

Abstract
X-linked intellectual disability is the most common form of cognitive disability in males. Syndromic intellectual disability encompasses cognitive deficits with other medical and behavioral manifestations. Recently, a large family with a novel form of syndromic X-linked intellectual disability was characterized. Eight of 24 members of the family are male and had cognitive dysfunction, short stature, aphasia, skeletal abnormalities, and minor anomalies. To identify the causative gene(s), we performed exome sequencing in three affected boys, both parents, and an unaffected sister. We identified a haplotype consisting of eight variants located in cis within the linkage region that segregated with affected members in the family. Of these variants, two were novel. The first was at the splice-donor site of intron 7 (c.974+1G>T) in the cullin-RING ubiquitin ligase (E3) gene, CUL4B. This variant is predicted to result in failure to splice and remove intron 7 from the primary transcript. The second variant mapped to the 3′-UTR region of the KAISO gene (c.1127T>G). Sanger sequencing validated the variants in these relatives as well as in three affected males and five carriers. The KAISO gene variant was predicted to create a binding site for the microRNAs miR-4999 and miR-4774; however, luciferase expression assays failed to validate increased targeting of these miRNAs to the variant 3′-UTR. This SNP may affect 3′-UTR structure leading to decreased mRNA stability. Our results suggest that the intellectual disability phenotype in this family is caused by aberrant splicing and removal of intron 7 from CUL4B gene primary transcript.
Keywords: intellectual disability, X-linked, exome sequencing
INTRODUCTION
Intellectual disability (ID) is characterized by limitations in intellectual function and developmental delays in motor, cognitive, and speech functioning [Lubs et al., 2012; Bassani et al., 2013] and affects 1–3% of the population [Bassani et al., 2013]. Intellectual disability can be caused by genetic and non-genetic factors such as infections or perinatal asphyxia. Genetic causes include large deletions, duplications, or aneuplodies affecting multiple genes [Vissers et al., 2003; Ravnan et al., 2006].
X-linked ID (XLID) accounts for 5–10% of all intellectual disability [Bassani et al., 2013] and is the most common cause of ID in males. X-linked ID can be categorized into two groups based on phenotype. Syndromic ID is associated with neurological or behavioral manifestations, structural anomalies where patients often manifest autism and/or seizures [Stevenson et al., 2012]. In contrast, non-syndromic ID is classified as those in which ID is the only characteristic. To date, more than 150 XLID syndromes have been described and over 100 mutated genes have been identified, accounting for 81 of these syndromes [Cabezas et al., 2000; Tarpey et al., 2009; Lubs et al., 2012; Stevenson et al., 2012; Bassani et al., 2013].
We previously reported a form of syndromic XLID associated with short stature in a large family [Vitale et al., 2001]. The 26-member family consisted of eight affected males with severe ID, lack of speech, short stature, microcephaly, coarse lineaments, short down-slanted palpebral fissure, bulbous nose, wide mouth, small ear lobes, small hands and feet, as well as short toes and fingers (Fig. 1). Carriers did not exhibit ID, but two female cousins had a minor learning disability. Linkage analysis in the family identified a 16 cm region on the X-chromosome with a LOD score of 3.61 [Vitale et al., 2001]. Additional families with XLID with some overlapping phenotypes have been reported [Cabezas et al., 2000; Tarpey et al., 2007; Zou et al., 2007; Badura-Stronka et al., 2010; Isidor et al., 2010] and in most cases, mutations in the CUL4B (NM_003588.3) gene were identified as causative in these families (Supplemental Table SI—see supporting information online). CUL4B is within the X-chromosome region of linkage in this family. Here, we report on the use of exome sequencing to identify the causative variant in this family.
FIG. 1.

Pedigree of the extended family with the XLID phenotype (A). The six members of the family for whom exome sequencing was performed are highlighted in the red box. Sanger sequencing was performed to validate the identified variants in the CUL4B gene (B) and the KAISO gene (C). Sanger sequencing of the two variants was performed also on members of the extended family, and those containing the two variants are highlighted (asterisk).
MATERIALS AND METHODS
Exome Sequencing and Data Analysis
Informed consent signed by legal guardians and/or patients for genetic studies was obtained during routine clinical care according to the World Medical Association Declaration of Helsinki. Exome sequencing was performed using the TargetSeq enrichment kit and sequenced on the SOLiD 5500×l (Life Technologies, Carlsbad, CA) platform. Paired-end sequence reads of 50 and 35 bp in length were generated and aligned to the hg19 reference genome using LifeScope software (Life Technologies). Only those sequence reads mapping uniquely to the genome and with no more than two mismatches were considered. Post-alignment processing including re-aligning around insertions and deletions (indels) and base quality recalibration were performed using the Genome Analysis Toolkit (GATK, Broad Institute, Cambridge, MA) [McKenna et al., 2010; DePristo et al., 2011]. The SNP and indel calls were made using the UnifiedGenotyper module of GATK. All variants were further annotated using the SNPeff analysis package [Cingolani et al., 2012]. Identified variants were maintained if they met the following criteria: (i) a minimum coverage ≥20×, (ii) base and strand mapping qualities ≥20, and (iii) ≥20% of the reads having the non-reference allele. Sanger sequencing was used to validate candidate variants on an Applied Biosystems 3730 DNA Sequencer (Life Technologies).
Luciferase Assay
A 333 bp sequence containing part of KAISO (NM_00118472.1) 3′-UTR was amplified from DNA samples of patients 102 (carrying SNP) and 103 (normal) and cloned into Psi-check2 dual-luciferase plasmid at the Xho I site located in the 3′-UTR of the renilla luciferase gene. As a positive control, a portion of the 3′-UTR (1,508 bp sequence; with miR-211 seed sequence) of a known miR-211 target gene, TCF4 (NM_001243230.1), was cloned into psi-check2. Luciferase constructs were transfected into HEK293 cells. Firefly and renilla luciferase activities were evaluated using the Dual-Glo® Luciferase Assay System (Promega, Madison, WI) according to manufacturer’s protocol were assayed 2-days post-transfection. The ratio of renilla/firefly luciferase luminescence signal was used to evaluate the effect of miR-4774 or miR-4999 on relative expression of the KAISO gene caused by increased targeting of the variant in the 3′-UTR. The effect of miR-4774 or miR-4999 on renilla luciferase intensity (normalized to firefly/luciferase intensity) was determined using the Dual-Glo® renilla/luciferase assay. Total RNA harvested from cells using Trizol reagent, and mature miR-4774 and miR-4999 expression in the samples were verified using TaqMan miRNA assay (Life Technologies). The qRT-PCR was performed using ABI PRISM® 7000 Sequence Detection System (Life Technologies) using settings indicated by the manufacturer.
RESULTS
Exome Sequencing Identifies a Haplotype of Two Novel Variants in an XLID Linkage Region
A total of 38,802 unique variants were identified (a summary of sequencing and read mapping results is shown in Supplemental Table SII—in supporting information online). As the phenotype and linkage studies indicated X-linked inheritance (Fig. 1A), we focused our analysis on X-chromosome variants (676 variants) within the linkage region (38 variants, Supplemental Table SIII—in supporting information online). Of these 38 variants, eight were identified as segregating with the phenotype in the family (i.e., all three affected boys had the variant, while the mother and daughter were heterozygous and the father did not have the variant). Included among these variants, were four synonymous mutations in the SEPT6 (NM_145799.3), LAMP2 (NM_002294.2), DOCK11 (NM_144658.3), and CXorf64 (NM_001122716.1) genes and two additional changes resulted in missense mutations in the UTP14A and BCORL1 genes, but both were predicted to be non-damaging and catalogued previously in dbSNP or the 1000 Genomes Project.
The remaining two variants were novel (not represented in dbSNP or the 1000 Genomes project). The first is in the 3′-UTR of the KAISO gene (c.1127T>G; transcript id NM_00118472.1). KAISO binds to methylated DNA, represses target genes in the Wnt signaling pathway and is highly expressed in the brain [Della Ragione et al., 2006; Defossez and Stancheva, 2011; Blattler et al., 2013]. The second is at the splice-donor site at the exon7/intron 7 splice donor site (c.974+1G>T; transcript ID NM_003588.3) in the CUL4B (NM_003588.3) gene, and is predicted to result in aberrant or no splicing of intron 7 from the primary transcript. CUL4B is a E3 ubiquitin ligase, which catalyzes polyubiquitinylation. Additionally, CUL4B has previously been associated with XLID (Table I). Sanger sequencing validated both of the identified variants in the six sequenced family members as well as for their presence in three additional affected members of the extended pedigree, and five carriers (indicated in the pedigree in Fig. 1B and C and Supplemental Fig. S1—in supporting information online).
TABLE I.
XLID Associated CUL4B Mutations
| CUL4B variant | Mutation class | Refs. |
|---|---|---|
| c.638C>T; p.T213I | Missense | Tarpey et al. AJHG [2007] |
| c.901-2A>G | Splice (exon 6/7 splice acceptor) | Tarpey et al. AJHG [2007] |
| c.974+1G>T | Splice (intron 7/8 splice donor) | This study |
| c.1007_1011delTTATA | Deletion | Tarpey et al. AJHG [2007] |
| c.1162C>T; p.R388X | Nonsense | Tarpey et al. AJHG [2007]; Zou et al. AJHG [2007] |
| c.1714C>T; p.R572C | Missense | Tarpey et al. AJHG [2007] |
| c.2107A>T; p.K703X | Nonsense | Badura-Stronka et al. Clin Genet [2010] |
| c.2234T>C; p.V745A | Missense | Tarpey et al. AJHG [2007] |
| c.2493G>A; p.T831T | Cryptic splice site | Tarpey et al. AJHG [2007] |
| c.2566C>T; p.R856X | Nonsense | Tarpey et al. AJHG [2007] |
| CUL4B gene deletion | Deletion of gene region | Isidor et al. Am J Med Genet Part A [2010];Ravn et al. Clin Genet [2012] |
The c.1127T>G KAISO 3′-UTR Variant is Predicted to Create a Binding site for miR-4999 and miR-4774
In contrast to the splice-donor site mutation of CUL4B, the effect of the 3′-UTR variant on gene function was much less obvious. While the mutation does not result in a protein change, the 3′-UTR contains regulatory regions, which influence post-transcriptional expression, including polyadenylation, translational efficiency, stability of the mRNA as well as harboring miRNA binding sites [Chatterjee and Pal, 2009; Barrett et al., 2012]. With this in mind, we hypothesized that this variant may affect the binding of microRNAs (miRNAs) within the 3′-UTR of the gene. To determine if the KAISO c.1127T>G variant functioned in this manner, both RNA22 [Miranda et al., 2006] and TargetScan [Lewis et al., 2005] algorithms were used to determine if this variant affected binding of miRNAs. Using both programs, we identified two miRNAs (miR-4999 and miR-4774) for which the variant was predicted to create a new binding site.
To test this hypothesis, the KAISO 3′-UTRs harboring either normal or variant allele was cloned into the 3′-UTR of the renilla luciferase gene within the psi-check2 dual-luciferase vector. This dual-luciferase vector contains both renilla and firefly cDNAs, so relative expression is readily determined and corrected for variation in transfection efficiencies. Each of these vectors was co-expressed with pre-miR-4774 or pre-miR-4999 in HEK293 cells, and renilla luciferase expression standardized to firefly expression was evaluated after 2 days. If the variant created a new 3′-UTR binding site for miRNA targeting, we would expect to see a decrease in relative renilla expression in these assays. As shown in Supplemental Fig. S2 (see supporting information online), expression of pre-miR-4774 or -4999 had no effect on relative expression of renilla luciferase containing normal or variant SNP. In a separate experiment, we verified that transfecting HEK293 cells with pre-miR-4774 or -4999 resulted in high expression of the mature form of the corresponding miRNAs in HEK293 cells. In addition, as a positive control, psi-check2 containing renilla luciferase gene with the 3′-UTR of a known miR-211 target gene (TCF4) was co-expressed with pre-miR-211 in HEK293 cells. Under the same experimental conditions, miR-211 suppressed expression of renilla luciferase gene containing TCF4 3′-UTR (Supplemental Fig. S2—in supporting information online). Taken together, our data indicated that the novel SNP in the ZBTB33 3′-UTR did not create a target site for miR-4774 or -4999, but nonetheless, the variant may still function to regulate KAISO gene function.
DISCUSSION
We performed exome sequencing in a large family with syndromic XLID. Previous linkage analysis of the family identified a 16 cm locus (lod score of 3.61) segregating with the phenotype [Vitale et al., 2001]. Our analysis identified two novel variants located within the region of linkage, which segregated (Supplemental Table SIII—in supporting information online) with the phenotype. The first variant was located within the 3′-UTR of the KAISO gene and the second was a splice donor mutation in the CUL4B gene. The combination of the previous linkage study and the segregation of the variants within the family as well as presence in additional affected members of the extended family implicate both changes as potentially causative for the XLID phenotype.
CUL4B is a scaffold protein of the Cullin4B-RING E3 ligase complex (CRL4B) that regulates degradation of cellular proteins, signals nucleotide excision repair, and DNA damage response control. One indel (c.1007_1011delTTATA), three nonsense (c.1162C>T, c.2107A>T and c.2566C>T), one exon 7 splice acceptor site (c.901-2A>G), one cryptic splice (c.2493G>A), three missense mutations (c.1714C>T, c.2234T>C, c.638C>T), and one deletion have been associated to date with ID (Fig. 2 and Table I). It should be noted that the families for which these mutations were identified shared overlapping phenotypic features with the family examined here including short stature, skeletal abnormalities, and dysmorphic features, facial dysmorphism [Zou et al., 2007; Tarpey et al., 2009; Isidor et al., 2010] (OMIN 300304) (Supplemental Table SI—in supporting information online). In addition, it has been shown that in β-thalassemia patients harboring a G to T change at the splice junction in the first nucleotide of intron 2 results in a β0 phenotype resulting in no β-globin chain production [Baird et al., 1981; Talerico and Berget, 1990]. With the high degree of overlapping phenotypic features in this family and the reported families with CUL4B mutations, the identified splice-donor mutation identified here (c.974+1G>T) is probably causing the XLID phenotype observed in this family. Furthermore, we believe that this variant results in aberrant or no splicing of the CUL4B gene ultimately leading to little or no protein produced.
FIG. 2.
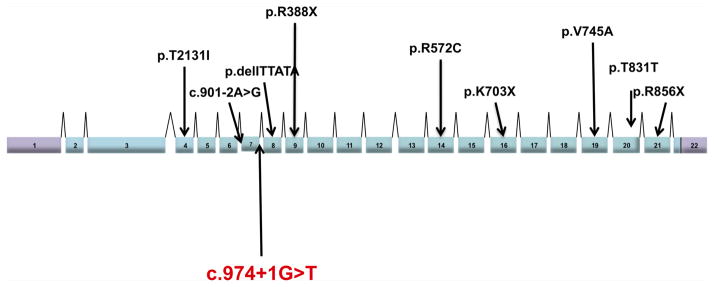
Shown is a schematic representation of the genomic structure of the CUL4B gene with all the locations of the previously characterized mutations. The splice-site mutation identified here is highlighted.
The second novel variant identified was in the 3′-UTR (c.1127T>G) of the KAISO gene. KAISO is a member of the zinc finger and BTB family of transcription factors [Della Ragione et al., 2006; Defossez and Stancheva, 2011; Blattler et al., 2013]. Functionally, KAISO is a methyl-CpG-binding protein, which binds to methylated DNA and recruits chromatin remodeling proteins, which facilitate transcriptional silencing. In Xenopus, inhibition of KAISO function results in premature transcription of many genes leading to developmental arrest and apoptosis [Ruzov et al., 2004; Defossez and Stancheva, 2011]. This dysfunction is similar to the effect of removal of the DNA methylating enzyme DNMT1, suggesting a role as a methylation-dependent repressor. In contrast, in the mouse embryo, KAISO knockout does not cause major phenotypic disturbances; however, minor phenotypic changes may occur [Della Ragione et al., 2006; Martin Caballero et al., 2009]. Additionally, its high expression in the brain is suggestive of some neuronal function for this protein [Della Ragione et al., 2006].
The role of methyl-binding proteins in the vertebrate nervous system has been studied in relation to X-linked syndromes, including ID. Effects observed in Xenopus embryos are similar to those when other methyl-deacylases such as MECP2 are inhibited. MECP2 is the cause of Rett syndrome, one of the most common causes of ID [Berdasco and Esteller, 2013]. Thus, it is plausible that KAISO is functioning in a similar manner.
The functional effect of this 3′-UTR mutation is less obvious than the splice-donor CUL4B mutation. Alterations in the 3′-UTR have been associated with disease. For example, dysregulation of AU-rich binding proteins by mutations can lead to hematopoietic malignancies and leukemogenesis [Khabar, 2010; Baou et al., 2011]. An expanded number of trinucleotide (CTG) repeats in the 3′-UTR of the dystrophia myotonica protein kinase gene causes muscular dystrophy [Udd and Krahe, 2012], while rare forms of non-deletional alpha thalassemia result from mutation of the polyadenylation signal in the adult alpha-globin gene [Prior et al., 2007]. While the predicted miRNA binding sites identified here did not alter the expression of the gene, the variant may still functionally regulate genes expression through means other than miRNA binding.
In summary, by using exome sequencing, we identified a haplo-type consisting of two variants in the CUL4B and KAISO genes conferring risk to XLID. The CUL4B splice-donor mutation will lead to aberrant or no splicing of intron 6 from the primary transcript and is most likely the major contributing factor to the phenotype. While the functional effect of the 3′-UTR KAISO variant is currently unknown, it may still play an additional contributing factor to the disease phenotype.
Supplementary Material
Acknowledgments
We thank Ms. Kathleen Delgrosso and Ms. Kathryn Scott for technical assistance in performing exome sequencing on the AB 5500×l instrument.
Footnotes
Conflict of interest: none.
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Badura-Stronka M, Jamsheer A, Materna-Kiryluk A, Sowinska A, Kiryluk K, Budny B, Latos-Bielenska A. A novel nonsense mutation in CUL4B gene in three brothers with X-linked mental retardation syndrome. Clin Genet. 2010;77:141–144. doi: 10.1111/j.1399-0004.2009.01331.x. [DOI] [PubMed] [Google Scholar]
- Baird M, Driscoll C, Schreiner H, Sciarratta GV, Sansone G, Niazi G, Ramirez F, Bank A. A nucleotide change at a splice junction in the human beta-globin gene is associated with beta 0-thalassemia. Proc Natl Acad Sci USA. 1981;78:4218–4221. doi: 10.1073/pnas.78.7.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baou M, Norton JD, Murphy JJ. AU-rich RNA binding proteins in hematopoiesis and leukemogenesis. Blood. 2011;118:5732–5740. doi: 10.1182/blood-2011-07-347237. [DOI] [PubMed] [Google Scholar]
- Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69:3613–3634. doi: 10.1007/s00018-012-0990-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani S, Zapata J, Gerosa L, Moretto E, Murru L, Passafaro M. The neurobiology of X-linked intellectual disability. The Neuroscientist. 2013;19:541–542. doi: 10.1177/1073858413493972. [DOI] [PubMed] [Google Scholar]
- Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;132:359–383. doi: 10.1007/s00439-013-1271-x. [DOI] [PubMed] [Google Scholar]
- Blattler A, Yao L, Wang Y, Ye Z, Jin VX, Farnham PJ. ZBTB33 binds unmethylated regions of the genome associated with actively expressed genes. Epigenetics Chromatin. 2013;6:13. doi: 10.1186/1756-8935-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas DA, Slaugh R, Abidi F, Arena JF, Stevenson RE, Schwartz CE, Lubs HA. A new X linked mental retardation (XLMR) syndrome with short stature, small testes, muscle wasting, and tremor localises to Xq24–q25. J Med Genet. 2000;37:663–668. doi: 10.1136/jmg.37.9.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Pal JK. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol Cell. 2009;101:251–262. doi: 10.1042/BC20080104. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defossez PA, Stancheva I. Biological functions of methyl-CpG-binding proteins. Progr Mol Biol Transl Sci. 2011;101:377–398. doi: 10.1016/B978-0-12-387685-0.00012-3. [DOI] [PubMed] [Google Scholar]
- Della Ragione F, Tiunova A, Vacca M, Strazzullo M, Gonzalez E, Armstrong J, Valero R, Campanile C, Pineda M, Hulten M, Monros E, D’Esposito M, Prokhortchouk E. The X-linked methyl binding protein gene Kaiso is highly expressed in brain but is not mutated in Rett syndrome patients. Gene. 2006;373:83–89. doi: 10.1016/j.gene.2006.01.015. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidor B, Pichon O, Baron S, David A, Le Caignec C. Deletion of the CUL4B gene in a boy with mental retardation, minor facial anomalies, short stature, hypogonadism, and ataxia. Am J Med Genet Part A. 2010;152A:175–180. doi: 10.1002/ajmg.a.33152. [DOI] [PubMed] [Google Scholar]
- Khabar KS. Post-transcriptional control during chronic inflammation and cancer: A focus on AU-rich elements. Cell Mol Life Sci. 2010;67:2937–2955. doi: 10.1007/s00018-010-0383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lubs HA, Stevenson RE, Schwartz CE. Fragile X and X-linked intellectual disability: four decades of discovery. Am J Hum Genet. 2012;90:579–590. doi: 10.1016/j.ajhg.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Caballero I, Hansen J, Leaford D, Pollard S, Hendrich BD. The methyl-CpG binding proteins Mecp2, Mbd2 and Kaiso are dispensable for mouse embryogenesis, but play a redundant function in neural differentiation. PloS ONE. 2009;4:e4315. doi: 10.1371/journal.pone.0004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Prior JF, Lim E, Lingam N, Raven JL, Finlayson J. A moderately severe alpha-thalassemia condition resulting from a combination of the alpha2 polyadenylation signal (AATAAA-->AATA--) mutation and a 3.7 Kb alpha gene deletion in an Australian family. Hemoglobin. 2007;31:173–177. doi: 10.1080/03630260701288997. [DOI] [PubMed] [Google Scholar]
- Ravnan JB, Tepperberg JH, Papenhausen P, Lamb AN, Hedrick J, Eash D, Ledbetter DH, Martin CL. Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet. 2006;43:478–489. doi: 10.1136/jmg.2005.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravn K, Lindquist SG, Nielsen K, Dahm TL, Tumer Z. Deletion of CUL4B leads to concordant phenotype in a monozygotic twin pair. Clin Genet. 2012;82:292–294. doi: 10.1111/j.1399-0004.2011.01839.x. [DOI] [PubMed] [Google Scholar]
- Ruzov A, Dunican DS, Prokhortchouk A, Pennings S, Stancheva I, Prokhortchouk E, Meehan RR. Kaiso is a genome-wide repressor of transcription that is essential for amphibian development. Development. 2004;131:6185–6194. doi: 10.1242/dev.01549. [DOI] [PubMed] [Google Scholar]
- Stevenson RE, Holden KR, Rogers RC, Schwartz CE. Seizures and X-linked intellectual disability. Eur J Med Genet. 2012;55:307–312. doi: 10.1016/j.ejmg.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, O’Meara S, Edkins S, Teague J, Butler A, Dicks E, Stevens C, Tofts C, Avis T, Barthorpe S, Buck G, Cole J, Gray K, Halliday K, Harrison R, Hills K, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Varian J, West S, Widaa S, Mallya U, Moon J, Luo Y, Holder S, Smithson SF, Hurst JA, Clayton-Smith J, Kerr B, Boyle J, Shaw M, Vandeleur L, Rodriguez J, Slaugh R, Easton DF, Wooster R, Bobrow M, Srivastava AK, Stevenson RE, Schwartz CE, Turner G, Gecz J, Futreal PA, Stratton MR, Partington M. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet. 2007;80:345–352. doi: 10.1086/511134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O’Meara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, Xue Y, Tyler-Smith C, Thompson D, Gray K, Andrews J, Barthorpe S, Buck G, Cole J, Dunmore R, Jones D, Maddison M, Mironenko T, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Teague J, Butler A, Jenkinson A, Jia M, Richardson D, Shepherd R, Wooster R, Tejada MI, Martinez F, Carvill G, Goliath R, de Brouwer AP, van Bokhoven H, Van Esch H, Chelly J, Raynaud M, Ropers HH, Abidi FE, Srivastava AK, Cox J, Luo Y, Mallya U, Moon J, Parnau J, Mohammed S, Tolmie JL, Shoubridge C, Corbett M, Gardner A, Haan E, Rujirabanjerd S, Shaw M, Vandeleur L, Fullston T, Easton DF, Boyle J, Partington M, Hackett A, Field M, Skinner C, Stevenson RE, Bobrow M, Turner G, Schwartz CE, Gecz J, Raymond FL, Futreal PA, Stratton MR. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- Vissers LE, de Vries BB, Osoegawa K, Janssen IM, Feuth T, Choy CO, Straatman H, van der Vliet W, Huys EH, van Rijk A, Smeets D, van Ravenswaaij-Arts CM, Knoers NV, van der Burgt I, de Jong PJ, Brunner HG, van Kessel AG, Schoenmakers EF, Veltman JA. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale E, Specchia C, Devoto M, Angius A, Rong S, Rocchi M, Schwalb M, Demelas L, Paglietti D, Manca S, Mastropaolo C, Serra G. Novel X-linked mental retardation syndrome with short stature maps to Xq24. Am J Med Genet. 2001;103:1–8. doi: 10.1002/ajmg.1495. [DOI] [PubMed] [Google Scholar]
- Talerico M, Berget SM. Effect of 5′ splice site mutations on splicing of the preceding intron. Mol Cell Biol. 1990;10:6299–6305. doi: 10.1128/mcb.10.12.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Liu Q, Chen B, Zhang X, Guo C, Zhou H, Li J, Gao G, Guo Y, Yan C, Wei J, Shao C, Gong Y. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet. 2007;80:561–566. doi: 10.1086/512489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.