survival actions[20]. Moreover, higher NO concentrations, induced by the inflammatory response, can cause oxidative and nitrosative stress, and apoptosis. These actions are partly responsible for cell death in chronic and degenerative diseases. Pharmacological treatment with high NO concentrations promote embryonic stem cell (ESC) differentiation[9,21-23]. However, the functional significance of high NO concentrations on differentiation in vivo has not yet been demonstrated.
It has been clearly shown that NO has an important role as regulator of many physiological functions, and has thus become a target of interest in the fields of metabolism, the hypoxia response, pluripotency, and stem cell differentiation. This review aims to describe the progress on understanding the role of NO in these interrelated biological processes.
MECHANISM OF NO ACTION IN STEM CELL BIOLOGY
The downstream effects of NO can be mediated in cyclic guanosine monophosphate (cGMP) dependent or independent ways[24-26]. When acting independently of cGMP, it has been shown that NO interacts with metal complexes, oxygen (O2), super-oxide anion (O2•-) and CcO[15]. These interactions have different effects depending on the amount of NO present. Protein nitrosylation and nitration can occur when NO interacts with oxygen species, which happens more frequently at high levels of NO[27]. On the other hand, cGMP-dependent effects are mediated by the NO receptor, soluble guanylylcyclase (sGC). sGC is a heterodimeric hemoprotein composed of alpha (sGCα) and beta (sGCβ) subunits[28]. NO activates sGC by interacting with its hemo group and catalysing the conversion of GTP into cGMP. cGMP controls a variety of physiological effects in several tissues by interacting with downstream effectors such as a family of cGMP-dependent protein kinases (PKG), cGMP-dependent phosphodiesterases, and cyclic nucleotide gated channels[24]. In addition, triple NOS knockout mice, where NO production is abolished, have been shown to have a reduced survival rate and lower numbers of offspring[29]. Thus, it was proposed that NO/cGMP signalling plays a significant role in embryonic development and cell differentiation. This hypothesis has gained additional support from studies that show differential expression and function of various NO signalling components in ESCs and differentiated cells[25,26,30]. Although it remains unclear whether the action of NO on stem cell biology is mediated via the cGMP pathway, it has been shown that the effects of NO on bone marrow stem cell potency and differentiation are independent of the sGC-cGMP pathway[31,32]. Recent studies have also shown that NO can regulate protein function via the nitrosylation of sGC cysteine residues[33]. Indeed, NO has been reported to modulate transcription factor function via Cys-nitrosylation[34]. It was concluded that the effects of NO on stem cell pluripotency and differentiation are independent of the sGC-cGMP pathway[5]. The mechanisms by which NO modulates the differentiation of ESCs remain unclear. Intriguingly, NO appears to act independently of the LIF/Stat3 pathway, as NO does not induce Stat3 phosphorylation in ESCs[8]. Moreover, the expression of Nanog is LIF/Stat3 independent, and Nanog over-expression in the absence of LIF is able to maintain ESC pluripotency[35].
NO REGULATES STEM CELL PLURIPOTENCY AND DIFFERENTIACION
The study of stem cells has gained considerable interest in recent years because they potentially offer an unlimited source of cells for therapeutic purposes. Many research groups have participated in efforts to expand and characterise populations of ESCs and induced-pluripotent stem cells (iPSCs), and to identify ways of directing their differentiation towards particular tissue types. In this context, the identification of small molecules that act on specific cell signalling pathways involved in embryonic development might be instrumental in the design of protocols that can efficiently control cell “stemness” and stem cell differentiation.
NO is, potentially, one such small molecule, with evidence supporting a dual role for it in the control of ESC differentiation and tissue morphogenesis. It is important to note that the effects of NO on differentiation and the maintenance of pluripotency are dose dependent. Moreover, while it is generally considered to be an inducer of apoptosis[36,37], it has also been shown to protect certain cell types from programmed cell death[38].
NO has been demonstrated to have the potential to influence the proliferation and differentiation cascades of certain cell types but its effect varies widely from cell to cell. For example, the proliferation and differentiation of human skin cells is modulated by NO[39], a finding that helps explain the pathophysiology of human skin diseases. NO causes growth inhibition of immortalised human oral keratinocytes and primary oral cancer cells (HN4) mainly via apoptotic cell death induced indirectly by stimulating the differentiation of both immortalised and malignant oral keratinocytes[2]. This suggests that the generation of NO in human skin diseases is not directly associated with local cell destruction, in contrast to observations in several other human diseases[39].
Our group has reported that the exposure of ESCs to low concentrations of the NO donor, diethylenetriamine NO adduct (DETA-NO), prevents the loss of expression of self-renewal genes and blocks mouse and human ESC differentiation. Similarly, constitutive NOS3 over-expression in cells grown under LIF withdrawal conditions maintained the expression of pluripotency markers[8]. On the other hand, exposure to high concentrations of DETA-NO promotes the differentiation of mouse ESCs (mESCs) by down-regulating Nanog and Oct4, the two master genes that control the pluripotent state[9].
Low concentrations of NO modulate pluripotency gene expression
It has been shown that low amounts of DETA-NO (2-20 μmol/L) delay the differentiation of human embryonic stem cells (hESCs), since the addition of NO induces an increase in the expression of Nanog, Oct4 and Sox2 to levels even higher than when cells are cultured with basic fibroblast growth factor (bFGF). Moreover, the cell surface antigen SSEA-4, which disappears following five passages under bFGF withdrawal, is retained when the growth medium is supplemented with NO. In addition, NO represses some differentiation markers (Brachyury, Gata4, Gata6, Fgf5 and Fgf8), which are present in the absence of bFGF. Similar observations were also seen in mESCs grown in the absence of LIF and supplemented with NO, which also induced Oct4 and Nanog protein expression, together with a decrease in the expression of the aforementioned early differentiation genes (Figure 1). Constitutive over-expression of NOS3 in cells cultured in the absence of LIF protected them from apoptosis and promoting cell survival[8].
Figure 1.
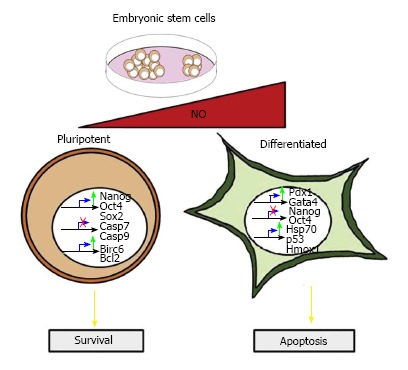
The dual roles of nitric oxide in stem cell pluripotency and apoptosis. Low amounts of nitric oxide (NO) induce the expression of Nanog, Oct4 and Sox2. This confers protection from apoptosis by down-regulating pro-apoptotic genes including Casp7, Casp9, as well as up-regulating the anti-apoptotic genes Bcl-2 and Birc6. High NO concentrations promote apoptosis markers, such as Poly (ADP-ribose) polymerase degradation, cleaved caspase-3, and increased p53 protein levels. Cells expressing Hsp70, a cyto-protective gene, are resistant to oxidative stress and enter into a differentiation program. Differentiation events are initiated by the down-regulation of Nanog and Oct4 and the expression of definitive endoderm markers, such as Pdx1 and Gata4.
NO has also been shown to play an important role in other cell types, such as multipotent resident cardiac stem cells and adult bone marrow cells (BMCs)[40,41]. NOS3-generated NO plays an important role in cardiomyocyte proliferation and maturation during early neonatal heart development[42]. Furthermore, a recent study using a porcine model has shown that activation of the NO pathway directs BMCs toward a preferential cardiomyogenic phenotype and also stimulates cell proliferation[43].
Genomic studies carried out by our group indicate that low concentrations of NO in mESCs regulate apoptosis, survival and the hypoxia response, areas that will be looked at more closely in the remainder of this review.
Regulation of differentiation by high concentrations of NO
NO plays an important role in development: It has been reported that NO may act as an essential negative regulator of cell proliferation during tissue differentiation and organ development in Xenopus and Drosophila. In addition, NO production is crucial for the establishment of ordered neuronal connections in the fly’s visual system, indicating that NO affects the acquisition of differentiated neural tissue[7]. Moreover, blocking NO production in neural precursor cells (NPCs) isolated from the ventricular zone of postnatal mice, resulted in increased proliferation[44]. In ESCs, it has been demonstrated that NO generation is required for cardiomyogenesis, since NOS inhibitors prevent the maturation of terminally-differentiated cardiomyocytes (CMs). Moreover, it has been shown that the differentiation of mESCs toward a cardiac phenotype is arrested by NO synthase inhibitors, which can in turn be readily rescued by NO donors[13]. NOS2 and NOS3 isoforms are prominently expressed during the early stages of cardiomyogenesis until NOS expression starts to decline around E14.5[13]. In the same context, undifferentiated ESCs are known to express high levels of NOS3. During differentiation, NOS3 levels decrease, whereas NOS2, NOS1, sGCα1, sGCβ1, and PKG levels significantly increase, suggesting that NO could be involved in the early differentiation events or physiological processes of ES cells or ES derived-cell lineages[25].
On the other hand, high DETA-NO concentrations (0.25-1.0 mmol/L) promote mESC differentiation by down-regulating Nanog and Oct4 expression. The repression of Nanog by NO is dependent on p53 activation associated with covalent modifications, such as Ser315 phosphorylation and Lys379 acetylation. In addition, exposure to high DETA-NO concentrations (0.5 mmol/L) increases the expression of definitive endoderm markers, such as FoxA2, Gata4, Hfn1-β and Sox 17 (Figure 1)[9].
The use of NO in differentiation protocols for the generation of different germinal lines: The first report of an efficient differentiation protocol using mESCs treated with NO was published by Kanno et al[45] in 2004. They demonstrated that NO promoted differentiation in part through its effects on cell survival, leading to the differentiation of mESCs into CMs[45]. Other groups have identified the role of cGMP signalling components and NO in cardiac development, as well as molecules that may regulate early cardiomyogenesis in ESCs via NO-dependent pathways, such as polyphenol curcumin and ascorbic acid[25,46-48]. In addition, it has been shown that other signalling pathways besides NO are involved in oxytocin-stimulated cardiomyogenesis[49].
It has been reported that NO is involved in vascular tissue differentiation. Because of the pivotal role played by NO in the biology of endothelial cells (ECs)[50] and its role in angiogenesis[51], it was hypothesised that it may also play a role in endothelial development. This was tested by studying the effect of differential expression of NO signalling components, or conversely of NOS inhibition, on endothelial differentiation. These studies indicate that NO is important for later endothelial development and function, although other signalling pathways appear to play a greater role in early development[52]. Although differentiation protocols for generating ECs from ESCs do not include NO-like active small molecules, the expression of endothelial NO synthase in these cell types is considered to be a characteristic of mature ECs[53,54]. Moreover, it has been demonstrated that NO/cGMP signalling molecules regulate neural lineage commitment and govern neural precursor differentiation, suggesting that NO/cGMP signalling contributes to the development of neural precursors and enhances the differentiation of precursors toward functional neurons[55,56]. Furthermore, it was reported that NO exposure to NPCs leads to a decrease in neuronal differentiation paralleled by an increase in astroglial differentiation. Neuron restrictive silencing factor1/repressor element-1 silencing transcription factor has been shown to play an essential role in the regulation of neurogenesis and is required for NO-mediated neural to glial fate induction[57].
It has been reported that NO modulates the growth and differentiation of human erythroid and myeloid cells from CD34+ BMCs in vivo[58], promotes bone and chondrocyte terminal differentiation[59], stimulates pre-adipocyte differentiation in rat[60] and mediates osteoblastic differentiation[61].
Bone marrow multipotent adult progenitor cells (MAPCs) can be purified and cultured from humans, rats and mice. MAPCs express Oct4 and are able to differentiate into multiple cell lineages including endothelial cells and neurons[62,63]. Importantly, NO appears to play a key role in both the maintenance of MAPC multipotency as well as in promoting their endothelial differentiation. In fact, it has been shown that NO inhibits the proliferation of MAPCs when treated with high concentrations of different NO donors. In addition, NO promotes the endothelial differentiation of MAPCs through a cGMP-independent-mechanism[5].
A protocol for inducing the differentiation of ESCs into definitive endoderm has been developed in our laboratory. Exposure of mESC to 0.5 mmol/L DETA-NO induces early differentiation events with the acquisition of an epithelial morphology and expression of definitive endoderm markers, such as FoxA2, Gata4, Hfn1-β and Sox 17. This phenotype was increased when cells were also treated with valproic acid for 10 d[9].
CONTROL OF APOPTOSIS AND SURVIVAL BY NO
Many reports have indicated that NO down-regulates pro-apoptotic response in mESCs and up-regulates anti-apoptotic responses by regulating Bcl2 family proteins[64]. mESCs over-expressing Bcl2 are characterised by long-term maintenance of an undifferentiated state and pluripotency. In addition, these cells maintain their potential to differentiate into mature cell types[65]. This role of NO depends on its local concentration. At low non-toxic levels, NO induces resistance to tumour necrosis factor-α (TNFα)-induced hepatotoxicity[66], inhibits Fas-induced apoptosis in B lymphocytes[67], and modulates CD95-induced apoptosis in T-lymphocytes[68]. Moreover, in carcinogenesis, low NO levels can promote the development and growth of tumour cells, while high levels may be toxic to them[2]. It has been proved that low concentrations of NO donors can protect murine bone marrow stromal cells against spontaneous apoptosis[32].
We have reported that NO generation by NOS3 overexpression and low levels of DETA-NO contribute to the survival of pancreatic beta cells through the activation of IGF-1 and insulin-induced survival pathways[23]. Our group has also reported that low level exposure (2-20 μmol/L) to DETA-NO protects ESCs from apoptosis. These NO actions involve: (1) decreased caspase-3 activity, combined with the degradation of Poly (ADP-ribose) polymerase; (2) decreased expression of pro-apoptotic genes, such as Casp7, Casp9, Bax and Bak1; and (3) increased expression of the anti-apoptotic genes, Bcl-2 and Birc6 (Figure 1). Similarly, constitutive NOS3 over-expression in cells deprived of LIF protects them against apoptosis[8].
On the other hand, several studies show that NO induces apoptosis in different cell types, including pancreatic beta cells[69,70], thymocytes[71], hepatocytes[72], among others[73,74]. It has been reported in ESCs that the nitrosative stress induced by exposure to high levels of DETA-NO, induces apoptotic events in part of the ESC population. Other parts of the ESC population are resistant to nitrosative stress and express the cytoprotective genes, heme-oxigenase-1 and Hsp70. Moreover, the resistant cells enter into a differentiation program (Figure 1)[9].
NITRIC OXIDE, HYPOXIA AND STEM CELL METABOLISM
As we have seen in the previous sections, there is very clear evidence indicating that NO can regulate cell pluripotency and differentiation, but, as mentioned at the beginning of this review, the molecular mechanisms behind these functions are still unclear[8]. The metabolic profile of pluripotent stem cells (PSCs) undergoes substantial changes during differentiation. Indeed, there is a shift from glycolytic to oxidative metabolism[75]. On the other hand, cellular response to hypoxia is also involved in the control of pluripotency. Hypoxia affects many physiological processes during the early stages of mammalian ontogeny, as it is a key feature of this stem cell niche. Moreover, it has been reported that low oxygen tension and low NO concentrations prevent the differentiation of hESC colonies and are required to maintain the majority of cells within a colony in a fully pluripotent state[8,76]. Therefore, in this review we will address the relationship between NO and the regulation of cellular respiration, as well as how they combine to effect cellular responses to hypoxia, energy metabolism and pluripotency.
PSC metabolism
PSCs have a short G1 cell cycle phase, which acts to limit the growth and differentiation potential of PSCs[77]. To facilitate rapid cell duplication, PSCs must balance energetic and biosynthetic demands, which is similar to what happens in cancer cells. Several studies have shown that mESCs, hESCs and iPSCs have an elevated dependence on glycolysis under aerobic conditions compared to differentiated cell types[78-81]. In cancer cells and human PSCs, a high glycolytic flux provides sufficient ATP and anabolic precursors for rapid proliferation, with the pentose-phosphate pathway generating ribose-5-phosphate for nucleotides and NADPH-reducing power for nucleotide and lipid biosynthesis[82,83]. Thus, aerobic glycolysis is a common feature of PSC and cancer cell metabolism in culture[84,85].
During PSC differentiation, energy production is mainly obtained from oxidative phosphorylation. Studies on mitochondrial morphology and mitochondrial DNA have shown that ESCs contain fewer mitochondria, in a less mature state than in differentiated cells[83]. The maximum human PSC respiration capacity is limited and it has been suggested that low expression of electron transport chain complex IV cytochrome c oxidase subunits, which donate electrons to oxygen, may be the cause of this decreased level of respiratory activity[86].
Efforts are being made to understand the molecular mechanisms that regulate energy metabolism in PSCs and the changes that occur during differentiation or reprogramming events. Glycolysis-regulating enzymes, including hexokinase and lactate dehydrogenase, are highly expressed in PSCs and maybe controlled by the mammalian target of rapamycin and phosphoinositide 3-kinase (PI3K) signalling pathways, which regulate glycolytic genes in other cell contexts. Glycolytic and Mitochondrial Oxidative Phosphorylation (OXPHOS) pathway gene expression and DNA methylation patterns also change during pluripotency reprogramming[80]. A number of possible mechanisms for the low levels of respiration and ATP production in PSCs have been reported, including limited pyruvate access to mitochondria due to an inactive pyruvate dehydrogenase (PDH) complex and the expression of uncoupling protein 2 (UCP2)[78,81]. The same mechanisms have also been found in cancer cells. The PDH-mediated conversion of pyruvate to acetyl-CoA and its entry into mitochondria is blocked in hypoxic cancer cells by hypoxia inducible factor-1α (HIF1α), which induces pyruvate dehydrogenase kinase 1 (PDK1) expression and inactivates PDH phosphorylation[87]. Also, several types of cancer overexpress UCP2[87]. In addition to these examples, other mechanisms have also been proposed for the hypoxia response of PSCs but all with the same end result, i.e., reduced energy production by OXPHOS and increased flux through the PSC glycolytic pathway.
HIF regulates pluripotency
PSCs have distinct metabolic requirements and cell reprogramming requires a shift from oxidative to glycolytic metabolism[78-81]. However, it is still unknown how stem cells activate mitochondrial oxidative phosphorylation pathways during differentiation.
Human iPSCs are usually reprogrammed from somatic cells and are metabolically similar to hESCs[78,80]. Therefore, a metabolic switch from an oxidative to a highly glycolytic state ought to take place during iPSC formation. The dependency of stem cells on glycolysis to produce ATP could be an adaptation to low oxygen tensions in vivo, given that hypoxia appears to be a key feature of “stemness”[88,89]. Supporting this idea, low oxygen levels have been shown to be beneficial for hESCs, adult stem cells[76,90-92] and cancer cells[93,94].
HIF-1α and HIF-2α have essential roles during development. Increasing evidence suggests that HIFs can activate factors involved in pluripotency and regulate stem-cell properties in both cancerous and normal cells. For example, it has been suggested that HIF-2α activates one of the core pluripotency master genes such as Oct4[76,93-98]. This and other evidence indicate that HIF is a regulator of stem cell properties[88,99-102]. Because of this, there is now a high degree of interest in understanding the hypoxia mechanism involved in cell reprogramming and the maintenance of pluripotent states.
NO regulates mitochondrial respiration and modulates metabolic changes
It has been reported that NO interacts with oxygen bound to CcO, the last enzyme of the electron transport chain[15]. CcO is located on the inner mitochondrial membrane and contains two heme and two copper centres. The cytochrome’s heme iron and copper molecules (in their reduced form) constitute the binding site for oxygen as it catalyses the reduction of oxygen to water. This process is related to the transport of protons into the mitochondrial inter-membrane space. In this last step, water is generated from oxygen and protons to drive the ATPase[103]. During normoxia the enzyme is predominantly in an oxidised state, while during hypoxia the reduced state predominates. CcO has a greater affinity for NO than for oxygen, suggesting that this interaction may be significant under certain physiological conditions. If true, it would make NO a physiological regulator that acts directly on the mitochondrial respiratory chain[14,15,104]. This reaction and its biological consequences are dependent on the redox state and turnover of CcO, the in situ oxygen concentration and the activity of NO synthase[105].
At high oxygen and NO concentrations, CcO is in its oxidised state and NO is metabolised into nitrite and nitrate; whereas at lower oxygen concentrations, CcO is mainly reduced and NO is not metabolised. Thus, under lower oxygen concentrations, NO accumulates in the intracellular microenvironment, with important implications for the balance between glycolysis and OXPHOS. Interaction between NO and CcO changes the availability of intracellular oxygen and the generation of reactive oxygen species (ROS). This results in changes to cell signalling pathways, especially oxygen-sensitive ones, which determine the nature of cellular hypoxia responses[106].
At low oxygen concentrations mitochondrial ATP generation decreases due to reduced respiratory activity. This results in increased levels of its precursor, AMP, and activation of AMP-activated kinase (AMPK), which is a critical regulator of cellular energy homeostasis and crucial for adapting to low oxygen (hypoxic) conditions[107]. The activation of AMPK promotes catabolic pathways, including glucose transport, gluconeogenesis, respiration, and the use of oxygen-independent energy sources. Moreover, it acts to down-regulate anabolic pathways.
NO inhibits mitochondrial respiration and increases glycolysis, thus maintaining normal cellular ATP levels (Figure 2). It has been reported that astrocyte energy production is maintained following NO-mediated inhibition of CcO by up-regulating glycolysis[19]. After the NO-induced inhibition of respiration, there is a cGMP-independent increase in the activity of 6-phosphofructo-1-kinase, a master regulator of glycolysis[108], and an increase in the concentration of its most powerful positive allosteric activator[109], fructose-2,6-bisphosphate (F2,6P2). Also, it has been shown that NO-induced glycolysis activation is dependent on the phosphorylation of AMPK resulting in increased 6-phosphofructo-2-kinase activity and protection against apoptosis[19]. On the other hand, it has also been described that NO regulates HIF-1α[18]. This activity of NO is an important additional mechanism by which NO might modulate cellular responses to hypoxia in mammalian cells (Figure 2).
Figure 2.
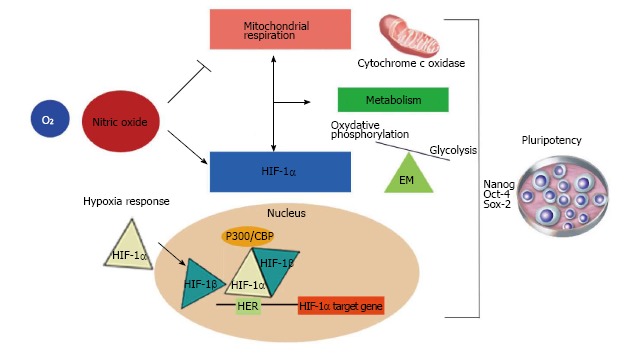
The role of nitric oxide as a regulator of mitochondrial function, metabolism and the hypoxia response. The wide range of functions controlled by nitric oxide (NO) is a consequence of its dose-dependent dual roles. Physiological concentrations of NO inhibit cytochrome c oxidase in a reversible manner in competition with O2. This interaction depends on NO and oxygen concentrations and the redox state of the enzyme. Because of this, it has been reported that NO might be a regulator of cell respiration, modulating metabolic changes responsible for an increase in glycolytic pathway flux to maintain energy homeostasis. On the other hand, NO can also regulate the hypoxia response through hypoxia inducible factor-1α (HIF-1α). In a normoxic microenvironment, NO induces HIF-1α accumulation in a mitochondrial-dependent and -independent manner. Hypoxia conditions promote delayed cell differentiation. These facts indicate that NO might maintain pluripotency via regulation of metabolism and the hypoxia response. EM: Energy metabolism.
NO and the hypoxia response
Most cells respond to exposure to low oxygen concentrations through the activation of hypoxia-responsive genes such as erythropoietin, NOS2 and glycolytic enzymes. This adaptive response to hypoxia is mediated by HIF-1α, which regulates the expression of a large number of genes that control cellular responses to reduced oxygen availability[110].
HIF is composed of two subunits, the constitutively-expressed beta-subunit (HIF-1β), and the alpha subunit (HIF-1α), whose expression is highly sensitive to oxygen concentration and accumulates rapidly under hypoxic conditions[111,112]. HIF-1α protein degradation is tightly regulated by oxygen levels through two mechanisms, both involving HIF-1α hydroxylation. The first consists of the prolyl hydroxylation of two HIF-1α proline residues, targeting the protein for E3-ubiquitin-ligase mediated ubiquitination. This complex contains the von-hippel-lindau tumour suppressor protein (VHL), which binds to hydroxylated HIF-1α. The second mechanism is mediated by asparagine hydroxylation of HIF-1α residues located in aprotein domain that is involved in interaction with the p-300 transcription activator. The result of this hydroxylation is the inhibition of target genes. Under hypoxic conditions, the hydroxylation of HIF-1α does not occur. This allows HIF-1α to dimerise with HIF-1β and translocate to the nucleus. Once in the nucleus, the dimer recruits the p300 and CBP co-activators, which induces the expression of its transcriptional targets, via binding to hypoxia-responsive elements (HERs), located in the promoter region of target genes[113,114].
Mitochondria play an important role in sensing cellular oxygen concentrations. Studies using pharmacological inhibitors have shown that the hypoxic regulation of HIF-1α is dependent on mitochondrial function through inhibition of the respiratory chain[115,116]. As NO inhibits CcO, it is possible that NO modulates the hypoxia response through HIF-1α.
NO regulates the accumulation of HIF-1α
Numerous studies have shown that NO affects HIF-1α accumulation. However, there is some controversy as to whether it promotes HIF-1α stabilisation[117-119] or HIF-1α destabilisation[120-122]. Moncada et al[6] demonstrated that NO-induced HIF-1α regulation occurs via both mitochondria-dependent and -independent pathways (Figure 2). This work was performed using the HEK-293 cell culture model where NO was generated inside cells in a finely controlled manner using a tetracycline-regulated inducible NOS2 expression system. They demonstrated that NO concentrations of 400 nmol/L prevented the accumulation of HIF-1α under hypoxic conditions, in a mitochondria-dependent manner. This study, also showed that intracellular high NO levels (> 1 µmol/L) always resulted in HIF-1α stabilization, under both hypoxic and non-hypoxic conditions. They found that activation of NOS2 sufficient to yield approximately 1 μmol/L NO, at elevated oxygen concentrations, resulted in significant HIF-1α accumulation, even when there was sufficient oxygen for prolyl hydroxylation-mediated HIF-1α degradation. This observation and those described by other authors[117-119,123], strongly suggest that HIF-1α accumulation by NO is independent of oxygen concentration. Moreover, this activity was shown to be independent of any action on the respiratory chain, since it was not affected by compounds that inhibit mitochondrial respiration at different stages and also occurred in cells lacking a functional respiratory chain. In this case, it is possible that the stabilisation of HIF-1α was the result of S-nitrosylation of thiol groups of the HIF-1α protein[119,123] or by the direct inactivation of prolyl hydroxylases (PHD) by NO as suggested by Metzen et al[124].
NO inhibits HIF-1α accumulation in a concentration-dependent manner in cells subject to a low oxygen concentration (3%O2) which would normally allow HIF-1α levels to increase[18]. Under hypoxia, endogenous NO (400 nmol/L) destabilises HIF-1α by inhibiting mitochondrial respiration, thus increasing the cytosolic oxygen concentration where PHD2 is mainly located[125,126].
Unlike the destabilising effect of NO on HIF-1α, its effect promoting HIF-1α accumulation was correlated with inhibition of cell respiration and could be mimicked by all mitochondrial respiration inhibitors, indicating that it is dependent on the respiratory chain. However, under normoxia (21%O2) and 1 mmol/L of DETA-NO, HIF-1α accumulates in a way that mimics the stabilising effect observed at high NO concentrations under hypoxia. Interestingly, this explains why, although all other inhibitors completely destabilise HIF-1α, NO does not, since its stabilising effect is apparent even at intermediate NO concentrations, something not seen for the other inhibitors. It has been proposed that the destabilising effect of NO on HIF-1α may be due to the inhibition of Complex I-dependent generation of super-oxide anion (O2•-), and consequently, peroxynitrite (ONOO−)[122]. In conclusion, at low concentrations, NO has the ability to destabilise HIF-1α under hypoxia, an effect that is mitochondria-dependent, while at high concentrations, it can stabilise HIF-1α in a mitochondria-independent manner. It is also possible that the effect of low NO concentrations is part of its physiological regulatory mechanism related to the inhibition of CcO. The stabilisation of HIF-1α by high NO concentrations and its synergy with the hypoxic stabilisation of HIF-1α may play a role in pathological conditions such as inflammation, degeneration and cancer, in which high concentrations of NO, hypoxia and HIF-1α stabilisation have been described[18,125,126].
There are various potential mechanisms by which NO regulates HIF-1α activity. First, NO donors, as well as other growth factors[127,128], stimulate HIF-1α translation by activating the PI3K/Akt pathway[129]. Second, the NO donors S-nitrosoglutathione (GSNO) and S-nitroso-N-acetylpenicillamine (SNAP) increase HIF-1α accumulation by inhibiting the interaction between VHL and hydroxylated HIF-1α, without blocking PHD2-mediated hydroxylation[124,130,131]. Third, biotin switch assays have reveal that endogenous and exogenous NO produce the S-nitrosylation of the HIF-1α trans-activation domain (amino acids 727-826)[132,133] and the oxygen-dependent degradation domain[123,134]. Fourth, direct inhibition of HIF-1α asparagine hydroxylation activity by SNAP permits interaction between HIF-1α and p300/CBP[130]. Fifth, the dependence of VHL recruitment on proline hydroxylation[124] is controversial, as some authors reported that GSNO and SNAP inhibit it, while others have shown that SNP, PAPA NONOate, and MAHMA NONOate increase it[135]. However, using a specific antibody, Li et al[134] were able to detect proline-hydroxylated HIF-1α. It was also reported that mouse HIF-1α Cys533 is nitrosylated and that this S-nitrosylation does not inhibit proline-hydroxylation[134].
Park et al[130] concluded that SNAP blocks VHL recruitment but not HIF-1α proline hydroxylation. This inhibitory effect is reversed by reducing agents such as vitamin C and Fe (II)[130]. They proposed that NO increases p300/CBP recruitment, not by HIF-1α Cys800 nitrosylation, but by inhibiting HIF-1α via asparagine hydroxylases. They also suggested that SNAP inhibited this enzyme by oxidation of the Fe (II) in its catalytic core or by nitrosylation of some cysteine residues[136].
CONCLUSION
In this review we have studied in detail the actions of NO on specific cell signalling pathways involved in embryonic development, placing emphasis on the regulation of the hypoxia response. We have extensively analysed the dual role of NO. Low NO concentrations delay stem cell differentiation, inducing the expression of Nanog, Oct4 and Sox2, and the activation of survival pathways. At high concentrations, NO induces differentiation events with the expression of early differentiation markers as well as promoting apoptosis.
Due to these activities, NO represents a promising tool for culturing pluripotent cell lines in an undifferentiated state, as it has been proved that it consistently promotes the maintenance of pluripotency and survival. Thus, NO could enhance the pluripotency-promoting activities of factors such as LIF or bFGF. Moreover, studies based in the generation of chemically-defined xeno-free media using NO to maintain pluripotency are being carried out by our group, which would not only reduce costs, but also would avoid animal contamination issues.
Stem cell culture under hypoxic conditions helps prevent spontaneous differentiation. This is relevant for the design of better strategies for directing differentiation towards specific cell types. The generation of hypoxic cell culture microenvironments can be costly, such as maintaining bioreactors with hypoxic atmospheres. Knowing how NO regulates cellular respiration opens up the possibility of using exposure to low NO levels to mimic the hypoxia response under aerobic conditions by inducing HIF-1α. Activation of this master hypoxia response regulator would up-regulate glycolytic genes and help maintain energy homeostasis.
On the other hand, high NO levels can be used to develop differentiation protocols for differentiating pluripotent cells into the three germinal lines (ectoderm, mesoderm and endoderm). In this context, NO has been used to differentiate cells to cardiomyocytes, vascular and endothelial tissues, neurons, astrocytes, erythroid and myeloid cells, bone, chondrocytes and definitive endoderm. To sum up, the studies reviewed here underline the diverse range of potential applications for NO in stem cell biotechnology.
Footnotes
P- Reviewer: Il Kim T, Saeki K S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Supported by Grants from Consejería de Igualdad, Salud y Politicas Sociales, Junta de Andalucía, No. PI105/2010; and Consejería de Economía, Innovación, Ciencia y Empleo, Junta de Andalucía, No. CTS-7127/2011 (to Bedoya FJ); Consejería de Igualdad, Salud y Políticas Sociales, Junta de Andalucía, ISCIII co-funded by Fondos FEDER (RED TERCEL), No. RD06/0010/0025, RD12/0019/0028 and PI10/00964; Consejería de Economía, Innovación, Ciencia y Empleo, No. P10.CTS.6505; and the Ministry of Health and Consumer Affairs (Advanced Therapies Program Grant TRA-120) (to Soria B); Consejería de Igualdad, Salud y Políticas Sociales, No. PI0022/2008; and Consejería de Economía, Innovación, Ciencia y Empleo, Junta de Andalucía (PAI, BIO311) (to Martín F); Servicio Andaluz de Salud (SAS 11245) and Ministerio de Economía y Competitividad-Secretaría de Estado de Investigación Desarrollo e Innovación, No. IPT-2011-1615-900000 (to Tejedo JR).
Conflict-of-interest: Gladys M Cahuana and all co-authors declare that they have no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 30, 2014
First decision: September 18, 2014
Article in press: December 17, 2014
References
- 1.Marletta MA. Nitric oxide synthase: aspects concerning structure and catalysis. Cell. 1994;78:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 2.Lee SK, Kim HS, Lee HJ, Lee J, Jeon BH, Jun CD, Lee SK, Kim EC. Dual effect of nitric oxide in immortalized and malignant human oral keratinocytes: induction of apoptosis and differentiation. J Oral Pathol Med. 2006;35:352–360. doi: 10.1111/j.1600-0714.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- 3.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 4.Kots AY, Bian K, Murad F. Nitric oxide and cyclic GMP signaling pathway as a focus for drug development. Curr Med Chem. 2011;18:3299–3305. doi: 10.2174/092986711796504646. [DOI] [PubMed] [Google Scholar]
- 5.Chu L, Jiang Y, Hao H, Xia Y, Xu J, Liu Z, Verfaillie CM, Zweier JL, Liu Z. Nitric oxide enhances Oct-4 expression in bone marrow stem cells and promotes endothelial differentiation. Eur J Pharmacol. 2008;591:59–65. doi: 10.1016/j.ejphar.2008.06.066. [DOI] [PubMed] [Google Scholar]
- 6.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 7.Enikolopov G, Banerji J, Kuzin B. Nitric oxide and Drosophila development. Cell Death Differ. 1999;6:956–963. doi: 10.1038/sj.cdd.4400577. [DOI] [PubMed] [Google Scholar]
- 8.Tejedo JR, Tapia-Limonchi R, Mora-Castilla S, Cahuana GM, Hmadcha A, Martin F, Bedoya FJ, Soria B. Low concentrations of nitric oxide delay the differentiation of embryonic stem cells and promote their survival. Cell Death Dis. 2010;1:e80. doi: 10.1038/cddis.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mora-Castilla S, Tejedo JR, Hmadcha A, Cahuana GM, Martín F, Soria B, Bedoya FJ. Nitric oxide repression of Nanog promotes mouse embryonic stem cell differentiation. Cell Death Differ. 2010;17:1025–1033. doi: 10.1038/cdd.2009.204. [DOI] [PubMed] [Google Scholar]
- 10.Davis KL, Martin E, Turko IV, Murad F. Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol. 2001;41:203–236. doi: 10.1146/annurev.pharmtox.41.1.203. [DOI] [PubMed] [Google Scholar]
- 11.Krumenacker JS, Hanafy KA, Murad F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain Res Bull. 2004;62:505–515. doi: 10.1016/S0361-9230(03)00102-3. [DOI] [PubMed] [Google Scholar]
- 12.Kojda G, Laursen JB, Ramasamy S, Kent JD, Kurz S, Burchfield J, Shesely EG, Harrison DG. Protein expression, vascular reactivity and soluble guanylate cyclase activity in mice lacking the endothelial cell nitric oxide synthase: contributions of NOS isoforms to blood pressure and heart rate control. Cardiovasc Res. 1999;42:206–213. doi: 10.1016/s0008-6363(98)00315-0. [DOI] [PubMed] [Google Scholar]
- 13.Bloch W, Fleischmann BK, Lorke DE, Andressen C, Hops B, Hescheler J, Addicks K. Nitric oxide synthase expression and role during cardiomyogenesis. Cardiovasc Res. 1999;43:675–684. doi: 10.1016/s0008-6363(99)00160-1. [DOI] [PubMed] [Google Scholar]
- 14.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 15.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 16.Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 17.Foster MW, Stamler JS. New insights into protein S-nitrosylation. Mitochondria as a model system. J Biol Chem. 2004;279:25891–25897. doi: 10.1074/jbc.M313853200. [DOI] [PubMed] [Google Scholar]
- 18.Mateo J, García-Lecea M, Cadenas S, Hernández C, Moncada S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem J. 2003;376:537–544. doi: 10.1042/BJ20031155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- 20.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, et al. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, Cabrales P, Fago A, Feelisch M, Ford PC, et al. Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol. 2009;5:865–869. doi: 10.1038/nchembio.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cahuana GM, Tejedo JR, Hmadcha A, Ramírez R, Cuesta AL, Soria B, Martin F, Bedoya FJ. Nitric oxide mediates the survival action of IGF-1 and insulin in pancreatic beta cells. Cell Signal. 2008;20:301–310. doi: 10.1016/j.cellsig.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Krumenacker JS, Murad F. NO-cGMP signaling in development and stem cells. Mol Genet Metab. 2006;87:311–314. doi: 10.1016/j.ymgme.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Krumenacker JS, Katsuki S, Kots A, Murad F. Differential expression of genes involved in cGMP-dependent nitric oxide signaling in murine embryonic stem (ES) cells and ES cell-derived cardiomyocytes. Nitric Oxide. 2006;14:1–11. doi: 10.1016/j.niox.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Mujoo K, Krumenacker JS, Murad F. Nitric oxide-cyclic GMP signaling in stem cell differentiation. Free Radic Biol Med. 2011;51:2150–2157. doi: 10.1016/j.freeradbiomed.2011.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 28.Murad F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006;355:2003–2011. doi: 10.1056/NEJMsa063904. [DOI] [PubMed] [Google Scholar]
- 29.Tsutsui M, Shimokawa H, Morishita T, Nakashima Y, Yanagihara N. Development of genetically engineered mice lacking all three nitric oxide synthases. J Pharmacol Sci. 2006;102:147–154. doi: 10.1254/jphs.cpj06015x. [DOI] [PubMed] [Google Scholar]
- 30.Mujoo K, Krumenacker JS, Wada Y, Murad F. Differential expression of nitric oxide signaling components in undifferentiated and differentiated human embryonic stem cells. Stem Cells Dev. 2006;15:779–787. doi: 10.1089/scd.2006.15.779. [DOI] [PubMed] [Google Scholar]
- 31.Napoli C, Paolisso G, Casamassimi A, Al-Omran M, Barbieri M, Sommese L, Infante T, Ignarro LJ. Effects of nitric oxide on cell proliferation: novel insights. J Am Coll Cardiol. 2013;62:89–95. doi: 10.1016/j.jacc.2013.03.070. [DOI] [PubMed] [Google Scholar]
- 32.Wong JC, Fiscus RR. Essential roles of the nitric oxide (no)/cGMP/protein kinase G type-Iα (PKG-Iα) signaling pathway and the atrial natriuretic peptide (ANP)/cGMP/PKG-Iα autocrine loop in promoting proliferation and cell survival of OP9 bone marrow stromal cells. J Cell Biochem. 2011;112:829–839. doi: 10.1002/jcb.22981. [DOI] [PubMed] [Google Scholar]
- 33.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 34.Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci USA. 2004;101:8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 36.Hajri A, Metzger E, Vallat F, Coffy S, Flatter E, Evrard S, Marescaux J, Aprahamian M. Role of nitric oxide in pancreatic tumour growth: in vivo and in vitro studies. Br J Cancer. 1998;78:841–849. doi: 10.1038/bjc.1998.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Bombeck CA, Yang S, Kim YM, Billiar TR. Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J Biol Chem. 1999;274:17325–17333. doi: 10.1074/jbc.274.24.17325. [DOI] [PubMed] [Google Scholar]
- 39.Krischel V, Bruch-Gerharz D, Suschek C, Kröncke KD, Ruzicka T, Kolb-Bachofen V. Biphasic effect of exogenous nitric oxide on proliferation and differentiation in skin derived keratinocytes but not fibroblasts. J Invest Dermatol. 1998;111:286–291. doi: 10.1046/j.1523-1747.1998.00268.x. [DOI] [PubMed] [Google Scholar]
- 40.Leri A, Kajstura J, Anversa P. Role of cardiac stem cells in cardiac pathophysiology: a paradigm shift in human myocardial biology. Circ Res. 2011;109:941–961. doi: 10.1161/CIRCRESAHA.111.243154. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Napoli C, Hayashi T, Cacciatore F, Casamassimi A, Casini C, Al-Omran M, Ignarro LJ. Endothelial progenitor cells as therapeutic agents in the microcirculation: an update. Atherosclerosis. 2011;215:9–22. doi: 10.1016/j.atherosclerosis.2010.10.039. [DOI] [PubMed] [Google Scholar]
- 42.Lepic E, Burger D, Lu X, Song W, Feng Q. Lack of endothelial nitric oxide synthase decreases cardiomyocyte proliferation and delays cardiac maturation. Am J Physiol Cell Physiol. 2006;291:C1240–C1246. doi: 10.1152/ajpcell.00092.2006. [DOI] [PubMed] [Google Scholar]
- 43.Ybarra N, del Castillo JR, Troncy E. Involvement of the nitric oxide-soluble guanylyl cyclase pathway in the oxytocin-mediated differentiation of porcine bone marrow stem cells into cardiomyocytes. Nitric Oxide. 2011;24:25–33. doi: 10.1016/j.niox.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 44.Matarredona ER, Murillo-Carretero M, Moreno-López B, Estrada C. Nitric oxide synthesis inhibition increases proliferation of neural precursors isolated from the postnatal mouse subventricular zone. Brain Res. 2004;995:274–284. doi: 10.1016/j.brainres.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 45.Kanno S, Kim PK, Sallam K, Lei J, Billiar TR, Shears LL. Nitric oxide facilitates cardiomyogenesis in mouse embryonic stem cells. Proc Natl Acad Sci USA. 2004;101:12277–12281. doi: 10.1073/pnas.0401557101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gassanov N, Jankowski M, Danalache B, Wang D, Grygorczyk R, Hoppe UC, Gutkowska J. Arginine vasopressin-mediated cardiac differentiation: insights into the role of its receptors and nitric oxide signaling. J Biol Chem. 2007;282:11255–11265. doi: 10.1074/jbc.M610769200. [DOI] [PubMed] [Google Scholar]
- 47.Mujoo K, Nikonoff LE, Sharin VG, Bryan NS, Kots AY, Murad F. Curcumin induces differentiation of embryonic stem cells through possible modulation of nitric oxide-cyclic GMP pathway. Protein Cell. 2012;3:535–544. doi: 10.1007/s13238-012-2053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartsch C, Bekhite MM, Wolheim A, Richter M, Ruhe C, Wissuwa B, Marciniak A, Müller J, Heller R, Figulla HR, et al. NADPH oxidase and eNOS control cardiomyogenesis in mouse embryonic stem cells on ascorbic acid treatment. Free Radic Biol Med. 2011;51:432–443. doi: 10.1016/j.freeradbiomed.2011.04.029. [DOI] [PubMed] [Google Scholar]
- 49.Danalache BA, Paquin J, Donghao W, Grygorczyk R, Moore JC, Mummery CL, Gutkowska J, Jankowski M. Nitric oxide signaling in oxytocin-mediated cardiomyogenesis. Stem Cells. 2007;25:679–688. doi: 10.1634/stemcells.2005-0610. [DOI] [PubMed] [Google Scholar]
- 50.Dulak J, Józkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, Florek I, Wójtowicz A, Szuba A, Cooke JP. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:659–666. doi: 10.1161/01.atv.20.3.659. [DOI] [PubMed] [Google Scholar]
- 51.Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, Geppetti P, Ledda F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest. 1994;94:2036–2044. doi: 10.1172/JCI117557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang NF, Fleissner F, Sun J, Cooke JP. Role of nitric oxide signaling in endothelial differentiation of embryonic stem cells. Stem Cells Dev. 2010;19:1617–1626. doi: 10.1089/scd.2009.0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malan D, Wenzel D, Schmidt A, Geisen C, Raible A, Bölck B, Fleischmann BK, Bloch W. Endothelial beta1 integrins regulate sprouting and network formation during vascular development. Development. 2010;137:993–1002. doi: 10.1242/dev.045377. [DOI] [PubMed] [Google Scholar]
- 54.McCloskey KE, Smith DA, Jo H, Nerem RM. Embryonic stem cell-derived endothelial cells may lack complete functional maturation in vitro. J Vasc Res. 2006;43:411–421. doi: 10.1159/000094791. [DOI] [PubMed] [Google Scholar]
- 55.Tao Li J, Somasundaram C, Bian K, Xiong W, Mahmooduddin F, Nath RK, Murad F. Nitric oxide signaling and neural stem cell differentiation in peripheral nerve regeneration. Eplasty. 2010;10:e42. [PMC free article] [PubMed] [Google Scholar]
- 56.Arnhold S, Fassbender A, Klinz FJ, Kruttwig K, Löhnig B, Andressen C, Addicks K. NOS-II is involved in early differentiation of murine cortical, retinal and ES cell-derived neurons-an immunocytochemical and functional approach. Int J Dev Neurosci. 2002;20:83–92. doi: 10.1016/s0736-5748(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 57.Bergsland M, Covacu R, Perez Estrada C, Svensson M, Brundin L. Nitric oxide-induced neuronal to glial lineage fate-change depends on NRSF/REST function in neural progenitor cells. Stem Cells. 2014;32:2539–2549. doi: 10.1002/stem.1749. [DOI] [PubMed] [Google Scholar]
- 58.Shami PJ, Weinberg JB. Differential effects of nitric oxide on erythroid and myeloid colony growth from CD34+ human bone marrow cells. Blood. 1996;87:977–982. [PubMed] [Google Scholar]
- 59.Teixeira CC, Ischiropoulos H, Leboy PS, Adams SL, Shapiro IM. Nitric oxide-nitric oxide synthase regulates key maturational events during chondrocyte terminal differentiation. Bone. 2005;37:37–45. doi: 10.1016/j.bone.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 60.Yan H, Aziz E, Shillabeer G, Wong A, Shanghavi D, Kermouni A, Abdel-Hafez M, Lau DC. Nitric oxide promotes differentiation of rat white preadipocytes in culture. J Lipid Res. 2002;43:2123–2129. doi: 10.1194/jlr.m200305-jlr200. [DOI] [PubMed] [Google Scholar]
- 61.Pan W, Quarles LD, Song LH, Yu YH, Jiao C, Tang HB, Jiang CH, Deng HW, Li YJ, Zhou HH, et al. Genistein stimulates the osteoblastic differentiation via NO/cGMP in bone marrow culture. J Cell Biochem. 2005;94:307–316. doi: 10.1002/jcb.20308. [DOI] [PubMed] [Google Scholar]
- 62.Liu Z, Jiang Y, Hao H, Gupta K, Xu J, Chu L, McFalls E, Zweier J, Verfaillie C, Bache RJ. Endothelial nitric oxide synthase is dynamically expressed during bone marrow stem cell differentiation into endothelial cells. Am J Physiol Heart Circ Physiol. 2007;293:H1760–H1765. doi: 10.1152/ajpheart.01408.2006. [DOI] [PubMed] [Google Scholar]
- 63.Ulloa-Montoya F, Kidder BL, Pauwelyn KA, Chase LG, Luttun A, Crabbe A, Geraerts M, Sharov AA, Piao Y, Ko MS, et al. Comparative transcriptome analysis of embryonic and adult stem cells with extended and limited differentiation capacity. Genome Biol. 2007;8:R163. doi: 10.1186/gb-2007-8-8-r163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun. 2001;282:1075–1079. doi: 10.1006/bbrc.2001.4670. [DOI] [PubMed] [Google Scholar]
- 65.Yamane T, Dylla SJ, Muijtjens M, Weissman IL. Enforced Bcl-2 expression overrides serum and feeder cell requirements for mouse embryonic stem cell self-renewal. Proc Natl Acad Sci USA. 2005;102:3312–3317. doi: 10.1073/pnas.0500167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim YM, de Vera ME, Watkins SC, Billiar TR. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-alpha-induced apoptosis by inducing heat shock protein 70 expression. J Biol Chem. 1997;272:1402–1411. doi: 10.1074/jbc.272.2.1402. [DOI] [PubMed] [Google Scholar]
- 67.Mannick JB, Miao XQ, Stamler JS. Nitric oxide inhibits Fas-induced apoptosis. J Biol Chem. 1997;272:24125–24128. doi: 10.1074/jbc.272.39.24125. [DOI] [PubMed] [Google Scholar]
- 68.Sciorati C, Rovere P, Ferrarini M, Heltai S, Manfredi AA, Clementi E. Autocrine nitric oxide modulates CD95-induced apoptosis in gammadelta T lymphocytes. J Biol Chem. 1997;272:23211–23215. doi: 10.1074/jbc.272.37.23211. [DOI] [PubMed] [Google Scholar]
- 69.Ankarcrona M, Dypbukt JM, Brüne B, Nicotera P. Interleukin-1 beta-induced nitric oxide production activates apoptosis in pancreatic RINm5F cells. Exp Cell Res. 1994;213:172–177. doi: 10.1006/excr.1994.1187. [DOI] [PubMed] [Google Scholar]
- 70.Kaneto H, Fujii J, Seo HG, Suzuki K, Matsuoka T, Nakamura M, Tatsumi H, Yamasaki Y, Kamada T, Taniguchi N. Apoptotic cell death triggered by nitric oxide in pancreatic beta-cells. Diabetes. 1995;44:733–738. doi: 10.2337/diab.44.7.733. [DOI] [PubMed] [Google Scholar]
- 71.Fehsel K, Kröncke KD, Meyer KL, Huber H, Wahn V, Kolb-Bachofen V. Nitric oxide induces apoptosis in mouse thymocytes. J Immunol. 1995;155:2858–2865. [PubMed] [Google Scholar]
- 72.Kim YM, Bergonia H, Lancaster JR. Nitrogen oxide-induced autoprotection in isolated rat hepatocytes. FEBS Lett. 1995;374:228–232. doi: 10.1016/0014-5793(95)01115-u. [DOI] [PubMed] [Google Scholar]
- 73.Suenobu N, Shichiri M, Iwashina M, Marumo F, Hirata Y. Natriuretic peptides and nitric oxide induce endothelial apoptosis via a cGMP-dependent mechanism. Arterioscler Thromb Vasc Biol. 1999;19:140–146. doi: 10.1161/01.atv.19.1.140. [DOI] [PubMed] [Google Scholar]
- 74.Battinelli E, Loscalzo J. Nitric oxide induces apoptosis in megakaryocytic cell lines. Blood. 2000;95:3451–3459. [PubMed] [Google Scholar]
- 75.Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11:589–595. doi: 10.1016/j.stem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA. 2005;102:4783–4788. doi: 10.1073/pnas.0501283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Singh AM, Dalton S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell. 2009;5:141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA, Ramalho-Santos J, Van Houten B, Schatten G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One. 2011;6:e20914. doi: 10.1371/journal.pone.0020914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264–271. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerías A, Batchelder EM, Plongthongkum N, Lutz M, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22:168–177. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30:4860–4873. doi: 10.1038/emboj.2011.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011;14:443–451. doi: 10.1016/j.cmet.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells. 2010;28:721–733. doi: 10.1002/stem.404. [DOI] [PubMed] [Google Scholar]
- 84.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 86.Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012;31:2103–2116. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 88.Mohyeldin A, Garzón-Muvdi T, Quiñones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7:150–161. doi: 10.1016/j.stem.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 89.Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 90.Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest. 2003;112:126–135. doi: 10.1172/JCI17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci. 2000;20:7370–7376. doi: 10.1523/JNEUROSCI.20-19-07370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Takubo K, Suda T. Roles of the hypoxia response system in hematopoietic and leukemic stem cells. Int J Hematol. 2012;95:478–483. doi: 10.1007/s12185-012-1071-4. [DOI] [PubMed] [Google Scholar]
- 94.Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, Hubaud A, Stadler B, Choi M, Bar M, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–4652. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mathieu J, Zhang Z, Nelson A, Lamba DA, Reh TA, Ware C, Ruohola-Baker H. Hypoxia induces re-entry of committed cells into pluripotency. Stem Cells. 2013;31:1737–1748. doi: 10.1002/stem.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8:399–411. doi: 10.1016/j.stem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Das B, Bayat-Mokhtari R, Tsui M, Lotfi S, Tsuchida R, Felsher DW, Yeger H. HIF-2α suppresses p53 to enhance the stemness and regenerative potential of human embryonic stem cells. Stem Cells. 2012;30:1685–1695. doi: 10.1002/stem.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Babcock GT, Wikström M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 104.Ojaimi C, Li W, Kinugawa S, Post H, Csiszar A, Pacher P, Kaley G, Hintze TH. Transcriptional basis for exercise limitation in male eNOS-knockout mice with age: heart failure and the fetal phenotype. Am J Physiol Heart Circ Physiol. 2005;289:H1399–H1407. doi: 10.1152/ajpheart.00170.2005. [DOI] [PubMed] [Google Scholar]
- 105.Cooper CE, Giulivi C. Nitric oxide regulation of mitochondrial oxygen consumption II: Molecular mechanism and tissue physiology. Am J Physiol Cell Physiol. 2007;292:C1993–C2003. doi: 10.1152/ajpcell.00310.2006. [DOI] [PubMed] [Google Scholar]
- 106.Taylor CT, Moncada S. Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia. Arterioscler Thromb Vasc Biol. 2010;30:643–647. doi: 10.1161/ATVBAHA.108.181628. [DOI] [PubMed] [Google Scholar]
- 107.Gabryel B, Kost A, Kasprowska D, Liber S, Machnik G, Wiaderkiewicz R, Labuzek K. AMP-activated protein kinase is involved in induction of protective autophagy in astrocytes exposed to oxygen-glucose deprivation. Cell Biol Int. 2014;38:1086–1097. doi: 10.1002/cbin.10299. [DOI] [PubMed] [Google Scholar]
- 108.Richards CS, Uyeda K. Changes in the concentration of activation factor for phosphofructokinase in hepatocytes in response to glucose and glucagon. Biochem Biophys Res Commun. 1980;97:1535–1540. doi: 10.1016/s0006-291x(80)80040-4. [DOI] [PubMed] [Google Scholar]
- 109.Van Schaftingen E, Lederer B, Bartrons R, Hers HG. A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur J Biochem. 1982;129:191–195. doi: 10.1111/j.1432-1033.1982.tb07039.x. [DOI] [PubMed] [Google Scholar]
- 110.Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993–998. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- 111.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 112.Jewell UR, Kvietikova I, Scheid A, Bauer C, Wenger RH, Gassmann M. Induction of HIF-1alpha in response to hypoxia is instantaneous. FASEB J. 2001;15:1312–1314. [PubMed] [Google Scholar]
- 113.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 114.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 115.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2002;283:L922–L931. doi: 10.1152/ajplung.00014.2002. [DOI] [PubMed] [Google Scholar]
- 117.Sandau KB, Faus HG, Brüne B. Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI 3K pathway. Biochem Biophys Res Commun. 2000;278:263–267. doi: 10.1006/bbrc.2000.3789. [DOI] [PubMed] [Google Scholar]
- 118.Sandau KB, Fandrey J, Brüne B. Accumulation of HIF-1alpha under the influence of nitric oxide. Blood. 2001;97:1009–1015. doi: 10.1182/blood.v97.4.1009. [DOI] [PubMed] [Google Scholar]
- 119.Palmer LA, Gaston B, Johns RA. Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: redox-dependent effect of nitrogen oxides. Mol Pharmacol. 2000;58:1197–1203. doi: 10.1124/mol.58.6.1197. [DOI] [PubMed] [Google Scholar]
- 120.Sogawa K, Numayama-Tsuruta K, Ema M, Abe M, Abe H, Fujii-Kuriyama Y. Inhibition of hypoxia-inducible factor 1 activity by nitric oxide donors in hypoxia. Proc Natl Acad Sci USA. 1998;95:7368–7373. doi: 10.1073/pnas.95.13.7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang LE, Willmore WG, Gu J, Goldberg MA, Bunn HF. Inhibition of hypoxia-inducible factor 1 activation by carbon monoxide and nitric oxide. Implications for oxygen sensing and signaling. J Biol Chem. 1999;274:9038–9044. doi: 10.1074/jbc.274.13.9038. [DOI] [PubMed] [Google Scholar]
- 122.Agani FH, Puchowicz M, Chavez JC, Pichiule P, LaManna J. Role of nitric oxide in the regulation of HIF-1alpha expression during hypoxia. Am J Physiol Cell Physiol. 2002;283:C178–C186. doi: 10.1152/ajpcell.00381.2001. [DOI] [PubMed] [Google Scholar]
- 123.Sumbayev VV, Budde A, Zhou J, Brüne B. HIF-1 alpha protein as a target for S-nitrosation. FEBS Lett. 2003;535:106–112. doi: 10.1016/s0014-5793(02)03887-5. [DOI] [PubMed] [Google Scholar]
- 124.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brüne B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 126.Palacios-Callender M, Quintero M, Hollis VS, Springett RJ, Moncada S. Endogenous NO regulates superoxide production at low oxygen concentrations by modifying the redox state of cytochrome c oxidase. Proc Natl Acad Sci USA. 2004;101:7630–7635. doi: 10.1073/pnas.0401723101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Karni R, Dor Y, Keshet E, Meyuhas O, Levitzki A. Activated pp60c-Src leads to elevated hypoxia-inducible factor (HIF)-1alpha expression under normoxia. J Biol Chem. 2002;277:42919–42925. doi: 10.1074/jbc.M206141200. [DOI] [PubMed] [Google Scholar]
- 128.Lauzier MC, Pagé EL, Michaud MD, Richard DE. Differential regulation of hypoxia-inducible factor-1 through receptor tyrosine kinase transactivation in vascular smooth muscle cells. Endocrinology. 2007;148:4023–4031. doi: 10.1210/en.2007-0285. [DOI] [PubMed] [Google Scholar]
- 129.Kasuno K, Takabuchi S, Fukuda K, Kizaka-Kondoh S, Yodoi J, Adachi T, Semenza GL, Hirota K. Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J Biol Chem. 2004;279:2550–2558. doi: 10.1074/jbc.M308197200. [DOI] [PubMed] [Google Scholar]
- 130.Park YK, Ahn DR, Oh M, Lee T, Yang EG, Son M, Park H. Nitric oxide donor, (+/-)-S-nitroso-N-acetylpenicillamine, stabilizes transactive hypoxia-inducible factor-1alpha by inhibiting von Hippel-Lindau recruitment and asparagine hydroxylation. Mol Pharmacol. 2008;74:236–245. doi: 10.1124/mol.108.045278. [DOI] [PubMed] [Google Scholar]
- 131.Berchner-Pfannschmidt U, Yamac H, Trinidad B, Fandrey J. Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J Biol Chem. 2007;282:1788–1796. doi: 10.1074/jbc.M607065200. [DOI] [PubMed] [Google Scholar]
- 132.Yasinska IM, Sumbayev VV. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003;549:105–109. doi: 10.1016/s0014-5793(03)00807-x. [DOI] [PubMed] [Google Scholar]
- 133.Cho H, Ahn DR, Park H, Yang EG. Modulation of p300 binding by posttranslational modifications of the C-terminal activation domain of hypoxia-inducible factor-1alpha. FEBS Lett. 2007;581:1542–1548. doi: 10.1016/j.febslet.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 134.Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, Vujaskovic Z, Dewhirst MW, Li CY. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. 2007;26:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang F, Sekine H, Kikuchi Y, Takasaki C, Miura C, Heiwa O, Shuin T, Fujii-Kuriyama Y, Sogawa K. HIF-1alpha-prolyl hydroxylase: molecular target of nitric oxide in the hypoxic signal transduction pathway. Biochem Biophys Res Commun. 2002;295:657–662. doi: 10.1016/s0006-291x(02)00729-5. [DOI] [PubMed] [Google Scholar]
- 136.Hewitson KS, Liénard BM, McDonough MA, Clifton IJ, Butler D, Soares AS, Oldham NJ, McNeill LA, Schofield CJ. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem. 2007;282:3293–3301. doi: 10.1074/jbc.M608337200. [DOI] [PubMed] [Google Scholar]