

Abstract
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease of the motor neurons without a known cure. Based on the possibility of cellular neuroprotection and early preclinical results, stem cells have gained widespread enthusiasm as a potential treatment strategy. Preclinical models demonstrate a protective role of engrafted stem cells and provided the basis for human trials carried out using various types of stem cells, as well as a range of cell delivery methods. To date, no trial has demonstrated a clear therapeutic benefit; however, results remain encouraging and are the basis for ongoing studies. In addition, stem cell technology continues to improve, and induced pluripotent stem cells may offer additional therapeutic options in the future. Improved disease models and clinical trials will be essential in order to validate stem cells as a beneficial therapy.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-015-0339-9) contains supplementary material, which is available to authorized users.
Key words: Amyotrophic lateral sclerosis, Stem cell therapy, Cell transplantation, Neural progenitor cell, Mesenchymal stem cell, Granulocyte-colony stimulating factor, Clinical trials
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder affecting motor neurons (MNs) in the cortex, brainstem, and spinal cord that causes weakness and atrophy of skeletal muscles [1]. While traditionally considered a purely motor disease, neuronal abnormalities in the prefrontal and temporal cortex may also lead to frontal executive dysfunction, with about 15 % of patients manifesting frontotemporal dementia [2]. The worldwide incidence of ALS is 2–4 cases per 100,000 persons, although there is some ethnic variation [3]. The disease is sporadic in about 85 % of cases and is familial in about 15 % of cases [4]. The average survival is 3–5 years from symptom onset [1]. Riluzole, the only Food and Drug Administration-approved medication for ALS, has at best modest effects [5]. Owing to the relentless nature of the disease, many therapeutics have been tested; however, most have been without success [6, 7]. Thus, interest in the potential of stem cell-based therapies has been increasing considerably in recent years.
The initial proposed use of stem cells as a therapy for ALS stemmed from the possibility of MN replacement and considered several stem cell types. All stem cells possess the capacity for self-renewal and undergo asymmetric division to give rise to a daughter cell that is capable of developing a phenotype other than that of the parent cell. Embryonic stem cells are totipotent and able to generate all cell types, whereas pluripotent stem cells give rise to a particular subset of cells [8]. Neural progenitor cells (NPCs) are pluripotent stem cells that possess an ability to achieve characteristics of neurons or glia in daughter cells [8, 9]. Given the versatility of embryonic and pluripotent stem cells, an opportunity arose to harness stem cells for the generation of new MNs for a disease like ALS with selective MN loss. Early attempts at MN replacement using NPCs and embryonic stem cells, however, were fraught with difficulty [10–12]. Although NPCs can successfully recapitulate normal MN development, stem cell-derived MNs must survive in a potentially diseased microenvironment, integrate into descending and local circuits of motor control, grow projection axons that travel over a meter in some cases, and form functional neuromuscular junctions [10, 12]. Thus, present studies have redirected focus away from MN replacement to a “neighborhood theory”, where stem cells offer a local neuroprotective role to prevent the degeneration of existing MNs.
Mechanisms by which stem cells may provide neuroprotective support include the paracrine expression of neurotrophic factors, differentiation into nondiseased supporting non-neuronal cells, including astrocytes and microglia, and differentiation into modulatory neurons that synapse on diseased MNs [13]. Sources of stem cells that continue to generate interest for therapeutic potential in ALS are embryonic stem cells, NPC lines derived from fetal or adult tissues, and non-neural progenitor cells that may moderate the MN microenvironment [14]. This has effectively translated into several human therapeutic trials, which have employed the induction of peripheral blood stem cells (PBSCs) by granulocyte colony-stimulating factor (G-CSF) treatment, autologous transplantation of mesenchymal stem cells (MSCs) derived from the bone marrow, transplantation of olfactory ensheathing cells (OECs), and, most recently, transplantation of fetal-derived human spinal cord stem cells (HSSCs) and human fetal cortex-derived NPCs modified to secrete glial-derived neurotrophic factor (GDNF).
This review will briefly touch on preclinical studies (Table 1) relevant to stem cell-based paradigms that have been successfully translated to clinical trials (Table 2). While the preclinical literature is vast regarding stem cells and their application in ALS, the relative paucity of clinical trials underscores both the challenge of our current in vitro and animal models, as well as the difficulty in conducting well-designed clinical trials for this disease. Still, many novel strategies are gaining traction and significant achievements in stem cell therapy for ALS are on the horizon.
Table 1.
Summary of preclinical stem cell studies in amyotrophic lateral sclerosis
| Reference | Donor spec. | Cell type | Modification | Experimental model | Delivery method | Treatment age | Adjunct therapy | Functional improvement | Survival benefit | Histologic change | Other comments |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lopez-Gonzales (2009) [11] | Mouse | ES | MN differentiated in vitro; GFP expressing | SOD1-G93A rat | 1 × 10^5 cells, intraspinal | 10 weeks | Ciclosporin A | Delay in motor decline by Rotarod | None | n/a | Degeneration of grafted cells at endstage |
| Deshpande (2006) [15] | Mouse | ES | MN differentiated in vitro; GFP expressing | Neuroadapted Sindbis virus-induced motor neuron death, rat | 6 × 10^4 cells, intraspinal | 5–7 weeks | Ciclosporin A; dibutyryl cyclic adenosine monophosphate, GDNF and/or rolipram | Recovery of grip strength in animals receiving all supplemental treatments | n/a | n/a | n/a |
| Harper (2004) [16] | Mouse | ES | MN differentiated in vitro; GFP expressing | Neuroadapted Sindbis virus-induced motor neuron death, rat | 6 × 10^4 cells, intraspinal | 5–7 weeks | Ciclosporin A; Rho kinase inhibitor (Y27632) or dibutyryl cyclic adenosine monophosphate | n/a | n/a | n/a | Axonal growth facilitated by dibutyryl cyclic adenosine monophosphate |
| Kerr (2003) [17] | Human | ES | none | Neuroadapted Sindbis virus-induced motor neuron death, rat | 3 × 10^5 cells, intrathecal | 3–4 weeks | Ciclosporin A or FK-506 | Recovery of motor function by BBB and grip strength testing | n/a | Increased motor neuron survival | Possibly mediated via TGF-α or BDNF |
| Corti (2004) [18] | Mouse | BM | None | SOD1-G93A mouse | 3 × 10^7 cells intraperitoneal | 4 weeks | Irradiation, 800 rad | Delay in motor decline by Rotarod | 10–13 days | Increased motor neurons and ventral root axons at 100 days | A large proportion of microglial cells formed |
| Solomon (2006) [19] | Mouse | BM | GFP expressing | SOD1-G93A mouse | 5 × 10^6 cells, intravenous | 6 weeks | Irradiation, 950–1100 rads | None | None | Large proportion of transplanted cells with microglial markers integrated into spinal cord | n/a |
| Ohnishi (2009) [20] | Mouse | BM | GFP expressing | SOD1-G93A mouse | 6 × 10^7 cells intra-bone marrow | 87.7–89 days | Irradiation, 6 Gy × 2 doses | Delay in motor decline by grip strength meter | 10–13 days | Increased motor neurons and ventral root axons in both eGFP and mutant SOD1 BM transplanted animals over wild type | Microglial markers noted in transplanted cells |
| Pastor (2013) [21] | Mouse | BM | GFP expressing or GDNF knockout | mdf mouse | 1 × 10^6 cells, intramuscular | 10 weeks | None | Improvement in motor function by rotarod and treadmill testing | n/a | Improvement in neuromuscular junction and muscle histology; increased motor neuron survival on the side of treated limb | Increased GDNF expression noted in spinal cord and corresponding cortex |
| Cabanes (2007) [22] | Mouse | BM | CD117+, GFP expressing | mdf mouse | 3 × 10^5 cells, intraspinal | 6 weeks | n/a | Improvement by footprint testing | n/a | Increase motor neuron survival | GDNF levels higher in grafted mice |
| Corti (2010) [23] | Mouse | BM | Lin-c-kit+ | SOD1-G93A mouse | 1 × 10^6 cells intravenous | 70 days | n/a | Delay in motor decline by Rotarod | 16–17 days | Increased motor neuron survival (~50 %) with preserved ventral nerve root axons | Mechanism of protection possibly mediated by expression of GLT1 and elaboration of VEGF and angiopoietin 2 |
| Pastor (2012) [24] | Mouse | BM, MSC | GFP expressing or GDNF knockout | mdf mouse | 0.5–1 × 10^6 cells, intraspinal | n/a | None | Greater improvements seen in BM transplants over MSC transplants | n/a | Transplanted BM proliferate and retain bone marrow phenotype; MSCs underwent apoptosis | Increased GDNF expression in BM transplanted animals, functional improvements abolished in GDNF knockout BM transplant |
| Garbuzova-Davis (2008) [25] | Human | UCB | none | SOD1-G93A mouse | 1–5 × 10^7 cells intravenous | 7–8 weeks | Ciclosporin A | Delay and improvement in hindlimb extension and Rotarod | ~13 days (2.5 × 10^7 cell group) | Reduced microglial density in spinal cord | n/a |
| Garbuzova-Davis (2003) [26] | Human | UCB | None | SOD1-G93A mouse | 1 × 10^6 cells intravenous | 9 weeks | Ciclosporin A | Delay in motor decline by extension reflex and footprint testing | None | n/a | Distributed throughout CNS and formed cells with astrocyte and neuronal markers |
| Souayah (2012) [27] | Human | UCB | None | SOD1-G93A mouse | 1 × 10^8 cells, intravenous | 5–6 weeks | None | n/a | n/a | n/a | Improved neuromuscular transmission by electrodiagnostic testing |
| Bigini (2011) [28] | Human | UCB | None | SOD1-G93A mouse; Wobbler mouse | 5 × 10^5 cells, intraventricular | 10 weeks (SOD1); 4 weeks (Wobbler) | Ciclosporin A | Slowed decline by stride length and rotarod (SOD1); slowed decline by running speed and grip strength (Wobbler) | 18 days | No difference in MN (SOD1); increased MN survival (Wobbler) | n/a |
| Knippenberg (2012) [29] | Human | UCB | CD34+ | SOD1-G93A mouse | 2 × 10^5 cells, intraspinal | 40 or 90 days | Ciclosporin A | Improvement in motor function by rotarod, stride length, and footprint analysis | 6 days (12 days in females) | Increased motor neuron survival (~50 %) | No detected changes in growth factor production |
| Rizvanov (2011) [30] | Human | UCB | VEGF and FGF2 expressing | SOD1-G93A mouse | 1 × 10^6 cells intravenous | 24–28 weeks | n/a | n/a | n/a | n/a | VEGF-FGF2 expressing cells demonstrated astrocyte markers |
| Rizvanov (2008) [31] | Human | UCB | VEGF and L1CAM expressing | SOD1-G93A mouse | 1 × 10^6 cells, intravenous | 22–25 weeks | None | n/a | n/a | Transplanted cells formed endothelial cells | n/a |
| Habisch (2007) [32] | Human | MSC, UCB | Neuroectodermal derivatives of each also used | SOD1-G93A mouse | 1 × 10^5 cells, intrathecal | 45 days | Ciclosporin A | None | None | Improved intraparenchymal incorporation with bone marrow-derived cells | n/a |
| Uccelli (2012) [33] | Mouse | MSC | Luciferase expressing | SOD1-G93A mouse | 1 × 10^6 cells, intravenous | 90 days | None | Improvement by rotarod, extension reflex, and motor score | 17 days | Reduced ubiquitin inclusions | Decreased activated astrocyte and microglial cells, improvement in profile of oxidative stress/antioxidant enzyme expression |
| Forostyak (2011) [34] | Rat | MSC | GFP expressing | SOD1-G93A rat | 1 × 10^5 cells intraspinal, 2 × 10^6 intravenous | 16 weeks | Ciclosporin A | Slowed decline in motor function by BBB and grip strength testing | 11 days | Increased motor neuron survival in treated group | n/a |
| Boucherie (2009) [35] | Rat | MSC | None | SOD1-G93A rat | 2 × 10^6 cells, intrathecal | 90 days | None | Delayed disease onset by motor score | 16 days | Increased motor neuron survival and significant proportion formed astrocyte-like cells, with decreased microglia | Reduced expression of inflammatory mediators |
| Zhao (2007) [36] | Human | MSC | None | SOD1-G93A mouse | 3 × 10^6 cells, intravenous | 8 weeks | Irradiation, 6 Gy | Improved motor function by rotarod testing | 18 days | Increased motor neuron survival at 16 and 20 weeks, improved CMAP amplitudes | n/a |
| Vercelli (2008) [37] | Human | MSC | None | SOD1-G93A mouse | 1 × 10^5 cells, intraspinal | 28 weeks | none | Improved motor function by rotarod testing | n/a | Increased motor neuron survival and decreased microglial activation | Motor improvement noted in male animals only |
| Kim (2010) [38] | Human | MSC | None | SOD1-G93A mouse | 0.1, 2.0, and 10 × 10^5 cells, intrathecal | 60 days | Ciclosporin A | Slowed decline by rotarod testing (10 × 10^5 cell group) | ~6–8 days (2 × 10^5 and 10 × 10^5 cell groups) | Increased motor neuron survival | n/a |
| Suzuki (2008) [39] | Human | MSC | GFP, GDNF expressing | SOD1-G93A rat | 3.6 × 10^5 cells, intramuscular split in 3 doses | 80 days | Bupivacaine; ciclosporin A | Delay in motor dysfunction by BBB rating | 18–28 days | Preservation of neuromuscular junctions and corresponding motor neurons | n/a |
| Knippenberg (2012) [40] | Human | MSC | Glucagon-like peptide 1 expressing, alginate matrix embedded | SOD1-G93A mouse | 30 alginate capsules | 40 days | None | Improvement in motor function by rotarod and footprint analysis | 13 days | No difference in MN, reduction in reactive astrocytes and microglia | Increased expression of heat shock protein 70 in treated mice |
| Choi (2010) [41] | Human | MSC | Retroviral transduction of Neurogenin 1 | SOD1-G93A mouse | 1 × 10^6 cells intravenous | 8, 14–16, or 13 and 15 weeks | Ciclosporin A | Delay in motor decline by rotarod | n/a | Improved motor neurons survival | n/a |
| Morita (2008) [42] | Mouse/Rat | OEC, MSC | GFP expressing | Leu126delTT mouse | 3 − 4 × 10^5 cells, intraventricular | 13–14 weeks | FK-506 | None | Difference seen only in MSC-treated females | n/a | n/a |
| Martin (2007) [43] | Mouse | OEC | GFP expressing | SOD1-G93A mouse | 0.4 − 120 × 10^5 cells, intraspinal | 70 days | None | Improved motor function by running wheel activity and inclined plane test | 22.3 days | Differentiated cells show markers for MNs, astrocytes, and oligodendrocytes | Axons seen to project to peripheral nerve but fail to make neuromuscular junctions |
| Lepore (2008) [44] | Rat | Glial restricted precursors | GLT1-overexpressing, also GLT1-null cells | SOD1-G93A rat | 9 × 10^5 cells, intraspinal | 90 days | Ciclosporin A | Delay in forelimb (site of transplantation) but not hindlimb function; slowed decline of phrenic nerve peak CMAP amplitudes | 16.9 days | Increased motor neuron survival (47 % increase) | Majority astrocyte differentiation, may be mediated by GLT1 expression |
| Lepore (2011) [45] | Human | Glial restricted precursors | None | SOD1-G93A mouse | 2 − 6 × 10^5 cells, intraspinal | 50–60 days | Ciclosporin A or FK-506/rapamycin | None | None | No difference | n/a |
| Corti (2007) [46] | Mouse | Primary NPC | GFP expressing; LewisX + −CXCR4+ | SOD1-G93A mouse | 2 × 10^4 cells intraspinal | 70 days | n/a | Motor decline delayed, no change in slope of disease progression | 22–23 days | Increased MN survival and preservation of ventral nerve roots at 110 days but not at end stage | A large proportion of neuronal cells formed, with a subset of motor neurons. Neuroprotection may be mediated by VEGF and IGF-1 |
| Mitrecic (2010) [47] | Rat | Primary NPC | GFP expressing | SOD1-G93A rat | 1 × 10^7 cells intravenous | 14 and 26 weeks | Nitroprusside; TNF in half of treatment group | n/a | n/a | n/a | Improvement in graft survival with TNF therapy at 7 days; dominant GFAP positive cells at 7 days |
| Hwang (2009) [48] | Human | NPC | VEGF expressing | SOD1-G93A mouse | 1 × 10^5 cells, intrathecal | 70 days | n/a | Delay in motor decline by rotarod, paw grip endurance and extension reflex testing | 12 days | n/a | Downregulation of apoptotic proteins in treated animals |
| Park (2009) [49] | Human | NPC | GFP with GDNF or multiple growth factors (BDNF, IGF-1, VEGF, NT-3, GDNF) expressing | SOD1-G93A mouse | Intrathecal | 75 days | Ciclosporin A | none | Survival decreased in treatment group | Increased motor neuron survival with GDNF expressing cells | Sexual dimorphism noted, with female animals with worse outcomes |
| Xu (2006) [50] | Human | NPC | None | SOD1-G93A rat | 4 × 10^5 cells, intraspinal | 62 day | FK-506 | Slowed progression by BBB tests | 11 days | Increased motor neuron survival (nearly 200 %) | Increased GDNF secretion in grafted animals |
| Xu (2009) [51] | Human | NPC | none | SOD1-G93A rat | 1.6 × 10^5 cells, intraspinal | 56 day | FK-506 | n/a | n/a | n/a | n/a |
| Yan (2006) [52] | Human | NPC | None | SOD1-G93A mouse | 8 × 10^5 cells, intraspinal | 8 weeks | FK-506 and/or rapamycin and/or mycophenolate mofetil; or anti CD4 antibodies | Improved motor function by modified Wrathall scale with combination immunosuppressants | ~3 week benefit with combination immunosuppressants | Prominently neuronal phenotype formed | Combination immunosuppression promotes survival of grafted cells, which imparts functional and survival benefit |
| Klein (2005) [53] | Human | NPC | GDNF expressing | SOD1-G93A rat | 4.8 × 10^5 cells, intraspinal | 90 days | Ciclosporin A | None | None | None | n/a |
| Suzuki (2007) [54] | Human | NPC | GDNF expressing | SOD1-G93A rat | 4.8 − 7.2 × 10^5 cells, intraspinal | 65 days | Ciclosporin A | none | none | Increased motor neuron survival by grafted cells; no change in neuromuscular junctions | n/a |
| Garbuzova-Davis (2002) [55] | Human | NPC | None | SOD1-G93A mouse | 7.5 × 10^4 cells, intraspinal | 53 days | Ciclosporin A | Suggestion of delayed motor decline by staircase testing, beam balance, open field and footprint testing | None | None | n/a |
| Gao (2005) [56] | Human | NPC | MN differentiated in vitro; GFP expressing | Neonatal sciatic axotomy rat | 1 × 10^5 cells, intraspinal | 2 months | Ciclosporin A | Improvement by sciatic function index | n/a | 51 % transplanted cells with MN markers, and 19 % taking up retrograde marker from muscle | n/a |
| Hwang (2009) [48] | Human | NPC | VEGF expressing | SOD1-G93A mouse | 1 × 10^5 cells, intrathecal | 70 days | None | Delayed disease onset by rotarod, paw grip endurance and extension reflex testing | 12 days | Some transplanted cells differentiated into MN-like cells | Reduced proapoptotic proteins and elevated antiapoptotic proteins |
| Xu (2011) [57] | Human | NPC | None | SOD1-G93A rat | 2.4 × 10^5 cells, intraspinal | 63 days | FK-506 | Delayed motor decline by BBB and inclined plane testing | 17 days | n/a | n/a |
| Hefferan (2012) [58] | Human | NPC | None | SOD1-G93A rat | 1.8 − 2.6 × 10^5 cells, intraspinal | 60–65 days | FK-506 and mycophenolate mofetil | Improvement in motor function by BBB scale and electrodiagnostic testing | None | Increased motor neuron survival (~43 %); decreased reactive astrocyte and microglial populations | n/a |
| Popescu (2013) [59] | Human | iPSC-NPC | None | SOD1-G93A rat | 1 × 10^5 cells, intraspinal | 3 months | Ciclosporin A | n/a | n/a | Cells formed neuronal phenotype, with MN-like morphology | n/a |
| Nizzardo (2014) [60] | Human | iPSC-NPC | ALDHhi-SSClo-VLA4+; GFP expressing | SOD1-G93A mouse | 1 × 10^6 cells per dose, intravenous (multiple doses) or intrathecal (3 doses) | 90 days | None | Improvement in motor function by rotarod | 10 days (i.t.); 23 days (i.v.) | Increased motor neuron survival (40 %) and preservation of ventral axons (50 %) | Decreased microglial activation and astrogliosis |
ALDH aldehyde dehydrogenase, BBB Basso-Beatti-Bresnahan scale, BDNF brain derived neurotrophic factor, BM bone marrow, CMAP compound muscle action potential, ES embryonic stem cell, FGF fibroblast growth factor, GDNF glial cell-line derived neurotrophic factor, GFP green fluorescent protein, GLT1 glutamate transporter 1, IGF-1 insulin-like growth factor 1, iPSC induced pluripotent stem cell, L1CAM L1 cell adhesion molecule, MN motor neuron, MSC mesenchymal stem cells, NPC neural progenitor cell, NT-3 neurotrophin 3, OEC olfactory ensheathing cell, SSC side scatter, TGF transforming growth factor, UCB umbilical cord blood cells, VEGF vascular endothelial growth factor, VLA4 very late antigen 4 (integrin alpha 4 beta 1) SOD1 superoxide dismutase; n/a not applicable; CNS central nervous system; i.t intrathecal; i.v. intravenous
Table 2.
Summary of clinical stem cell trials in amyotrophic lateral sclerosis
| Stem cell type | Delivery method (target) | Dose | Patient eligibility | Patients (controls) | Planned outcomes | Results | Reference(s) |
|---|---|---|---|---|---|---|---|
| PBSC | G-CSF 2 μg/kg/day SC × 5 days | Not provided | 13 (0) | Primary: ALSFRS-R Secondary: CMAP amplitude (muscle(s) not specified) |
Reduction in slope of decline of ALSFRS-R and CMAP amplitude postprocedure | Zhang (2009) [61] | |
| G-CSF 5 μg/kg/day SC × 4 days repeated at months 0, 3, 6, 9 | Age: 18–85 years EEC: D, Pr Symptom duration: <6 years FVC: >50 % FALS: Excluded Cognition: Normal |
19 (20), but only those completing 6 months of study included in analysis Patients assessed at each month 0: 19 (20) 3: 17 (18) 6: 12 (16) 9: 9 (12) |
Primary: ALSFRS-R Secondary: FVC, MMT megascore, CMAP megascore, NI, QoL; tracheostomy, death |
No difference in clinical measures between treatment and placebo group No safety concerns G-CSF resulted in increased WBC count & circulating CD34+ cells |
Nefussy (2010) [62] | ||
| G-CSF 5 μg/kg/day SC every 12 h × 4 days at months 0, 3, 6, 9 with 125 ml 18 % mannitol IV 4 times per day × 5 days starting on day 3 of G-CSF | Age: 40–65 years EEC: D, Pr, Pr-LS Symptom duration: <12 months FVC: >80 % FALS: Excluded Cognition: FTD excluded |
24 (0) Patients assessed at each month 0: 24 3: 24 6: 21 9: 20 |
Primary: safety and tolerability, clinical progression, and changes in chemokine and cytokine levels | No safety concerns No change in disease progression ↑ WBC & CD34+ cells in blood ↓MCP-1 in CSF/serum ↓IL-17 in CSF ↑ IP-10 in serum |
Tarella (2010) [63] Chio (2011) [64] |
||
| IV | G-CSF 300–600 μg/day SC × 5–6 days Leukapheresis for target of 2.0 × 106 CD34+ cells/kg |
Not clearly specified | 8 (0) | Not clearly specified | No safety concerns No chance in disease progression No change in MRS NAA/Cr ratio |
Cashman (2008) [65] | |
| Intrathecal | G-CSF administration followed by leukopheresis to isolate CD34+ cells | Case series | 3 (0) Patient 1: 100 million cells via lumbar intrathecal catheter over 2 days Patient 2: 20 million cells over minutes intrathecal at L3/4 and cisterna magna Patient 3: 100 million cells intrathecal at C1/2 and lumbar region |
Patient 1: 2 h loss of sensation in lower limbs, subjective speech improvements Patient 2: no change in disease Patient 3: gain in leg and neck strength |
Janson (2001) [66] | ||
| IV | Donor-mobilized CD34+ cells generated by G-CSF following total body radiation, fludarabine, and horse ATG Tacrolimus and methotrexate for GVHD prophylaxis |
Age: 20–65 years EEC: D FVC: >60 % HLA-identical related donor |
6 (matched historical controls) | Primary: Donor engraftment, clinical measures | Engraftment successful Cases of cutaneous and limited GVHD |
Appel (2008) [67] | |
| Intracortical | G-CSF 300 μg/daily SC × 3 days followed by isolation of CD133 cells by leukapheresis | EEC: Any FVC: Any Severe bulbar involvement excluded |
10 (13) Control patients were those that did not accept treatment but met inclusion criteria, or those that applied after study completed recruitment |
Primary: survival rate Secondary: ALSFRS-R |
No safety concerns Improved survival in treatment group (disease duration 30.1 months in treatment group vs 14.3 in controls) |
Martinez (2009) [68] | |
| Intracortical | G-CSF 300 μg/daily SC × 3 days followed by isolation of CD133+ cells by leukapheresis Bilateral injections into frontal motor cortex 3–4 cm from midline |
EEC: Any FVC: >30 % |
67 (0) Appears to have included patients previously reported in Martinez 2009 [68] |
Primary: Safety (not clearly specified) | 2 subject deaths in postoperative period (respiratory failure, MI/SDH); otherwise procedure was well tolerated | Martinez (2012) [69] | |
| Bone marrow derived MSCs | Intrathecal (10 patients) Intrathecal + Intravenous (9 patients) |
EEC: D Age: 25–65 years At least 5 point decline in ALSFRS in 1 year |
19 (0) | Primary: Safety analysis | No safety concerns | Karussis (2010) [70] | |
| Intrathecal | Intrathecal administration at L2-3 or L3-4 | EED: D, Pr, Pr-LS Age: >18 |
10 (0) | Primary: ALSFRS-R at day 90, 180, 270, 365 Secondary: ALSFRS-R subscores, time to 4-point worsening, survival |
Trend towards stabilization of ALSFRS-R (individual patient characteristics are not reported such as disease duration, which could impact interpretation) No safety concerns |
Prabhakar (2012) [71] | |
| Intramuscular, Intrathecal | MSCs induced to secrete NTFs | EEC: D, Pr, Pr-LS, Po Age: 18–75 years Disease duration <24 months ALSFRS-R > 30 SVC > 65 % |
48 estimated (0) | Primary: Safety Secondary: change in ALSFRS-R, change in SVC |
Study in process, results not yet reported | Clinical Trial | |
| Intraventricular | 1 × 107 cells/kg | Case report of 63-year-old with definite ALS by EEC | 1 (0) | Authors state ALS was too advanced to assess efficacy | No safety concerns | Baek (2012) [72] | |
| Intraspinal | Cells injected into thoracic cord (central part of spinal cord) 1 mm apart in 3 rows spaced by 3 mm | EEC: D Age: 21–75 years Disease duration: 6–96 months Mild-to-severe functional impairment at spinal level No or mild bulbar involvement No respiratory failure |
7 (0) (+2 patients under compassionate use) | Primary: ALSFRS-R, Norris score, FVC every 3 months following 6-month lead in period | 4 patients showed a reduction in the ALSFRS-R and FVC decline No safety concerns |
Mazzini (2006) [73] Mazzini (2008) [74] Mazzini (2012) [75] |
|
| Intraspinal | Same methods as 2006 study [73] | EEC: D, Pr Age: 20–65 years Disease Duration: <3 years FVC: >50 % FALS: excluded Onset: spinal |
10 (0) | Primary: ALSFRS-R, MRC, respiratory assessment, MUNE, neurophysiological index, MRI, DTI, safety | No change in the rate of decline of clinical measures No safety concerns |
Mazzini (2010) [76] Mazzini (2012) [75] |
|
| Intraspinal | T3-4 injections 1–2.5 mm from midline at depth of 6 mm | EEC: D Age: 20–65 years Disease duration: 6–36 months FVC: >50 % Onset: Spinal |
11 (0) | Primary: Safety Secondary: FVC, ALSRFS-R, MRC, Norris scale |
No safety concerns No changes in disease progression |
Blanquer (2010) [77] Blanquer (2012) [78] |
|
| Intraspinal | C1-2 laminectomy and multiple injections at these levels | Disease duration: >6 months FALS: Excluded Rapid decline, FVC in terminal period (on mechanical ventilator or unable to speak) |
13 (0) | Primary: Not specified | No safety concerns Authors report 7/13 patients improved postprocedure (no clear criteria for assessment) |
Deda (2009) [79] | |
| Intraarterial | T-cell vaccination every 28 days for 10 doses Bone marrow harvest, following purification, one aliquot given to patient on same day “by selective intralesion infusion into the feeding artery” MSCs differentiated into NSCs and given intra-arterial |
Disease duration 3–5 years | 7 (0), only 5 completed full regimen | Primary: ALSFRS-R | No safety concerns Results not well reported, no apparent change in disease progression |
Moviglia (2012) [80] | |
| OECs | Intracortical | OECs extracted from human fetal olfactory bulb tissue 2 million OECs injected into bilateral corona radiata |
EEC: D, Pr Age: 20–70 years ALSFRS-R ≥ 15 |
15 (20) Controls not randomized, rather first 15 patients served as cases and next 20 as controls; no formal matching No patients share nationality of the study (China) |
Primary: ALSFRS-R at 4 months provided by patient, caregiver, and family member | Rate of decline between months 3 and 4 was slower for the treatment group compared with control Authors do not report presence/absence of adverse events; however, additional safety and efficacy outcomes reported in references [81–83] (see text) |
Huang (2008) [84] |
| Intracortical, Intraspinal | Intraspinal injections not standardized, reported to occur at impaired segments Authors suggest all patients received intraspinal injections, although the 2007 report suggest some patients only received intracortical injections |
EEC; D, Pr Age: >18 years |
507 (0) Intracortical only: 35 patients second injection; 5 patients 3 injections; 1 patient 4 injections; 1 patient 5 injections |
ALSFRS-R, Norris scale, video recordings of patients, EMG, PFT | Authors report improved ALSFRS-R and respiratory measures after each treatment, although a progressive decline in ALSFRS-R continued | Chen (2007) [85] Chen (2012) [86] |
|
| NPCs | Intraspinal (phase I) | Dose escalation, see Table 3 for details Patients immunosuppressed with basiliximab mycophenolate mofetil, and tacrolimus and received tapering steroid dose following injections |
Disease severity changed during trial to enroll less severely affected patients in later groups | 15 (0) | Primary: Safety | No safety concerns Possible slowing of disease progression in patients without bulbar symptoms early in disease course; however, number of subjects fulfilling this criteria is small |
Riley (2012) [87] Glass (2012) [88] Riley (2014) [89] Tadesse (2014) [90] Feldman (2014) [91] |
| Intraspinal (phase II) | Dose escalation, see Table 3 for details Same immunosuppression regimen as phase I |
EEC: D, Pr, Pr-LS Age: >18 years FVC: >60 % seated, >50 % supine SALS or FALS |
15 (0) | Primary: Safety Secondary: attenuation of motor function loss, maintenance of respiratory capacity, stabilization of ALSFRS-R, reduction of spasticity/rigidity, graft survival |
Study in process, results not yet reported | Clinical trial NCT01730716 |
ALSRFS-R ALS Functional Rating Scale Revised, CMAP compound muscle action potential, Cr creatine, D Definite, EEC El Escorial Criteria, FALS familial ALS, FVC forced vital capacity, HLA human leukocyte antigen, IV intravenous, MMT manual muscle strength; MRS magnetic resonance spectroscopy, MSC mesenchymal stromal cells, NAA N-acetylasparate, NI neurophysiolocal index (NI = CMAP amplitude × F-wave persistence / distal motor latency in ulnar nerves), NPC neural progenitor cell, NSC neural stem cells, NTF neurotrophic factors, OECs olfactory ensheathing cells, Po Possible, Pr Probable, Pr-LS Probable-Laboratory Supported, QoL quality of life, SALS sporadic ALS, SVC slow vital capacity; PBSC peripheral blood stem cell; G-CSF granulocyte colony stimulating factor; SC stem cell; WBC white blood cell; FTD frontotemporal dementia; MCP-1 monocyte chemoattractant protein-1; CSF cerebrospinal fluid; IL-17 interleukin-17; IP-10 interferon-induced protein-10; ATG ATGAM; GVHD graft versus host disease; MI myocardial infarction; SDH subdural hematoma; MUNE motor unit number estimation; MRI magnetic resonance imaging; DTI diffusion tensor imaging; MRC Medical Research Council; EMG electromyographt; PFT pulmonary function test
Transitioning from Early Preclinical Studies to Current Transplantation Paradigms
The mutant Cu2+/Zn2+ superoxide dismutase (SOD1)-G93A transgenic mouse and rat have served as the basis for much of the preclinical work in ALS stem cell therapy. These animals are based on the first identified gene underlying familial ALS [92], and recapitulate the progressive weakness and muscle wasting associated with selective MN loss characteristic of the disease. In the earliest studies, a survival benefit was demonstrated in irradiated SOD1-G93A mice treated with human umbilical cord blood [93, 94]. Follow-up studies also showed that transplanted human cord blood along with immunosuppression with cyclosporine delayed disease progression and that the transplanted cells were detected in the brain and spinal cord [25, 26]. From these beginnings arose many strategies to harness the potential of stem cells for ALS.
Given that the goal of early stem cell therapies for ALS were directed at MN replacement, the finding that MNs derived from mouse embryonic stem cells could be grafted into a chick spinal cord and synapse with muscles was exciting [95]; however, results of similar studies in rodent models of ALS were not met with the same success, likely related to features of both ALS, as well as the challenges of reconstructing the motor system as mentioned above. For example, SOD1-G93A rats that underwent grafting of mouse embryonic stem cells into the spinal cord only exhibited a transient motor improvement that may have been due to trophic support provided by the grafted MNs to the degenerating endogenous MNs [11]. Thus, this transient improvement instead served as a springboard for studies focused on the neighborhood theory, which promotes beneficial neuronal synaptic interactions and the creation of a microenvironment that is supportive of existing MNs [10, 12]. This concept is important, especially considering the notion that ALS may not be a cell autonomous disorder, and that, in at least the mutant SOD1 form of the disease, toxicity is not limited to the MNs but also affects surrounding microglia and astrocytes, which can be manipulated with stem cell therapy [30, 96–98].
Evidence supporting the potential therapeutic benefit of altering the MN microenvironment was the focus of a number of studies that aimed to modify non-neuronal cells and offer neurotrophic factor support. Injections of hematopoietic stem cells into a mouse model that undergoes selective MN degeneration did not result in the formation of primary neural tissue, but did result in functional improvements thought to be related to a neuroprotective effect from GDNF produced by the grafted cells [22]. Further support of the microenvironment was demonstrated in chimeric mice produced by injecting wild-type embryonic stem cells into SOD1-G85R or G37R mutant mouse blastocysts. In this chimeric model, it was discovered that mutant SOD1-expressing MNs exhibited prolonged survival when surrounded by wild-type non-neuronal cells [99]. Other studies have focused on specific non-neuronal cells. In a study of astrocytes, SOD1-G37R mice with reduced mutant SOD1 expression in astrocytes exhibited delayed microglial activation that resulted in slowed disease progression [100]. Further animal models supporting the therapeutic benefit of wild-type astrocytes involved the transplantation of glial-restricted precursors into the cervical spine of SOD1-G93A mice. The result of this intervention was prolonged survival with reduced MN loss and slowed progression of motor functional declines [44]. Benefits in survival are also noted when cells modified to secrete GDNF are injected in mutant SOD1 rodent models. Interestingly, implantation of human MSCs engineered to secrete GDNF into skeletal muscle were also able to support MN survival in the spinal cord [39], with synergistic effects attributed to vascular endothelial growth factor (VEGF) production [101]. Therefore, the ability of surrounding non-neuronal cells in the spinal cord and at the neuromuscular junction appears to play an important role in ALS pathogenesis, and the properties of stem cells are thereby ideally suited to achieve the goal of modulating the MN microenvironment.
Several stem cell types have since been examined for their potential efficacy in preclinical ALS models, including stem cells obtained directly from bone marrow. Direct implantation of nondiseased bone marrow into bone marrow of SOD1-G93A mice improved survival, an effect linked to the presence of non-neuronal cells derived from the transplanted tissue in the spinal cord [20]. Likewise, intraperitoneal transplantation of wild-type bone marrow into SOD1-G93A mice resulted in extensive incorporation of transplanted cells as microglia with improved animal survival [18]. Enriching the population of transplanted cells for c-kit+ stem cells allowed the infusion of cells peripherally into SOD1-G93A mice, with again improved function and survival associated with non-neuronal cells that migrated to the spinal cord [23]. Similarly, crossing SOD1-G93A mice with mice unable to generate myeloid or lymphoid cells slowed the disease course following subsequent bone marrow transplantation with wild-type SOD1-expressing cells, and this study further demonstrated that microglia in the mice receiving wild-type bone marrow transplants produced less superoxide, nitrite, and nitrate, and were therefore less neurotoxic [102]. Thus, these experiments again showed that MN pathology is not necessarily intrinsic to the MN, but may additionally rely on interactions with surrounding glial tissue.
Another potential source of stem cells are OECs isolated from the olfactory bulb, as they represent a relatively accessible source of endogenous NPCs. These cells showed promise in early models of spinal cord injury, where they promoted axonal regrowth and remyelination [103, 104]. Interestingly, transplantation of OECs isolated from the olfactory bulb of green fluorescent protein-expressing C57BL/6 mice into SOD1-G93A animals prolonged survival, and this effect was not associated with the formation of neuromuscular junctions in SOD1-G93A mice [43], suggesting again that the formation of interneurons, astrocytes, and even oligodendroglia may provide support for diseased MNs.
Studies using NPCs derived from elsewhere in the nervous system have lent further credence to a strategy of supporting existing MNs via trophic support. In one study, mouse-derived NPCs selected for those expressing the Lewis X surface marker and the chemokine receptor CXCR4 supported native MNs via VEGF and insulin-like growth factor-I (IGF-I)-mediated neuroprotective pathways [46]. Subsequent use of human NPCs modified to overexpress VEGF also favored antiapoptotic pathways over proapoptotic pathways in native MNs [48], and human NPCs modified to secret GDNF were able to confer protection when transplanted into the spinal cords of SOD1-G93A rats [53], although this did not result in continued muscle innervation [54]. Similarly, experiments with transplanted GDNF or IGF-1-secreting human NPCs rescued MNs but again did not result in improvement of motor performance or lifespan in SOD1-G93A rats [49]. Taken together, these experiments demonstrated that NPCs are able to rescue native MNs via local trophic signaling rather than replacing MNs, although more work to attain a more robust effect will be required to observe functional benefit.
Along the lines of creating a neuroprotective neighborhood, the human fetal spinal cord stem cell line NSI-566RSC has been studied for use in cellular therapy [14]. This cell line was derived from an 8-week fetal spinal cord and has the advantage of reduced teratoma formation risk compared with embryonic stem cells, as they are partially differentiated NPCs. When grafted into the SOD1-G93A rat spinal cord, these cells form both neurons and glia, synapse with native MNs, and elaborate a range of neurotrophic factors [14]. Furthermore, grafting of these cells resulted in a delay in onset and progression of disease symptoms and increased the lifespan of experimental animals [50]. Notably, the grafted cells formed gamma-aminobutyric acid-ergic neurons that synapsed with neighboring MNs in the ventral horn, although they did not form synaptic connections outside the spinal cord [51, 58]. Therefore, with the ability to form glial cells and a diversity of neuronal synaptic contacts around diseased MNs, NPCs such as NSI-566RSC have the advantage of being able to tackle the multifaceted local physiologic derangements seen in ALS.
Overall, the preclinical studies (Table 1; reviewed in [105]) underscore the potential for stem cells to modulate the microenvironment in ALS. Along with the many detailed mechanistic studies that have been performed using the various cell types described above, these preclinical data have formed the foundation for early clinical trials that seek to harness the unique attributes of stem cells to rescue diseased MNs.
Clinical Studies
Translation of stem cell therapy from the laboratory to the clinical realm requires 1) propagation of an easily accessible source of progenitor cells; 2) efficient delivery of cells to the affected areas; and 3) the ability of the cells to survive and integrate into local circuits, such that degenerating cell populations can be replaced or aberrant physiology reversed. In terms of the first requirement for suitable stem cell sources, recent studies have focused on the use of G-CSF-induced PBSCs, bone marrow-derived MSCs, OECs, and NPCs. The second point has also been addressed by a number of techniques for the collection and delivery for each stem cell population, with delivery methods including intravenous, intra-arterial, intrathecal, intracerebral, and intraspinal routes. And, finally, to the third requirement, stem cell integration must be quantifiable on a clinical scale. For ALS, this includes measurements of function such as the ALS Functional Rating Scale-Revised (ALSFRS-R), respiratory parameters, and survival, as well as the enrollment of adequate numbers to detect improvements in comparison with control treatments. To date, most studies are proof-of-concept or safety trials, and, as such, are conducted without placebo control groups. Likewise, the study sizes are small and are not powered to determine clinical efficacy. Thus, none of the reviewed studies make firm conclusions regarding improvements in the ALS disease course; however, these clinical studies represent an important first step in the development of stem cell therapies for ALS. In the following sections, the published studies involving the application of stem cells for the treatment of ALS in humans (summarized in Table 2) will be reviewed.
Granulocyte-Colony Stimulating Factor and Peripheral Blood Stem Cells
G-CSF is a hematopoietic growth factor that can mobilize CD34+ hematopoietic stem cells from the bone marrow to the peripheral blood, resulting in a population of PBSCs that can be collected for later use [63, 65, 106]. These CD34+ PBSCs were initially identified after cancer patients received hematotoxic chemotherapy [106], and it was later shown that these circulating hematopoietic progenitor cells could migrate into the central nervous system (CNS) and provide support for diseased MNs [107]. G-CSF itself is suggested to have a neuroprotective effect [108]. Thus, a number of strategies were implemented using G-CSF to mobilize or collect and redistribute PBSCs to the CNS in ALS.
Based on the presumption that PBSCs can migrate into the CNS, subcutaneous G-CSF was given to 13 patients, resulting in a slowing of disease progression evidenced by ALSFRS-R, as well as maintenance of compound muscle action potential amplitudes [61]. Another study in 17 patients comparing subcutaneous G-CSF with a placebo control arm showed elevated CD34+ PBSCs, but no difference in disease progression [62]. Similarly, the STEMALS trial utilized subcutaneous G-CSF along with a 5-day course of mannitol with the hopes of increasing permeability across the blood–brain barrier in 26 patients [63, 64]. Again, there was an increase in circulating CD34+ PBSCs, and in this trial a decrease in the proinflammatory cytokines monocyte chemoattractant protein-1 and interleukin-17, but no change in ALSFRS-R. Enriching the number of circulating PBSCs by first collecting and then readministering CD34+ cells induced by G-CSF has also been attempted. One study utilized subcutaneous G-CSF in 8 patients prior to PBSC isolation and peripheral infusion, and while no adverse effects were reported, clinical and imaging measures did not seem to be significantly affected [65]. Finally, an aggressive strategy was attempted in 6 patients with ALS who received total body radiation followed by peripheral infusion of G-CSF-primed PBSCs from human leukocyte antigen-matched siblings [67]. Patients were immunosuppressed with methotrexate and tacrolimus, and subsequent graft-versus-host disease occurred in half of the patients. Although donor hematopoietic stem cells entered the CNS at sites of MN degeneration and engrafted as immunomodulatory cells, no clinical benefit was detected and the study was halted owing to a lack of benefit and impaired quality of life.
In contrast to the above G-CSF studies, others have examined more invasive procedures for PBSC delivery in order to bypass the blood–brain barrier. An early study tested 3 patients using a protocol of subcutaneous G-CSF therapy and isolation of CD34+ stem cells followed by intrathecal administration of the collected stem cells and showed minimal adverse effects [66]. Alternatively, a separate group focused on CD133+ cells mobilized by G-CSF followed by direct injection of the cells into cortical motor areas of the brain utilizing a frame-based or frame-less stereotactically guided needle [68, 69]. In the initial study, 10 patients were enrolled and compared with 10 control patients not accepting treatment or who applied after the study period [68]. One patient died within 10 days of surgery from a myocardial infarction. The treatment group showed an improvement in baseline ALSFRS-R scores; however, the control group had a higher ALSFRS-R score at baseline limiting a comparison. An additional 67 patients were then evaluated after having undergone the same procedure [69]. Two postoperative deaths were reported, and no outcome data were reported, but these serious adverse events suggest that further use of frontal cortex injection should be approached cautiously to minimize patient risk.
Overall, these G-CSF studies appear to demonstrate clinical safety, with a suggestion of clinical efficacy in some cases as well, and, based on preclinical models, this is an area that demonstrates therapeutic potential. It is clear, however, that these trials struggle with the technique of stem cell delivery, balancing widespread yet inefficient distribution of blood-borne PBSCs against expedient yet risky surgical methods. Hence, moving forward, this method of therapy will benefit from some agreement on G-CSF delivery strategies, as well as good clinical trial design utilizing large numbers of well-defined patient populations and standardized outcome measures.
Bone Marrow-Derived Hematopoietic Progenitor Stem Cells
Bone marrow-derived MSCs are another source of therapeutic potential in ALS [74]. MSCs may exert neuroprotective effects via paracrine, or “bystander”, mechanisms, such as the release of anti-inflammatory, anti apoptotic, and neurotrophic factors, and by influencing other cell types to take on a protective phenotype [109, 110]. MSCs further offer the advantages of 1) being an easily obtainable source; 2) possessing the ability for expansion in vitro; 3) lacking a requirement for immunosuppressive therapy to prevent rejections; and 4) having a reduced risk of malignant transformation [70]. Of note, it has also been suggested that MSCs are able to differentiate into neuron-like [111, 112] and glia-like [112, 113] lineages, although some have questioned this ability [114].
A number of studies have attempted to harness the potential of MSCs as a treatment for ALS. One study intrathecally administered MSCs obtained by bone marrow aspiration in 19 patients, and while all patients received MSCs via intrathecal lumbar puncture, 9 of the 19 patients also received intravenous MSCs [70]. No serious adverse events and a 6-month period of disease stability was reported following the procedure. In another study, 10 patients underwent isolation of bone marrow-derived MSCs, which were then administered intrathecally via lumbar puncture [71]. Again, the procedure was without serious adverse events; however, while some patients demonstrated stability of ALSFRS-R scores, others showed a decline from baseline. In addition, bone marrow-derived MSCs are the focus of an ongoing phase II clinical trial at the Mayo Clinic, Massachusetts General Hospital, and University of Massachusetts sponsored by Brainstorm-cell Therapeutics (Clinic Trial NCT02017912). In this study, patients are randomized to receive an intramuscular and intrathecal injection of autologous bone marrow-derived MSCs that are propagated ex vivo and induced to secrete neurotrophic factors. The study plans to enroll 48 patients and will evaluate safety and efficacy of the intervention.
Many other groups have also attempted more invasive methods of introducing MSCs in to the CNS. A case in which an Ommaya reservoir was utilized for intraventricular delivery of bone marrow-derived MSCs reported no adverse events [72]. Consecutive phase I studies in Italy in which bone marrow was obtained from the iliac crest and expanded in vitro prior to direct surgical implantation of the cells into the dorsal spinal cord involved 9 patients initially [73, 74], followed by an additional 10 patients [76], for a total of 19 patients [75]. In these studies, the procedure was well-tolerated and led to a slowed disease course in 6 patients; 4 of these patients were the youngest in the trials, and 2 subjects had a lower MN-predominant form of the disease. Thus, the authors suggested that slowing of the disease course in these subjects may have been a reflection of the disease phenotype and not the therapy. Another group, in Spain, evaluated 11 patients following injection of autologous bone marrow-derived MSCs into the spinal cord [77, 78]. No serious adverse events were reported nor were there any reported changes in the disease course; however, MNs near the areas of grafting showed fewer degenerative signs on histopathology. Finally, a study in Turkey assessed patients following various routes of bone marrow-derived MSC administration and indicated that the procedure was safe, and in some cases motor improvement was also reported. [79].
Alternatively, the combination of MSCs from the bone marrow combined with T-cell vaccination has also been studied [80]. In this procedure, MSCs that were obtained from 7 patients were differentiated into NPCs and infused intra-arterially 48 h after a third T-cell vaccination dose. No serious adverse events were reported and some patients experienced a transient improvement in symptoms.
In summary, bone marrow-derived MSCs remain a viable source of cells that confer the advantage of easy expansion and manipulation in vitro for subsequent autologous transplantation. While the currently reported approaches for grafting bone marrow-derived MSCs have tended to be more invasive, these strategies circumvent some of the limitations of intravenous administration seen in the G-CSF studies. Moving forward, however, it will be important to determine therapeutic differences between G-CSF-induced PBSCs and bone marrow-derived MSCs, as well as to determine the optimum collection and delivery procedures.
OECs
OECs have been studied in China based on the preclinical evidence studying NPCs obtained from olfactory tissue [84]. In 1 study, fetal OECs were injected into the bilateral corona radiata in a nonrandomized, nonblinded cohort of patients [84]. At 4 months, reporting by the patients or caregivers indicated a reduction in disease progression rates. In a larger study involving 507 patients receiving intraspinal and intracortical injections (with many undergoing multiple intracortical injections), short- and long-term outcomes were reported [85, 86], reflecting a statistically significant increase in ALSFRS-R scores following the injections, but no improvement in pulmonary function tests. Notably, 7 Dutch patients with ALS who underwent the procedure in China were evaluated at their local institution and showed no improvement in symptoms [81]. Similarly, a patient followed in the USA who underwent the procedure had an acceleration in disease progression and also suffered from a possible brain hemorrhage and vasogenic edema at the injection site [82]. Moreover, a postmortem study examining brain tissue from 2 patients who underwent the procedure showed graft encasement and did not show evidence of axonal regeneration, neuronal differentiation, or myelination to suggest an alteration of ALS neuropathology [83].
Based on preclinical models, while the studies involving OECs may deserve further attention, these studies have not utilized good clinical study design, especially given the large number of patients enrolled. More objective measures aside from the ALSFRS-R did not show improvements and these nonblinded studies may be influenced by a number of types of bias. Thus, this therapy should continue to be evaluated with close scrutiny, with a need for further support from well-designed clinical studies.
NPCs
Based on the promising preclinical data mentioned above, a phase I, first-in-human, Food and Drug Administration-approved clinical trial utilizing NSI-566RSC has recently been completed [14, 87–89, 91]. In this study, 12 patients underwent unilateral or bilateral lumbar intraspinal transplantation surgeries and 6 patients received unilateral cervical intraspinal transplantation surgeries following a risk-escalation design. Of note, 3 patients received both lumbar and cervical transplants; therefore, the phase I study involved a total of 15 patients (Table 3). The study demonstrated safety and tolerability of the surgical procedure using a customized injection platform with a floating cannula designed to reduce risk of injury to the spinal cord given cardiorespiratory motion [89, 91], and anatomical injection accuracy to the ventral horn was determined using presurgical magnetic resonance imaging evaluation (Fig. 1; [10]). As of the final outcome reporting in early 2014 [91], 6 patients died owing to disease progression, and 1 died from an unrelated congenital heart defect. Moreover, while the study was not designed to evaluate efficacy, preliminary analysis of disease monitoring in a majority of patients demonstrated slowed disease progression in multiple clinical measures, with the greatest effect on disease progression seen in those patients who received the highest number of injections/cells (Fig. 2; [91]). Briefly, ALSFRS-R measurements for the cohort of patients receiving both lumbar and cervical injections (upper panel, Fig. 2a) were converted into data points reflecting the change in ALSFRS-R per year for various 9-month windows (lower panel, Fig. 2a). For example, the presurgical window reflected disease progression rates prior to the initial lumbar surgery (green window), and windows following the transplantation surgery reflect changes in ALSFRS-R over set time frames postsurgery (see representative blue windows). Of note, the timing of the second transplantation surgery in which cells were delivered into the cervical targets are indicated by the vertical dashed lines. Taken together, this analysis reflects an improvement in the rate of decline in ALSFRS-R following both the lumbar and cervical stem cell transplants [as demonstrated by positive slope peaks (Fig. 2a)]; however, this benefit decreases over time [as noted by the trough in the plot (Fig. 2a)], suggesting that there are apparent windows of benefit, which result following cellular transplantation (Fig. 2b).
Table 3.
Phase I and II trial design for the first-in-human, Food and Drug Administration-approved clinical trial utilizing NSI-566RSC in patients with amyotrophic lateral sclerosis
| Phase I trial design | |||||
| Group | Number of patients | Subject details | Injection target | Injection details | Final cell dose |
| A1 | 3 | Nonambulatory | Lumbar | 5 unilateral | 5 × 105 |
| A2 | 3 | Nonambulatory | Lumbar | 10 bilateral | 1 × 106 |
| B | 3 | Ambulatory | Lumbar | 5 unilateral | 5 × 105 |
| C/E | 3 | Ambulatory | Lumbar | 10 bilateral | 1 × 106 |
| Cervical | 5 unilateral | 5 × 105 | |||
| D | 3 | Ambulatory | Cervical | 5 unilateral | 5 × 105 |
| Phase II trial design | |||||
| Group | Number of patients | Injection target | Number of injections | Final cell dose | |
| A | 3 | Cervical | 10 bilateral | 2 × 106 | |
| B | 3 | Cervical | 20 bilateral | 4 × 106 | |
| C | 3 | Cervical | 20 bilateral | 6 × 106 | |
| D | 3 | Cervical | 20 bilateral | 8 × 106 | |
| E | 3 | Lumbar and cervical | 20 bilateral for each target | 16 × 106 | |
Fig. 1.
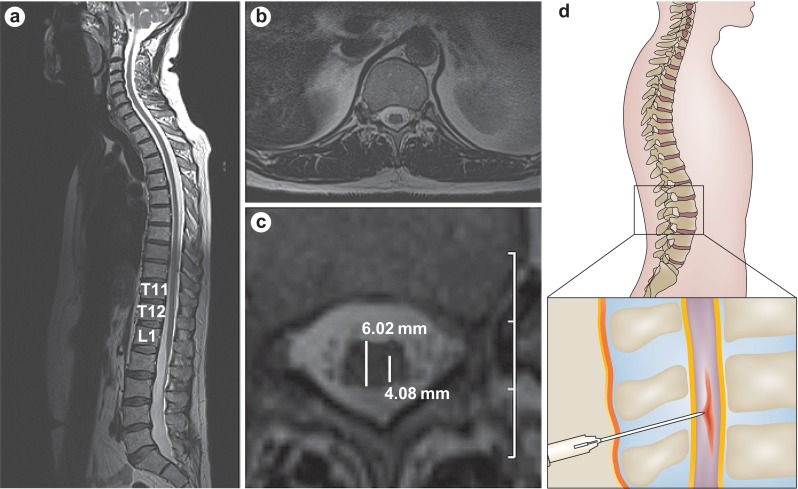
Accurate anatomical targeting of stem cell delivery. a. T2-weighted magnetic resonance imaging scan showing a sagittal view of the spinal cord and the position of the conus medullaris and lumbar enlargement. b. Axial view of the spinal cord at the level of T12. c. Precise needle placement into the ventral horn of the spinal cord is calculated from a magnified image of part b. Estimated measurements of spinal cord diameter (6.02 mm) and distance from the dorsal root entry zone to the ventral horn (4.08 mm) are shown. Scale: 1 cm per grid division. d. Schematic of targeted injection of stem cells into the spinal cord. Reproduced from Boulis et al., Nat Rev Neurol 2011;8:172–6, [10]
Fig. 2.

Preliminary analysis of potential windows of human spinal stem cell (HSSC) biological activity in subjects 10–12. To identify the most biologically active period of the injected HSSCs, postsurgery data points for group E subjects were divided into a series of 9-month windows, beginning each month postsurgery, and slopes were calculated across each window. Slopes were also calculated using Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R) data points for the presurgical window. a The top panel demonstrates ALSFRS-R scores for group E subjects during the presurgical period (green) and representative ranges associated with the various sliding postsurgical 9-month windows (dark blue). The bottom panel demonstrates the slopes obtained for each sliding window, with the x-axis corresponding to the first month for each 9-month window (i.e., window 1 corresponds to months 1–10 postsurgery, window 2 corresponds to months 2–11 postsurgery, window 3 corresponds to months 3–12 postsurgery, etc.). The first plotted slope for each subject corresponds to their presurgical progression rate. Slope values higher than the presurgical slope at baseline represent improved or attenuated progression rates during the designated window. Note that the starting month of the final sliding window for each patient coincides with the dates of the second surgery, which occur at 17.5, 19.0, and 16.6 months after the initial cohort C surgery (time 0) for subjects 10, 11, and 12, respectively. (b) The presurgical slope and postsurgical slopes associated with the window correlating to the peak benefit windows for both the lumbar and cervical postsurgery time frames are summarized. Reproduced from Feldman et al., Ann Neurol 2014;75:363–73 [91]
The promise of this trial has been further underscored by the recent in-depth postmortem analyses available for 6 patients [90]. DNA from transplanted cells was detected in the spinal cord of all samples near the injection sites, and nests of stem cells could also be visualized on histology (Fig. 3; [90]). Notably, the 5 patients who demonstrated a slowed progression or stabilization of disease in this phase I trial were all treated within approximately 2 years of symptom onset and had no bulbar features, suggesting that early intervention may provide a better response to this modality of stem cell treatment [91]. Again, this highlights the potential for stem cells to rescue native MNs, although the window for neuroprotection closes as the disease progresses.
Fig. 3.

Gross and histological analysis of male amyotrophic lateral sclerosis (ALS) spinal cord. Gross image of the spinal cord shows the cord surface at the site of human spinal cord stem cell (HSSC) transplant (a, b). The vascular anatomy between intraoperative videos (a) corresponds to the postmortem tissue (b). Cross-section of the cord shows no visible tissue disruption (c). Histological staining with hematoxylin and eosin (d, g), Luxol fast blue (e, h), and immunohistochemistry for glial fibrillary acidic protein (f, i) of 8-lm spinal cord sections from patient 4 are shown. Nest of putative HSSCs are outlined in (d–f). Scale bars: 1 mm (d–f); 50 lm (g–i). Reproduced from Tadesse et al., Ann Clin Transl Neurol, in press [90]
Given the safety and feasibility established in the phase I trial, a phase II study began in September 2013 and ended in July 2014. The phase II trial was designed to identify the maximum tolerated dose of stem cells coupled with the maximum tolerated number of cervical and lumbar injections (Table 3). There were 5 treatment groups with 3 patients per group, whereby Group A received a dose of 2 million cells via 5 bilateral cervical injections (10 total), Group B progressed to 4 million cells injected in 10 bilateral cervical injections (20 total), and Groups C and D received 6 and 8 million cells, respectively, in 20 cervical injections. The final group, group E, then received 20 injections of 8 million cells into both cervical and lumbar regions, for a total of 16 million cells. All surgeries have been completed and final outcome monitoring and data review are underway; it is anticipated that an initial safety report is forthcoming, and planning is ongoing for future trials with this therapy to assess efficacy.
Induced Pluripotent Stem Cells
The ability to reprogram differentiated cells into an embryonic-like state, thus generating induced pluripotent stem (iPS) cells, was first demonstrated in 2006 [115], and has opened the door for attractive disease modeling and therapeutic strategies in ALS. Notably, the possibility of generating pluripotent iPS cells as a therapeutic option eliminates ethical concerns, as well as the risk of tissue rejection. Early preclinical studies demonstrated the successful grafting of human iPS cells into rat spinal cords, which gave rise to NPCs and, in particular, to astrocyte-like cells [116]. Given the relatively early state of this technology, however, no clinical trials using iPS cells are underway, although it is likely that iPS cells will play an increasing role in ALS research in the near future. Additionally, as it has been shown that iPS cells can be generated from human dermal fibroblasts [117, 118] and many groups are capitalizing on this chance to produce embryonic-like iPS cells from patients with MN disease such as ALS [119–122]. Thus, the clinical potential of patient-derived iPS lies in the ability for patient-specific disease modeling, high-throughput drug screening, and perhaps eventually gene editing and cell replacement therapy [122, 123].
Conclusions and Future Directions
The results of preclinical studies supported the utilization of stem cell therapies as a means to modify the disease course in ALS. As previously mentioned, although early goals were set on MN replacement, preclinical models now suggest that the mechanistic benefits of stem cells in ALS favor a neighborhood theory, where secreted factors from stem cells support diseased MNs. Many results from animal models, however, have yet to be confirmed in human studies, and in order to realize the full potential of stem cells, further advancements are needed in terms of both therapeutic optimization and clinical trial design.
Moving forward, while stem cell therapeutic trials in humans are currently using a variety of cell types, state-of-the-art advances in disease modeling will advance our ability to determine which strategies represent viable treatment strategies. Notably, the ability to model ALS using iPS cells is a promising frontier because, at this time, our preclinical models mainly focus on animal models based on SOD1-G93A. Given that mutations in SOD1 only represent a very small fraction of all ALS cases, and that pathologic features may vary from familial and more frequent sporadic forms of the disease, SOD1-G93A models may not allow us to understand the full risks and benefits of potential stem cell therapies. By establishing iPS cell lines derived directly from patients with ALS, physiologically relevant and high-throughput in vitro evaluation of treatments will allow us to further characterize ALS disease mechanisms and further understand how stem cells act as modifiers.
Next, although unrelated to stem cell therapies directly, the diagnostic delays of around 1 year typical of ALS also hinder therapeutic windows [124]. It is likely that the microenvironment is already compromised at the time symptoms develop and even more so when ALS is diagnosed; therefore, improving the time to diagnosis as a means to achieve earlier institution of therapy is essential in order to confer the best possible opportunity for MN rescue. The development of more robust biomarkers, including electrodiagnostic testing, transcranial magnetic stimulation, and advanced imaging techniques will complement advances in stem cell therapy by allowing earlier detection of disease, as well as more detailed assessments of therapeutic benefits [125, 126].
Notably, the selection of optimal outcome measures is also a challenge not unique to stem cell studies. As highlighted above and in Table 2, most studies to date, including our own, have focused on the safety and tolerability of the stem cell therapy, with efficacy remaining a secondary end point. As these trials mature and safety is established, efficacy outcomes will be expected. Assessment of MNs, graft survival, and migration of stem cells in vivo, however, is currently not directly possible, making surrogate endpoints essential. The topic of disease measurements has been the subject of a recent review [125], and is also covered elsewhere in this issue of Neurotherapeutics. Hence, while a full discussion of outcome measures is outside the scope of this review, a few points deserve attention. First, unlike drug trials, pharmacokinetic and pharmacodynamic markers are not available. Some groups have assessed the presence of circulating cells in the blood, although this type of evaluation is not applicable to all stem cell delivery mechanisms. In upcoming trials with cellular therapies enhanced to secrete growth factors, functional measures of these cells may be appropriate. As in many ALS trials, ALSFRS-R and survival remain viable endpoints but do not have specificity for evaluating therapy-specific efficacy measures. In other words, while ALSFRS-R can provide a good measure of patient function, it does not provide specific insight into the MN microenvironment. This highlights the importance of an autopsy to assess, in appropriate cases, graft survival and pathologic alterations in response to the therapy. The drawback is that an analysis of the native MN survival may only reflect those MNs that would have survived regardless of the intervention. Some groups have also tried to demonstrate stem cell migration via tagging procedures, and while this can show the location of these stem cells, it does not prove efficacy. As a result, we must still rely on surrogate markers. In our trial we are particularly excited about the potential insight that spinal cord MRI could provide, and certainly the modalities of diffusion tensor imaging and MR spectroscopy may be useful for other varying stem cell delivery methods as well.
Finally, good clinical trial designs will be essential to fully understand the effects and mechanisms of stem cell therapies. To date, only rare stem cell trials have advanced to a point where control groups are integrated into the study design or efficacy is being evaluated. Double-blinded, randomized, placebo-controlled studies may be possible with certain paradigms, but, with increasing invasiveness of procedures, the ethics and utility of sham procedures/surgeries becomes problematic. Still, for stem cell therapy to remain a legitimate treatment avenue, rigorous adherence to principles of clinical trial design must remain paramount. Now is a time when scientific innovation, bioengineering technologies, and medical expertise have reached a threshold, thus allowing stem cell therapy for ALS to be realized. Opportunities for new discovery remain close, and a time when stem cell therapy may turn the tide against ALS hopefully remains just over the horizon.
Electronic supplementary material
(PDF 515 kb)
Acknowledgments
We wish to thank Stacey A. Sakowski, PhD, for her critical review of the manuscript. Funding support during the preparation of this review was received from the Program for Neurology Research & Discovery, the A. Alfred Taubman Medical Research Institute, and the National Institutes of Health (University of Michigan Clinician Scientist Training Program, NINDS R25NS089450 to K.S.C.).
References
- 1.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14(4):248–64. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 2.Ringholz GM, et al. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65(4):586–590. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 3.Goutman SA, et al. Minorities, men, and unmarried amyotrophic lateral sclerosis patients are more likely to die in an acute care facility. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(5–6):440–3. doi: 10.3109/21678421.2014.924143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585–91. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 6.Sorenson EJ, et al. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2008;71(22):1770–5. doi: 10.1212/01.wnl.0000335970.78664.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller RG, et al. Phase III randomized trial of gabapentin in patients with amyotrophic lateral sclerosis. Neurology. 2001;56(7):843–8. doi: 10.1212/wnl.56.7.843. [DOI] [PubMed] [Google Scholar]
- 8.Gage FH. Mammalian neural stem cells. Science. 2000;287(5457):1433–8. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 9.Temple S. The development of neural stem cells. Nature. 2001;414(6859):112–7. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- 10.Boulis NM, et al. Translational stem cell therapy for amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;8(3):172–6. doi: 10.1038/nrneurol.2011.191. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Gonzalez R, Kunckles P, Velasco I. Transient recovery in a rat model of familial amyotrophic lateral sclerosis after transplantation of motor neurons derived from mouse embryonic stem cells. Cell Transplant. 2009;18(10):1171–81. doi: 10.3727/096368909X12483162197123. [DOI] [PubMed] [Google Scholar]
- 12.Maragakis NJ. Stem cells and the ALS neurologist. Amyotroph Lateral Scler. 2010;11(5):417–23. doi: 10.3109/17482968.2010.489116. [DOI] [PubMed] [Google Scholar]
- 13.Lunn JS, Sakowski SA, Feldman EL. Concise review: Stem cell therapies for amyotrophic lateral sclerosis: recent advances and prospects for the future. Stem Cells. 2014;32(5):1099–109. doi: 10.1002/stem.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo X, et al. Characterization of a human fetal spinal cord stem cell line, NSI-566RSC, and its induction to functional motoneurons. J Tissue Eng Regen Med. 2010;4(3):181–193. doi: 10.1002/term.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deshpande DM, et al. Recovery from paralysis in adult rats using embryonic stem cells. Ann Neurol. 2006;60(1):32–44. doi: 10.1002/ana.20901. [DOI] [PubMed] [Google Scholar]
- 16.Harper JM, et al. Axonal growth of embryonic stem cell-derived motoneurons in vitro and in motoneuron-injured adult rats. Proc Natl Acad Sci U S A. 2004;101(18):7123–8. doi: 10.1073/pnas.0401103101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerr DA, et al. Human embryonic germ cell derivatives facilitate motor recovery of rats with diffuse motor neuron injury. J Neurosci. 2003;23(12):5131–40. doi: 10.1523/JNEUROSCI.23-12-05131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corti S, et al. Wild-type bone marrow cells ameliorate the phenotype of SOD1-G93A ALS mice and contribute to CNS, heart and skeletal muscle tissues. Brain. 2004;127(Pt 11):2518–32. doi: 10.1093/brain/awh273. [DOI] [PubMed] [Google Scholar]
- 19.Solomon JN, et al. Origin and distribution of bone marrow-derived cells in the central nervous system in a mouse model of amyotrophic lateral sclerosis. Glia. 2006;53(7):744–53. doi: 10.1002/glia.20331. [DOI] [PubMed] [Google Scholar]
- 20.Ohnishi S, et al. Intra-bone marrow-bone marrow transplantation slows disease progression and prolongs survival in G93A mutant SOD1 transgenic mice, an animal model mouse for amyotrophic lateral sclerosis. Brain Res. 2009;1296:216–24. doi: 10.1016/j.brainres.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Pastor D, et al. Bone marrow transplantation in hindlimb muscles of motoneuron degenerative mice reduces neuronal death and improves motor function. Stem Cells Dev. 2013;22(11):1633–44. doi: 10.1089/scd.2012.0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabanes C, et al. Neuroprotective effect of adult hematopoietic stem cells in a mouse model of motoneuron degeneration. Neurobiol Dis. 2007;26(2):408–18. doi: 10.1016/j.nbd.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 23.Corti S, et al. Systemic transplantation of c-kit+ cells exerts a therapeutic effect in a model of amyotrophic lateral sclerosis. Hum Mol Genet. 2010;19(19):3782–96. doi: 10.1093/hmg/ddq293. [DOI] [PubMed] [Google Scholar]
- 24.Pastor D, et al. Comparative effects between bone marrow and mesenchymal stem cell transplantation in GDNF expression and motor function recovery in a motorneuron degenerative mouse model. Stem Cell Rev. 2012;8(2):445–58. doi: 10.1007/s12015-011-9295-x. [DOI] [PubMed] [Google Scholar]
- 25.Garbuzova-Davis S, et al. Human umbilical cord blood treatment in a mouse model of ALS: optimization of cell dose. PLoS One. 2008;3(6):e2494. doi: 10.1371/journal.pone.0002494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garbuzova-Davis S, et al. Intravenous administration of human umbilical cord blood cells in a mouse model of amyotrophic lateral sclerosis: distribution, migration, and differentiation. J Hematother Stem Cell Res. 2003;12(3):255–70. doi: 10.1089/152581603322022990. [DOI] [PubMed] [Google Scholar]
- 27.Souayah N, et al. Defective neuromuscular transmission in the SOD1 G93A transgenic mouse improves after administration of human umbilical cord blood cells. Stem Cell Rev. 2012;8(1):224–8. doi: 10.1007/s12015-011-9281-3. [DOI] [PubMed] [Google Scholar]
- 28.Bigini P, et al. Intracerebroventricular administration of human umbilical cord blood cells delays disease progression in two murine models of motor neuron degeneration. Rejuvenation Res. 2011;14(6):623–39. doi: 10.1089/rej.2011.1197. [DOI] [PubMed] [Google Scholar]
- 29.Knippenberg S, et al. Intraspinal injection of human umbilical cord blood-derived cells is neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis. 2012;9(3):107–20. doi: 10.1159/000331327. [DOI] [PubMed] [Google Scholar]
- 30.Rizvanov AA, et al. Genetically modified human umbilical cord blood cells expressing vascular endothelial growth factor and fibroblast growth factor 2 differentiate into glial cells after transplantation into amyotrophic lateral sclerosis transgenic mice. Exp Biol Med (Maywood) 2011;236(1):91–8. doi: 10.1258/ebm.2010.010172. [DOI] [PubMed] [Google Scholar]
- 31.Rizvanov AA, et al. Human umbilical cord blood cells transfected with VEGF and L(1)CAM do not differentiate into neurons but transform into vascular endothelial cells and secrete neuro-trophic factors to support neuro-genesis-a novel approach in stem cell therapy. Neurochem Int. 2008;53(6–8):389–94. doi: 10.1016/j.neuint.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 32.Habisch HJ, et al. Intrathecal application of neuroectodermally converted stem cells into a mouse model of ALS: limited intraparenchymal migration and survival narrows therapeutic effects. J Neural Transm. 2007;114(11):1395–406. doi: 10.1007/s00702-007-0748-y. [DOI] [PubMed] [Google Scholar]
- 33.Uccelli A, et al. Intravenous mesenchymal stem cells improve survival and motor function in experimental amyotrophic lateral sclerosis. Mol Med. 2012;18:794–804. doi: 10.2119/molmed.2011.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forostyak S, et al. Mesenchymal stromal cells prolong the lifespan in a rat model of amyotrophic lateral sclerosis. Cytotherapy. 2011;13(9):1036–46. doi: 10.3109/14653249.2011.592521. [DOI] [PubMed] [Google Scholar]
- 35.Boucherie C, et al. Chimerization of astroglial population in the lumbar spinal cord after mesenchymal stem cell transplantation prolongs survival in a rat model of amyotrophic lateral sclerosis. J Neurosci Res. 2009;87(9):2034–46. doi: 10.1002/jnr.22038. [DOI] [PubMed] [Google Scholar]
- 36.Zhao CP, et al. Human mesenchymal stromal cells ameliorate the phenotype of SOD1-G93A ALS mice. Cytotherapy. 2007;9(5):414–26. doi: 10.1080/14653240701376413. [DOI] [PubMed] [Google Scholar]
- 37.Vercelli A, et al. Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2008;31(3):395–405. doi: 10.1016/j.nbd.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 38.Kim H, et al. Dose-dependent efficacy of ALS-human mesenchymal stem cells transplantation into cisterna magna in SOD1-G93A ALS mice. Neurosci Lett. 2010;468(3):190–4. doi: 10.1016/j.neulet.2009.10.074. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki M, et al. Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Mol Ther. 2008;16(12):2002–10. doi: 10.1038/mt.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knippenberg S, et al. Intracerebroventricular injection of encapsulated human mesenchymal cells producing glucagon-like peptide 1 prolongs survival in a mouse model of ALS. PLoS One. 2012;7(6):e36857. doi: 10.1371/journal.pone.0036857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan-Il C, et al. Neural induction with neurogenin 1 enhances the therapeutic potential of mesenchymal stem cells in an amyotrophic lateral sclerosis mouse model. Cell Transplant. 2013;22(5):855–70. doi: 10.3727/096368912X637019. [DOI] [PubMed] [Google Scholar]
- 42.Morita E, et al. A novel cell transplantation protocol and its application to an ALS mouse model. Exp Neurol. 2008;213(2):431–8. doi: 10.1016/j.expneurol.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 43.Martin LJ, Liu Z. Adult olfactory bulb neural precursor cell grafts provide temporary protection from motor neuron degeneration, improve motor function, and extend survival in amyotrophic lateral sclerosis mice. J Neuropathol Exp Neurol. 2007;66(11):1002–18. doi: 10.1097/nen.0b013e318158822b. [DOI] [PubMed] [Google Scholar]
- 44.Lepore AC, et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11(11):1294–301. doi: 10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lepore AC, et al. Human glial-restricted progenitor transplantation into cervical spinal cord of the SOD1 mouse model of ALS. PLoS One. 2011;6(10):e25968. doi: 10.1371/journal.pone.0025968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corti S, et al. Neural stem cells LewisX+ CXCR4+ modify disease progression in an amyotrophic lateral sclerosis model. Brain. 2007;130(Pt 5):1289–305. doi: 10.1093/brain/awm043. [DOI] [PubMed] [Google Scholar]
- 47.Mitrecic D, et al. Distribution, differentiation, and survival of intravenously administered neural stem cells in a rat model of amyotrophic lateral sclerosis. Cell Transplant. 2010;19(5):537–48. doi: 10.3727/096368910X498269. [DOI] [PubMed] [Google Scholar]
- 48.Hwang DH, et al. Intrathecal transplantation of human neural stem cells overexpressing VEGF provide behavioral improvement, disease onset delay and survival extension in transgenic ALS mice. Gene Ther. 2009;16(10):1234–44. doi: 10.1038/gt.2009.80. [DOI] [PubMed] [Google Scholar]
- 49.Park S, et al. Growth factor-expressing human neural progenitor cell grafts protect motor neurons but do not ameliorate motor performance and survival in ALS mice. Exp Mol Med. 2009;41(7):487–500. doi: 10.3858/emm.2009.41.7.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu L, et al. Human neural stem cell grafts ameliorate motor neuron disease in SOD-1 transgenic rats. Transplantation. 2006;82(7):865–75. doi: 10.1097/01.tp.0000235532.00920.7a. [DOI] [PubMed] [Google Scholar]
- 51.Xu L, et al. Human neural stem cell grafts in the spinal cord of SOD1 transgenic rats: differentiation and structural integration into the segmental motor circuitry. J Comp Neurol. 2009;514(4):297–309. doi: 10.1002/cne.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan J, et al. Combined immunosuppressive agents or CD4 antibodies prolong survival of human neural stem cell grafts and improve disease outcomes in amyotrophic lateral sclerosis transgenic mice. Stem Cells. 2006;24(8):1976–85. doi: 10.1634/stemcells.2005-0518. [DOI] [PubMed] [Google Scholar]
- 53.Klein SM, et al. GDNF delivery using human neural progenitor cells in a rat model of ALS. Hum Gene Ther. 2005;16(4):509–21. doi: 10.1089/hum.2005.16.509. [DOI] [PubMed] [Google Scholar]
- 54.Suzuki M, et al. GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One. 2007;2(8):e689. doi: 10.1371/journal.pone.0000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garbuzova-Davis S, et al. Positive effect of transplantation of hNT neurons (NTera 2/D1 cell-line) in a model of familial amyotrophic lateral sclerosis. Exp Neurol. 2002;174(2):169–80. doi: 10.1006/exnr.2002.7860. [DOI] [PubMed] [Google Scholar]
- 56.Gao J, et al. Human neural stem cell-derived cholinergic neurons innervate muscle in motoneuron deficient adult rats. Neuroscience. 2005;131(2):257–62. doi: 10.1016/j.neuroscience.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 57.Xu L, et al. Dual transplantation of human neural stem cells into cervical and lumbar cord ameliorates motor neuron disease in SOD1 transgenic rats. Neurosci Lett. 2011;494(3):222–6. doi: 10.1016/j.neulet.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hefferan MP, et al. Human neural stem cell replacement therapy for amyotrophic lateral sclerosis by spinal transplantation. PLoS One. 2012;7(8):e42614. doi: 10.1371/journal.pone.0042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Popescu IR, et al. Neural progenitors derived from human induced pluripotent stem cells survive and differentiate upon transplantation into a rat model of amyotrophic lateral sclerosis. Stem Cells Transl Med. 2013;2(3):167–74. doi: 10.5966/sctm.2012-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nizzardo M, et al. Minimally invasive transplantation of iPSC-derived ALDHhiSSCloVLA4+ neural stem cells effectively improves the phenotype of an amyotrophic lateral sclerosis model. Hum Mol Genet. 2014;23(2):342–54. doi: 10.1093/hmg/ddt425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, et al. Preliminary investigation of effect of granulocyte colony stimulating factor on amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(5–6):430–1. doi: 10.3109/17482960802588059. [DOI] [PubMed] [Google Scholar]
- 62.Nefussy B, et al. Recombinant human granulocyte-colony stimulating factor administration for treating amyotrophic lateral sclerosis: A pilot study. Amyotroph Lateral Scler. 2010;11(1–2):187–93. doi: 10.3109/17482960902933809. [DOI] [PubMed] [Google Scholar]
- 63.Tarella C, et al. Consistent bone marrow-derived cell mobilization following repeated short courses of granulocyte-colony-stimulating factor in patients with amyotrophic lateral sclerosis: results from a multicenter prospective trial. Cytotherapy. 2010;12(1):50–9. doi: 10.3109/14653240903300682. [DOI] [PubMed] [Google Scholar]
- 64.Chio A, et al. Repeated courses of granulocyte colony-stimulating factor in amyotrophic lateral sclerosis: clinical and biological results from a prospective multicenter study. Muscle Nerve. 2011;43(2):189–95. doi: 10.1002/mus.21851. [DOI] [PubMed] [Google Scholar]
- 65.Cashman N, et al. Pilot study of granulocyte colony stimulating factor (G-CSF)-mobilized peripheral blood stem cells in amyotrophic lateral sclerosis (ALS) Muscle Nerve. 2008;37(5):620–5. doi: 10.1002/mus.20951. [DOI] [PubMed] [Google Scholar]
- 66.Janson CG, et al. Human intrathecal transplantation of peripheral blood stem cells in amyotrophic lateral sclerosis. J Hematother Stem Cell Res. 2001;10(6):913–5. doi: 10.1089/152581601317211015. [DOI] [PubMed] [Google Scholar]
- 67.Appel SH, et al. Hematopoietic stem cell transplantation in patients with sporadic amyotrophic lateral sclerosis. Neurology. 2008;71(17):1326–34. doi: 10.1212/01.wnl.0000327668.43541.22. [DOI] [PubMed] [Google Scholar]
- 68.Martinez HR, et al. Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy. 2009;11(1):26–34. doi: 10.1080/14653240802644651. [DOI] [PubMed] [Google Scholar]
- 69.Martinez HR, et al. Stem cell transplantation in amyotrophic lateral sclerosis patients: methodological approach, safety, and feasibility. Cell Transplant. 2012;21(9):1899–907. doi: 10.3727/096368911X582769. [DOI] [PubMed] [Google Scholar]
- 70.Karussis D, et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67(10):1187–94. doi: 10.1001/archneurol.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prabhakar S, et al. Autologous bone marrow-derived stem cells in amyotrophic lateral sclerosis: a pilot study. Neurol India. 2012;60(5):465–9. doi: 10.4103/0028-3886.103185. [DOI] [PubMed] [Google Scholar]
- 72.Baek W, et al. Stem cell transplantation into the intraventricular space via an Ommaya reservoir in a patient with amyotrophic lateral sclerosis. J Neurosurg Sci. 2012;56(3):261–3. [PubMed] [Google Scholar]
- 73.Mazzini L, et al. Autologous mesenchymal stem cells: clinical applications in amyotrophic lateral sclerosis. Neurol Res. 2006;28(5):523–6. doi: 10.1179/016164106X116791. [DOI] [PubMed] [Google Scholar]
- 74.Mazzini L, et al. Stem cell treatment in Amyotrophic Lateral Sclerosis. J Neurol Sci. 2008;265(1–2):78–83. doi: 10.1016/j.jns.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 75.Mazzini L, et al. Mesenchymal stromal cell transplantation in amyotrophic lateral sclerosis: a long-term safety study. Cytotherapy. 2012;14(1):56–60. doi: 10.3109/14653249.2011.613929. [DOI] [PubMed] [Google Scholar]
- 76.Mazzini L, et al. Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: A Phase I clinical trial. Exp Neurol. 2010;223(1):229–37. doi: 10.1016/j.expneurol.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 77.Blanquer M, et al. A surgical technique of spinal cord cell transplantation in amyotrophic lateral sclerosis. J Neurosci Methods. 2010;191(2):255–7. doi: 10.1016/j.jneumeth.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 78.Blanquer M, et al. Neurotrophic Bone Marrow Cellular Nests Prevent Spinal Motoneuron Degeneration in Amyotrophic Lateral Sclerosis Patients: A Pilot Safety Study. STEM CELLS. 2012;30(6):1277–1285. doi: 10.1002/stem.1080. [DOI] [PubMed] [Google Scholar]
- 79.Deda H, et al. Treatment of amyotrophic lateral sclerosis patients by autologous bone marrow-derived hematopoietic stem cell transplantation: a 1-year follow-up. Cytotherapy. 2009;11(1):18–25. doi: 10.1080/14653240802549470. [DOI] [PubMed] [Google Scholar]
- 80.Moviglia GA, et al. Feasibility, safety, and preliminary proof of principles of autologous neural stem cell treatment combined with T-cell vaccination for ALS patients. Cell Transplant. 2012;21(Suppl 1):S57–63. doi: 10.3727/096368912X633770. [DOI] [PubMed] [Google Scholar]
- 81.Piepers S, van den Berg LH. No benefits from experimental treatment with olfactory ensheathing cells in patients with ALS. Amyotroph Lateral Scler. 2010;11(3):328–30. doi: 10.3109/17482961003663555. [DOI] [PubMed] [Google Scholar]
- 82.Chew S, et al. Olfactory ensheathing glia injections in Beijing: misleading patients with ALS. Amyotroph Lateral Scler. 2007;8(5):314–6. doi: 10.1080/17482960701419315. [DOI] [PubMed] [Google Scholar]
- 83.Giordana MT, et al. Neuropathology of olfactory ensheathing cell transplantation into the brain of two amyotrophic lateral sclerosis (ALS) patients. Brain Pathol. 2010;20(4):730–7. doi: 10.1111/j.1750-3639.2009.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang H, et al. Fetal olfactory ensheathing cells transplantation in amyotrophic lateral sclerosis patients: a controlled pilot study. Clin Transplant. 2008;22(6):710–8. doi: 10.1111/j.1399-0012.2008.00865.x. [DOI] [PubMed] [Google Scholar]
- 85.Chen L, et al. Short-term outcome of olfactory ensheathing cells transplantation for treatment of amyotrophic lateral sclerosis. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2007;21(9):961–6. [PubMed] [Google Scholar]
- 86.Chen L, et al. Olfactory ensheathing cell neurorestorotherapy for amyotrophic lateral sclerosis patients: benefits from multiple transplantations. Cell Transplant. 2012;21(Suppl 1):S65–77. doi: 10.3727/096368912X633789. [DOI] [PubMed] [Google Scholar]
- 87.Riley J, et al. Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: a phase I safety trial, technical note, and lumbar safety outcomes. Neurosurgery. 2012;71(2):405–16. doi: 10.1227/NEU.0b013e31825ca05f. [DOI] [PubMed] [Google Scholar]
- 88.Glass JD, et al. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells. 2012;30(6):1144–51. doi: 10.1002/stem.1079. [DOI] [PubMed] [Google Scholar]
- 89.Riley J, et al. Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: a phase I trial, cervical microinjection, and final surgical safety outcomes. Neurosurgery. 2014;74(1):77–87. doi: 10.1227/NEU.0000000000000156. [DOI] [PubMed] [Google Scholar]
- 90.Tadesse T, et al. Analysis of graft survival in a trial of stem cell transplant in ALS. Ann Clin Transl Neurol 2014; p. n/a-n/a [DOI] [PMC free article] [PubMed]
- 91.Feldman EL, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann Neurol. 2014;75(3):363–73. doi: 10.1002/ana.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 93.Ende N, et al. Human umbilical cord blood effect on sod mice (amyotrophic lateral sclerosis) Life Sci. 2000;67(1):53–9. doi: 10.1016/s0024-3205(00)00602-0. [DOI] [PubMed] [Google Scholar]
- 94.Chen R, Ende N. The potential for the use of mononuclear cells from human umbilical cord blood in the treatment of amyotrophic lateral sclerosis in SOD1 mice. J Med. 2000;31(1–2):21–30. [PubMed] [Google Scholar]
- 95.Wichterle H, et al. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110(3):385–97. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 96.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187(6):761–72. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boillee S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312(5778):1389–92. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 98.Borchelt DR. Amyotrophic lateral sclerosis–are microglia killing motor neurons? N Engl J Med. 2006;355(15):1611–3. doi: 10.1056/NEJMcibr064358. [DOI] [PubMed] [Google Scholar]
- 99.Clement AM, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302(5642):113–7. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 100.Yamanaka K, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–3. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Krakora D, et al. Synergistic effects of GDNF and VEGF on lifespan and disease progression in a familial ALS rat model. Mol Ther. 2013;21(8):1602–10. doi: 10.1038/mt.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beers DR, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103(43):16021–6. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Y, Field PM, Raisman G. Repair of adult rat corticospinal tract by transplants of olfactory ensheathing cells. Science. 1997;277(5334):2000–2. doi: 10.1126/science.277.5334.2000. [DOI] [PubMed] [Google Scholar]
- 104.Ramon-Cueto A, et al. Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron. 2000;25(2):425–35. doi: 10.1016/s0896-6273(00)80905-8. [DOI] [PubMed] [Google Scholar]
- 105.Lunn JS, et al. Stem cell technology for the study and treatment of motor neuron diseases. Regen Med. 2011;6(2):201–13. doi: 10.2217/rme.11.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hubel K, Engert A. Clinical applications of granulocyte colony-stimulating factor: an update and summary. Ann Hematol. 2003;82(4):207–13. doi: 10.1007/s00277-003-0628-y. [DOI] [PubMed] [Google Scholar]
- 107.Deng J, et al. Bone marrow mesenchymal stem cells can be mobilized into peripheral blood by G-CSF in vivo and integrate into traumatically injured cerebral tissue. Neurol Sci. 2011;32(4):641–51. doi: 10.1007/s10072-011-0608-2. [DOI] [PubMed] [Google Scholar]
- 108.Tanaka M, et al. Intrathecal upregulation of granulocyte colony stimulating factor and its neuroprotective actions on motor neurons in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2006;65(8):816–25. doi: 10.1097/01.jnen.0000232025.84238.e1. [DOI] [PubMed] [Google Scholar]
- 109.Uccelli A, et al. Neuroprotective features of mesenchymal stem cells. Best Pract Res Clin Haematol. 2011;24(1):59–64. doi: 10.1016/j.beha.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 110.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8(9):726–36. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 111.Woodbury D, et al. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res. 2000;61(4):364–70. doi: 10.1002/1097-4547(20000815)61:4<364::AID-JNR2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 112.Sanchez-Ramos J, et al. Adult bone marrow stromal cells differentiate into neural cells in vitro. Exp Neurol. 2000;164(2):247–56. doi: 10.1006/exnr.2000.7389. [DOI] [PubMed] [Google Scholar]
- 113.Bossolasco P, et al. Neuro-glial differentiation of human bone marrow stem cells in vitro. Exp Neurol. 2005;193(2):312–25. doi: 10.1016/j.expneurol.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 114.Lu P, Blesch A, Tuszynski MH. Induction of bone marrow stromal cells to neurons: differentiation, transdifferentiation, or artifact? J Neurosci Res. 2004;77(2):174–91. doi: 10.1002/jnr.20148. [DOI] [PubMed] [Google Scholar]
- 115.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 116.Sareen D, et al. Human induced pluripotent stem cells are a novel source of neural progenitor cells (iNPCs) that migrate and integrate in the rodent spinal cord. J Comp Neurol. 2014;522(12):2707–28. doi: 10.1002/cne.23578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 118.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 119.Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–21. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 120.Burkhardt MF, et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 2013;56:355–64. doi: 10.1016/j.mcn.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chestkov IV, et al. Patient-Specific Induced Pluripotent Stem Cells for SOD1-Associated Amyotrophic Lateral Sclerosis Pathogenesis Studies. Acta Naturae. 2014;6(1):54–60. [PMC free article] [PubMed] [Google Scholar]
- 122.Egawa N, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med 2012; 4(145): p. 145ra104 [DOI] [PubMed]
- 123.Faravelli I, et al. Motor neuron derivation from human embryonic and induced pluripotent stem cells: experimental approaches and clinical perspectives. Stem Cell Res Ther. 2014;5(4):87. doi: 10.1186/scrt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Paganoni S, et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS) Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(5–6):453–6. doi: 10.3109/21678421.2014.903974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Simon, N.G., et al., Quantifying disease progression in amyotrophic lateral sclerosis. Ann Neurol, 2014. 76(5): p. 643–57. [DOI] [PMC free article] [PubMed]
- 126.Foerster BR, Welsh RC, Feldman EL. 25 years of neuroimaging in amyotrophic lateral sclerosis. Nat Rev Neurol. 2013;9(9):513–24. doi: 10.1038/nrneurol.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 515 kb)